

Abstract
The brain tumor microenvironment (TME) is emerging as a critical regulator of cancer progression in primary and metastatic brain malignancies. The unique properties of this organ require a specific framework for designing TME-targeted interventions. Here we discuss a number of these distinct features, including brain-resident cell types, the blood-brain barrier, and various aspects of the immune-suppressive environment. We also highlight recent advances in therapeutically targeting the brain TME in cancer. By developing a comprehensive understanding of the complex and interconnected microenvironmental landscape of brain malignancies we will greatly expand the range of therapeutic strategies available to target these deadly diseases.
INTRODUCTION
The tumor microenvironment (TME) contains many different non-cancerous cell types in addition to cancer cells, including endothelial cells, pericytes, fibroblasts and immune cells (Quail and Joyce, 2013). While several of these cell types are also prevalent in brain tumors, there are some important features that distinguish the normal brain from other tissues; the composition of the extracellular matrix (ECM) is distinctive, there are unique tissue-resident cell types including microglia, astrocytes and neurons, and it is physically protected from inflammation by the blood-brain barrier (BBB). Indeed, the normal brain was long considered to be one of the ‘immune privileged’ organs in the body that must be sheltered from immune cell entry and/or attack for a number of reasons (Engelhardt et al., 2017; Medawar, 1948). For instance, activated immune cells produce inflammatory factors that can be cytotoxic and cause neurodegeneration. Additionally, the skull provides a physical barrier to the swelling that often coincides with inflammatory reactions, and thus interactions with the immune system need to be exquisitely regulated within the brain.
However, this viewpoint of immune privilege has been revised as a functional lymphatic vasculature was recently reported along the dural sinuses in mice (Aspelund et al., 2015; Louveau et al., 2015b), and central nervous system (CNS)-derived antigens have been shown to induce an immune response in the cervical lymph nodes (reviewed in (Louveau et al., 2015a)). Moreover, in certain brain tumors, the BBB is often compromised and there can be a robust infiltration of multiple immune cell types from the peripheral circulation (Weiss et al., 2009). Nonetheless, the microenvironment of the normal brain and some early-stage brain tumors is generally immunosuppressive, with essentially no trafficking or patrolling by peripheral immune cells, presenting a formidable challenge to overcome for the successful application of immune-oncology strategies.
In this Perspective, we will review how the brain TME regulates cancer progression and therapeutic response across several tumor types. We will focus on gliomas, including high-grade glioblastoma, the most aggressive primary brain tumor in adults; brain metastases, the most common brain malignancy overall, frequently arising from non-small cell lung cancer (40–50%), breast cancer (15–25%) and melanoma (5–20%) (Eichler et al., 2011); and medulloblastoma, the most common pediatric brain cancer. While these tumor types all share a common tissue of residence (i.e. the brain), they are subject to many different inputs from their microenvironment due to variance in evolutionary history (e.g. primary brain tumors versus metastases), cell-of-origin (e.g. medulloblastomas versus gliomas), and niche maturity (e.g. immature brain in pediatric tumors versus the adult brain). Not surprisingly, the unique and highly complex properties of this organ indicate that we will also need to construct a specific framework for understanding and therapeutically targeting TME cells across distinct brain malignancies.
The immune cell landscape of brain tumors
Tumor-associated macrophages and microglia (TAMs)
The majority of immune cells within brain tumors are macrophages (Graeber et al., 2002), often comprising up to ~30% of the tumor mass. Ontogenetically distinct macrophage populations exist within the brain TME, including both tissue-resident microglia and bone marrow-derived macrophages (Bowman et al., 2016; Hambardzumyan et al., 2016)(Figure 1A). Microglia develop from embryonic yolk sac progenitor cells (Ginhoux et al., 2010; Gomez Perdiguero et al., 2015), and are not replenished postnatally through peripheral mononuclear hematopoiesis (Ajami et al., 2007). Therefore, the maintenance of microglia in the normal adult brain is thought to occur through prolonged cellular longevity and local proliferation (Ajami et al., 2007; Elmore et al., 2014; Gomez Perdiguero et al., 2015). Similarly, non-parenchymal macrophages within the CNS arise during embryonic development, and are largely stable populations during adult life (Goldmann et al., 2016). By contrast, in response to perturbations of tissue homeostasis or pathological conditions, circulating monocytes are recruited to the brain parenchyma and give rise to bone marrow-derived macrophages (BMDMs) (Shi and Pamer, 2011). These cells are replenished through monocytosis, particularly during tumor progression where the integrity of the BBB can be compromised and permissive to inflammation (Weiss et al., 2009). Whether microglia and BMDMs have distinct functions in the brain TME has been controversial, and is a topic of active investigation.
Figure 1. Cellular components of the brain tumor microenvironment.
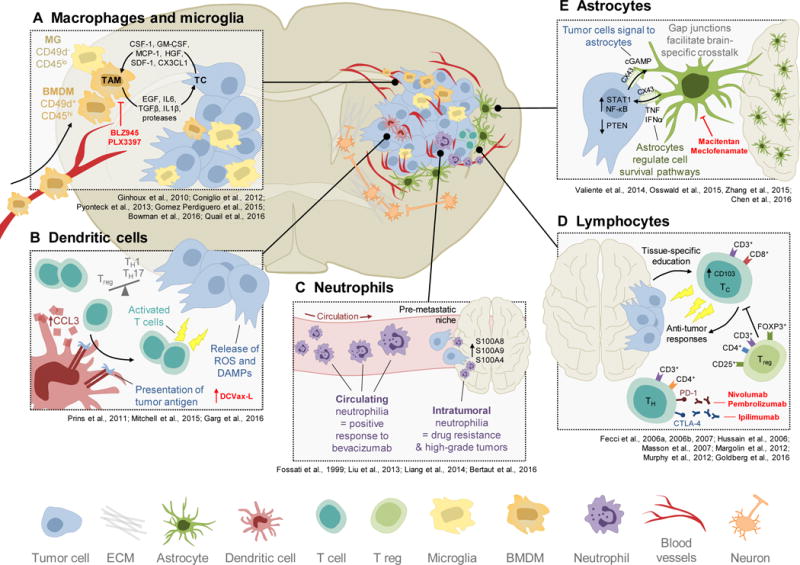
Brain tumors are composed of diverse cellular players, ranging from peripherally-derived immune cells to various specialized organ-resident cell types. Each of these cell types contributes to brain tumor biology in unique ways. (A) Tumor-associated macrophages and microglia (TAMs) arise from two distinct sources, including the periphery (bone marrow-derived macrophages; BMDM; CD49d+) or the yolk sac (microglia; MG; CD49d−). TAMs engage in significant bidirectional cross-talk with tumor cells (TC) in the brain, whereby brain tumor cells release cytokines and chemoattractants to recruit TAMs to the microenvironment, and TAMs in turn supply pro-tumorigenic, pro-survival factors. One of the therapeutic strategies used to target TAMs is via CSF-1R inhibitors, including BLZ945 and PLX3397. (B) The potent antigen-presenting ability of dendritic cells is being harnessed clinically for brain tumors, in the form of vaccines (e.g. DCVax-L). Dendritic cells can present tumor antigen to T cells to elicit an anti-tumor immune response. These responses can be further enhanced by factors that are released into the microenvironment by tumor cells, such as reactive oxygen species (ROS) or danger-associated molecular patterns (DAMPs). (C) Neutrophils have prognostic value for brain cancers. In the circulation, a high neutrophil count (i.e. neutrophilia) is associated with positive response to anti-VEGF-A therapy (bevacizumab). However, neutrophilia within tumor tissue in the brain is associated with development of drug resistance to bevacizumab, and high-grade glioma. This suggests there may be tissue-specific reprogramming of neutrophils following their extravasation into tissues. Furthermore, neutrophils can play a role during brain metastasis, where they may seed the pre-metastatic niche in response to S100 inflammatory proteins, to assist in tumor cell colonization. (D) During effector phases of anti-tumor immunity, T lymphocytes are further reprogrammed by the brain microenvironment to extend their retention. This can occur via changes in integrin expression, for example through induction of CD103. T regulatory (T reg) cells can also suppress cytotoxic T cells, leading to an immunosuppressive microenvironment that is permissive to tumor outgrowth. T cells can be activated via multiple immune checkpoint inhibitors, such as those targeting CTLA-4 (e.g. ipilimumab) or PD-1 (e.g. nivolumab, pembrolizumab). (E) Astrocytes are unique to the central nervous system and play important roles in mediating tissue-specific communication in the brain, including in brain tumors. For example, several studies have demonstrated in both primary gliomas and in brain metastases that astrocytes form functional gap junctions with tumor cells, which serve as a physical conduit for transferring signaling molecules in a heterotypic manner. Efforts to disrupt this communication axis pharmacologically have been made, for example with macitentan (an endothelin receptor antagonist) or meclofenamate (a cyclooxygenase inhibitor that modulates gap junctions).
Until recently, it has not been possible to definitively distinguish between brain macrophage populations without the use of genetic lineage tracing models in mice, which obviously cannot be directly translated to humans. Most studies that do not utilize lineage tracing have instead relied on differential CD45 expression levels to distinguish microglia (CD45low) from BMDMs (CD45high). However, while this approach is reasonably accurate for mouse models, it does not seem to hold true in human brain tumors (Bowman et al., 2016). In recent years, more accurate cell surface markers for flow cytometry that discriminate microglia and BMDMs have emerged. For example, Tmem119 is enriched on microglia, but not BMDMs, in human brain tissue and mouse models of inflammation (Bennett et al., 2016). Cx3cr1 and Siglec-H are also expressed by normal microglia, but not by macrophages derived from lung, peritoneum or spleen (Gautier et al., 2012). However, in brain tumors, subsequent studies have demonstrated that peripherally-derived macrophages can assimilate to the local TME upon entry into the brain, by upregulating ‘microglia-specific’ genes including Cx3cr1 (Bowman et al., 2016). In the context of brain malignancy, CD49D/ITGA4 was recently identified as a marker for BMDMs but not microglia, thus enabling accurate discrimination and isolation of these distinct cell types in both patient and murine tumors (Bowman et al., 2016). Collectively, these studies represent an invaluable resource as we can now explore how macrophage ontogeny translates to function in brain tumors and other neuropathologies, and definitively determine the relative contributions of BMDMs versus microglia to different brain tumor phenotypes.
Prior to these developments, the majority of literature on functional contributions of TAMs to brain tumors has focused on bulk cell populations. Importantly, the dogma that macrophages exist along a simple linear M1–M2 phenotypic continuum has been disputed. Many groups are instead focused on defining context-specific macrophage activation and phenotype as a measure of functional diversity (Ginhoux et al., 2015; Qian and Pollard, 2010). TAMs within the brain tend to be pro-tumorigenic and accumulate with higher tumor grade (Hambardzumyan et al., 2016; Komohara et al., 2008). Furthermore, analysis of patient glioma tissue indicates that TAMs produce low levels of pro-inflammatory cytokines and lack expression of key molecules involved in T cell co-stimulation (e.g. CD86, CD80, CD40), suggesting that they are poor inducers of T cell responses in glioma (Hussain et al., 2006).
While TAM depletion strategies in several types of cancer can provide a survival advantage, the effects of targeting TAMs in brain tumors seem to be more context-dependent. Recent studies have described an important role for colony stimulating factor-1 receptor (CSF-1R) in brain TAM biology, such that inhibition of CSF-1R either depletes (Coniglio et al., 2012) or depolarizes TAMs (Pyonteck et al., 2013; Quail et al., 2016), depending on the preclinical model tested, leading to reduced glioma growth and invasion. Interestingly, in studies that show TAM population persistence in the face of CSF-1R inhibition due to the production of specific survival factors (including GM-CSF and IFNγ) (Pyonteck et al., 2013), the resultant changes in TAM gene expression and phenotype (e.g. enhanced phagocytosis of glioma cells) resulted in a far more pronounced effect on glioma regression compared to TAM depletion. In regression trials of high-grade lesions this led to a substantial survival effect (0% survival by 17 weeks in control mice versus 44% survival at the 26 week endpoint in CSF-1R inhibitor-treated mice) (Quail et al., 2016).
Given the robust effects of TAM inhibition on blocking gliomagenesis (Pyonteck et al., 2013), it is not surprising that activated TAMs have been shown to regulate glioma stem cell pools within the brain (Sarkar et al., 2014; Wu et al., 2010; Zhou et al., 2015). In addition, TAMs have been implicated in brain tumor angiogenesis and resistance to anti-angiogenic therapies (De Palma et al., 2005; Lu-Emerson et al., 2013; Piao et al., 2012), and may also contribute to the colonization and outgrowth of brain metastases (Pukrop et al., 2010; Sevenich et al., 2014), in part through their ability to modulate vessel integrity and function. In light of their plasticity, increasing advances are being made to develop strategies that re-educate macrophages to specifically adopt anti-tumor phenotypes in cancer, including brain tumors, which are likely to be more efficacious than collectively depleting all TAM populations (Bowman and Joyce, 2014; Quail and Joyce, 2013). Whether CSF-1R inhibitors ultimately represent the most effective means to achieve this goal remains to be seen, as different preclinical glioma models have shown that acquired resistance to CSF-1R inhibition ultimately develops in approximately half of the treated animals (Quail et al., 2016).
Dendritic cells
Dendritic cells (DCs) are myeloid-derived, highly potent antigen-presenting cells (APCs) that stimulate T cell responses (Figure 1B). In the brain, early studies demonstrated that microglia are the predominant APCs, and that DCs play a less-prevalent role (Hart and Fabre, 1981; Hickey and Kimura, 1988; Lowe et al., 1989; Ulvestad et al., 1994). Therefore, application of DC biology within the brain TME field has largely taken a different tack; as interest for immune checkpoint inhibitors in cancer therapy continues to expand, DC vaccines are likewise gaining significant clinical attention as an alternative strategy to stimulate T cell responses (Anguille et al., 2014; Palucka and Banchereau, 2012). DCVax-L, an autologous tumor lysate-pulsed DC vaccine, is furthest along in this regard, with early clinical trials reporting a median survival of 31.4 months in glioblastoma patients (Prins et al., 2011) – an improvement over historical controls of 14.6 months for the standard of care comprising radiation and temozolomide chemotherapy (Stupp et al., 2005). As a result, Phase III trials have now been initiated (ClinicalTrials.gov identifier: NCT00045968) (Table 1).
