

Abstract
Eukaryotic cilia are organelles that project from the surface of cells to fulfill motility and sensory functions. In vertebrates, the functions of both motile and immotile cilia are critical for embryonic development and adult tissue homeostasis. Importantly, a multitude of human diseases is caused by abnormal cilia biogenesis and functions which rely on the compartmentalization of the cilium and the maintenance of its protein composition. The transition zone (TZ) is a specialized ciliary domain present at the base of the cilium and is part of a gate that controls protein entry and exit from this organelle. The relevance of the TZ is highlighted by the fact that several of its components are coded by ciliopathy genes. Here we review recent developments in the study of TZ proteomes, the mapping of individual components to the TZ structure and the establishment of the TZ as a lipid gate.
Keywords: centriole, centrosome, cilia, transition zone
INTRODUCTION
Eukaryotic cilia/flagella are evolutionarily conserved microtu-bule (MT)-based organelles that protrude from the cell surface and play sensory and motility roles. In vertebrates, multiple cilia types are critical for embryonic development and homeostasis of adult tissues. Consequently, absence of cilia or their malfunction contributes to a plethora of human diseases (e.g. Primary cilia dyskinesis, Meckel-Gruber syndrome and Nephronophthisis) often presenting overlapping clinical manifestations such as: infertility, blindness, obesity, cognitive impairment, polydactyly, and polycystic kidneys (reviewed by Mitchison and Valente, 2017). The basic ciliary structure consists of an axoneme made of 9 MT doublets templated by a centriole/basal body (BB) attached to the cell membrane by transition fibers, and covered by a specialized membrane domain (Fig. 1). Motile cilia which promote cell movement or fluid flow generation typically have an additional MT central pair (9+2 axoneme) and accessory structures required for ciliary movement (e.g. axonemal dyneins and radial spokes). Immotile (primary) cilia fulfil sensory/signaling functions (e.g. Shh pathway in vertebrates) and usually lack a MT central pair (9+0 axoneme) and the machinery required for movement. Cilia assembly and maintenance rely on the intraflagellar transport (IFT) machinery which uses MT motors to transport cargo from the cell body to the ciliary tip and back (reviewed by Ishikawa and Marshall, 2017). In this review we focus on a ciliary sub-domain, the TZ, which corresponds to the proximal portion of the axoneme, distal to the BB. The TZ is required for the compartmentalization of the cilium, functioning as a gate that strictly controls the protein composition of the ciliary compartment (reviewed by Reiter et al., 2012). The TZ is characterized by MT-ciliary membrane connectors, usually y-shaped (y-links), and the ciliary necklace, a specialized membrane domain typified by rows of membrane particles encircling the base of the axoneme. This ciliary domain gained considerable attention in recent years since most of its known components are associated with human diseases. Here we review recent findings concerning the molecular composition of the TZ, the function and localization of TZ proteins, and the role of the TZ as a lipid gate.
Fig. 1.
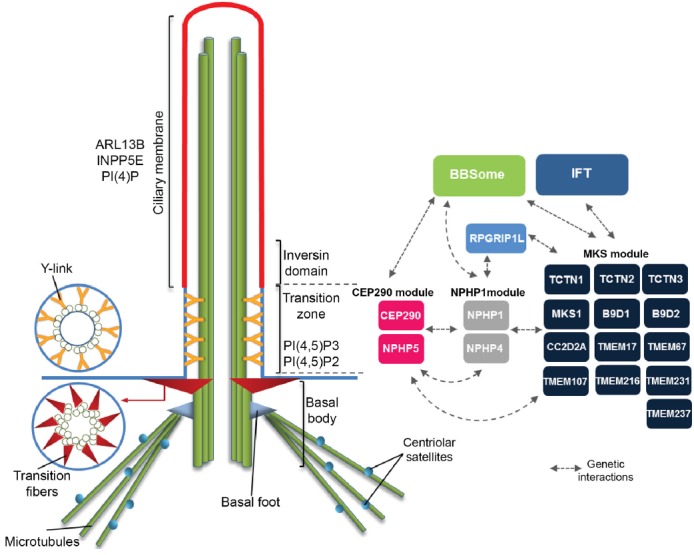
Cilia structure.
The scheme depicts the structure of a primary cilium. At their base eukaryotic cilia present a centriole/basal body, from which the axoneme microtubules elongate, and accessory structures such as the basal foot and the transition fibers. The ciliary membrane is a specialized membrane domain enriched in specific proteins (e.g. ARL13B) and lipid species (e.g. PI(4)P). At the proximal region of the axoneme is the transition zone characterized by microtubule-membrane connectors. Distal to the transition zone is localized the inversin domain which lacks y-links and has a distinct protein composition from the transition zone. The figure shows the protein modules present at the transition zone largely as they were described by Chih et al. (2011), Garcia-Gonzalo et al. (2011), and Sang et al. (2011). Indicated are also genetic interactions between components of the transition zone modules and components of the BBSome and IFT.
COMPOSITION OF THE CILIARY TRANSITION ZONE
Genetic interaction studies in C. elegans (discussed below), as well as the biochemical characterization of protein-protein interactions in mammalian systems revealed the existence of three main TZ modules, MKS, NPHP and CEP290 (Table 1; Fig. 1) which are composed of soluble and membrane-associated proteins that collaborate for the assembly and gating function of the TZ (Chih et al., 2011; Garcia-Gonzalo et al., 2011; Sang et al., 2011). Each of these complexes is composed of multiple proteins that can be co-purified and are interdependent for their localization to the TZ (Chih et al., 2011; Garcia-Gonzalo et al., 2011; Huang et al., 2011; Sang et al., 2011). More recently, additional proteomic studies were done to define the protein composition of the TZ in different organisms. Studies on TZs isolated from Chlamydomonas identified known components of the TZ such as homologs of MKS and NPHP module proteins. Also of note was the identification of six subunits of the endosomal sorting complexes required for transport (ESCRT) system, one of which, VPS4, was confirmed to localize at the TZ. These data suggest ESCRT has a ciliary role likely related to ciliary membrane dynamics and processes such as the formation of ciliary ectosomes (Diener et al., 2015; Long et al., 2016). A subsequent study determined the proteome of affinity purified Trypanosome’s TZs and identified kinetoplastids-specific proteins as well as proteins with Chlamydomonas or human orthologues. Among these were several components of the MKS complex, BBSome (protein complex involved in protein trafficking to cilia; Jin et al., 2010) proteins and components of the inversin domain (Fig. 1). RNAi studies showed that a subset of the identified proteins have important roles in flagellar biogenesis some of them being required to build axonemal structures like the MT central pair (Dean et al., 2016). Finally, BioID (proximity-dependent biotin identification) was used in human cells to screen for interactors of most known TZ components and proteins localizing to the centriole, centriolar appendages, centriolar satellites and the inversin domain (Gupta et al., 2015). The BioID technique is based on the expression of a protein of interest fused to a promiscuous biotin ligase (BirA). Upon incubation with biotin, the BirA tag promotes the proximity-dependent biotinylation of proteins in close proximity to the fusion protein in their proper in vivo context. The biotinylated proteins can then be purified and identified by mass spectrometry (Roux et al., 2012). TZ proteins shared extensive proximity interactors with components of the other centrosomal and ciliary sub-structures, which determined their clustering into different groups. TZ proteins known to localize to the centrosome (e.g., RPGRIP1L, AHI1) and centriolar satellites (e.g. CEP290) clustered with other centrosomal and satellite baits, whereas TZ proteins associated with membranes (e.g. TMEM17, TMEM67, TMEM237, TCTNs) formed a distinct group. Moreover, BioID was conducted in non-ciliated cells and serum-starved ciliated cells. This allowed for the observation of differences in the interaction profiles between the two conditions, which might reflect the ciliogenesis program. Known interactions between TZ components were detected even in non-ciliated cells (e.g. B9D1–B9D2; NPHP1–NPHP4; CEP290-NPHP5) suggesting that the known TZ modules are assembled, at least in part, prior to their incorporation into the TZ which is in agreement with previous studies (Chih et al., 2011; Garcia-Gonzalo et al., 2011; Sang et al., 2011). Importantly, under ciliated conditions there was an increase in shared interactors between the TZ proteins present in different clusters (centrosome and membrane-associated), most of which corresponding to cytoskeletal and membrane trafficking related proteins. Co-immunoprecipitation experiments confirmed the interaction between TZ components such as CEP162, RPGRIP1L, AHI1 and LCA5 with centriolar satellite proteins like PCM1 and KIAA0753. These results suggest that, like CEP290, other TZ components also rely on centriolar satellites for their delivery to the TZ (Gupta et al., 2015; Klinger et al., 2014). In accordance with other studies suggesting TZ proteins cooperate with the IFT machinery for correct ciliary protein targeting (Goetz et al., 2017; Zhao and Malicki, 2011) a component of the IFT-B complex (IFT74), was detected as a prey for RPGRIP1, RPGRIP1L and CEP162 (Gupta et al., 2015). These studies will certainly contribute to further our understanding on the composition of the different TZ modules, and on how they cooperate with different machineries (e.g. IFT, BBsome, ESCRT) to properly assemble and maintain the ciliary compartment. Moreover, techniques such as BioID will allow us to profile different types of cilia and compare the cell type specific ciliary interaction networks.
