

Abstract
Evidence suggests that the role of autophagy in tumorigenesis is context dependent. Using genetically engineered mouse models (GEMMs) for human non-small-cell lung cancer (NSCLC), we found that deletion of the essential autophagy gene, Atg7, in KRASG12D-driven NSCLC inhibits tumor growth and converts adenomas and adenocarcinomas to benign oncocytomas characterized by the accumulation of respiration-defective mitochondria. Atg7 is required to preserve mitochondrial fatty acid oxidation (FAO) to maintain lipid homeostasis upon additional loss of Trp53 in NSCLC. Furthermore, cell lines derived from autophagy-deficient tumors depend on glutamine to survive starvation. This suggests that autophagy is essential for the metabolism, growth, and fate of NSCLC.
Keywords: autophagy, KRAS, NSCLC, metabolism, mitochondria, oncocytoma, p53, fatty acid oxidation
The role of autophagy in cancer is thought to be context dependent. Autophagy can act as tumor suppressor via its quality control function, which removes damaged organelles and protein aggregates to suppress oxidative stress, tissue damage, and genomic instability that can promote tumor initiation. In contrast, in established tumors, autophagy is upregulated in hypoxic regions to support tumor cell survival via recycling cargos and generating substrates such as amino acids, fatty acids (FAs) and nucleotides as well as ATP. In support of the prosurvival role for autophagy in cancer, in our previous work we found that RAS activation upregulates basal autophagy, and human cancer cells with activated RAS have high basal autophagy and depend on autophagy for survival. The mechanism behind this “autophagy addiction” is that cancer cells require autophagy to maintain the pool of functioning mitochondria and cellular metabolism that are necessary for tumor cell growth and survival. Autophagy is particularly critical to the survival of cancer cells during metabolic stress, which is a common feature of the tumor microenvironment.
To further understand the role of autophagy in RAS-driven cancers in a physiological setting, we examined the consequence of autophagy ablation in spontaneously arising RAS-driven cancers in immune-competent mice, the LSL-KRASG12D GEMM for NSCLC. Mice were also engineered to additionally delete the Trp53 tumor suppressor gene in tumor cells, which alters metabolism and accelerates progression to adenocarcinoma, potentially influencing autophagy.
Autophagy is Required to Maintain KRASG12D-Driven NSCLC Tumor Growth and Fate
To test if autophagy blockade suppressed tumorigenesis in spontaneously arising RAS-driven cancers, we crossed the LSL-KRASG12D GEMMs for human NSCLC to Atg7flox/flox mice possessing conditional deficiency in Atg7 in the presence or absence of Trp53. Initiation of tumorigenesis by KRASG12D activation, without or with Trp53 deletion, without or with Atg7 deletion, was achieved by intranasal delivery of adenovirus-Cre to the mice. Atg7 wild-type, KRASG12D-driven NSCLC tumor progression begins with hyperplasia that gradually progresses to adenomas and then to adenocarcinomas after acquisition of Trp53 mutations. However, Atg7 deficiency converts adenomas and adenocarcinomas to low-grade, papillary, oncocytic neoplasms or oncocytomas composed of tumor cells with a swollen, grainy, eosinophilic cytoplasm (oncocytes). Importantly, Atg7-deficient tumors accumulate respiration-defective mitochondria, as demonstrated by loss of enzymatic activity for mitochondrial cytochrome c oxidase. The conversion of adenomas and carcinomas to oncocytomas upon Atg7 deletion in NSCLC results in a drastic reduction in tumor cell proliferation that dramatically decreases tumor burden. Thus, autophagy is required for KRAS-driven tumor mitochondrial metabolism, growth, and fate (Fig. 1A and B). These findings suggest that mutations in essential autophagy genes may be the genetic basis for the development of human oncocytomas, and that converting adenomas and carcinomas to more benign oncocytomas by inhibiting autophagy or mitophagy might be a potential therapy for the treatment of NSCLC.
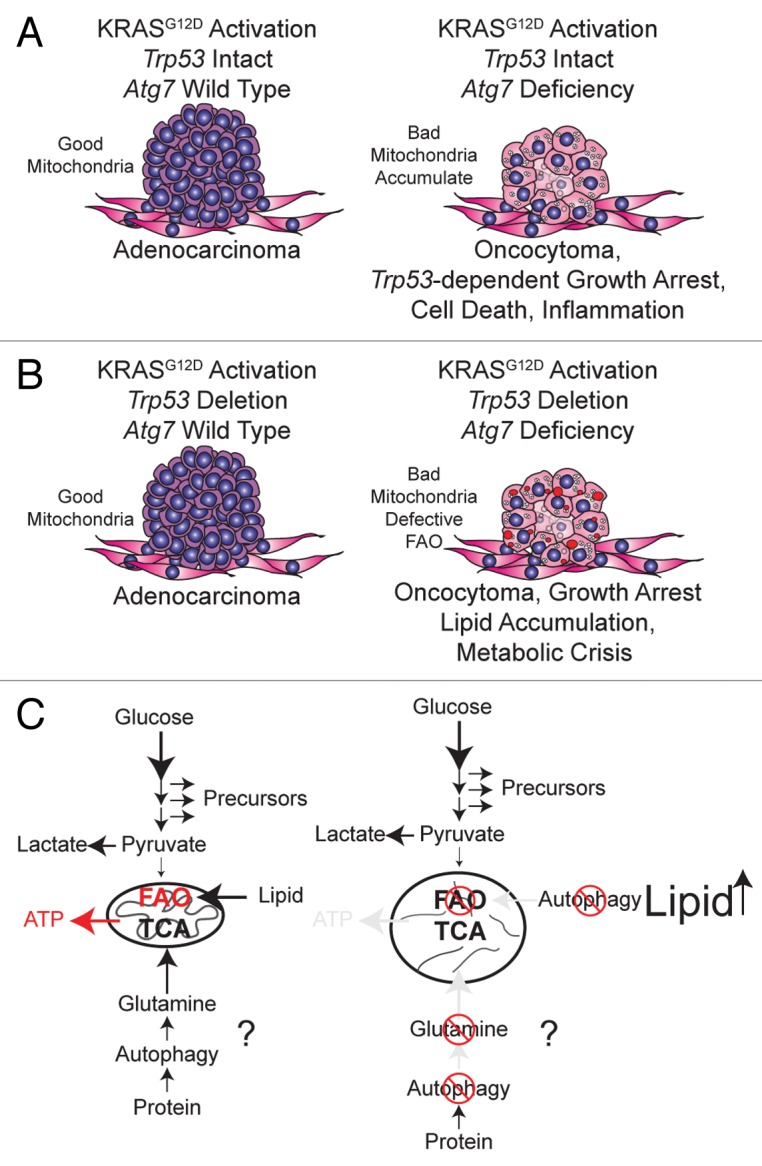