Table 1.
Clinical use of microenvironment-targeted therapies in brain tumors
| Drug | Function | Phase | Tumor types | Refs |
|---|---|---|---|---|
| PLX3397 | CSF-1R inhibitor | II | Glioma | (Butowski et al., 2015) |
| BLZ945 | CSF-1R inhibitor | I/II | Advanced solid tumors (including glioblastoma) and metastases | First in human (with PDR001 anti-PD-1) |
| DCVax-L | Dendritic cell vaccine | III | Glioma | (Prins et al., 2011) |
| Bevacizumab | anti-VEGF-A | Approved I/II |
Recurrent glioma Brain metastases |
(Cohen et al., 2009; Friedman et al., 2009; Gilbert et al., 2014; Kreisl et al., 2009; Levy et al., 2014) |
| Ipilimumab Pembrolizumab Nivolumab |
anti-CTLA-4 anti-PD-1 anti-PD-1 |
II/III I/II II/III |
Glioma and brain metastasis (melanoma and NSCLC) | (Berghoff et al., 2016; Bouffet et al., 2016; Brahmer et al., 2015; Cohen and Kluger, 2016; Goldberg et al., 2016; Hutchinson, 2016; Le et al., 2015; Margolin et al., 2012; Preusser et al., 2015; Restifo et al., 2016) |
Recent studies have also attempted to identify factors that dictate and/or enhance DC vaccine efficacy. For example, pre-conditioning the vaccination site with an inflammatory stimulus (e.g. tetanus toxoid) significantly increased DC homing to nearby draining lymph nodes, leading to prolonged progression-free and overall survival in glioblastoma patients (Mitchell et al., 2015). Murine models demonstrated that this phenomenon was dependent on the chemokine CCL3, as pre-conditioning efficacy was blunted in Ccl3−/− mice (Mitchell et al., 2015). In another study, DC vaccination was combined with induction of immunogenic cell death (ICD) via hypericin photodynamic therapy to generate enhanced anti-tumor responses in preclinical glioma models (Garg et al., 2016). This treatment regimen caused cancer cells to become highly immunogenic through the release of danger signals that induced potent Th1 immunity, leading to a substantial improvement in overall survival (0% survival at ~30 days in control mice versus 70% survival at 100 days in ICD-based DC vaccine mice) (Garg et al., 2016). Together these studies, along with initial results from clinical trials, demonstrate the anti-tumor potential of DC vaccines in glioblastoma, which are largely untreatable tumors in the clinical setting.
Neutrophils
Granulopoiesis occurs in the bone marrow and gives rise to multiple granulocytic immune cell subsets, which are mobilized into the blood during inflammation (Coffelt et al., 2016; Nicolas-Avila et al., 2017). The most abundant granulocyte in humans is the neutrophil, which can comprise up to 70% of total leukocytes in the body (Coffelt et al., 2016). Most studies to date on neutrophils and brain tumors have focused on the impact of these cells on the response to anti-angiogenic therapy and vascularity – a hallmark feature of high-grade glioma. Clinically, while it has been shown that high peripheral neutrophil count prior to treatment correlates with positive initial response to the anti-VEGF-A antibody bevacizumab (Bertaut et al., 2016), enhanced neutrophil infiltration into tumor tissue is associated with acquired resistance and higher glioma grade at later stages (Fossati et al., 1999; Liang et al., 2014) (Figure 1C). These studies and others suggest that neutrophils may have prognostic value in glioblastoma (Bertaut et al., 2016; Fossati et al., 1999; Gabrusiewicz et al., 2016; Liang et al., 2014). Indeed, preclinical studies have shown that neutrophils contribute to glioblastoma progression by supporting the expansion of the glioma stem cell pool in a manner that is dependent on S100 proteins (specifically, S100A4) (Liang et al., 2014).
S100 proteins have also been strongly connected to secondary dissemination, particularly for breast cancer (Bresnick et al., 2015). While a comprehensive characterization of the brain pre-metastatic niche remains very limited compared to other common secondary organs such as the lung, it has been shown that S100A8 and S100A9 are upregulated in the pre-metastatic brain leading to recruitment of neutrophils, which support subsequent metastatic seeding (Liu et al., 2013). These observations are reminiscent of what has been shown in other metastatic organs that are not subject to immune privilege (Casbon et al., 2015; Coffelt et al., 2015; Liu et al., 2016; Wculek and Malanchi, 2015), supporting the concept that in the context of a tumor, the brain may be aberrantly exposed to pathologic inflammation that influences disease progression. Moreover, from a prognostic standpoint, a high ratio of neutrophils to lymphocytes in the peripheral blood is associated with reduced survival time, including following surgical resection, for both brain metastasis and glioblastoma patients (Bambury et al., 2013; McNamara et al., 2014; Mitsuya et al., 2016). It will therefore be imperative going forward to develop a more complete understanding of how the brain microenvironment changes in response to a primary tumor outside the CNS, in an analogous manner to what has been shown for other secondary organs such as lung or liver.
Lymphoid cells
The lymphoid arm of the immune system is primarily composed of T cells, B cells and NK cells. Lymphopoiesis involves the bone marrow and the thymus, from which mature T cells are released to peripheral lymphoid organs where they are primed by engaging with professional APCs. This priming phase leads to clonal expansion of T cells with antigen-specific effector functions, which then infiltrate effector tissues (Chen and Flies, 2013) (Figure 1D). In patients, it is unclear whether further adaptation to specific microenvironments occurs during effector phases of anti-tumor immunity. This question has been explored using syngeneic murine brain tumor models, whereby adoptive transfer of antigen-experienced tumor-specific CD8+ T cells exhibited proliferation and differentiation within the brain, leading to enhanced retention within the effector microenvironment (Masson et al., 2007). Mechanistically, the authors observed that interaction with the brain TME caused re-education of CD8+ T cells (for example, through induction of αE (CD103) β7 integrin) in a manner that was recapitulated in human glioma samples (Masson et al., 2007). This study critically demonstrated that the local microenvironment can alter T cell effector function during anti-tumor immunity, even within the brain, where T cell-mediated inflammatory responses are minimal under normal physiological contexts.
Enhancing T cell activation by enabling co-stimulation, e.g. through the use of checkpoint inhibitors, is an emerging field of cancer therapy, and is currently under clinical investigation in patients with brain tumors (including primary gliomas or brain metastases). These clinical studies are being conducted either in combination with standard of care treatment or in recurrent late-stage disease (Cohen and Kluger, 2016; Fecci et al., 2014). In mouse glioma models, blockade of the checkpoint molecule CTLA-4 leads to prolonged survival and enhanced CD4+ helper T (Th) cell activity (Fecci et al., 2007). Consistently, administrating tumor lysate vaccines in combination with an Fc-OX40L fusion protein (a T cell co-stimulatory molecule) results in a 50% cure rate via enhanced tumoricidal lymphocytes, and long-term immunologic memory against glioma cells (Murphy et al., 2012). Adding temozolomide to this treatment regimen yields an even more pronounced effect on survival (Murphy et al., 2012). Finally, in the metastatic setting in patients, studies in melanoma are beginning to demonstrate efficacy of the anti-CTLA-4 antibody ipilimumab for patients with CNS metastases (Konstantinou et al., 2014; Margolin et al., 2012; Queirolo et al., 2014). Whether this also applies to patients with non-small cell lung cancer, the most frequent source of brain metastases, is now under active investigation, and early results have been promising (Dudnik et al., 2016; Goldberg et al., 2016).
An alternative means to boost T cell responses is by blocking immunosuppressive lymphocytes, such as CD4+CD25+FOXP3+ T regulatory cells (Tregs; Figure 1D). Glioma patients exhibit a decrease in absolute numbers of circulating Th cells; however, there is an increased proportion of immunosuppressive Tregs within the remaining CD4+ cell pool in blood (Fecci et al., 2006a), and a prominent infiltrating Treg population within tumor tissues (Hussain et al., 2006). Similarly, in murine glioma models, it has been shown that while Th cell numbers are reduced overall, immunosuppressive Tregs comprise a large fraction of the remaining CD4+ cell fraction in blood, lymph nodes, and spleen (Fecci et al., 2006b). Treating mice with neutralizing antibodies against CD25 eliminated the suppressive function of Tregs, thereby increasing cytotoxic T cell anti-tumor functions. Furthermore, when combined with DC vaccinations, neutralization of CD25 resulted in 100% glioma cell rejection in animal models (Fecci et al., 2006b). These studies demonstrate that reprogramming of immunosuppressive T cell subsets in the context of glioma, and potentially other brain tumors, may be sufficient to boost anti-tumor immune responses.
The brain vasculature
Blood-brain barrier (BBB)
The presence of the BBB uniquely distinguishes the brain from other organs, by providing a selective barrier between the systemic circulation and the brain. The BBB thus protects this essential organ from infection and toxic substances, though it simultaneously limits the delivery of many therapeutic agents. Indeed, the BBB is estimated to block the transport of ~98% of molecules, thus representing a major challenge for treating brain tumors. However, various brain pathologies including cancer can display a loss of BBB integrity resulting in leakiness (Abbott et al., 2006; Davies, 2002; Papadopoulos et al., 2001). This may in part explain why several BBB-impenetrable agents have nevertheless shown clinical efficacy; for example, as discussed below, antibodies against CTLA-4 in patients with small, asymptomatic melanoma-to-brain metastases have shown clinical benefit akin to their efficacy for other sites of non-CNS metastasis (Margolin et al., 2012).
The BBB is composed of specialized endothelial cells, pericytes and astrocytic foot processes, which dictate junctional integrity (Abbott, 2013; Abbott et al., 2006). Microglia can additionally contribute to regulating BBB integrity (da Fonseca et al., 2014) (Figure 2A), and specifically repair the BBB following injury in a purinergic receptor P2RY12-dependent manner (Lou et al., 2016). Interestingly, the composition and integrity of the BBB can be dictated by the molecular subtype of tumors, as shown for pediatric medulloblastoma (Phoenix et al., 2016). Medulloblastoma can be classified into four different subtypes, with WNT-driven tumors showing the best response to therapy. This was recently attributed to the absence of a functional BBB specifically in the WNT subtype, in which an aberrant and extensively fenestrated vasculature allows the accumulation of high levels of chemotherapeutic agents in the brain (Phoenix et al., 2016). The SHH-driven subtype, by contrast, has an intact BBB which renders it impermeable to chemotherapy. However, by specifically manipulating the levels of tumor-endothelial cell paracrine signaling in SHH-medulloblastomas, the authors could improve brain penetration of the chemotherapy vincristine, thereby increasing survival. These results have important clinical implications for treatment of medulloblastoma, as all patients currently receive vincristine regardless of subtype. Thus, administration of vincristine in combination with agents that transiently open the BBB, or nanoparticle formulations for example (Abbott, 2013; O’Keeffe and Campbell, 2016), might be necessary to improve drug delivery in this tumor and other brain malignancies in which the BBB remains intact.
Figure 2. Vascular and lymphatic vessels in the brain.
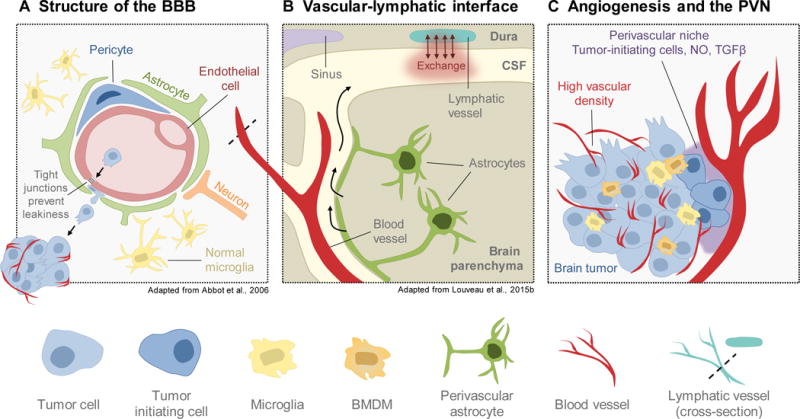
(A) The blood-brain barrier (BBB) serves to protect the brain from inflammation and systemic insults. It is composed of endothelial cells, pericytes, and astrocytes, which tightly seal the endothelium to regulate permeability. Microglia and neurons can additionally contribute to regulation of BBB integrity. Breakdown of junctional integrity can increase permissiveness to seeding of brain-metastatic cancer cells. (B) Recent studies have led to the discovery of lymphatic vasculature in the brain along the dural sinuses in mouse. These vessels exchange fluid with the cerebral spinal fluid (CSF) that surrounds the brain parenchyma, explaining longstanding questions about how immune cells are trafficked into and out of the brain. (C) Brain tumors, particularly gliomas, exhibit extremely high vascularity and angiogenesis. Furthermore, the perivascular niche (PVN) serves as a reservoir for tumor-initiating cells within the brain, which supports tumor outgrowth and aggressive behavior. As such, disrupting the brain vasculature and/or the PVN is of interest for clinical management of brain tumors.
While the BBB restricts the entry of many molecules and cells, it is not an impenetrable barrier to transmigration of metastasizing cancer cells. Indeed, in this regard, it has been proposed that the brain can function as a sanctuary site for metastatic cells that effectively breach the BBB, where they are subsequently protected from the effects of chemotherapy and other agents that cannot penetrate the brain (Eichler et al., 2011; Steeg et al., 2011). Metastatic cells utilize different approaches to cross the multiple cell layers that constitute the BBB, including proteolysis of junctional adhesion molecules, such as JAM-B (Sevenich et al., 2014). Interestingly, in the case of breast-to-brain metastasis, proteases that are typically expressed by immune cells, such as cathepsin S, are upregulated in metastatic cells which successfully penetrate and seed within the brain (Sevenich et al., 2014). This phenomenon may indicate a type of ‘leukocytic mimicry’ through which metastatic cells could execute immune expression programs that promote cell migration and invasion. In other studies, additional factors have been identified as potential site-specific mediators of BBB transmigration, including PTGS2, HB-EGF, ST6GALNAC5 (Bos et al., 2009), PLEKHA5 (Jilaveanu et al., 2015), and the serum factor PLGF (Li et al., 2013). It is also becoming apparent that BBB integrity is heterogeneously affected within brain tumors (Lockman et al., 2010; Morikawa et al., 2015). For example, in brain metastases, composition variance of pericyte subpopulations is associated with differential permeability (Lyle et al., 2016), and correspondingly only drugs designed to fully penetrate the BBB are therapeutically efficacious (Osswald et al., 2016). Together, these emerging studies indicate that different components of the brain TME can fundamentally regulate the properties of the BBB, necessitating an integrated analysis of these cellular and extracellular mediators in the future to improve the efficacy of brain-targeted therapies.