Table 1.
TZ proteins – associated diseases and loss of function phenotypes in vertebrate systems
| Protein | Associated disease | Sub-cellular localization | Loss of function phenotypes in vertebrate systems |
|---|---|---|---|
| MKS1 | MKS, JBTS, BBS | Centrosome, TZ (Chih et al., 2011; Dawe et al., 2007; Garcia-Gonzalo., 2011) |
KO Mouse/RNAi/Patient fibroblasts - Tissue-specific ciliation defects; disfunctional ciliary gate and Shh signaling; affected ciliation in spheroids (Cui et al., 2011; Dawe et al., 2007; Weatherbee et al., 2012; Slaats et al., 2016; Goetz et al., 2017) |
| B9D1 | MKS, JBTS, BBS | Basal body, TZ, axoneme (Chih et al., 2011; Garcia-Gonzalo., 2011) |
KO Mouse/RNAi - Tissue-specific ciliation defects; disfunctional ciliary gate and Shh signaling (Chih et al., 2011; Dowdle et al., 2011; Garcia-Gonzalo et al., 2011) |
| B9D2 | MKS | Basal body, TZ, nucleus (Chih et al., 2011; Dowdle et al., 2011) |
KO Mouse - Tissue-specific ciliation defects; disfunctional ciliary gate and Shh signaling (Garcia-Gonzalo et al., 2011); KD Zebrafish - cilia-related developmental problems (Zhao and Malicki, 2011) |
| TCTN1 | JBTS | Basal body, TZ (Garcia-Gonzalo., 2011) |
KO Mouse -Tissue-specific ciliation defects; disfunctional ciliary gate and Shh signaling (Garcia-Gonzalo et al., 2011) |
| TCTN2 | MKS, JBTS | Basal body, TZ, axoneme (Garcia-Gonzalo et al., 2011) |
KO mouse -Tissue-specific ciliation defects; disfunctional ciliary gate and Shh signaling (Garcia-Gonzalo et al., 2011; Sang et al., 2011) |
| TCTN3 | JBTS, OFD4 | Basal body, TZ, axoneme (Garcia-Gonzalo., 2011) |
Patient fibroblasts - Disfunctional Shh signaling (Thomas et al., 2012) |
| CC2D2A | MKS, JBTS, COACH | Centrosome, TZ (Chih et al., 2011; Gorden et al., 2008) |
KO Mouse/RNAi - Tissue-specific ciliation defects; disfunctional ciliary gate and Shh signaling (Chih et al, 2011; Garcia-Gonzalo et al., 2011); KD Zebrafish - cilia-related developmental problems (Bachmann-Gagescu et al., 2011; Gorden et al., 2008) |
| TMEM17 | TZ (Chih et al., 2011) |
RNAi - mild ciliation phenotype; perturbed ciliary protein composition (Chih et., 2011); Patient fibroblasts - ciliation failure (Li et al., 2016) | |
| TMEM67 | MKS, JBTS, COACH, NPHP, BBS | Basal body, TZ, axoneme (Garcia-Gonzalo et al., 2011) |
KO Mouse -Tissue-specific ciliation defects; disfunctional ciliary gate and Shh signaling (Abdelhamed et al., 2015; Garcia-Gonzalo et al., 2011) |
| TMEM107 | MKS, JBTS, OFD | TZ (Lambacher et al., 2016) |
Mutant Mouse - Tissue-specific ciliation defects and disfunctional Shh signaling (Christopher et al., 2012); RNAi/Patient fibroblasts- affected ciliation, ciliary gate, Shh signaling and spheroid formation (Lambacher et al., 2016; Shaheen et al., 2015; Shylo et al., 2016) |
| TMEM216 | MKS, JBTS | Basal body, TZ, axoneme, Golgi (Lee et al., 2012) |
Patient fibroblasts/RNAi - affected ciliation; KD zebrafish - cilia and PCP-related phenotypes (Lee et al., 2012; Valente et al., 2010) |
| TMEM231 | MKS, JBTS | TZ (Chih et al., 2011) |
KO Mouse/RNAi -Tissue-specific ciliation defects; disfunctional ciliary gate and Shh signaling (Chih et al., 2011) |
| TMEM237 | JBTS | TZ (Huang et al., 2011) |
Patient fibroblasts/RNAi -affected ciliation and WNT and PCP signal- ing; KD Zebrafish - developmental problems (Huang et al., 2011) |
| NPHP1 | JBTS, NPHP, SLSN | TZ, cell junctions (Garcia-Gonzalo et al., 2011; Sang et al., 2011) |
KO Mouse - retinal degeneration (Louie et al., 2010); KD Zebrafish - pronephric problems (Slanchev et al., 2011); RNAi - defective formation of spheroids (Delous et al., 2009; Sang et al., 2011) |
| NPHP4 | NPHP, SLSN | Centrosome, TZ, cilium, cell junctions (Garcia-Gonzalo et al., 2011; Mollet et al., 2005; Sang et al., 2011) |
KO Mice - retinal degeneration; KD Zebrafish - cilia-related developmental problems (Slanchev et al., 2011); RNAi - defective formation of spheroids (Delous et al., 2009; Sang et al., 2011) |
| CEP290 | MKS, JBTS, LCA, BBS, SLS | Centriolar satellites, centrosome, TZ (Garcia-Gonzalo et al., 2011; Kim et al., 2008; Sang et al., 2011) |
KO mouse -Tissue-specific ciliation defects (Rachel et al., 2015); KD Zebrafish - cilia related developmental problems (Sayer et al., 2006; Schäfer et al., 2008) |
| NPHP5 | SLS | Centrosome, cilium (Otto et al., 2005; Sang et al., 2011) |
KO Mouse - Tissue-specific ciliation defects; retinal degeneration; failure to assemble TZ in photoreceptors (Ronquillo et al., 2016); KD Zebrafish - cilia-related developmental problems (Schäfer et al., 2008) |
| RPGRIP1L | MKS, JBTS, COACH | Centrosome, basal body, TZ (Arts et al., 2007; Delous et al.,2007; Garcia-gonzalo et al., 2011) |
KO Mouse - Tissue-specific ciliation defects; disturbed Shh signaling (Vierkotten, et al., 2007); KD Zebrafish - cilia positioning and planar cell polarity phenotypes (Mahuzier et al., 2012) |
| RPGRIP1 | LCA | Centrioles, TZ, axoneme (Hong et al., 2001; Shu et al., 2005) |
KO Mouse - retinal degeneration; required for the localization of other TZ proteins (Patil et al., 2012; Won et al., 2009; Zhao et al., 2003) |
| RPGR | CORD, MC, RP | Centrosome, TZ (Khanna et al., 2005; Shu et al., 2005) |
KO Mouse - retinal degeneration (Hong et al., 2000); RNAi - ciliation defect; alterations in the actin cytoskeleton (Gakovic et al., 2011); KD Zebrafish - cilia related developmental problems (Gerner et al., 2010) |
| LCA5 | LCA | Centrosome, TZ, MTs (den Hollander et al., 2007) |
KO Mouse - retinal degeneration; defect in IFT (Boldt et al., 2011) |
| AHI1 | JBTS | Mother centriole, basal body, TZ (Hsiao et al., 2009) |
KO Mouse - defective photoreceptors (Louie et al., 2010); RNAi - affected ciliation (Hsiao et al., 2009); Zebrafish Mutant - tissue specific ciliogenesis problems (Lessieur et al., 2017) |
| CEP162 | TZ, MTs (Wang et al., 2013) |
RNAi - failure to assemble the TZ; KD Zebrafish - cilia-related developmental problems (Wang et al., 2013) | |
| TMEM138 | JBTS | Basal body, TZ, axoneme (Lee et al., 2012) |
Patient fibroblasts/RNAi - ciliation defects; KD Zebrafish - cilia-related developmental phenotypes (Lee et al., 2012) |
| JBTS17 | JBTS | TZ (Damerla et al., 2015) |
Mutant Mouse/Mutant MEFS/Patient fibroblasts - ciliation defects; disfunctional ciliary gate and Shh signaling (Damerla et al., 2015) |
| TMEM80 | TZ (Li et al., 2016) |