Figure 1. Autophagy maintains functioning mitochondria to support KRASG12D-driven NSCLC tumor metabolism, growth, and fate. (A) When Trp53 is intact in KRASG12D-driven NSCLC, Atg7 deficiency causes accumulation of dysfunctional mitochondria and converts adenomas and adenocarcinomas to oncocytomas, which results in tumor growth arrest and tumor atrophy. (B) With the additional loss of Trp53, Atg7 deficiency still converts adenocarcinomas to oncocytomas, but also causes defective mitochondrial FAO, leading to tumor cell lipid accumulation and metabolic impairment. (C) Tumor cell metabolism regulated by autophagy is oncogene- and tumor suppressor gene-dependent: with the additional loss of Trp53, autophagy preserves mitochondrial FAO and may provide metabolic substrates such as glutamine from protein degradation. RAS-driven cancer cells increase glucose metabolism to support their high energetic and biosynthetic demands. Increased glycolysis enhances conversion of pyruvate to lactate, resulting in insufficient pyruvate to support mitochondrial tricarboxylic acid (TCA) cycle metabolism upon nutrient deprivation. In this case, activated autophagy will degrade proteins and organelles to supply glutamine to maintain mitochondrial metabolism and FAO. When autophagy is blocked, however, depletion of the metabolic substrate glutamine, important for TCA cycle anaplerosis and defective FAO, will lead to tumor cell metabolic catastrophe.
Autophagy is Required for Lipid Homeostasis with Loss of Trp53 in NSCLC
Trp53 deficiency accelerates NSCLC and regulates cancer metabolism by increasing glucose uptake, directing glucose to lipid storage, and suppressing FAO. We found that in the KRASG12D-driven GEMMs for NSCLC, lipid accumulation was observed only in Atg7-deficient tumors with Trp53 deletion, and not in Atg7-deficient or wild-type tumors with intact Trp53. This suggests that Atg7 is required for lipid homeostasis in NSCLC specifically in the absence of Trp53 (Fig. 1B).
To elucidate the mechanism by which autophagy maintains lipid homeostasis in Trp53-deficient NSCLC, we generated tumor-derived cell lines (TDCLs) from the GEMM lung tumors and found that atg7−/− TDCLs were extremely sensitive to starvation-induced cell death and were defective for tumor growth as we expected. As we reported previously for autophagy-deficient HRAS or KRAS-transformed baby mouse kidney cells, the atg7−/− TDCLs were also defective for oxygen consumption in starvation. Similar to Atg7-deficient tumors lacking Trp53, atg7−/− TDCLs accumulate neutral lipids and FAs. Importantly, atg7−/− TDCLs fail to respire when palmitate is provided during starvation, indicative of defective mitochondrial FAO. This suggests that Atg7 deficiency causes NSCLC cells to become defective for mitochondrial FAO identifying the major reason for impaired mitochondria metabolism that leads to lipid and FA accumulation (Fig. 1C). Additionally, atg7−/− TDCLs are more sensitive to further inhibition of FAO during starvation than Atg7+/+ TDCLs, indicating combination therapy involving autophagy and FAO inhibition is a strategy for KRAS-driven lung cancers.
Upon metabolic stress, autophagy is activated and degrades intracellular components to provide fuel to support cell metabolism, and amino acids are one important fuel. We found that glutamine, but not glucose, supplementation successfully rescues survival of atg7−/− TDCLs during Hank’s Balanced Salt Solution induced-starvation, suggesting that in addition to preserving FAO, autophagy may also supply metabolic substrates such as glutamine from protein degradation to maintain metabolism (Fig. 1C).
Although the detailed mechanism behind the role of autophagy in tumorigenesis still needs to be further investigated, we provide the first evidence that autophagy is required to maintain the pool of functional mitochondria for KRAS-driven tumor growth in a physiological setting. We are also the first to report that autophagy is required to maintain NSCLC fate, and lipid and glutamine metabolism, and that functions of autophagy are oncogene- and tumor suppressor gene-specific (Fig. 1).
Glossary
Abbreviations:
- FAO
fatty acid oxidation
- FAs
fatty acids
- GEMMs
genetically engineered mouse models
- NSCLC
non-small-cell lung cancer
- TDCLs
tumor-derived cell lines
- TCA
tricarboxylic acid
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
This work was supported by the NIH grants R37CA53370, RC1CA147961, R01CA147961, and RO1CA130893, the Department of Defense (W81XWH-09-01-0394), and the Val Skinner Foundation.