Angiogenesis
High vascularity is a hallmark feature of glioblastoma. In contrast to the specialized heterotypic BBB in normal brain tissue (Abbott et al., 2006), the tumor-associated vasculature within the brain exhibits aberrant organization and poor structural integrity (Jain et al., 2007). As a consequence of this leakiness, gliomas exhibit deregulated permselectivity and perfusion, high interstitial fluid pressure, extensive hypoxia and necrosis, and edema (Jain et al., 2007). Not surprisingly, capitalizing on the dependence of primary brain tumors and metastases on a robust vasculature by targeting proangiogenic mediators (e.g. via bevacizumab, sorafinib, sunitinib, etc.) is actively being investigated. However, these efforts have yielded limited successes – and in some cases they have even had detrimental effects. Recent results from a randomized clinical trial of bevacizumab in glioblastoma patients with newly diagnosed disease showed a modest increase in progression-free survival compared to placebo controls (Gilbert et al., 2014). However, there were no differences in overall survival, and treatment with bevacizumab caused complications that impaired quality of life over time, including hypertension, thrombosis, neutropenia, and reduced cognitive function (Gilbert et al., 2014). These findings were in part recapitulated by another Phase III study in glioblastoma, which reported no improvement in overall survival and increased incidence of adverse events with the addition of bevacizumab to radiotherapy-temozolomide treatment (Chinot et al., 2014). However, there were improvements in progression-free survival with bevacizumab treatment, potentially justifying its use as a combination therapy with standard of care (Chinot et al., 2014). Overall, these results have led to some controversy amongst oncologists, as there are still many unknowns with respect to vascular targeting as a treatment strategy for brain tumors (Fine, 2014; Malkki, 2014).
Vascular normalization – arguably opposite to the goal of anti-angiogenic therapies and one potential explanation for their clinical shortcomings after long-term treatment (Jain, 2001; Jain, 2005; Jain et al., 2007) – has been proposed as an alternative strategy to modulate the tumor vasculature in order to reduce hypoxia, improve perfusion and drug delivery, and ultimately support physiological angiogenesis rather than pathologic angiogenesis (Jain, 2005; Tong et al., 2004; Winkler et al., 2004). Indirect evidence that this concept could be applicable to glioblastoma patients stems from a clinical trial that tested cediranib monotherapy (a pan-VEGF receptor inhibitor) in patients with recurrent glioblastoma (Batchelor et al., 2010; Batchelor et al., 2007; Sorensen et al., 2009). By imaging the vasculature via MRI, it was shown that cediranib rapidly normalized vessel size and permeability, leading to a diminution in edema in patients. Whether vascular normalization strategies in brain tumors will be more efficient than vascular depletion, for example through suboptimal dosing or scheduling of anti-angiogenic therapies that are already being applied clinically (Jain, 2005), remains an ongoing area of investigation.
It is also possible that anti-angiogenic therapies have limited clinical benefit due to insufficient understanding of the unique mechanisms underlying vascularization of brain tumors. Several hypotheses have been put forward to shed light on this issue. First, in contrast to other tissues, it has been shown that normal neural stem cells (NSCs) within the brain are able to differentiate into an endothelial-like phenotype (Wurmser et al., 2004). More recently, it has been suggested that glioma stem cells retain this ability, as endothelial cells within the glioma vasculature have genomic alterations that recapitulate those of the tumor, such as amplification of EGFR (Ricci-Vitiani et al., 2010; Wang et al., 2010). It has also been demonstrated that glioma stem cells harbor a transcriptional program that mimics endothelial cells, which correlates with enhanced endothelial-glioma stem cell associations in peritumoral satellite tumor foci (Hu et al., 2016). Corroborating these findings, several groups have shown that the perivascular niche (PVN) in brain tumors serves as a glioma stem cell reservoir, where endothelial cells secrete factors (e.g. nitric oxide) that maintain the tumor-initiating cell pool (Calabrese et al., 2007; Charles and Holland, 2010; Charles et al., 2010; Gilbertson and Rich, 2007; Hambardzumyan et al., 2008; Lathia et al., 2010; Lathia et al., 2015; Pietras et al., 2014), and similar results have been shown in the NSC-rich subventricular zone of normal brain tissue (Ramirez-Castillejo et al., 2006; Shen et al., 2004; Tavazoie et al., 2008) (Figure 2C). In a recent study, it was shown that osteopontin, possibly derived from astrocytes in the PVN, supports CD44+ glioma stem cells leading to enhanced tumor outgrowth and radiation resistance (Pietras et al., 2014). Taken together, these studies collectively demonstrate a critical relationship between the endothelium and normal/glioma stem cells within the brain that may be imperative for understanding how to optimize potential benefit of vascular-targeted therapies.
A second hypothesis that could explain in part the limited clinical success of anti-angiogenic therapy in brain malignancies is vascular co-option (Carmeliet and Jain, 2011), a non-angiogenic mechanism of tumor vascularization that makes use of preexisting vessels, which has been shown to occur in brain tumors. Indeed, in a syngeneic mouse model of melanoma-to-brain metastasis, although treatment with a VEGFR2 inhibitor reduced metastases detectable by MRI, histological analyses revealed residual disease that was supported by vessel co-option which compensated for angiogenesis blockade (Leenders et al., 2004). In support of these findings, mouse models of breast and lung cancer metastasis to brain have shown that tumor-supplied serpins (e.g. neuroserpin, serpin B2) block the anti-metastatic effects of plasmin to promote vascular co-option, creating a microenvironment that is conducive to migration and colonization (Valiente et al., 2014). Similarly, another recent study used high-resolution imaging techniques to show that melanoma metastases within the brain exhibit angiotropism and vascular co-option to facilitate cancer cell spreading within the brain parenchyma (Bentolila et al., 2016). Whether these processes also occur in primary brain tumors is less well understood; however, vascular co-option as a potential compensatory mechanism for angiogenesis has major implications for patients with highly vascularized tumors (e.g. glioblastomas) that are eligible for anti-angiogenic therapy.
Lymphatic vessels
The lymphatic system connects the circulatory and immune systems, working in concert with blood vessels to exchange fluid, waste, debris, and immune cells within tissues. Until recently, it was commonly believed that the brain lacked a lymphatic drainage system, in part explaining immune privilege. However, challenging this dogma was the observation that the brain is subject to immune surveillance under normal homeostatic conditions, particularly within the meninges (Ransohoff and Engelhardt, 2012; Shechter et al., 2013). Furthermore, detection of lymphocyte and tumor cell trafficking from the brain to the cervical lymph nodes suggests that there could be a direct route of passage into the lymphatic system (Goldmann et al., 2006; Mondin et al., 2010).
Indeed, two recent paradigm-shifting studies led to the discovery of a lymphatic system in mice along the draining cerebral sinuses (Aspelund et al., 2015; Louveau et al., 2015b). Initial efforts by these authors were focused on identifying mechanisms of CNS immune surveillance and T cell trafficking in and out of the meningeal region, which envelops the brain and spinal cord, when functional lymphatic vessels expressing Lyve-1, VEFGR3, podoplanin, and Prox1, were unexpectedly found along the dural sinuses and meningeal arteries (Aspelund et al., 2015; Louveau et al., 2015b). Explaining earlier observations of brain-derived cells within distant lymph nodes, both studies demonstrated that these lymphatic vessels serve as conduits to the cervical lymph nodes to exchange fluid and immune cells with the cerebral spinal fluid (Figure 2B). Although the existence of these vessels has recently been reported in mice, whether they are present in humans is an important unanswered question. Furthermore, how dural lymphatic vessels might contribute to brain tumor progression, neurodegeneration, or other brain pathologies is completely unknown (Iliff et al., 2015). For example, it is possible that brain tumors would benefit from therapies targeted against these vessels, via either conventional (e.g. anti-VEGF-C) or brain-specific mechanisms of lymphangiogenesis. Going forward, these open questions will be critical to address.
Astrocyte contributions beyond the BBB
A site-specific role for astrocytes during brain metastases
Astrocytes are one of the most abundant cell types within and unique to the brain microenvironment; therefore, it is likely that metastases that preferentially grow within the brain must find ways to adapt and favorably interact with these unfamiliar cellular players (Massague and Obenauf, 2016; Quail and Joyce, 2013). In support of this notion, a recent study reported that expression of the tumor suppressor gene PTEN is specifically downregulated in established brain metastases compared to primary tumors and metastases from other common secondary organs (e.g. bone and lung), using mouse models and patient samples (Zhang et al., 2015). Mechanistic co-culture experiments revealed that microRNAs from astrocyte-derived exosomes were capable of suppressing PTEN expression within brain metastatic cells, leading to enhanced PI3K signaling and increased outgrowth. This astrocyte-driven phenotype was not recapitulated by other microenvironmental cell types, such as fibroblasts, explaining the organ specificity of the reported observations. Furthermore, intracarotid injection of cancer cells harboring a Pten shRNA knockdown yielded no change in size or incidence of brain metastasis compared to control shRNA cells, suggesting that this effect was dependent on transcriptional changes that were driven by the brain microenvironment (Zhang et al., 2015).
In another study, it was shown that cancer cells within established brain metastases form functional gap junctions with astrocytes in the adjacent microenvironment, thereby establishing a conduit for bidirectional communication to support outgrowth (Chen et al., 2016). Through these gap junctions, cancer cells reprogram astrocytes by providing cGAMP to induce a pro-inflammatory program, characterized by the production of a variety of cytokines (e.g. IFNα and TNF). In turn, these cytokines support outgrowth of metastases by activating STAT1 and NF-κB signaling within cancer cells. By interfering with the formation of gap junctions through pharmacological intervention, the authors demonstrated that this heterotypic signaling loop can be blocked, thus mitigating brain metastasis outgrowth (Chen et al., 2016) (Figure 1E). The finding that astrocyte-associated gap junctions support brain metastasis progression is consistent with reports of primary astrocytomas being uniquely capable of forming functional gap junctions via connexin 43 to support growth and invasion (Osswald et al., 2015). This phenotype is not observed in oligodendroglial brain tumors, suggesting that astrocytes are particularly adept at establishing gap junction communication networks with surrounding cells in their microenvironment. While these examples highlight the growth-promoting effects of astrocytes on established brain metastases, the opposite has been shown for earlier steps in the brain metastasis process, whereby astrocytes reduce the survival of newly arriving metastatic cells within the brain (Valiente et al., 2014). Taken together, these studies demonstrate how astrocytes may play temporally-distinct roles in organ tropism of metastasis to the brain, by providing tumor-stroma interactions that are unique to the brain microenvironment.
Gliosis and wound healing in the CNS: Potential impact on cancer
In response to brain injury, astrocytes become activated to form a protective barrier that limits the extent of tissue damage within the delicate brain tissue, and cooperate with microglia to supply factors that promote axonal regrowth. This process, termed reactive gliosis, is the mechanism of wound healing in the brain, and is the source of a wide variety of growth factors and cytokines that support viability and repair of brain tissue during different forms of injury (Robel et al., 2011; Silver and Miller, 2004; Sofroniew, 2015; Zamanian et al., 2012). Analogous to the effect of an aberrant wound healing program in epithelial cancers, a provocative question is whether gliosis similarly provides a cytokine-rich niche that stimulates outgrowth of brain tumor cells and confers resistance to therapy. Tangential preclinical evidence supports this notion; for example, during early colonization of brain metastases, the reactive stroma produces the protease plasmin to release astrocyte-bound FasL, which acts as a death signal to mediate cancer cell clearance. Eventually however, this contributes to a strong selective pressure for outgrowth of metastatic cells that produce high levels of serpins, which are protease inhibitors that counteract the effects of plasmin (Valiente et al., 2014). In glioblastoma, it has been proposed that resistance to macrophage-targeted therapy via CSF-1R blockade may initially be triggered by glial scarring as a result of massive tumor regression, ultimately leading to induction of aberrant IGF-1/PI3K signaling that drives recurrent disease (Quail et al., 2016). Consequently, dual inhibition of CSF-1R with either IGF-1R or PI3K signaling blockade resulted in significantly improved long-term responses in preclinical trials (Quail et al., 2016). These studies demonstrate the important contributions of reactive astrocytes in supporting cancer progression and resistance to targeted therapy, as discussed further below, across a variety of different types of brain tumors. Furthermore, although the direct effects of gliosis on brain tumors are currently unknown, the evident parallels between wound healing and cancer more generally suggest that these phenomena should be explored in more detail in order to improve our understanding of tumor cell-microenvironment interactions within the brain.
Neurons
In addition to astrocytes, neurons are a highly-specialized cell type that may also contribute to organ-specific mechanisms of tumor initiation and progression. It is known that neurons can provide mitogenic signals within the brain microenvironment to stimulate growth of neuronal and oligodendroglial precursor cells. Given the interest in stem/progenitor cells as putative cells of origin for gliomas (Liu et al., 2011; Wang et al., 2009), these studies give insight into important aspects of the brain tumor niche composition. In a recent study, optogenetic stimulation of neuronal activity in patient-derived xenograft glioma models promoted proliferation of cancer cells through upregulation of neuroligin-3 (NLGN3) in post-synaptic neurons (Venkatesh et al., 2015). Upon release, soluble NLGN3 subsequently induced PI3K signaling in glioma cells, increased FOS and feedforward-NLGN3 gene expression, and ultimately enhanced proliferative activity. In human glioblastoma, NLGN3 was inversely associated with survival (Venkatesh et al., 2015). In breast cancer, it has been shown that brain metastases mimic neurons by displaying key elements of neurotransmitter signaling, including upregulated expression of GABA receptors and transporters, and increased GABA catabolism (Neman et al., 2014). Together these studies suggest that neuronal-specific processes involved in synaptic transmission may have underappreciated mitogenic effects on brain tumors, and that neurotransmitters may inadvertently act as oncometabolites. These provocative questions warrant further investigation given the indispensable role for neurons within the brain.