 MKS, Meckel syndrome; JBTS, Joubert syndrome; BBS, Bardet-Biedl syndrome; NPHP, Nephronophthisis; OFD4, Orofaciodigital syndrome IV; COACH, COACH syndrome; SLSN, Senior-Loken syndrome
MKS, Meckel syndrome; JBTS, Joubert syndrome; BBS, Bardet-Biedl syndrome; NPHP, Nephronophthisis; OFD4, Orofaciodigital syndrome IV; COACH, COACH syndrome; SLSN, Senior-Loken syndrome
 LCA, Leber congenital amaurosis; CORD, Cone-rod dystrophy; MC, Macular degeneration; RP, Retinitis pigmentosa
LCA, Leber congenital amaurosis; CORD, Cone-rod dystrophy; MC, Macular degeneration; RP, Retinitis pigmentosa
 KO, Knock-out; KD, Knock-down; MEFS, mouse embryonic fibroblasts
KO, Knock-out; KD, Knock-down; MEFS, mouse embryonic fibroblasts
MAPPING INDIVIDUAL COMPONENTS WITHIN THE TZ STRUCTURE
With the increasing number of known TZ components, attention is now focused on finely mapping the location of these proteins within the TZ. Recent studies in multiple model organisms used different imaging approaches to show that TZ proteins occupy distinct domains within this compartment. Stimulated emission depletion (STED) super resolution microscopy combined with electron microscopy (EM) were used to study the localization of the TZ proteins CEP290, RPGRIP1L, MKS1, TCTN2 and TMEM67 in human RPE-1 cells (Yang et al., 2015). CEP290’s signal had a width close to that of the axoneme, and occupied a proximal position close to the BB and transition fiber markers centrin and CEP164 respectively. RPGRIP1L was distal to CEP290 and presented a similar width. The MKS complex components MKS1, TCTN2 and TMEM67 were present at the same axial level as RPGRIP1L but occupied wider areas. TCNT2 and TMEM67, which are transmembrane proteins, were the most peripheral (Fig. 2). Consistent with these findings, in Chlamydomonas CEP290 localizes to the TZ proximal end and NPHP4 is distal to it (Awata et al., 2014). In C. elegans CEP290 also occupies a core position within the TZ whereas MKSR-2/B9D2 and NPHP4 localize to the periphery of the TZ (Schouteden et al., 2015). Supporting the evolutionary conservation of the TZ, 3D super-resolution structured illumination microscopy, revealed a similar organization of TZ proteins in mature Drosophila spermatocytes where CEP290 also seemed to overlap with axonemal MTs. CBY partially overlapped with CEP290 whereas MKS complex components (Tectonic, MKS1, TMEM216, B9D1 and B9D2) localized between the axonemal MTs and the ciliary membrane (Fig. 2; Pratt et al., 2016). DILA was found inside the centriole and TZ lumens being capped by CBY (Vieillard et al., 2016). The core localization of CEP290 in multiple organisms, its detection by EM at the MT-membrane linkers in Chlamydomonas and the connecting cilium of mouse photoreceptors, and its requirement for y-links assembly in C. elegans, suggest CEP290 is a component of these structures (Craige et al., 2010; Li et al., 2016; Rachel et al., 2015; Schouteden et al., 2015). Finally, the use of fluorescence microscopy and EM also revealed distinct domains within the Trypanosome TZ. For instance, components of the MKS complex (MKS1, MKS1-R1/B9D1, MKS1-R2/B9D2), localize to the distal half of the TZ corresponding to the region containing y-links. On the other hand, the kinetoplastid-specific protein TZP292 localized to the proximal region of the TZ (Fig. 2; Dean et al., 2016). These studies, supported by the proximity mapping of the ciliary base (Gupta et al., 2015), show that the different TZ modules occupy distinct domains within the TZ. Moreover, despite the similarities in protein organization within the TZ of different cilia there are clear differences between them. In the future, it will be important to map the localization of TZ proteins at the base of different cilia types of the same organism. Moreover, it will be interesting to study how disease mutations may affect the localization of these proteins in different cilia types.
Fig. 2.
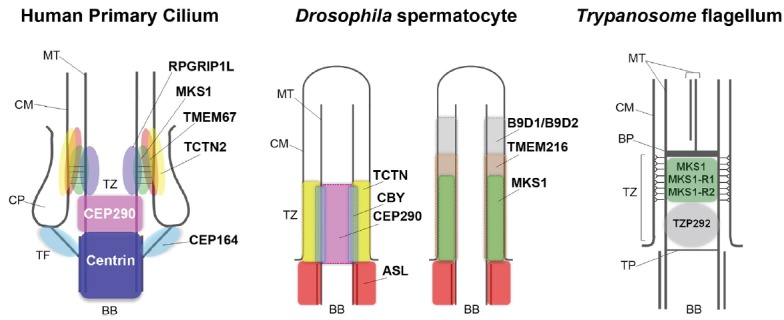
Localization of transition zone proteins.
The schemes represent the localization of transition zone and basal body components in human, Drosophila and Trypanosome cilia and flagella, and were adapted from Dean et al. (2016), Pratt et al. (2016), and Yang et al. (2015). MT – microtubules; CM – ciliary membrane; CP – ciliary pocket; TF – transition fibers; BB – basal body; TZ – transition zone; BP – basal plate; TP – terminal plate.
GENETIC INTERACTIONS GOVERNING TZ ASSEMBLY AND FUNCTION
Genetic interaction studies in C. elegans helped to identify distinct TZ protein modules with overlapping functions in the formation of TZ structures. These studies showed that single mutants of components of the MKS complex (MKS1, MKSR-1/B9D1, MKSR-2/B9D2, MKS3/TMEM67, MKS-6/CC2D2A, TMEM107, TMEM237, TMEM218) or the NPHP complex (NPHP1, NPHP4) presented very mild or no ciliary phenotypes. Mutants with multiple disrupted MKS or NPHP components are largely indistinguishable from corresponding single mutants. However, combining the disruption of a MKS complex component with the disruption of NPHP1 or NPHP4 severely disrupted ciliogenesis, and ciliary functions (Huang et al., 2011; Lambacher et al., 2016; Li et al., 2016; Williams et al., 2008; 2011). The TZ structure is severely disrupted in double mutants, which lack y-links and consequently the connection between MTs and the ciliary membrane. The TZ gating function was compromised, which was evidenced by the failure to exclude proteins such as RPI-2 and TRAM-1a from the cilium. A strong genetic interaction was also observed between MKS5/RPGRIP1L and components of both the MKS and NPHP modules which rely on it for their recruitment to the TZ (Huang et al., 2011; Jensen et al., 2015; Li et al., 2016; Williams et al., 2011). The C. elegans CEP290 homolog also plays a core role in TZ assembly being required for the localization of MKS module proteins (Li et al., 2016; Schouteden et al., 2015). Moreover, CEP290 interacts genetically with NPHP1 and NPHP4, but not with MKSR-2/B9D2, placing it in the MKS module in the worm (Li et al., 2016; Schouteden et al., 2015). Two genes coding for novel TZ proteins, CDKL1 and TMEM138, interact genetically with CEP290 but not with MKS or NPHP module components suggesting they are part of a new complex (Li et al., 2016). Finally, NPHP4 and BBS5 interacted genetically, with double mutants presenting more severe structural problems such as a reduction in y-link number than the individual mutants (Yee et al., 2015).