Brain matrix
The brain extracellular matrix (ECM) differs substantially from other organs in multiple aspects. ECM proteins that are abundantly located throughout the stroma in non-CNS tissues, including collagens, laminins and fibronectin, are typically only found in association with vascular basement membranes within the brain. The predominant ECM constituents of the normal brain parenchyma instead include proteoglycans, glycoproteins and glycosaminoglycans, such as heparan sulfate proteoglycans (HSPGs) and hyaluronic acid (HA). Many of these macromolecules are concentrated in NSC niches (Reinhard et al., 2016) or vascular niches where they contribute to providing a supportive local environment and regulate normal stem cell homeostasis within the brain.
Because of the unique composition of the brain ECM and the associated relative proportions of ECM constituents, the mechanisms regulating tumor cell niches, angiogenesis, and invasion are likely to be somewhat different from other tissues. However, to date, comprehensive analyses of the ECM across different brain tumors, which could provide important insights into tumorigenic processes, have been limited. Nonetheless, from candidate-based analyses of individual ECM components, it is known that significant increases in HSPG production occur in gliomas (Lemjabbar-Alaoui et al., 2015; Wade et al., 2013). These macromolecules can serve as a reservoir for heparin-binding angiogenic growth factors, such as fibroblast growth factors (FGFs) and VEGFs, which are subsequently released in a localized manner by heparanase (Kundu et al., 2016). Vessel-associated macromolecules, which are upregulated on the glioma vasculature include tenascin C (TNC) and periostin (Brosicke and Faissner, 2015; Mustafa et al., 2012), which interestingly are also critical niche factors for promoting cancer cell survival (Oskarsson et al., 2014). Moreover, periostin can be secreted by glioma stem cells to promote recruitment of tumor-promoting M2-like macrophage progenitors from the peripheral circulation (Zhou et al., 2015).
Conversely, stroma/ECM-regulated mechanisms can contribute to physical exclusion of T cells across different tumor types, presenting a major challenge to immunotherapy delivery and efficacy (Joyce and Fearon, 2015), and this likely also contributes to immune suppression in brain tumors. For example, high concentrations of TNC in glioma-associated blood vessels apparently ‘traps’ T cells and prevents their transmigration into the brain parenchyma (Huang et al., 2010), A recent study, which provided some intriguing insights into the importance of the ECM and tissue mechanics in regulating glioma cell biology, showed that brain ECM stiffness correlated with increasing tumor grade (Miroshnikova et al., 2016). Interestingly, this was linked to increased levels of TNC and HA, regulated in a HIF1α-dependent manner. The authors also found that the mutational status of the glioma cells had consequences for the extent of ECM stiffness as mutations in the metabolic regulator isocitrate dehydrogenase 1 (IDH1), which are associated with better patient prognosis, decreased TNC expression, ECM stiffness and mechanosignaling (Miroshnikova et al., 2016). Comparable analyses of how genetic mutations influence other components of the TME in gliomas and additional brain malignancies should provide critical insights and potentially novel therapeutic targets for personalized medicine in the future.
Cancer therapy and the brain TME
Targeting the microenvironment in brain tumors
As discussed throughout this Perspective, several approaches to targeting the microenvironment in brain tumors are ongoing in preclinical and clinical studies (see Table 1 for a summary of clinical trials discussed here). CSF-1R inhibitors are currently in clinical trials to target macrophages in glioblastoma in the recurrent setting and/or in combination with standard of care (e.g. BLZ945 with PRD001 anti-PD-1 in solid tumors including recurrent glioblastoma, NCT02829723, Phase I/II; PLX3397 with temozolomide and radiotherapy in newly-diagnosed glioblastoma, NCT01790503, Phase Ib/2; Table 1) (Butowski et al., 2015). DC vaccinations such as DCVax-L have shown promising results, and as a consequence are in advanced clinical trials (newly-diagnosed glioblastoma, NCT00045968, Phase III) (Prins et al., 2011). Neutrophils also hold potential for their prognostic value in patients with primary brain tumors (Bertaut et al., 2016; Fossati et al., 1999), and metastatic disease (Koh et al., 2016; Mitsuya et al., 2016; Serdarevic et al., 2016). Targeting the vasculature through anti-angiogenic strategies is also relevant in glioblastoma patients given that this tumor type is highly vascularized; however, the current evidence seems to suggest that these strategies may be optimally used in the recurrent setting in combination with additional therapies (Friedman et al., 2009; Kreisl et al., 2009), rather than as frontline monotherapy on newly diagnosed, untreated disease (Gilbert et al., 2014). Targeting astrocytes with macitentan (an endothelin receptor antagonist; NCT01499251) or meclofenamate (a cyclooxygenase inhibitor that modulates gap junctions; NCT02429570) has also received clinical interest following important preclinical findings in both primary brain tumors (Kim et al., 2015) and brain metastases (Chen et al., 2016; Lee et al., 2016); however, these initiatives are still in their infancy, and early Phase I trials with macitentan showed no benefit for recurrent glioblastoma patients (Rosano et al., 2013). Finally, as we will discuss below, immune checkpoint inhibitors are gaining significant clinical attention for treatment of both primary brain tumors (e.g. nivolumab with radiotherapy in newly-diagnosed glioblastoma, NCT02617589, Phase III; nivolumab and/or ipilimumab versus bevacizumab in recurrent glioblastoma, NCT02017717, Phase III) (Preusser et al., 2015) and brain metastases (e.g. ipilimumab with either nivolumab or fotemustine in brain metastasis, NCT02460068, Phase III) (Berghoff et al., 2016). These studies collectively underscore the clinical potential of targeting the microenvironment, either through monotherapy or via rational combinations, as an alternative, integrated strategy for the management of different types of brain tumors.
The effects of standard of care treatment on the brain tissue landscape
Thoughtful consideration about how different treatments might impact critical structures or functions of the brain is imperative, as the goal of cancer therapy is to kill tumor cells while leaving healthy tissue unharmed. This is of particular concern given that the brain is a vulnerable and non-regenerative organ that is indispensable to survival. Current standard of care treatment for glioma, for example, is surgical resection, radiation, and temozolomide chemotherapy for high-grade disease (Stupp et al., 2005), and procarbazine, lomustine, and/or vincristine chemotherapy for low-grade disease (Buckner et al., 2016). Adverse effects from these treatment regimens range in severity between individuals, and can include loss of BBB integrity, cytokine deregulation, and ultimately, changes in neuronal integrity and cognitive function, particularly in children (Ahles and Saykin, 2007). Of particular concern is the potential for significant neurological damage, often with only modest improvements in overall survival. For example, a recent study showed no improvement in quality of life or overall survival after whole brain radiation therapy for patients with lung-to-brain metastases (Mulvenna et al., 2016). Given the high degree of cytotoxicity of these therapies on healthy brain tissue (Weiss et al., 2016), these findings strongly suggest that supplementation of optimal supportive care with whole brain radiotherapy ought to be reconsidered for the clinical management of brain metastases. Additionally, they indicate that clinical assessment of benefit versus risk in a quantifiable manner is particularly important to minimize additional unnecessary harm to this patient population.
During early phases of tumor regression in response to treatment, standard of care therapies can have positive, immune-supportive effects on the TME. For example, in mouse models of glioma, it has been shown that both temozolomide and radiation treatment cause the release of HMGB1 from dying cancer cells into the microenvironment, where it signals via TLR2 on DCs to enhance anti-tumor immunity (Curtin et al., 2009). In primary glioma models, treatment with temozolomide also increases cell surface expression of calreticulin, leading to enhanced cross-priming of T cells (Kim et al., 2010). Molecular changes such as these ultimately lead to treatment efficacy during early phases of tumor regression. In light of the divergent effects of therapy, however, understanding how to balance the positive and negative effects of treatment on the brain TME in order to optimize efficacy and minimize risk for patients will be important for translational studies going forward.
Microenvironment-mediated resistance to standard of care therapy
Tissue-specific cells within the CNS whose inherent function is to protect the brain from damage, such as astrocytes (Silver and Miller, 2004), have been shown to also play a role in blunting cytotoxic effects of therapy. For example, in melanoma-to-brain metastasis, tumors within the brain become surrounded by astrocytes, which suppress apoptosis in response to several types of chemotherapy including 5-FU, cisplatin and paclitaxel (Lin et al., 2010). Consistent with other findings discussed here, astrocytes achieve this protective effect by establishing gap junctions with cancer cells to sequester cytoplasmic calcium (Lin et al., 2010), akin to their neuroprotection functions (Burda and Sofroniew, 2014; Sofroniew, 2015). In a subsequent study from the same group, it was shown that co-culture of astrocytes with brain metastasis cells from breast or lung induces a pro-survival program in cancer cells, marked by upregulation of GSTA5, BCL2L1 and TWIST1. The induction of these genes supported the protective effects of astrocytes in response to taxol, whereby simultaneous genetic silencing of all three genes reversed this phenotype (Kim et al., 2011). Corroborating these studies, co-culture of astrocytes with glioma cells protects against apoptosis in response to temozolomide and vincristine, by establishing heterotypic gap junctions via connexin 43 (Chen et al., 2015). In metastases, astrocyte gap junctions are established in a PCDH7-dependent manner (Chen et al., 2016); it would therefore be of interest to determine whether blockade of these interactions (for example through meclofenamate or tonabersat) in gliomas could improve cytotoxic drug responses. Collectively, these data suggest that cancer cells are able to hijack normal neuroprotective interactions with astrocytes within the brain that are intended to be beneficial, and not detrimental, to the host.
Enhancing immunoreactivity of brain tumors via checkpoint inhibition
Checkpoint immunotherapy is currently under clinical evaluation for patients with brain tumors, including advanced metastases and glioblastoma. Groundbreaking work by several groups in melanoma patients have demonstrated that high mutation load is associated with increased frequency of neoantigens, and as a consequence, improved response to checkpoint inhibition (Restifo et al., 2016). These findings are now being translated to other types of cancer, including glioblastoma where some patients exhibit a hypermutator phenotype and DNA-damaging agents (e.g. temozolomide) are part of the current standard of care. A recent clinical study in pediatric gliomas showed that nivolumab treatment of recurrent hypermutant glioblastoma in two siblings with biallelic mismatch repair deficiency led to a significant and sustained therapeutic response (Bouffet et al., 2016). Similar results have also been reported in metastatic cancers harboring mismatch repair deficiencies and high frequency of somatic mutations (Le et al., 2015). As a consequence of these findings, combining DNA damaging agents (e.g. chemotherapy, radiotherapy) with checkpoint inhibitors to increase mutation frequency and neoantigen formation, with the ultimate goal of improving treatment response and durability, is now being actively investigated in glioblastoma, brain metastases, and other types of cancer (Restifo et al., 2016).
Interestingly, a recent study evaluated local versus systemic chemotherapy in combination with anti-programed cell death-1 (PD-1) in glioma models (Mathios et al., 2016). While local chemotherapy, delivered via carmustine-impregnated polymers, increased tumor-associated DC infiltration and clonal expansion of antigen-specific T cells, systemic delivery of carmustine via intraperitoneal injection caused intratumoral and systemic lymphodepletion. Consequently, anti-PD-1 plus local chemotherapy resulted in a significant survival advantage compared to the combination with systemic chemotherapy (Mathios et al., 2016), indicating that consideration will also need to be given to dosing route as well as schedule for immunotherapy-combination strategies. Another critical point of consideration will be to evaluate how the tumor subtype/molecular driver influences the immune cell composition, and consequently the efficacy of immunotherapy approaches. Some recent insights into this question have come from preclinical models of medulloblastoma in which group 3 and SHH-driven tumors were compared. Group 3 medulloblastomas had higher percentages of PD-1+ CD8+ cells, and correspondingly, a more pronounced response to PD-1 blockade (Pham et al., 2016).
Many of the earlier clinical trials for immunotherapy in the context of metastatic disease excluded patients with brain lesions due to poor survival outcomes and caveats pertaining to ability to cross the BBB. However, given that melanoma has a particularly high propensity for brain metastasis, and has been a frontline disease for immunotherapy development, this is an area of research that is now entering active clinical investigation. Indeed, many clinical studies have been initiated to study the effects of different checkpoint inhibitors (e.g. ipilimumab, nivolumab, pembrolizumab, etc.) in combination with chemotherapy and/or radiotherapy in melanoma patients with brain metastases (clinical trials and published results are summarized in Table 1) (Cohen and Kluger, 2016). Comparison of results from these immunotherapy trials indicates that patients with brain metastases may respond similarly to patients with advanced cancer without brain metastases (i.e. patients with metastases in other organs) (Hodi et al., 2010; Margolin et al., 2012). This suggests that either these antibodies are able to cross the BBB in areas where integrity is compromised to have biological impact, or that reprogramming of immune cells systemically is sufficient to impact disease course within the brain. Regardless of the mechanism of action, these results have promising implications for patients that have previously had extremely limited options for improving prognosis, and raise new insights about whether we should consider treating brain malignancies with agents that cannot (in principle) cross the BBB.
CONCLUSIONS AND FUTURE PERSPECTIVES
From multiple studies over the past decade and more, including representative advances discussed here, it has become clear that the brain TME is a fundamental regulator of cancer progression and therapeutic efficacy in primary and metastatic brain malignancies. Mechanistic insights into the tumor-promoting roles of individual components of the brain TME have led to the identification of multiple potential therapeutic targets, several which are now under clinical evaluation. While there is certainly reason for optimism in targeting and/or re-educating the TME in brain malignancies, as evidenced by encouraging initial results with chimeric antigen receptor (CAR) T cell therapy in a glioma patient case study (Brown et al., 2016), we nonetheless have considerable progress to make and many open questions to address in the coming years.