In the mouse, knocking-out genes coding for components of the MKS, NPHP and CEP290 modules affects ciliogenesis in a tissue-specific way (Chih et al, 2011; Cui et al., 2011; Garcia-Gonzalo et al., 2011; Goetz et al., 2017; Rachel et al., 2015; Ronquillo et al., 2016; Sang et al., 2011; Vierkotten et al., 2007; Weatherbee et al., 2009; Yee et al., 2015). Knocking-out MKS module genes (e.g. MKS1, TCTN1, TCTN2, CC2D2A, TMEM67) phenocopies Meckel-Gruber syndrome phenotypes such as abnormal patterning of the neural tube and limbs buds and left-right asymmetry problems. Consistently, cilia in the embryonic node, required for left-right symmetry breaking, are often lost in these mutants. On the other hand cilia in the limb buds can form but are reduced in number and present abnormal protein composition and response to Shh. Nevertheless, differences between each individual mutant suggest distinct roles for individual MKS module proteins. For instance, TCNT1 or TCTN2 null MEFS (mouse embryonic fibroblasts) can ciliate (Garcia-Gonzalo et al., 2011; Yee et al., 2015) whereas CC2D2A null MEFS fail to do so likely due to the lack of sub-distal appendages (Veleri et al., 2014). Regarding the NPHP module, an NPHP4 mutant mouse developed retinal degeneration but not kidney cysts nor severe ciliogenesis defects; males were infertile and presented sperm with reduced motility (Won et al., 2011). Similarly, NPHP1 null mice also presented retinal degeneration, a phenotype that was enhanced by the combined loss of AHI1 (Louie et al., 2010). CEP290 knock-out mice lack connecting cilia in photoreceptors and fail to mature motile ependymal cilia, which is consistent with their retinal degeneration and hydrocepahalus phenotypes (Rachel et al., 2015). NPHP5 null mice also fail to assemble connecting cilia whereas ciliogenesis is not affected in other tissues like the kidney (Ronquillo et al., 2016).
As in C. elegans, the MKS and NPHP modules interact genetically in the mouse. Mutating single components of the MKS complex (TCTN1, TCNT2, TMEM67 and CC2D2A), or the NPHP complex (NPHP1 and NPHP4), results in multiple developmental problems and abnormal cilia-dependent signaling. Combining mutations in two components of the same complex had no synergistic effect (Garcia-Gonzalo et al., 2011; Yee et al., 2015) but mutating a component of both complexes greatly exacerbated the ciliogenesis and cilia function defects (Yee et al., 2015). For example, TCTN1 null mice presented single digit polydactyly in the hindlimbs whereas NPHP1 and NPHP4 mutants showed no polydactyly. On the other hand, TCTN1 and NPHP1 or NPHP4 double mutants presented polydactyly in both fore and hindlimbs and higher numbers of extra digits. In accordance, limb fibroblasts derived from single TCTN1 mutant mice could ciliate in contrast to double TCTN1 and NPHP4 mutant cells which lacked primary cilia despite the BBs being able to anchor to the cell membrane. Moreover, ARL13B was absent from cilia in TCNT1 but not NPHP4 single mutant cells. Consistently, single NPHP4 null cells were able to respond to SAG (Smoothened agonist) by localizing SMO (smoothened) to the cilium and activating Shh signaling-regulated genes whereas TCTN1 mutant cells were not. These data show that although both MKS and NPHP complexes cooperate to assemble the TZ and build the cilium they have distinct roles in controlling the protein composition of the ciliary compartment (Yee et al., 2015). Genetic interactions were also detected between TZ and BBSome components in the mouse. Specifically, TCTN1 interacted with BBS1 and MKS1 with BBS4, with double mutants having exacerbated developmental problems largely due to the deregulation of Shh signaling (Goetz et al., 2017; Yee et al., 2015). Also, TCTN1 and BBS1 double mutant forelimb fibroblasts failed to ciliate (Yee et al., 2015). Interestingly, MKS1 interacted genetically with components of the IFT complex (IFT72) and the dynein subunit DYNC2H1. These interactions resulted in enhanced ciliogenesis failure and stronger Shh associated phenotypes (Goetz et al., 2017). Supporting the cooperation between the TZ and the IFT machinery, the combined silencing of B9D2 and IFT components Fleer or IFT52 in Danio rerio lead to enhanced cilia and planar cell polarity-related phenotypes (Zhao and Malicki, 2011). Several genetic interactions have also been reported for CEP290. A CEP290 hypomorphic allele enhances the severity of retinal degeneration caused by RPGR loss in the mouse (Rao et al., 2016). Likewise, CEP290 and RPGR interact genetically in zebrafish (Gerner et al., 2010). Also in the mouse, CEP290 null phenotypes were exacerbated by the loss of MKKS/BBS6, a chaperone required for BBSome assembly. Moreover, a CEP290 hypomorphic allele interacted genetically with BBS4 with double mutants presenting faster retinal degeneration and increased weight gain than single mutants (Zhang et al., 2014). In accordance, NPHP5 and CEP290 interact physically with several BBSome subunits and are required to maintain BBSome integrity and its trafficking, as well as of its cargos, to the cilium compartment (Barbelanne et al., 2015; Zhang et al., 2014). Furthermore, CEP290 interacts physically and genetically with NPHP5 and CC2D2A in the zebrafish. Specifically, the combined loss of function of CEP290 and CC2D2A exacerbates pronephric cyst formation (Gorden et al., 2008; Schäfer et al., 2008). Finally, CC2D2A interacts genetically and physically with NINL in the zebrafish. The combined depletion of NINL and CC2D2A increased the severity of the multiple cilia related phenotypes caused by individual knock-downs (photoreceptor outer segment loss, mis-localization of opsins, problems with vesicular trafficking and pronephric cysts). This study proposed a model in which CC2D2A provides a docking site at the ciliary base for cargo transported by NINL through the action of its interactors dynein and MICAL3, which works with RAB8 in vesicular trafficking (Bachmann-Gagescu et al., 2015).
Interestingly, despite the conservation in the TZ structure and core molecular composition, there are differences between species. For example, Drosophila seems to lack both MKS5/RPGRIP1L and the NPHP module components NPHP1 and NPHP4. In Drosophila single MKS1 and B9D1 mutants, as well as in B9D2 and TCTN double mutants, the remaining components of the MKS module fail to localize to the TZ whereas the localizations of CEP290 and CBY are unperturbed (Pratt et al., 2016; Vieillard et al., 2016). These data support the evolutionarily conserved interdependence of MKS module proteins for their localization to the TZ. Interestingly, the loss of the MKS module from the TZ in these mutants did not severely affect the biogenesis and functions of sensory cilia and flagella (Pratt et al., 2016; Vieillard et al., 2016). On the other hand, CEP290 and CBY mutants are uncoordinated and have reduced fertility. Importantly, y-links are present in the TZ of MKS1 and B9D2/TCTN mutants suggesting the MKS complex is not required for their assembly in the fly (Pratt et al., 2016; Vieillard et al., 2016). Also in Drosophila, a strong genetic interaction was observed between CBY and DILA in the formation of the TZ and ciliogenesis. Single CBY and DILA mutants are uncoordinated due to a disorganization of sensory cilia. Nevertheless BB docking was unaffected and TZs had little (in the case of CBY; Enjolras et al., 2012) or no ultrastructual problems (in the case of DILA; Ma and Jarman, 2011). In double CBY and DILA mutants, however, ciliogenesis was severely disrupted. In sensory neurons, centrioles failed to dock at the membrane, the TZ did not form and its components such as CEP290 and MKS1 were absent from dendrite tips. In males, sperm flagella formation was also severely affected. In spermatocytes and spermatids MKS1 and B9D1 were lost from the tip of BBs and CEP290 was severely reduced. These data show that CBY and DILA cooperate for the recruitment of key proteins to the TZ and for its formation both in primary and motile cilia in the fly (Vieillard et al., 2016).
Collectively, these studies support the data discussed in the previous sections showing that the different TZ complexes cooperate with each other for the assembly of the TZ and the cilium, as well as with multiple pathways for the correct delivery of cargo to the cilium. Moreover, genetic interaction studies will continue to significantly further our knowledge on the clinical severity spectra caused by ciliopathy mutations.
CILIARY LIPID GATE
Multiple studies have established that the constitutive and regulated localization of proteins to the cilium compartment is highly dependent on a TZ gating function (reviewed in Reiter et al., 2012). Recent studies have also shown that the TZ acts as a lipid gate. Indeed, the TZ fulfils a conserved role in maintaining the ciliary membrane phosphoinositide (PI) composition. In mammalian cells the inositol polyphosphate-5-phosphatase E (INPP5E) localizes in the cilium where it hydrolyses its substrates PI(4,5)P3 and PI(4,5)P2 generating PI(4)P (Fig. 1; Garcia-Gonzalo et al., 2015). As a result, the activity of INPP5E restricts PI(4,5)P3 and PI(4,5)P2 to the TZ where they co-localize with TCTN1. On the other hand, PI(4)P is present throughout the ciliary membrane determining the localization of proteins involved in the Shh signaling pathway (Chávez et al., 2015; Dyson et al., 2016; Garcia-Gonzalo et al., 2015). Lack of INPP5E activity leads to the accumulation of PI(4,5)P2 as well as that of the PI(4,5)P2-binding protein TULP3 and its interactors IFT122, IFT139, IFT140 and GPR161 (negative regulators of the Shh pathway) in the cilium (Chávez et al., 2015; Garcia-Gonzalo et al., 2015). GPR161 accumulation leads to an increase in the production of cAMP with the consequent activation of PKA, a repressor of Shh signaling, and GLI3 repressor formation (Chávez et al., 2015). In INPP5E null cells, SMO still localizes to the cilium, albeit reduced, upon activation of the Shh pathway with SAG. However, GLI2 and GLI3 fail to accumulate at the ciliary tip and Shh target genes are not activated (Chávez et al., 2015; Dyson et al., 2016; Garcia-Gonzalo et al., 2015). INPP5E mutations cause Joubert and MORM (mental retardation, truncal obesity, retinal dystrophy and micropenis) syndromes and knocking out Inpp5e in mice phenocopies the effect of Inpp5e mutations in Joubert syndrome. These mice present phenotypes such as polydactyly, which are consistent with INPP5E’s role in Shh signaling regulation (Dyson et al., 2016). Importantly, the ciliary localization of INPP5E depends on the MKS complex proteins TCTN1, TMEM231, B9D1, and MKS1 (Garcia-Gonzalo et al., 2011; Goetz et al., 2017; Roberson et al., 2015; Slaats et al., 2015). Moreover, the activation of the Shh pathway modulates the levels of PI(4,5)P3 and PI(4,5)P2 at the TZ. Upon SAG treatment the levels of these PIs increases both in WT and INPP5E null MEFS being the levels higher in the null background. Also, the TZ localization of MKS1, TCTN1, B9D1 and TMEM231 was significantly reduced in INPP5E null cells upon SAG treatment. This might be associated with the reduction of SEPT2 at the ciliary base upon SAG treatment. Septins are required for the localization of MKS module proteins (Chih et al., 2011) and interact with PIs such as PI(4,5)P3 and PI(4,5)P2 which regulate their polymerization (Dyson et al., 2016). Similar results were observed in Drosophila where dINPP5E localizes to the TZ and regulates the PI composition at the ciliary base and protein trafficking to the ciliary membrane. Loss of dINPP5e increased PI(4,5)P2 levels at the ciliary base of chordotonal cilia, the accumulation of dTULP (TULP3 homolog) in these cilia and the abnormal localization of certain proteins such as IAV and NOMPC, both required for hearing. As a consequence, dINPP5e mutant flies have impaired hearing (Park et al., 2015). In C. elegans, PI(4,5)P2 is enriched at the ciliary base (periciliary membrane and BB regions) but it is significantly reduced at the TZ and excluded from the cilium compartment. In MKS5/RPGRIP1L mutants which have a disrupted TZ, PI(4,5)P2 is no longer excluded from the ciliary membrane, again suggesting a role for the TZ in maintaining its lipid composition (Jensen et al., 2015). This exclusion of PI(4,5)P2 from the cilium is expected to have implications for protein trafficking to this organelle also in worms.
Multiple studies conducted decades ago in different model organisms have shown the ciliary membrane lipid composition to be different from that of the plasma membrane, and similar to that of lipid rafts (reviewed in Emmer et al., 2010). This special lipid composition, as evidenced by the studies discussed here, has an impact on protein targeting to the cilium and consequently on cilia-related signaling. We are still beginning to understand how the specialized ciliary lipid content is achieved and maintained. Moreover, further studies will be required to fully grasp how the regulated modulation of lipid species at the TZ and ciliary membrane is determined by and impacts on TZ proteins.
CONCLUSION
The TZ is a specialized domain present at the base of the axoneme and is characterized by structures of poorly characterized composition that connect the axonemal MTs to the ciliary membrane. Together with the transition fibers and septins, the TZ works as a gate that regulates the protein and lipid composition of the ciliary compartment. How this gate is formed and fulfills its functions is not entirely understood. Given the TZ roles in ciliary functions, and its involvement in human disease, significant efforts have been made to identify its components and understand how they work together to assemble the TZ and the cilium. Multiple studies led to identification of at least three conserved protein modules (MKS, NPHP, CEP290) composed of multiple proteins that cooperate for TZ formation and function, occupy distinct domains within it and have distinct roles in terms of protein sorting to the cilium compartment. Moreover, TZ proteins can localize to different structures (e.g. centrosome, centriolar satellites, cell junctions; Table 1) and interact physically and genetically with components of different machineries (e.g. ESCRT, BBSome, IFT) with which they seem to work for the efficient delivery of cargo to the cilium and other ciliary processes. In the future it will be important to understand the tissue-specific roles of the TZ and its components, what are their interactors and where they localize in the context of different cilia types which might have different gates. Excitingly, the recent technology developments in terms of genetic manipulation, protein interaction screening, and super-resolution imaging will allow us to study cilia and its sub-compartments, such as the TZ, in different ciliated cell types. Genetic interaction studies, protein-protein interaction screens, and protein localization mapping in different genetic backgrounds and conditions, will considerably contribute to elucidate the mechanisms of assembly, maintenance and function of different cilia types.
ACKNOWLEDGMENTS
We would like to thank Dr. Johnny Tkach, Dr. Mikhail Bashkurov and Dr. Ladan Gheiratmand and Dr. Helena Soares for the critical reading of the manuscript. We apologise to our colleagues whose work we could not discuss due to space limitations.
REFERENCES
- Abdelhamed Z.A., Natarajan S., Wheway G., Inglehearn C.F., Toomes C., Johnson C.A., Jagger D.J. The Meckel-Gruber syndrome protein TMEM67 controls basal body positioning and epithelial branching morphogenesis in mice via the non-canonical Wnt pathway. Dis Model Mech. 2015;8:527–541. doi: 10.1242/dmm.019083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arts H.H., Doherty D., van Beersum S.E., Parisi M.A., Letteboer S.J., Gorden N.T., Peters T.A., Märker T., Voesenek K., Kartono A., et al. Mutations in the gene encoding the basal body protein RPGRIP1L, a nephrocystin-4 interactor, cause Joubert syndrome. Nat Genet. 2007;39:882–888. doi: 10.1038/ng2069. [DOI] [PubMed] [Google Scholar]