It will be essential to advance our current knowledge of individual brain TME components, as addressed herein, into a more complex microenvironmental landscape in which we analyze these cellular and non-cellular components as part of an integrated whole. Similarly, we expect that major insights will come from a detailed comparison of how distinct molecular subtypes or genetic drivers in cancer cells may differentially sculpt their microenvironment during the course of cancer progression, as already indicated from several studies discussed herein (Miroshnikova et al., 2016; Osswald et al., 2015; Phoenix et al., 2016; Unruh et al., 2016). Indeed, there are genetic subsets of glioma that do display efficacy to standard of care cytotoxic therapy, for example, those with mutant IDH, or co-deletion of 1p and 19q in oligodendrogliomas. Although as a field it is widely appreciated that there are differences in tumor evolution and response to therapy by virtue of different molecular subtypes such as these, appropriate dissection of the different TME determinants of therapeutic response is still in its infancy, and largely untapped clinically. Going forward, it will therefore be critical to determine the many differences in microenvironmental composition between different brain tumor subtypes in order to achieve a comprehensive understanding of tumor biology, including consideration of matrix stiffness, tumor-stromal interactions, and immune cell landscapes.
Moreover, it will be important to globally address how all aspects of the TME are affected by both standard of care therapy and new investigational therapies across all brain tumors and their respective molecular subtypes. From a practical perspective, we need to engage actively with medicinal chemists to continue to improve drug delivery into the brain; a perennial challenge for all brain-targeted therapies, including those that target the TME. Finally, if we cannot take a ‘one size fits all’ approach for targeting the TME in the brain, we will need to determine where the vulnerable points are to attack at a more personalized level. Given the current advances being made in the immunotherapy and TME fields, however, we can also expect an exciting time ahead for basic research and clinical translation in brain TME biology.
Acknowledgments
We thank members of the Joyce lab for stimulating discussions on this topic. Research in J.A.J.’s lab is supported by the Ludwig Institute for Cancer Research, University of Lausanne, Swiss League for Cancer Research, US National Cancer Institute (R01CA181355), and the Breast Cancer Research Foundation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abbott NJ. Blood-brain barrier structure and function and the challenges for CNS drug delivery. J Inherit Metab Dis. 2013;36:437–449. doi: 10.1007/s10545-013-9608-0. [DOI] [PubMed] [Google Scholar]
- Abbott NJ, Ronnback L, Hansson E. Astrocyte-endothelial interactions at the blood-brain barrier. Nat Rev Neurosci. 2006;7:41–53. doi: 10.1038/nrn1824. [DOI] [PubMed] [Google Scholar]
- Ahles TA, Saykin AJ. Candidate mechanisms for chemotherapy-induced cognitive changes. Nat Rev Cancer. 2007;7:192–201. doi: 10.1038/nrc2073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ajami B, Bennett JL, Krieger C, Tetzlaff W, Rossi FM. Local self-renewal can sustain CNS microglia maintenance and function throughout adult life. Nat Neurosci. 2007;10:1538–1543. doi: 10.1038/nn2014. [DOI] [PubMed] [Google Scholar]
- Anguille S, Smits EL, Lion E, van Tendeloo VF, Berneman ZN. Clinical use of dendritic cells for cancer therapy. Lancet Oncol. 2014;15:e257–267. doi: 10.1016/S1470-2045(13)70585-0. [DOI] [PubMed] [Google Scholar]
- Aspelund A, Antila S, Proulx ST, Karlsen TV, Karaman S, Detmar M, Wiig H, Alitalo K. A dural lymphatic vascular system that drains brain interstitial fluid and macromolecules. J Exp Med. 2015;212:991–999. doi: 10.1084/jem.20142290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bambury RM, Teo MY, Power DG, Yusuf A, Murray S, Battley JE, Drake C, O’Dea P, Bermingham N, Keohane C, et al. The association of pre-treatment neutrophil to lymphocyte ratio with overall survival in patients with glioblastoma multiforme. J Neurooncol. 2013;114:149–154. doi: 10.1007/s11060-013-1164-9. [DOI] [PubMed] [Google Scholar]
- Batchelor TT, Duda DG, di Tomaso E, Ancukiewicz M, Plotkin SR, Gerstner E, Eichler AF, Drappatz J, Hochberg FH, Benner T, et al. Phase II study of cediranib, an oral pan-vascular endothelial growth factor receptor tyrosine kinase inhibitor, in patients with recurrent glioblastoma. J Clin Oncol. 2010;28:2817–2823. doi: 10.1200/JCO.2009.26.3988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batchelor TT, Sorensen AG, di Tomaso E, Zhang WT, Duda DG, Cohen KS, Kozak KR, Cahill DP, Chen PJ, Zhu M, et al. AZD2171, a pan-VEGF receptor tyrosine kinase inhibitor, normalizes tumor vasculature and alleviates edema in glioblastoma patients. Cancer Cell. 2007;11:83–95. doi: 10.1016/j.ccr.2006.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett ML, Bennett FC, Liddelow SA, Ajami B, Zamanian JL, Fernhoff NB, Mulinyawe SB, Bohlen CJ, Adil A, Tucker A, et al. New tools for studying microglia in the mouse and human CNS. Proc Natl Acad Sci U S A. 2016;113:E1738–1746. doi: 10.1073/pnas.1525528113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bentolila LA, Prakash R, Mihic-Probst D, Wadehra M, Kleinman HK, Carmichael TS, Peault B, Barnhill RL, Lugassy C. Imaging of Angiotropism/Vascular Co-Option in a Murine Model of Brain Melanoma: Implications for Melanoma Progression along Extravascular Pathways. Sci Rep. 2016;6:23834. doi: 10.1038/srep23834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berghoff AS, Venur VA, Preusser M, Ahluwalia MS. Immune Checkpoint Inhibitors in Brain Metastases: From Biology to Treatment. Am Soc Clin Oncol Educ Book. 2016;35:e116–122. doi: 10.1200/EDBK_100005. [DOI] [PubMed] [Google Scholar]
- Bertaut A, Truntzer C, Madkouri R, Kaderbhai CG, Derangere V, Vincent J, Chauffert B, Aubriot-Lorton MH, Farah W, Mourier KL, et al. Blood baseline neutrophil count predicts bevacizumab efficacy in glioblastoma. Oncotarget. 2016;7:70948–70958. doi: 10.18632/oncotarget.10898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bos PD, Zhang XH, Nadal C, Shu W, Gomis RR, Nguyen DX, Minn AJ, van de Vijver MJ, Gerald WL, Foekens JA, Massague J. Genes that mediate breast cancer metastasis to the brain. Nature. 2009;459:1005–1009. doi: 10.1038/nature08021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouffet E, Larouche V, Campbell BB, Merico D, de Borja R, Aronson M, Durno C, Krueger J, Cabric V, Ramaswamy V, et al. Immune Checkpoint Inhibition for Hypermutant Glioblastoma Multiforme Resulting From Germline Biallelic Mismatch Repair Deficiency. J Clin Oncol. 2016;34:2206–2211. doi: 10.1200/JCO.2016.66.6552. [DOI] [PubMed] [Google Scholar]
- Bowman RL, Joyce JA. Therapeutic targeting of tumor-associated macrophages and microglia in glioblastoma. Immunotherapy. 2014;6:663–666. doi: 10.2217/imt.14.48. [DOI] [PubMed] [Google Scholar]
- Bowman RL, Klemm F, Akkari L, Pyonteck SM, Sevenich L, Quail DF, Dhara S, Simpson K, Gardner EE, Iacobuzio-Donahue CA, et al. Macrophage Ontogeny Underlies Differences in Tumor-Specific Education in Brain Malignancies. Cell reports. 2016;17:2445–2459. doi: 10.1016/j.celrep.2016.10.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brahmer J, Reckamp KL, Baas P, Crino L, Eberhardt WE, Poddubskaya E, Antonia S, Pluzanski A, Vokes EE, Holgado E, et al. Nivolumab versus Docetaxel in Advanced Squamous-Cell Non-Small-Cell Lung Cancer. N Engl J Med. 2015;373:123–135. doi: 10.1056/NEJMoa1504627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bresnick AR, Weber DJ, Zimmer DB. S100 proteins in cancer. Nat Rev Cancer. 2015;15:96–109. doi: 10.1038/nrc3893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brosicke N, Faissner A. Role of tenascins in the ECM of gliomas. Cell Adh Migr. 2015;9:131–140. doi: 10.1080/19336918.2014.1000071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown CE, Alizadeh D, Starr R, Weng L, Wagner JR, Naranjo A, Ostberg JR, Blanchard MS, Kilpatrick J, Simpson J, et al. Regression of Glioblastoma after Chimeric Antigen Receptor T-Cell Therapy. N Engl J Med. 2016;375:2561–2569. doi: 10.1056/NEJMoa1610497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckner JC, Shaw EG, Pugh SL, Chakravarti A, Gilbert MR, Barger GR, Coons S, Ricci P, Bullard D, Brown PD, et al. Radiation plus Procarbazine, CCNU, and Vincristine in Low-Grade Glioma. N Engl J Med. 2016;374:1344–1355. doi: 10.1056/NEJMoa1500925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burda JE, Sofroniew MV. Reactive gliosis and the multicellular response to CNS damage and disease. Neuron. 2014;81:229–248. doi: 10.1016/j.neuron.2013.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butowski N, Colman H, De Groot JF, Omuro AM, Nayak L, Wen PY, Cloughesy TF, Marimuthu A, Haidar S, Perry A, et al. Orally administered colony stimulating factor 1 receptor inhibitor PLX3397 in recurrent glioblastoma: an Ivy Foundation Early Phase Clinical Trials Consortium phase II study. Neuro Oncol. 2015;18:557–564. doi: 10.1093/neuonc/nov245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calabrese C, Poppleton H, Kocak M, Hogg TL, Fuller C, Hamner B, Oh EY, Gaber MW, Finklestein D, Allen M, et al. A perivascular niche for brain tumor stem cells. Cancer Cell. 2007;11:69–82. doi: 10.1016/j.ccr.2006.11.020. [DOI] [PubMed] [Google Scholar]
- Carmeliet P, Jain RK. Molecular mechanisms and clinical applications of angiogenesis. Nature. 2011;473:298–307. doi: 10.1038/nature10144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casbon AJ, Reynaud D, Park C, Khuc E, Gan DD, Schepers K, Passegue E, Werb Z. Invasive breast cancer reprograms early myeloid differentiation in the bone marrow to generate immunosuppressive neutrophils. Proc Natl Acad Sci U S A. 2015;112:E566–575. doi: 10.1073/pnas.1424927112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charles N, Holland EC. The perivascular niche microenvironment in brain tumor progression. Cell Cycle. 2010;9:3012–3021. doi: 10.4161/cc.9.15.12710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charles N, Ozawa T, Squatrito M, Bleau AM, Brennan CW, Hambardzumyan D, Holland EC. Perivascular nitric oxide activates notch signaling and promotes stem-like character in PDGF-induced glioma cells. Cell Stem Cell. 2010;6:141–152. doi: 10.1016/j.stem.2010.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Flies DB. Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat Rev Immunol. 2013;13:227–242. doi: 10.1038/nri3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Q, Boire A, Jin X, Valiente M, Er EE, Lopez-Soto A, Jacob LS, Patwa R, Shah H, Xu K, et al. Carcinoma-astrocyte gap junctions promote brain metastasis by cGAMP transfer. Nature. 2016;533:493–498. doi: 10.1038/nature18268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W, Wang D, Du X, He Y, Chen S, Shao Q, Ma C, Huang B, Chen A, Zhao P, et al. Glioma cells escaped from cytotoxicity of temozolomide and vincristine by communicating with human astrocytes. Med Oncol. 2015;32:43. doi: 10.1007/s12032-015-0487-0. [DOI] [PubMed] [Google Scholar]
- Chinot OL, Wick W, Mason W, Henriksson R, Saran F, Nishikawa R, Carpentier AF, Hoang-Xuan K, Kavan P, Cernea D, et al. Bevacizumab plus radiotherapy-temozolomide for newly diagnosed glioblastoma. N Engl J Med. 2014;370:709–722. doi: 10.1056/NEJMoa1308345. [DOI] [PubMed] [Google Scholar]
- Coffelt SB, Kersten K, Doornebal CW, Weiden J, Vrijland K, Hau CS, Verstegen NJ, Ciampricotti M, Hawinkels LJ, Jonkers J, de Visser KE. IL-17-producing gammadelta T cells and neutrophils conspire to promote breast cancer metastasis. Nature. 2015;522:345–348. doi: 10.1038/nature14282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coffelt SB, Wellenstein MD, de Visser KE. Neutrophils in cancer: neutral no more. Nat Rev Cancer. 2016;16:431–446. doi: 10.1038/nrc.2016.52. [DOI] [PubMed] [Google Scholar]
- Cohen JV, Kluger HM. Systemic Immunotherapy for the Treatment of Brain Metastases. Front Oncol. 2016;6:49. doi: 10.3389/fonc.2016.00049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen MH, Shen YL, Keegan P, Pazdur R. FDA drug approval summary: bevacizumab (Avastin) as treatment of recurrent glioblastoma multiforme. Oncologist. 2009;14:1131–1138. doi: 10.1634/theoncologist.2009-0121. [DOI] [PubMed] [Google Scholar]