- Awata J., Takada S., Standley C., Lechtreck K.F., Bellvé K.D., Pazour G.J., Fogarty K.E., Witman G.B. NPHP4 controls ciliary trafficking of membrane proteins and large soluble proteins at the transition zone. J Cell Sci. 2014;127:4714–4727. doi: 10.1242/jcs.155275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachmann-Gagescu R., Phelps I.G., Stearns G., Link B.A., Brockerhoff S.E., Moens C.B., Doherty D. The ciliopathy gene cc2d2a controls zebrafish photoreceptor outer segment development through a role in Rab8-dependent vesicle trafficking. Hum Mol Genet. 2011;20:4041–4055. doi: 10.1093/hmg/ddr332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachmann-Gagescu R., Dona M., Hetterschijt L., Tonnaer E., Peters T., de Vrieze E., Mans D.A., van Beersum S.E., Phelps I.G., Arts H.H., et al. The ciliopathy protein CC2D2A associates with NINL and functions in RAB8-MICAL3-regulated vesicle trafficking. PLoS Genet. 2015;11:e1005575. doi: 10.1371/journal.pgen.1005575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbelanne M., Hossain D., Chan D.P., Peränen J., Tsang W.Y. Nephrocystin proteins NPHP5 and Cep290 regulate BBSome integrity, ciliary trafficking and cargo delivery. Hum Mol Genet. 2015;24:2185–2200. doi: 10.1093/hmg/ddu738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boldt K., Mans D.A., Won J., van Reeuwijk J., Vogt A., Kinkl N., Letteboer S.J., Hicks W.L., Hurd R.E., Naggert J.K., et al. Disruption of intraflagellar protein transport in photoreceptor cilia causes Leber congenital amaurosis in humans and mice. J Clin Invest. 2011;121:2169–2180. doi: 10.1172/JCI45627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chávez M., Ena S., Van Sande J., de Kerchove d’Exaerde A., Schurmans S., Schiffmann S.N. Modulation of ciliary phosphoinositide content regulates trafficking and sonic hedgehog signaling output. Dev Cell. 2015;34:338–350. doi: 10.1016/j.devcel.2015.06.016. [DOI] [PubMed] [Google Scholar]
- Chih B., Liu P., Chinn Y., Chalouni C., Komuves L.G., Hass P.E., Sandoval W., Peterson A.S. A ciliopathy complex at the transition zone protects the cilia as a privileged membrane domain. Nat Cell Biol. 2011;14:61–72. doi: 10.1038/ncb2410. [DOI] [PubMed] [Google Scholar]
- Christopher K.J., Wang B., Kong Y., Weatherbee S.D. Forward genetics uncovers transmembrane protein 107 as a novel factor required for ciliogenesis and sonic hedgehog signaling. Dev Biol. 2012;368:382–392. doi: 10.1016/j.ydbio.2012.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craige B., Tsao C.C., Diener D.R., Hou Y., Lechtreck K.F., Rosenbaum J.L., Witman G.B. CEP290 tethers flagellar transition zone microtubules to the membrane and regulates flagellar protein content. J Cell Biol. 2010;190:927–940. doi: 10.1083/jcb.201006105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui C., Chatterjee B., Francis D., Yu Q., SanAgustin J.T., Francis R., Tansey T., Henry C., Wang B., Lemley B., et al. Disruption of Mks1 localization to the mother centriole causes cilia defects and developmental malformations in Meckel-Gruber syndrome. Dis Model Mech. 2011;4:43–56. doi: 10.1242/dmm.006262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damerla R.R., Cui C., Gabriel G.C., Liu X., Craige B., Gibbs B.C., Francis R., Li Y., Chatterjee B., San Agustin J.T., et al. Novel Jbts17 mutant mouse model of Joubert syndrome with cilia transition zone defects and cerebellar and other ciliopathy related anomalies. Hum Mol Genet. 2015;24:3994–4005. doi: 10.1093/hmg/ddv137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawe H.R., Smith U.M., Cullinane A.R., Gerrelli D., Cox P., Badano J.L., Blair-Reid S., Sriram N., Katsanis N., Attie-Bitach T., et al. The Meckel-Gruber Syndrome proteins MKS1 and meckelin interact and are required for primary cilium formation. Hum Mol Genet. 2007;16:173–186. doi: 10.1093/hmg/ddl459. [DOI] [PubMed] [Google Scholar]
- Dean S., Moreira-Leite F., Varga V., Gull K. Cilium transition zone proteome reveals compartmentalization and differential dynamics of ciliopathy complexes. Proc Natl Acad Sci USA. 2016;113:E5135–5143. doi: 10.1073/pnas.1604258113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delous M., Baala L., Salomon R., Laclef C., Vierkotten J., Tory K., Golzio C., Lacoste T., Besse L., Ozilou C., et al. The ciliary gene RPGRIP1L is mutated in cerebello-oculo-renal syndrome (Joubert syndrome type B) and Meckel syndrome. Nat Genet. 2007;39:875–881. doi: 10.1038/ng2039. [DOI] [PubMed] [Google Scholar]
- Delous M., Hellman N.E., Gaudé H.M., Silbermann F., Le Bivic A., Salomon R., Antignac C., Saunier S. Nephrocystin-1 and nephrocystin-4 are required for epithelial morphogenesis and associate with PALS1/PATJ and Par6. Hum Mol Genet. 2009;18:4711–4723. doi: 10.1093/hmg/ddp434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- den Hollander A.I., Koenekoop R.K., Mohamed M.D., Arts H.H., Boldt K., Towns K.V., Sedmak T., Beer M., Nagel-Wolfrum K., McKibbin M., et al. Mutations in LCA5, encoding the ciliary protein lebercilin, cause Leber congenital amaurosis. Nat Genet. 2007;39:889–895. doi: 10.1038/ng2066. [DOI] [PubMed] [Google Scholar]
- Diener D.R., Lupetti P., Rosenbaum J.L. Proteomic analysis of isolated ciliary transition zones reveals the presence of ESCRT proteins. Curr Biol. 2015;25:379–384. doi: 10.1016/j.cub.2014.11.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dowdle W.E., Robinson J.F., Kneist A., Sirerol-Piquer M.S., Frints S.G., Corbit K.C., Zaghloul N.A., van Lijnschoten G., Mulders L., Verver D.E., et al. Disruption of a ciliary B9 protein complex causes Meckel syndrome. Am J Hum Genet. 2011;89:94–110. doi: 10.1016/j.ajhg.2011.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dyson J.M., Conduit S.E., Feeney S.J., Hakim S., Di Tommaso T., Fulcher A.J., Sriratana A., Ramm G., Horan K.A., Gurung R., et al. INPP5E regulates phosphoinositide-dependent cilia transition zone function. J Cell Biol. 2016;216:247–263. doi: 10.1083/jcb.201511055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emmer B.T., Maric D., Engman D.M. Molecular mechanisms of protein and lipid targeting to ciliary membranes. J Cell Sci. 2010;123:529–536. doi: 10.1242/jcs.062968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enjolras C., Thomas J., Chhin B., Cortier E., Duteyrat J.L., Soulavie F., Kernan M.J., Laurençon A., Durand B. Drosophila chibby is required for basal body formation and ciliogenesis but not for Wg signaling. J Cell Biol. 2012;197:313–325. doi: 10.1083/jcb.201109148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Gonzalo F.R., Corbit K.C., Sirerol-Piquer M.S., Ramaswami G., Otto E.A., Noriega T.R., Seol A.D., Robinson J.F., Bennett C.L., Josifova D.J., et al. A transition zone complex regulates mammalian ciliogenesis and ciliary membrane composition. Nat Genet. 2011;43:776–784. doi: 10.1038/ng.891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Gonzalo F.R., Phua S.C., Roberson E.C., Garcia G., 3rd, Abedin M., Schurmans S., Inoue T., Reiter J.F. Phosphoinositides regulate ciliary protein trafficking to modulate hedgehog signaling. Dev Cell. 2015;34:400–409. doi: 10.1016/j.devcel.2015.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gakovic M., Shu X., Kasioulis I., Carpanini S., Moraga I., Wright A.F. The role of RPGR in cilia formation and actin stability. Hum Mol Genet. 2011;20:4840–4850. doi: 10.1093/hmg/ddr423. [DOI] [PubMed] [Google Scholar]
- Gerner M., Haribaskar R., Pütz M., Czerwitzki J., Walz G., Schäfer T. The retinitis pigmentosa GTPase regulator interacting protein 1 (RPGRIP1) links RPGR to the nephronophthisis protein network. Kidney Int. 2010;77:891–896. doi: 10.1038/ki.2010.27. [DOI] [PubMed] [Google Scholar]
- Goetz S.C., Bangs F., Barrington C.L., Katsanis N., Anderson K.V. The Meckel syndrome- associated protein MKS1 functionally interacts with components of the BBSome and IFT complexes to mediate ciliary trafficking and hedgehog signaling. PLoS One. 2017;12:e0173399. doi: 10.1371/journal.pone.0173399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorden N.T., Arts H.H., Parisi M.A., Coene K.L., Letteboer S.J., van Beersum S.E., Mans D.A., Hikida A., Eckert M., Knutzen D., et al. CC2D2A is mutated in Joubert syndrome and interacts with the ciliopathy-associated basal body protein CEP290. Am J Hum Genet. 2008;83:559–571. doi: 10.1016/j.ajhg.2008.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta G.D., Coyaud É, Gonçalves J., Mojarad B.A., Liu Y., Wu Q., Gheiratmand L., Comartin D., Tkach J.M., Cheung S.W., et al. A dynamic protein interaction landscape of the human centrosome-cilium interface. Cell. 2015;163:1484–1499. doi: 10.1016/j.cell.2015.10.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong D.H., Pawlyk B.S., Shang J., Sandberg M.A., Berson E.L., Li T. A retinitis pigmentosa GTPase regulator (RPGR)-deficient mouse model for X-linked retinitis pigmentosa (RP3) Proc Natl Acad Sci USA. 2000;97:3649–3654. doi: 10.1073/pnas.060037497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong D.H., Yue G., Adamian M., Li T. Retinitis pigmentosa GTPase regulator (RPGRr)-interacting protein is stably associated with the photoreceptor ciliary axoneme and anchors RPGR to the connecting cilium. J Biol Chem. 2001;276:12091–12099. doi: 10.1074/jbc.M009351200. [DOI] [PubMed] [Google Scholar]
- Hsiao Y.C., Tong Z.J., Westfall J.E., Ault J.G., Page-McCaw P.S., Ferland R.J. Ahi1, whose human ortholog is mutated in Joubert syndrome, is required for Rab8a localization, ciliogenesis and vesicle trafficking. Hum Mol Genet. 2009;18:3926–3941. doi: 10.1093/hmg/ddp335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang L., Szymanska K., Jensen V.L., Janecke A.R., Innes A.M., Davis E.E., Frosk P., Li C., Willer J.R., Chodirker B.N., et al. TMEM237 is mutated in individuals with a Joubert syndrome related disorder and expands the role of the TMEM family at the ciliary transition zone. Am J Hum Genet. 2011;89:713–730. doi: 10.1016/j.ajhg.2011.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikawa H., Marshall W.F. Intraflagellar transport and ciliary dynamics. Cold Spring Harb Perspect Biol. 2017;9 doi: 10.1101/cshperspect.a021998. pii: a021998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen V.L., Li C., Bowie R.V., Clarke L., Mohan S., Blacque O.E., Leroux M.R. Formation of the transition zone by Mks5/Rpgrip1L establishes a ciliary zone of exclusion (CIZE) that compartmentalises ciliary signalling proteins and controls PIP2 ciliary abundance. EMBO J. 2015;34:2537–56. doi: 10.15252/embj.201488044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin H., White S.R., Shida T., Schulz S., Aguiar M., Gygi S.P., Bazan J.F., Nachury M.V. The conserved Bardet-Biedl syndrome proteins assemble a coat that traffics membrane proteins to cilia. Cell. 2010;141:1208–1219. doi: 10.1016/j.cell.2010.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khanna H., Hurd T.W., Lillo C., Shu X., Parapuram S.K., He S., Akimoto M., Wright A.F., Margolis B., Williams D.S., et al. RPGR-ORF15, which is mutated in retinitis pigmentosa, associates with SMC1, SMC3, and microtubule transport proteins. J Biol Chem. 2005;280:33580–33587. doi: 10.1074/jbc.M505827200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J., Krishnaswami S.R., Gleeson J.G. CEP290 interacts with the centriolar satellite component PCM-1 and is required for Rab8 localization to the primary cilium. Hum Mol Genet. 2008;17:3796–3805. doi: 10.1093/hmg/ddn277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klinger M., Wang W., Kuhns S., Bärenz F., Dräger-Meurer S., Pereira G., Gruss O.J. The novel centriolar satellite protein SSX2IP targets Cep290 to the ciliary transition zone. Mol Biol Cell. 2014;25:495–507. doi: 10.1091/mbc.E13-09-0526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambacher N.J., Bruel A.L., van Dam T.J., Szymańska K., Slaats G.G., Kuhns S., McManus G.J., Kennedy J.E., Gaff K., Wu K.M., et al. TMEM107 recruits ciliopathy proteins to subdomains of the ciliary transition zone and causes Joubert syndrome. Nat Cell Biol. 2016;18:122–131. doi: 10.1038/ncb3273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lessieur E.M., Fogerty J., Gaivin R.J., Song P., Perkins B.D. The ciliopathy gene ahi1 is required for zebrafish cone photoreceptor outer segment morphogenesis and survival. Invest Ophthalmol Vis Sci. 2017;58:448–460. doi: 10.1167/iovs.16-20326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C., Jensen V.L., Park K., Kennedy J., Garcia-Gonzalo F.R., Romani M., De Mori R., Bruel A.L., Gaillard D., Doray B., et al. MKS5 and CEP290 dependent assembly pathway of the ciliary transition zone. PLoS Biol. 2016;14:e1002416. doi: 10.1371/journal.pbio.1002416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J.H., Silhavy J.L., Lee J.E., Al-Gazali L., Thomas S., Davis E.E., Bielas S.L., Hill K.J., Iannicelli M., Brancati F., et al. Evolutionarily assembled cis-regulatory module at a human ciliopathy locus. Science. 2012;335:966–999. doi: 10.1126/science.1213506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long H., Zhang F., Xu N., Liu G., Diener D.R., Rosenbaum J.L., Huang K. Comparative analysis of ciliary membranes and ectosomes. Curr Biol. 2016;26:3327–3335. doi: 10.1016/j.cub.2016.09.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louie C.M., Caridi G., Lopes V.S., Brancati F., Kispert A., Lancaster M.A., Schlossman A.M., Otto E.A., Leitges M., Gröne H.J., et al. AHI1 is required for photoreceptor outer segment development and is a modifier for retinal degeneration in nephronophthisis. Nat Genet. 2010;42:175–180. doi: 10.1038/ng.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma L., Jarman A.P. Dilatory is a Drosophila protein related to AZI1 (CEP131) that is located at the ciliary base and required for cilium formation. J Cell Sci. 2011;124:2622–2630. doi: 10.1242/jcs.084798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahuzier A., Gaudé H.M., Grampa V., Anselme I., Silbermann F., Leroux-Berger M., Delacour D., Ezan J., Montcouquiol M., Saunier S., et al. Dishevelled stabilization by the ciliopathy protein Rpgrip1l is essential for planar cell polarity. J Cell Biol. 2012;198:927–940. doi: 10.1083/jcb.201111009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchison H.M., Valente E.M. Motile and non-motile cilia in human pathology: from function to phenotypes. J Pathol. 2017;241:294–309. doi: 10.1002/path.4843. [DOI] [PubMed] [Google Scholar]
- Mollet G., Silbermann F., Delous M., Salomon R., Antignac C., Saunier S. Characterization of the nephrocystin/nephrocystin-4 complex and subcellular localization of nephrocystin-4 to primary cilia and centrosomes. Hum Mol Genet. 2005;14:645–656. doi: 10.1093/hmg/ddi061. [DOI] [PubMed] [Google Scholar]
- Otto E.A., Loeys B., Khanna H., Hellemans J., Sudbrak R., Fan S., Muerb U., O’Toole J.F., Helou J., Attanasio M., et al. Nephrocystin-5, a ciliary IQ domain protein, is mutated in Senior-Loken syndrome and interacts with RPGR and calmodulin. Nat Genet. 2005;37:282–288. doi: 10.1038/ng1520. [DOI] [PubMed] [Google Scholar]
- Park J., Lee N., Kavoussi A., Seo J.T., Kim C.H., Moon S.J. Ciliary Phosphoinositide Regulates Ciliary Protein Trafficking in Drosophila. Cell Rep. 2015;13:2808–2816. doi: 10.1016/j.celrep.2015.12.009. [DOI] [PubMed] [Google Scholar]
- Patil H., Tserentsoodol N., Saha A., Hao Y., Webb M., Ferreira P.A. Selective loss of RPGRIP1-dependent ciliary targeting of NPHP4, RPGR and SDCCAG8 underlies the degeneration of photoreceptor neurons. Cell Death Dis. 2012;3:e355. doi: 10.1038/cddis.2012.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pratt M.B., Titlow J.S., Davis I., Barker A.R., Dawe H.R., Raff J.W., Roque H. Drosophila sensory cilia lacking MKS proteins exhibit striking defects in development but only subtle defects in adults. J Cell Sci. 2016;129:3732–3743. doi: 10.1242/jcs.194621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rachel R.A., Yamamoto E.A., Dewanjee M.K., May-Simera H.L., Sergeev Y.V., Hackett A.N., Pohida K., Munasinghe J., Gotoh N., Wickstead B., et al. CEP290 alleles in mice disrupt tissue-specific cilia biogenesis and recapitulate features of syndromic ciliopathies. Hum Mol Genet. 2015;24:3775–3791. doi: 10.1093/hmg/ddv123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao K.N., Zhang W., Li L., Ronquillo C., Baehr W., Khanna H. Ciliopathy-associated protein CEP290 modifies the severity of retinal degeneration due to loss of RPGR. Hum Mol Genet. 2016;25:2005–2012. doi: 10.1093/hmg/ddw075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiter J.F., Blacque O.E., Leroux M.R. The base of the cilium: roles for transition fibres and the transition zone in ciliary formation, maintenance and compartmentalization. EMBO Rep. 2012;13:608–618. doi: 10.1038/embor.2012.