- Coniglio SJ, Eugenin E, Dobrenis K, Stanley ER, West BL, Symons MH, Segall JE. Microglial stimulation of glioblastoma invasion involves epidermal growth factor receptor (EGFR) and colony stimulating factor 1 receptor (CSF-1R) signaling. Mol Med. 2012;18:519–527. doi: 10.2119/molmed.2011.00217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtin JF, Liu N, Candolfi M, Xiong W, Assi H, Yagiz K, Edwards MR, Michelsen KS, Kroeger KM, Liu C, et al. HMGB1 mediates endogenous TLR2 activation and brain tumor regression. PLoS Med. 2009;6:e10. doi: 10.1371/journal.pmed.1000010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- da Fonseca AC, Matias D, Garcia C, Amaral R, Geraldo LH, Freitas C, Lima FR. The impact of microglial activation on blood-brain barrier in brain diseases. Front Cell Neurosci. 2014;8:362. doi: 10.3389/fncel.2014.00362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies DC. Blood-brain barrier breakdown in septic encephalopathy and brain tumours. J Anat. 2002;200:639–646. doi: 10.1046/j.1469-7580.2002.00065.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Palma M, Venneri MA, Galli R, Sergi Sergi L, Politi LS, Sampaolesi M, Naldini L. Tie2 identifies a hematopoietic lineage of proangiogenic monocytes required for tumor vessel formation and a mesenchymal population of pericyte progenitors. Cancer Cell. 2005;8:211–226. doi: 10.1016/j.ccr.2005.08.002. [DOI] [PubMed] [Google Scholar]
- Dudnik E, Yust-Katz S, Nechushtan H, Goldstein DA, Zer A, Flex D, Siegal T, Peled N. Intracranial response to nivolumab in NSCLC patients with untreated or progressing CNS metastases. Lung Cancer. 2016;98:114–117. doi: 10.1016/j.lungcan.2016.05.031. [DOI] [PubMed] [Google Scholar]
- Eichler AF, Chung E, Kodack DP, Loeffler JS, Fukumura D, Jain RK. The biology of brain metastases-translation to new therapies. Nat Rev Clin Oncol. 2011;8:344–356. doi: 10.1038/nrclinonc.2011.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elmore MR, Najafi AR, Koike MA, Dagher NN, Spangenberg EE, Rice RA, Kitazawa M, Matusow B, Nguyen H, West BL, Green KN. Colony-stimulating factor 1 receptor signaling is necessary for microglia viability, unmasking a microglia progenitor cell in the adult brain. Neuron. 2014;82:380–397. doi: 10.1016/j.neuron.2014.02.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelhardt B, Vajkoczy P, Weller RO. The movers and shapers in immune privilege of the CNS. Nat Immunol. 2017;18:123–131. doi: 10.1038/ni.3666. [DOI] [PubMed] [Google Scholar]
- Fecci PE, Heimberger AB, Sampson JH. Immunotherapy for primary brain tumors: no longer a matter of privilege. Clin Cancer Res. 2014;20:5620–5629. doi: 10.1158/1078-0432.CCR-14-0832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fecci PE, Mitchell DA, Whitesides JF, Xie W, Friedman AH, Archer GE, Herndon JE, 2nd, Bigner DD, Dranoff G, Sampson JH. Increased regulatory T-cell fraction amidst a diminished CD4 compartment explains cellular immune defects in patients with malignant glioma. Cancer Res. 2006a;66:3294–3302. doi: 10.1158/0008-5472.CAN-05-3773. [DOI] [PubMed] [Google Scholar]
- Fecci PE, Ochiai H, Mitchell DA, Grossi PM, Sweeney AE, Archer GE, Cummings T, Allison JP, Bigner DD, Sampson JH. Systemic CTLA-4 blockade ameliorates glioma-induced changes to the CD4+ T cell compartment without affecting regulatory T-cell function. Clin Cancer Res. 2007;13:2158–2167. doi: 10.1158/1078-0432.CCR-06-2070. [DOI] [PubMed] [Google Scholar]
- Fecci PE, Sweeney AE, Grossi PM, Nair SK, Learn CA, Mitchell DA, Cui X, Cummings TJ, Bigner DD, Gilboa E, Sampson JH. Systemic anti-CD25 monoclonal antibody administration safely enhances immunity in murine glioma without eliminating regulatory T cells. Clin Cancer Res. 2006b;12:4294–4305. doi: 10.1158/1078-0432.CCR-06-0053. [DOI] [PubMed] [Google Scholar]
- Fine HA. Bevacizumab in glioblastoma–still much to learn. N Engl J Med. 2014;370:764–765. doi: 10.1056/NEJMe1313309. [DOI] [PubMed] [Google Scholar]
- Fossati G, Ricevuti G, Edwards SW, Walker C, Dalton A, Rossi ML. Neutrophil infiltration into human gliomas. Acta Neuropathol. 1999;98:349–354. doi: 10.1007/s004010051093. [DOI] [PubMed] [Google Scholar]
- Friedman HS, Prados MD, Wen PY, Mikkelsen T, Schiff D, Abrey LE, Yung WK, Paleologos N, Nicholas MK, Jensen R, et al. Bevacizumab alone and in combination with irinotecan in recurrent glioblastoma. J Clin Oncol. 2009;27:4733–4740. doi: 10.1200/JCO.2008.19.8721. [DOI] [PubMed] [Google Scholar]
- Gabrusiewicz K, Rodriguez B, Wei J, Hashimoto Y, Healy LM, Maiti SN, Thomas G, Zhou S, Wang Q, Elakkad A, et al. Glioblastoma-infiltrated innate immune cells resemble M0 macrophage phenotype. JCI Insight. 2016;1:e85841. doi: 10.1172/jci.insight.85841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garg AD, Vandenberk L, Koks C, Verschuere T, Boon L, Van Gool SW, Agostinis P. Dendritic cell vaccines based on immunogenic cell death elicit danger signals and T cell-driven rejection of high-grade glioma. Sci Transl Med. 2016;8:328ra327. doi: 10.1126/scitranslmed.aae0105. [DOI] [PubMed] [Google Scholar]
- Gautier EL, Shay T, Miller J, Greter M, Jakubzick C, Ivanov S, Helft J, Chow A, Elpek KG, Gordonov S, et al. Gene-expression profiles and transcriptional regulatory pathways that underlie the identity and diversity of mouse tissue macrophages. Nat Immunol. 2012;13:1118–1128. doi: 10.1038/ni.2419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert MR, Dignam JJ, Armstrong TS, Wefel JS, Blumenthal DT, Vogelbaum MA, Colman H, Chakravarti A, Pugh S, Won M, et al. A randomized trial of bevacizumab for newly diagnosed glioblastoma. N Engl J Med. 2014;370:699–708. doi: 10.1056/NEJMoa1308573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbertson RJ, Rich JN. Making a tumour’s bed: glioblastoma stem cells and the vascular niche. Nat Rev Cancer. 2007;7:733–736. doi: 10.1038/nrc2246. [DOI] [PubMed] [Google Scholar]
- Ginhoux F, Greter M, Leboeuf M, Nandi S, See P, Gokhan S, Mehler MF, Conway SJ, Ng LG, Stanley ER, et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science. 2010;330:841–845. doi: 10.1126/science.1194637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginhoux F, Schultze JL, Murray PJ, Ochando J, Biswas SK. New insights into the multidimensional concept of macrophage ontogeny, activation and function. Nat Immunol. 2015;17:34–40. doi: 10.1038/ni.3324. [DOI] [PubMed] [Google Scholar]
- Goldberg SB, Gettinger SN, Mahajan A, Chiang AC, Herbst RS, Sznol M, Tsiouris AJ, Cohen J, Vortmeyer A, Jilaveanu L, et al. Pembrolizumab for patients with melanoma or non-small-cell lung cancer and untreated brain metastases: early analysis of a non-randomised, open-label, phase 2 trial. Lancet Oncol. 2016;17:976–983. doi: 10.1016/S1470-2045(16)30053-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldmann J, Kwidzinski E, Brandt C, Mahlo J, Richter D, Bechmann I. T cells traffic from brain to cervical lymph nodes via the cribroid plate and the nasal mucosa. J Leukoc Biol. 2006;80:797–801. doi: 10.1189/jlb.0306176. [DOI] [PubMed] [Google Scholar]
- Goldmann T, Wieghofer P, Jordao MJ, Prutek F, Hagemeyer N, Frenzel K, Amann L, Staszewski O, Kierdorf K, Krueger M, et al. Origin, fate and dynamics of macrophages at central nervous system interfaces. Nat Immunol. 2016;17:797–805. doi: 10.1038/ni.3423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez Perdiguero E, Klapproth K, Schulz C, Busch K, Azzoni E, Crozet L, Garner H, Trouillet C, de Bruijn MF, Geissmann F, Rodewald HR. Tissue-resident macrophages originate from yolk-sac-derived erythro-myeloid progenitors. Nature. 2015;518:547–551. doi: 10.1038/nature13989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graeber MB, Scheithauer BW, Kreutzberg GW. Microglia in brain tumors. Glia. 2002;40:252–259. doi: 10.1002/glia.10147. [DOI] [PubMed] [Google Scholar]
- Hambardzumyan D, Becher OJ, Rosenblum MK, Pandolfi PP, Manova-Todorova K, Holland EC. PI3K pathway regulates survival of cancer stem cells residing in the perivascular niche following radiation in medulloblastoma in vivo. Genes Dev. 2008;22:436–448. doi: 10.1101/gad.1627008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hambardzumyan D, Gutmann DH, Kettenmann H. The role of microglia and macrophages in glioma maintenance and progression. Nat Neurosci. 2016;19:20–27. doi: 10.1038/nn.4185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hart DN, Fabre JW. Demonstration and characterization of Ia-positive dendritic cells in the interstitial connective tissues of rat heart and other tissues, but not brain. J Exp Med. 1981;154:347–361. doi: 10.1084/jem.154.2.347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hickey WF, Kimura H. Perivascular microglial cells of the CNS are bone marrow-derived and present antigen in vivo. Science. 1988;239:290–292. doi: 10.1126/science.3276004. [DOI] [PubMed] [Google Scholar]
- Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, Gonzalez R, Robert C, Schadendorf D, Hassel JC, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363:711–723. doi: 10.1056/NEJMoa1003466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu B, Wang Q, Wang YA, Hua S, Sauve CG, Ong D, Lan ZD, Chang Q, Ho YW, Monasterio MM, et al. Epigenetic Activation of WNT5A Drives Glioblastoma Stem Cell Differentiation and Invasive Growth. Cell. 2016;167:1281–1295 e1218. doi: 10.1016/j.cell.2016.10.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang JY, Cheng YJ, Lin YP, Lin HC, Su CC, Juliano R, Yang BC. Extracellular matrix of glioblastoma inhibits polarization and transmigration of T cells: the role of tenascin-C in immune suppression. J Immunol. 2010;185:1450–1459. doi: 10.4049/jimmunol.0901352. [DOI] [PubMed] [Google Scholar]
- Hussain SF, Yang D, Suki D, Aldape K, Grimm E, Heimberger AB. The role of human glioma-infiltrating microglia/macrophages in mediating antitumor immune responses. Neuro Oncol. 2006;8:261–279. doi: 10.1215/15228517-2006-008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutchinson L. Immunotherapy: Exploiting mismatch repair in GBM. Nat Rev Clin Oncol. 2016;13:264. doi: 10.1038/nrclinonc.2016.56. [DOI] [PubMed] [Google Scholar]
- Iliff JJ, Goldman SA, Nedergaard M. Implications of the discovery of brain lymphatic pathways. Lancet Neurol. 2015;14:977–979. doi: 10.1016/S1474-4422(15)00221-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain RK. Normalizing tumor vasculature with anti-angiogenic therapy: a new paradigm for combination therapy. Nat Med. 2001;7:987–989. doi: 10.1038/nm0901-987. [DOI] [PubMed] [Google Scholar]
- Jain RK. Normalization of tumor vasculature: an emerging concept in antiangiogenic therapy. Science. 2005;307:58–62. doi: 10.1126/science.1104819. [DOI] [PubMed] [Google Scholar]
- Jain RK, di Tomaso E, Duda DG, Loeffler JS, Sorensen AG, Batchelor TT. Angiogenesis in brain tumours. Nat Rev Neurosci. 2007;8:610–622. doi: 10.1038/nrn2175. [DOI] [PubMed] [Google Scholar]
- Jilaveanu LB, Parisi F, Barr ML, Zito CR, Cruz-Munoz W, Kerbel RS, Rimm DL, Bosenberg MW, Halaban R, Kluger Y, Kluger HM. PLEKHA5 as a Biomarker and Potential Mediator of Melanoma Brain Metastasis. Clin Cancer Res. 2015;21:2138–2147. doi: 10.1158/1078-0432.CCR-14-0861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joyce JA, Fearon DT. T cell exclusion, immune privilege, and the tumor microenvironment. Science. 2015;348:74–80. doi: 10.1126/science.aaa6204. [DOI] [PubMed] [Google Scholar]
- Kim SJ, Kim JS, Park ES, Lee JS, Lin Q, Langley RR, Maya M, He J, Kim SW, Weihua Z, et al. Astrocytes upregulate survival genes in tumor cells and induce protection from chemotherapy. Neoplasia. 2011;13:286–298. doi: 10.1593/neo.11112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SJ, Lee HJ, Kim MS, Choi HJ, He J, Wu Q, Aldape K, Weinberg JS, Yung WK, Conrad CA, et al. Macitentan, a Dual Endothelin Receptor Antagonist, in Combination with Temozolomide Leads to Glioblastoma Regression and Long-term Survival in Mice. Clin Cancer Res. 2015;21:4630–4641. doi: 10.1158/1078-0432.CCR-14-3195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim TG, Kim CH, Park JS, Park SD, Kim CK, Chung DS, Hong YK. Immunological factors relating to the antitumor effect of temozolomide chemoimmunotherapy in a murine glioma model. Clin Vaccine Immunol. 2010;17:143–153. doi: 10.1128/CVI.00292-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh YW, Choi JH, Ahn MS, Choi YW, Lee HW. Baseline neutrophil-lymphocyte ratio is associated with baseline and subsequent presence of brain metastases in advanced non-small-cell lung cancer. Sci Rep. 2016;6:38585. doi: 10.1038/srep38585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komohara Y, Ohnishi K, Kuratsu J, Takeya M. Possible involvement of the M2 anti-inflammatory macrophage phenotype in growth of human gliomas. J Pathol. 2008;216:15–24. doi: 10.1002/path.2370. [DOI] [PubMed] [Google Scholar]
- Konstantinou MP, Dutriaux C, Gaudy-Marqueste C, Mortier L, Bedane C, Girard C, Thellier S, Jouary T, Grob JJ, Richard MA, et al. Ipilimumab in melanoma patients with brain metastasis: a retro-spective multicentre evaluation of thirty-eight patients. Acta Derm Venereol. 2014;94:45–49. doi: 10.2340/00015555-1654. [DOI] [PubMed] [Google Scholar]