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberson E.C., Dowdle W.E., Ozanturk A., Garcia-Gonzalo F.R., Li C., Halbritter J., Elkhartoufi N., Porath J.D., Cope H., Ashley-Koch A., et al. TMEM231, mutated in orofaciodigital and Meckel syndromes, organizes the ciliary transition zone. J Cell Biol. 2015;209:129–142. doi: 10.1083/jcb.201411087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronquillo C.C., Hanke-Gogokhia C., Revelo M.P., Frederick J.M., Jiang L., Baehr W. Ciliopathy-associated IQCB1/NPHP5 protein is required for mouse photoreceptor outer segment formation. FASEB J. 2016;30:3400–3412. doi: 10.1096/fj.201600511R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roux K.J., Kim D.I., Raida M., Burke B. A promiscuous biotin ligase fusion protein identifies proximal and interacting proteins in mammalian cells. J Cell Biol. 2012;196:801–810. doi: 10.1083/jcb.201112098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sang L., Miller J.J., Corbit K.C., Giles R.H., Brauer M.J., Otto E.A., Baye L.M., Wen X., Scales S.J., Kwong M., et al. Mapping the NPHP-JBTS-MKS protein network reveals ciliopathy disease genes and pathways. Cell. 2011;145:513–528. doi: 10.1016/j.cell.2011.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sayer J.A., Otto E.A., O’Toole J.F., Nurnberg G., Kennedy M.A., Becker C., Hennies H.C., Helou J., Attanasio M., Fausett B.V., et al. The centrosomal protein nephrocystin-6 is mutated in Joubert syndrome and activates transcription factor ATF4. Nat Genet. 2006;38:674–681. doi: 10.1038/ng1786. [DOI] [PubMed] [Google Scholar]
- Schäfer T., Pütz M., Lienkamp S., Ganner A., Bergbreiter A., Ramachandran H., Gieloff V., Gerner M., Mattonet C., Czarnecki P.G., et al. Genetic and physical interaction between the NPHP5 and NPHP6 gene products. Hum Mol Genet. 2008;17:3655–3662. doi: 10.1093/hmg/ddn260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schouteden C., Serwas D., Palfy M., Dammermann A. The ciliary transition zone functions in cell adhesion but is dispensable for axoneme assembly in C. elegans. J Cell Biol. 2015;210:35–44. doi: 10.1083/jcb.201501013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaheen R., Almoisheer A., Faqeih E., Babay Z., Monies D., Tassan N., Abouelhoda M., Kurdi W., Al Mardawi E., Khalil M.M., et al. Identification of a novel MKS locus defined by TMEM107 mutation. Hum Mol Genet. 2015;24:5211–5218. doi: 10.1093/hmg/ddv242. [DOI] [PubMed] [Google Scholar]
- Shu X., Fry A.M., Tulloch B., Manson F.D., Crabb J.W., Khanna H., Faragher A.J., Lennon A., He S., Trojan P., et al. RPGR ORF15 isoform co-localizes with RPGRIP1 at centrioles and basal bodies and interacts with nucleophosmin. Hum Mol Genet. 2005;14:1183–1197. doi: 10.1093/hmg/ddi129. [DOI] [PubMed] [Google Scholar]
- Shylo N.A., Christopher K.J., Iglesias A., Daluiski A., Weatherbee S.D. TMEM107 is a critical regulator of ciliary protein composition and is mutated in Orofaciodigital syndrome. Hum Mutat. 2016;37:155–159. doi: 10.1002/humu.22925. [DOI] [PubMed] [Google Scholar]
- Slaats G.G., Isabella C.R., Kroes H.Y., Dempsey J.C., Gremmels H., Monroe G.R., Phelps I.G., Duran K.J., Adkins J., Kumar S.A., et al. MKS1 regulates ciliary INPP5E levels in Joubert syndrome. J Med Genet. 2016;53:62–72. doi: 10.1136/jmedgenet-2015-103250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slanchev K., Pütz M., Schmitt A., Kramer-Zucker A., Walz G. Nephrocystin-4 is required for pronephric duct-dependent cloaca formation in zebrafish. Hum Mol Genet. 2011;20:3119–3128. doi: 10.1093/hmg/ddr214. [DOI] [PubMed] [Google Scholar]
- Thomas S., Legendre M., Saunier S., Bessières B., Alby C., Bonnière M., Toutain A., Loeuillet L., Szymanska K., Jossic F., et al. TCTN3 mutations cause Mohr-Majewski syndrome. Am J Hum Genet. 2012;91:372–378. doi: 10.1016/j.ajhg.2012.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valente E.M., Logan C.V., Mougou-Zerelli S., Lee J.H., Silhavy J.L., Brancati F., Iannicelli M., Travaglini L., Romani S., Illi B., et al. Mutations in TMEM216 perturb ciliogenesis and cause Joubert, Meckel and related syndromes. Nat Genet. 2010;42:619–625. doi: 10.1038/ng.594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veleri S., Manjunath S.H., Fariss R.N., May-Simera H., Brooks M., Foskett T.A., Gao C., Longo T.A., Liu P., Nagashima K., et al. Ciliopathy-associated gene Cc2d2a promotes assembly of subdistal appendages on the mother centriole during cilia biogenesis. Nat Commun. 2014;5:4207. doi: 10.1038/ncomms5207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vierkotten J., Dildrop R., Peters T., Wang B., Rüther U. Ftm is a novel basal body protein of cilia involved in Shh signalling. Development. 2007;134:2569–2577. doi: 10.1242/dev.003715. [DOI] [PubMed] [Google Scholar]
- Vieillard J., Paschaki M., Duteyrat J.L., Augière C., Cortier E., Lapart J.A., Thomas J., Durand B. Transition zone assembly and its contribution to axoneme formation in Drosophila male germ cells. J Cell Biol. 2016;214:875–889. doi: 10.1083/jcb.201603086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W.J., Tay H.G., Soni R., Perumal G.S., Goll M.G., Macaluso F.P., Asara J.M., Amack J.D., Tsou M.F. CEP162 is an axoneme-recognition protein promoting ciliary transition zone assembly at the cilia base. Nat Cell Biol. 2013;15:591–601. doi: 10.1038/ncb2739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weatherbee S.D., Niswander L.A., Anderson K.V. A mouse model for Meckel syndrome reveals Mks1 is required for ciliogenesis and Hedgehog signaling. Hum Mol Genet. 2009;18:4565–4575. doi: 10.1093/hmg/ddp422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams C.L., Winkelbauer M.E., Schafer J.C., Michaud E.J., Yoder B.K. Functional redundancy of the B9 proteins and nephrocystins in Caenorhabditis elegans ciliogenesis. Mol Biol Cell. 2008;19:2154–2168. doi: 10.1091/mbc.E07-10-1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams C.L., Li C., Kida K., Inglis P.N., Mohan S., Semenec L., Bialas N.J., Stupay R.M., Chen N., Blacque O.E., et al. MKS and NPHP modules cooperate to establish basal body/transition zone membrane associations and ciliary gate function during ciliogenesis. J Cell Biol. 2011;192:1023–1041. doi: 10.1083/jcb.201012116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Won J., Gifford E., Smith R.S., Yi H., Ferreira P.A., Hicks W.L., Li T., Naggert J.K., Nishina P.M. RPGRIP1 is essential for normal rod photoreceptor outer segment elaboration and morphogenesis. Hum Mol Genet. 2009;18:4329–4339. doi: 10.1093/hmg/ddp385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Won J., Marín de Evsikova C., Smith R.S., Hicks W.L., Edwards M.M., Longo-Guess C., Li T., Naggert J.K., Nishina P.M. NPHP4 is necessary for normal photoreceptor ribbon synapse maintenance and outer segment formation, and for sperm development. Hum Mol Genet. 2011;20:482–496. doi: 10.1093/hmg/ddq494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang T.T., Su J., Wang W.J., Craige B., Witman G.B., Tsou M.F., Liao J.C. Superresolution pattern recognition reveals the architectural map of the ciliary transition zone. Sci Rep. 2015;5:14096. doi: 10.1038/srep14096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yee L.E., Garcia-Gonzalo F.R., Bowie R.V., Li C., Kennedy J.K., Ashrafi K., Blacque O.E., Leroux M.R., Reiter J.F. Conserved genetic interactions between ciliopathy complexes cooperatively support ciliogenesis and ciliary signaling. PLoS Genet. 2015;11:e1005627. doi: 10.1371/journal.pgen.1005627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y., Seo S., Bhattarai S., Bugge K., Searby C.C., Zhang Q., Drack A.V., Stone E.M., Sheffield V.C. BBS mutations modify phenotypic expression of CEP290-related ciliopathies. Hum Mol Genet. 2014;23:40–51. doi: 10.1093/hmg/ddt394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao C., Malicki J. Nephrocystins and MKS proteins interact with IFT particle and facilitate transport of selected ciliary cargos. EMBO J. 2011;30:2532–2544. doi: 10.1038/emboj.2011.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y., Hong D.H., Pawlyk B., Yue G., Adamian M., Grynberg M., Godzik A., Li T. The retinitis pigmentosa GTPase regulator (RPGR)- interacting protein: subserving RPGR function and participating in disk morphogenesis. Proc Natl Acad Sci USA. 2003;100:3965–3970. doi: 10.1073/pnas.0637349100. [DOI] [PMC free article] [PubMed] [Google Scholar]