- Kreisl TN, Kim L, Moore K, Duic P, Royce C, Stroud I, Garren N, Mackey M, Butman JA, Camphausen K, et al. Phase II trial of single-agent bevacizumab followed by bevacizumab plus irinotecan at tumor progression in recurrent glioblastoma. J Clin Oncol. 2009;27:740–745. doi: 10.1200/JCO.2008.16.3055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kundu S, Xiong A, Spyrou A, Wicher G, Marinescu VD, Edqvist PD, Zhang L, Essand M, Dimberg A, Smits A, et al. Heparanase Promotes Glioma Progression and Is Inversely Correlated with Patient Survival. Mol Cancer Res. 2016;14:1243–1253. doi: 10.1158/1541-7786.MCR-16-0223. [DOI] [PubMed] [Google Scholar]
- Lathia JD, Gallagher J, Heddleston JM, Wang J, Eyler CE, Macswords J, Wu Q, Vasanji A, McLendon RE, Hjelmeland AB, Rich JN. Integrin alpha 6 regulates glioblastoma stem cells. Cell Stem Cell. 2010;6:421–432. doi: 10.1016/j.stem.2010.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lathia JD, Mack SC, Mulkearns-Hubert EE, Valentim CL, Rich JN. Cancer stem cells in glioblastoma. Genes Dev. 2015;29:1203–1217. doi: 10.1101/gad.261982.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, Skora AD, Luber BS, Azad NS, Laheru D, et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N Engl J Med. 2015;372:2509–2520. doi: 10.1056/NEJMoa1500596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HJ, Hanibuchi M, Kim SJ, Yu H, Kim MS, He J, Langley RR, Lehembre F, Regenass U, Fidler IJ. Treatment of experimental human breast cancer and lung cancer brain metastases in mice by macitentan, a dual antagonist of endothelin receptors, combined with paclitaxel. Neuro Oncol. 2016;18:486–496. doi: 10.1093/neuonc/now037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leenders WP, Kusters B, Verrijp K, Maass C, Wesseling P, Heerschap A, Ruiter D, Ryan A, de Waal R. Antiangiogenic therapy of cerebral melanoma metastases results in sustained tumor progression via vessel co-option. Clin Cancer Res. 2004;10:6222–6230. doi: 10.1158/1078-0432.CCR-04-0823. [DOI] [PubMed] [Google Scholar]
- Lemjabbar-Alaoui H, McKinney A, Yang YW, Tran VM, Phillips JJ. Glycosylation alterations in lung and brain cancer. Adv Cancer Res. 2015;126:305–344. doi: 10.1016/bs.acr.2014.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy C, Allouache D, Lacroix J, Dugue AE, Supiot S, Campone M, Mahe M, Kichou S, Leheurteur M, Hanzen C, et al. REBECA: a phase I study of bevacizumab and whole-brain radiation therapy for the treatment of brain metastasis from solid tumours. Ann Oncol. 2014;25:2351–2356. doi: 10.1093/annonc/mdu465. [DOI] [PubMed] [Google Scholar]
- Li B, Wang C, Zhang Y, Zhao XY, Huang B, Wu PF, Li Q, Li H, Liu YS, Cao LY, et al. Elevated PLGF contributes to small-cell lung cancer brain metastasis. Oncogene. 2013;32:2952–2962. doi: 10.1038/onc.2012.313. [DOI] [PubMed] [Google Scholar]
- Liang J, Piao Y, Holmes L, Fuller GN, Henry V, Tiao N, de Groot JF. Neutrophils promote the malignant glioma phenotype through S100A4. Clin Cancer Res. 2014;20:187–198. doi: 10.1158/1078-0432.CCR-13-1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Q, Balasubramanian K, Fan D, Kim SJ, Guo L, Wang H, Bar-Eli M, Aldape KD, Fidler IJ. Reactive astrocytes protect melanoma cells from chemotherapy by sequestering intracellular calcium through gap junction communication channels. Neoplasia. 2010;12:748–754. doi: 10.1593/neo.10602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C, Sage JC, Miller MR, Verhaak RG, Hippenmeyer S, Vogel H, Foreman O, Bronson RT, Nishiyama A, Luo L, Zong H. Mosaic analysis with double markers reveals tumor cell of origin in glioma. Cell. 2011;146:209–221. doi: 10.1016/j.cell.2011.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Gu Y, Han Y, Zhang Q, Jiang Z, Zhang X, Huang B, Xu X, Zheng J, Cao X. Tumor Exosomal RNAs Promote Lung Pre-metastatic Niche Formation by Activating Alveolar Epithelial TLR3 to Recruit Neutrophils. Cancer Cell. 2016;30:243–256. doi: 10.1016/j.ccell.2016.06.021. [DOI] [PubMed] [Google Scholar]
- Liu Y, Kosaka A, Ikeura M, Kohanbash G, Fellows-Mayle W, Snyder LA, Okada H. Premetastatic soil and prevention of breast cancer brain metastasis. Neuro Oncol. 2013;15:891–903. doi: 10.1093/neuonc/not031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lockman PR, Mittapalli RK, Taskar KS, Rudraraju V, Gril B, Bohn KA, Adkins CE, Roberts A, Thorsheim HR, Gaasch JA, et al. Heterogeneous blood-tumor barrier permeability determines drug efficacy in experimental brain metastases of breast cancer. Clin Cancer Res. 2010;16:5664–5678. doi: 10.1158/1078-0432.CCR-10-1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lou N, Takano T, Pei Y, Xavier AL, Goldman SA, Nedergaard M. Purinergic receptor P2RY12-dependent microglial closure of the injured blood-brain barrier. Proc Natl Acad Sci U S A. 2016;113:1074–1079. doi: 10.1073/pnas.1520398113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louveau A, Harris TH, Kipnis J. Revisiting the Mechanisms of CNS Immune Privilege. Trends Immunol. 2015a;36:569–577. doi: 10.1016/j.it.2015.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louveau A, Smirnov I, Keyes TJ, Eccles JD, Rouhani SJ, Peske JD, Derecki NC, Castle D, Mandell JW, Lee KS, et al. Structural and functional features of central nervous system lymphatic vessels. Nature. 2015b;523:337–341. doi: 10.1038/nature14432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe J, MacLennan KA, Powe DG, Pound JD, Palmer JB. Microglial cells in human brain have phenotypic characteristics related to possible function as dendritic antigen presenting cells. J Pathol. 1989;159:143–149. doi: 10.1002/path.1711590209. [DOI] [PubMed] [Google Scholar]
- Lu-Emerson C, Snuderl M, Kirkpatrick ND, Goveia J, Davidson C, Huang Y, Riedemann L, Taylor J, Ivy P, Duda DG, et al. Increase in tumor-associated macrophages after antiangiogenic therapy is associated with poor survival among patients with recurrent glioblastoma. Neuro Oncol. 2013;15:1079–1087. doi: 10.1093/neuonc/not082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyle LT, Lockman PR, Adkins CE, Mohammad AS, Sechrest E, Hua E, Palmieri D, Liewehr DJ, Steinberg SM, Kloc W, et al. Alterations in Pericyte Subpopulations Are Associated with Elevated Blood-Tumor Barrier Permeability in Experimental Brain Metastasis of Breast Cancer. Clin Cancer Res. 2016;22:5287–5299. doi: 10.1158/1078-0432.CCR-15-1836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malkki H. Neuro-oncology: Bevacizumab prolongs progression-free survival but not overall survival in newly diagnosed glioblastoma. Nat Rev Neurol. 2014;10:179. doi: 10.1038/nrneurol.2014.47. [DOI] [PubMed] [Google Scholar]
- Margolin K, Ernstoff MS, Hamid O, Lawrence D, McDermott D, Puzanov I, Wolchok JD, Clark JI, Sznol M, Logan TF, et al. Ipilimumab in patients with melanoma and brain metastases: an open-label, phase 2 trial. Lancet Oncol. 2012;13:459–465. doi: 10.1016/S1470-2045(12)70090-6. [DOI] [PubMed] [Google Scholar]
- Massague J, Obenauf AC. Metastatic colonization by circulating tumour cells. Nature. 2016;529:298–306. doi: 10.1038/nature17038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masson F, Calzascia T, Di Berardino-Besson W, de Tribolet N, Dietrich PY, Walker PR. Brain microenvironment promotes the final functional maturation of tumor-specific effector CD8+ T cells. J Immunol. 2007;179:845–853. doi: 10.4049/jimmunol.179.2.845. [DOI] [PubMed] [Google Scholar]
- Mathios D, Kim JE, Mangraviti A, Phallen J, Park CK, Jackson CM, Garzon-Muvdi T, Kim E, Theodros D, Polanczyk M, et al. Anti-PD-1 antitumor immunity is enhanced by local and abrogated by systemic chemotherapy in GBM. Sci Transl Med. 2016;8:370ra180. doi: 10.1126/scitranslmed.aag2942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNamara MG, Lwin Z, Jiang H, Templeton AJ, Zadeh G, Bernstein M, Chung C, Millar BA, Laperriere N, Mason WP. Factors impacting survival following second surgery in patients with glioblastoma in the temozolomide treatment era, incorporating neutrophil/lymphocyte ratio and time to first progression. J Neurooncol. 2014;117:147–152. doi: 10.1007/s11060-014-1366-9. [DOI] [PubMed] [Google Scholar]
- Medawar PB. Immunity to homologous grafted skin; the fate of skin homografts transplanted to the brain, to subcutaneous tissue, and to the anterior chamber of the eye. Br J Exp Pathol. 1948;29:58–69. [PMC free article] [PubMed] [Google Scholar]
- Miroshnikova YA, Mouw JK, Barnes JM, Pickup MW, Lakins JN, Kim Y, Lobo K, Persson AI, Reis GF, McKnight TR, et al. Tissue mechanics promote IDH1-dependent HIF1alpha-tenascin C feedback to regulate glioblastoma aggression. Nat Cell Biol. 2016 doi: 10.1038/ncb3429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell DA, Batich KA, Gunn MD, Huang MN, Sanchez-Perez L, Nair SK, Congdon KL, Reap EA, Archer GE, Desjardins A, et al. Tetanus toxoid and CCL3 improve dendritic cell vaccines in mice and glioblastoma patients. Nature. 2015;519:366–369. doi: 10.1038/nature14320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitsuya K, Nakasu Y, Kurakane T, Hayashi N, Harada H, Nozaki K. Elevated preoperative neutrophil-to-lymphocyte ratio as a predictor of worse survival after resection in patients with brain metastasis. J Neurosurg. 2016:1–5. doi: 10.3171/2016.8.JNS16899. [DOI] [PubMed] [Google Scholar]
- Mondin V, Ferlito A, Devaney KO, Woolgar JA, Rinaldo A. A survey of metastatic central nervous system tumors to cervical lymph nodes. Eur Arch Otorhinolaryngol. 2010;267:1657–1666. doi: 10.1007/s00405-010-1357-1. [DOI] [PubMed] [Google Scholar]
- Morikawa A, Peereboom DM, Thorsheim HR, Samala R, Balyan R, Murphy CG, Lockman PR, Simmons A, Weil RJ, Tabar V, et al. Capecitabine and lapatinib uptake in surgically resected brain metastases from metastatic breast cancer patients: a prospective study. Neuro Oncol. 2015;17:289–295. doi: 10.1093/neuonc/nou141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulvenna P, Nankivell M, Barton R, Faivre-Finn C, Wilson P, McColl E, Moore B, Brisbane I, Ardron D, Holt T, et al. Dexamethasone and supportive care with or without whole brain radiotherapy in treating patients with non-small cell lung cancer with brain metastases unsuitable for resection or stereotactic radiotherapy (QUARTZ): results from a phase 3, non-inferiority, randomised trial. Lancet. 2016;388:2004–2014. doi: 10.1016/S0140-6736(16)30825-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy KA, Lechner MG, Popescu FE, Bedi J, Decker SA, Hu P, Erickson JR, O’Sullivan MG, Swier L, Salazar AM, et al. An in vivo immunotherapy screen of costimulatory molecules identifies Fc-OX40L as a potent reagent for the treatment of established murine gliomas. Clin Cancer Res. 2012;18:4657–4668. doi: 10.1158/1078-0432.CCR-12-0990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mustafa DA, Dekker LJ, Stingl C, Kremer A, Stoop M, Sillevis Smitt PA, Kros JM, Luider TM. A proteome comparison between physiological angiogenesis and angiogenesis in glioblastoma. Mol Cell Proteomics. 2012;11:M111 008466. doi: 10.1074/mcp.M111.008466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neman J, Termini J, Wilczynski S, Vaidehi N, Choy C, Kowolik CM, Li H, Hambrecht AC, Roberts E, Jandial R. Human breast cancer metastases to the brain display GABAergic properties in the neural niche. Proc Natl Acad Sci U S A. 2014;111:984–989. doi: 10.1073/pnas.1322098111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolas-Avila JA, Adrover JM, Hidalgo A. Neutrophils in Homeostasis, Immunity, and Cancer. Immunity. 2017;46:15–28. doi: 10.1016/j.immuni.2016.12.012. [DOI] [PubMed] [Google Scholar]
- O’Keeffe E, Campbell M. Modulating the paracellular pathway at the blood-brain barrier: current and future approaches for drug delivery to the CNS. Drug Discov Today Technol. 2016;20:35–39. doi: 10.1016/j.ddtec.2016.07.008. [DOI] [PubMed] [Google Scholar]
- Obenauf AC, Zou Y, Ji AL, Vanharanta S, Shu W, Shi H, Kong X, Bosenberg MC, Wiesner T, Rosen N, et al. Therapy-induced tumour secretomes promote resistance and tumour progression. Nature. 2015;520:368–372. doi: 10.1038/nature14336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oskarsson T, Batlle E, Massague J. Metastatic stem cells: sources, niches, and vital pathways. Cell Stem Cell. 2014;14:306–321. doi: 10.1016/j.stem.2014.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osswald M, Blaes J, Liao Y, Solecki G, Gommel M, Berghoff AS, Salphati L, Wallin JJ, Phillips HS, Wick W, Winkler F. Impact of Blood-Brain Barrier Integrity on Tumor Growth and Therapy Response in Brain Metastases. Clin Cancer Res. 2016;22:6078–6087. doi: 10.1158/1078-0432.CCR-16-1327. [DOI] [PubMed] [Google Scholar]
- Osswald M, Jung E, Sahm F, Solecki G, Venkataramani V, Blaes J, Weil S, Horstmann H, Wiestler B, Syed M, et al. Brain tumour cells interconnect to a functional and resistant network. Nature. 2015;528:93–98. doi: 10.1038/nature16071. [DOI] [PubMed] [Google Scholar]
- Palucka K, Banchereau J. Cancer immunotherapy via dendritic cells. Nat Rev Cancer. 2012;12:265–277. doi: 10.1038/nrc3258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papadopoulos MC, Saadoun S, Davies DC, Bell BA. Emerging molecular mechanisms of brain tumour oedema. Br J Neurosurg. 2001;15:101–108. doi: 10.1080/02688690120036775. [DOI] [PubMed] [Google Scholar]
- Pham CD, Flores C, Yang C, Pinheiro EM, Yearley JH, Sayour EJ, Pei Y, Moore C, McLendon RE, Huang J, et al. Differential Immune Microenvironments and Response to Immune Checkpoint Blockade among Molecular Subtypes of Murine Medulloblastoma. Clin Cancer Res. 2016;22:582–595. doi: 10.1158/1078-0432.CCR-15-0713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phoenix TN, Patmore DM, Boop S, Boulos N, Jacus MO, Patel YT, Roussel MF, Finkelstein D, Goumnerova L, Perreault S, et al. Medulloblastoma Genotype Dictates Blood Brain Barrier Phenotype. Cancer Cell. 2016;29:508–522. doi: 10.1016/j.ccell.2016.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piao Y, Liang J, Holmes L, Zurita AJ, Henry V, Heymach JV, de Groot JF. Glioblastoma resistance to anti-VEGF therapy is associated with myeloid cell infiltration, stem cell accumulation, and a mesenchymal phenotype. Neuro Oncol. 2012;14:1379–1392. doi: 10.1093/neuonc/nos158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pietras A, Katz AM, Ekstrom EJ, Wee B, Halliday JJ, Pitter KL, Werbeck JL, Amankulor NM, Huse JT, Holland EC. Osteopontin-CD44 signaling in the glioma perivascular niche enhances cancer stem cell phenotypes and promotes aggressive tumor growth. Cell Stem Cell. 2014;14:357–369. doi: 10.1016/j.stem.2014.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preusser M, Lim M, Hafler DA, Reardon DA, Sampson JH. Prospects of immune checkpoint modulators in the treatment of glioblastoma. Nat Rev Neurol. 2015;11:504–514. doi: 10.1038/nrneurol.2015.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prins RM, Soto H, Konkankit V, Odesa SK, Eskin A, Yong WH, Nelson SF, Liau LM. Gene expression profile correlates with T-cell infiltration and relative survival in glioblastoma patients vaccinated with dendritic cell immunotherapy. Clin Cancer Res. 2011;17:1603–1615. doi: 10.1158/1078-0432.CCR-10-2563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pukrop T, Dehghani F, Chuang HN, Lohaus R, Bayanga K, Heermann S, Regen T, Van Rossum D, Klemm F, Schulz M, et al. Microglia promote colonization of brain tissue by breast cancer cells in a Wnt-dependent way. Glia. 2010;58:1477–1489. doi: 10.1002/glia.21022. [DOI] [PubMed] [Google Scholar]
- Pyonteck SM, Akkari L, Schuhmacher AJ, Bowman RL, Sevenich L, Quail DF, Olson OC, Quick ML, Huse JT, Teijeiro V, et al. CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat Med. 2013;19:1264–1272. doi: 10.1038/nm.3337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian BZ, Pollard JW. Macrophage diversity enhances tumor progression and metastasis. Cell. 2010;141:39–51. doi: 10.1016/j.cell.2010.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quail DF, Bowman RL, Akkari L, Quick ML, Schuhmacher AJ, Huse JT, Holland EC, Sutton JC, Joyce JA. The tumor microenvironment underlies acquired resistance to CSF-1R inhibition in gliomas. Science. 2016;352:aad3018. doi: 10.1126/science.aad3018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quail DF, Joyce JA. Microenvironmental regulation of tumor progression and metastasis. Nat Med. 2013;19:1423–1437. doi: 10.1038/nm.3394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Queirolo P, Spagnolo F, Ascierto PA, Simeone E, Marchetti P, Scoppola A, Del Vecchio M, Di Guardo L, Maio M, Di Giacomo AM, et al. Efficacy and safety of ipilimumab in patients with advanced melanoma and brain metastases. J Neurooncol. 2014;118:109–116. doi: 10.1007/s11060-014-1400-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez-Castillejo C, Sanchez-Sanchez F, Andreu-Agullo C, Ferron SR, Aroca-Aguilar JD, Sanchez P, Mira H, Escribano J, Farinas I. Pigment epithelium-derived factor is a niche signal for neural stem cell renewal. Nat Neurosci. 2006;9:331–339. doi: 10.1038/nn1657. [DOI] [PubMed] [Google Scholar]
- Ransohoff RM, Engelhardt B. The anatomical and cellular basis of immune surveillance in the central nervous system. Nat Rev Immunol. 2012;12:623–635. doi: 10.1038/nri3265. [DOI] [PubMed] [Google Scholar]
- Reinhard J, Brosicke N, Theocharidis U, Faissner A. The extracellular matrix niche microenvironment of neural and cancer stem cells in the brain. Int J Biochem Cell Biol. 2016;81:174–183. doi: 10.1016/j.biocel.2016.05.002. [DOI] [PubMed] [Google Scholar]
- Restifo NP, Smyth MJ, Snyder A. Acquired resistance to immunotherapy and future challenges. Nat Rev Cancer. 2016;16:121–126. doi: 10.1038/nrc.2016.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricci-Vitiani L, Pallini R, Biffoni M, Todaro M, Invernici G, Cenci T, Maira G, Parati EA, Stassi G, Larocca LM, De Maria R. Tumour vascularization via endothelial differentiation of glioblastoma stem-like cells. Nature. 2010;468:824–828. doi: 10.1038/nature09557. [DOI] [PubMed] [Google Scholar]
- Robel S, Berninger B, Gotz M. The stem cell potential of glia: lessons from reactive gliosis. Nat Rev Neurosci. 2011;12:88–104. doi: 10.1038/nrn2978. [DOI] [PubMed] [Google Scholar]
- Rosano L, Spinella F, Bagnato A. Endothelin 1 in cancer: biological implications and therapeutic opportunities. Nat Rev Cancer. 2013;13:637–651. doi: 10.1038/nrc3546. [DOI] [PubMed] [Google Scholar]
- Sarkar S, Doring A, Zemp FJ, Silva C, Lun X, Wang X, Kelly J, Hader W, Hamilton M, Mercier P, et al. Therapeutic activation of macrophages and microglia to suppress brain tumor-initiating cells. Nat Neurosci. 2014;17:46–55. doi: 10.1038/nn.3597. [DOI] [PubMed] [Google Scholar]
- Serdarevic M, Kukulj S, Nikolic I, Taradi I, Romic Z, Samarzija M. 203P: Could neutrophil-to-lymphocyte ratio be predictor of brain metastases in non small cell lung cancer? J Thorac Oncol. 2016;11:S145. [Google Scholar]
- Sevenich L, Bowman RL, Mason SD, Quail DF, Rapaport F, Elie BT, Brogi E, Brastianos PK, Hahn WC, Holsinger LJ, et al. Analysis of tumour- and stroma-supplied proteolytic networks reveals a brain-metastasis-promoting role for cathepsin S. Nat Cell Biol. 2014;16:876–888. doi: 10.1038/ncb3011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shechter R, London A, Schwartz M. Orchestrated leukocyte recruitment to immune-privileged sites: absolute barriers versus educational gates. Nat Rev Immunol. 2013;13:206–218. doi: 10.1038/nri3391. [DOI] [PubMed] [Google Scholar]
- Shen Q, Goderie SK, Jin L, Karanth N, Sun Y, Abramova N, Vincent P, Pumiglia K, Temple S. Endothelial cells stimulate self-renewal and expand neurogenesis of neural stem cells. Science. 2004;304:1338–1340. doi: 10.1126/science.1095505. [DOI] [PubMed] [Google Scholar]
- Shi C, Pamer EG. Monocyte recruitment during infection and inflammation. Nat Rev Immunol. 2011;11:762–774. doi: 10.1038/nri3070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silver J, Miller JH. Regeneration beyond the glial scar. Nat Rev Neurosci. 2004;5:146–156. doi: 10.1038/nrn1326. [DOI] [PubMed] [Google Scholar]
- Sofroniew MV. Astrocyte barriers to neurotoxic inflammation. Nat Rev Neurosci. 2015;16:249–263. doi: 10.1038/nrn3898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorensen AG, Batchelor TT, Zhang WT, Chen PJ, Yeo P, Wang M, Jennings D, Wen PY, Lahdenranta J, Ancukiewicz M, et al. A “vascular normalization index” as potential mechanistic biomarker to predict survival after a single dose of cediranib in recurrent glioblastoma patients. Cancer Res. 2009;69:5296–5300. doi: 10.1158/0008-5472.CAN-09-0814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steeg PS, Camphausen KA, Smith QR. Brain metastases as preventive and therapeutic targets. Nat Rev Cancer. 2011;11:352–363. doi: 10.1038/nrc3053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, Belanger K, Brandes AA, Marosi C, Bogdahn U, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352:987–996. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- Tavazoie M, Van der Veken L, Silva-Vargas V, Louissaint M, Colonna L, Zaidi B, Garcia-Verdugo JM, Doetsch F. A specialized vascular niche for adult neural stem cells. Cell Stem Cell. 2008;3:279–288. doi: 10.1016/j.stem.2008.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong RT, Boucher Y, Kozin SV, Winkler F, Hicklin DJ, Jain RK. Vascular normalization by vascular endothelial growth factor receptor 2 blockade induces a pressure gradient across the vasculature and improves drug penetration in tumors. Cancer Res. 2004;64:3731–3736. doi: 10.1158/0008-5472.CAN-04-0074. [DOI] [PubMed] [Google Scholar]
- Ulvestad E, Williams K, Bjerkvig R, Tiekotter K, Antel J, Matre R. Human microglial cells have phenotypic and functional characteristics in common with both macrophages and dendritic antigen-presenting cells. J Leukoc Biol. 1994;56:732–740. doi: 10.1002/jlb.56.6.732. [DOI] [PubMed] [Google Scholar]
- Unruh D, Schwarze SR, Khoury L, Thomas C, Wu M, Chen L, Chen R, Liu Y, Schwartz MA, Amidei C, et al. Mutant IDH1 and thrombosis in gliomas. Acta Neuropathol. 2016;132:917–930. doi: 10.1007/s00401-016-1620-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valiente M, Obenauf AC, Jin X, Chen Q, Zhang XH, Lee DJ, Chaft JE, Kris MG, Huse JT, Brogi E, Massague J. Serpins promote cancer cell survival and vascular co-option in brain metastasis. Cell. 2014;156:1002–1016. doi: 10.1016/j.cell.2014.01.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkatesh HS, Johung TB, Caretti V, Noll A, Tang Y, Nagaraja S, Gibson EM, Mount CW, Polepalli J, Mitra SS, et al. Neuronal Activity Promotes Glioma Growth through Neuroligin-3 Secretion. Cell. 2015;161:803–816. doi: 10.1016/j.cell.2015.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wade A, Robinson AE, Engler JR, Petritsch C, James CD, Phillips JJ. Proteoglycans and their roles in brain cancer. FEBS J. 2013;280:2399–2417. doi: 10.1111/febs.12109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R, Chadalavada K, Wilshire J, Kowalik U, Hovinga KE, Geber A, Fligelman B, Leversha M, Brennan C, Tabar V. Glioblastoma stem-like cells give rise to tumour endothelium. Nature. 2010;468:829–833. doi: 10.1038/nature09624. [DOI] [PubMed] [Google Scholar]
- Wang Y, Yang J, Zheng H, Tomasek GJ, Zhang P, McKeever PE, Lee EY, Zhu Y. Expression of mutant p53 proteins implicates a lineage relationship between neural stem cells and malignant astrocytic glioma in a murine model. Cancer Cell. 2009;15:514–526. doi: 10.1016/j.ccr.2009.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wculek SK, Malanchi I. Neutrophils support lung colonization of metastasis-initiating breast cancer cells. Nature. 2015;528:413–417. doi: 10.1038/nature16140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss N, Miller F, Cazaubon S, Couraud PO. The blood-brain barrier in brain homeostasis and neurological diseases. Biochim Biophys Acta. 2009;1788:842–857. doi: 10.1016/j.bbamem.2008.10.022. [DOI] [PubMed] [Google Scholar]
- Weiss T, Weller M, Roth P. Immunological effects of chemotherapy and radiotherapy against brain tumors. Expert Rev Anticancer Ther. 2016;16:1087–1094. doi: 10.1080/14737140.2016.1229600. [DOI] [PubMed] [Google Scholar]
- Winkler F, Kozin SV, Tong RT, Chae SS, Booth MF, Garkavtsev I, Xu L, Hicklin DJ, Fukumura D, di Tomaso E, et al. Kinetics of vascular normalization by VEGFR2 blockade governs brain tumor response to radiation: role of oxygenation, angiopoietin-1, and matrix metalloproteinases. Cancer Cell. 2004;6:553–563. doi: 10.1016/j.ccr.2004.10.011. [DOI] [PubMed] [Google Scholar]
- Wu A, Wei J, Kong LY, Wang Y, Priebe W, Qiao W, Sawaya R, Heimberger AB. Glioma cancer stem cells induce immunosuppressive macrophages/microglia. Neuro Oncol. 2010;12:1113–1125. doi: 10.1093/neuonc/noq082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wurmser AE, Nakashima K, Summers RG, Toni N, D’Amour KA, Lie DC, Gage FH. Cell fusion-independent differentiation of neural stem cells to the endothelial lineage. Nature. 2004;430:350–356. doi: 10.1038/nature02604. [DOI] [PubMed] [Google Scholar]
- Zamanian JL, Xu L, Foo LC, Nouri N, Zhou L, Giffard RG, Barres BA. Genomic analysis of reactive astrogliosis. J Neurosci. 2012;32:6391–6410. doi: 10.1523/JNEUROSCI.6221-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Zhang S, Yao J, Lowery FJ, Zhang Q, Huang WC, Li P, Li M, Wang X, Zhang C, et al. Microenvironment-induced PTEN loss by exosomal microRNA primes brain metastasis outgrowth. Nature. 2015;527:100–104. doi: 10.1038/nature15376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou W, Ke SQ, Huang Z, Flavahan W, Fang X, Paul J, Wu L, Sloan AE, McLendon RE, Li X, et al. Periostin secreted by glioblastoma stem cells recruits M2 tumour-associated macrophages and promotes malignant growth. Nat Cell Biol. 2015;17:170–182. doi: 10.1038/ncb3090. [DOI] [PMC free article] [PubMed] [Google Scholar]