

Abstract
Salmonella enterica Pullorum is one of the leading causes of mortality in poultry. Understanding the molecular response in chickens in response to the infection by S. enterica is important in revealing the mechanisms of pathogenesis and disease progress. There have been studies on identifying genes associated with Salmonella infection by differential expression analysis, but the relationships among regulated genes have not been investigated. In this study, we employed weighted gene coexpression network analysis (WGCNA) and differential coexpression analysis (DCEA) to identify coexpression modules by exploring microarray data derived from chicken splenic tissues in response to the S. enterica infection. A total of 19 modules from 13,538 genes were associated with the Jak-STAT signaling pathway, the extracellular matrix, cytoskeleton organization, the regulation of the actin cytoskeleton, G-protein coupled receptor activity, Toll-like receptor signaling pathways, and immune system processes; among them, 14 differentially coexpressed modules (DCMs) and 2,856 differentially coexpressed genes (DCGs) were identified. The global expression of module genes between infected and uninfected chickens showed slight differences but considerable changes for global coexpression. Furthermore, DCGs were consistently linked to the hubs of the modules. These results will help prioritize candidate genes for future studies of Salmonella infection.
1. Introduction
Chickens are an important component in the global agricultural economy by serving as one of the primary sources of proteins for humans. However, the poultry industry has been consistently threatened by various diseases, including those caused by viral, bacterial, and parasitic infections. Salmonella enterica serovar Pullorum (S. Pullorum) is one of the most important pathogens of poultry causing severe systemic disease [1, 2]. To prevent and control S. Pullorum in chickens, the host responses against this pathogen have been studied for decades. Although significant advances have been made, especially in the identification of molecules and genes involved in the host immune response [3, 4] and mucosal inflammation [5, 6], as well as their differential expression during infection [7–10], the precise pathways regulating immunity to Salmonella infection using a systems biology approach have not been investigated. Although gene differential expression analysis (DEA) provides important information, such as identification of genes that are expressed at different times during infection, which inform our understanding of pathogenesis, identifying gene interactions using a systems biology approach greatly enhances our knowledge at the mechanistic and regulatory levels. A large amount of information regarding gene interactions is available in microarray datasets and by applying network approaches the gap between individual genes and systems can be bridged [11–13]. The modularity in biological systems allows for both the study of independent components and identification of gene relationships within modules. Modern approaches, such as weighted gene coexpression network analysis (WGCNA) [14], can identify modules with expression levels that are highly correlated across samples and have been used to identify new candidate regulatory molecules and networks in Salmonella-infected pigs [15]. Differentially coexpressed modules (DCMs) can also be identified [16]. The holistic changes in modules would be reflected in transcriptional and coexpression changes for individual genes. In general, gene expression levels change during disease or infection, but some have reported that seemingly nonsignificant DEGs may also play a key role in a disease because their interactions with other genes change considerably [17]. These genes can be identified via differential coexpression analysis (DCEA), which can mine individual genes using a holistic approach [17–19]. Hence, combining the WGCNA and DCEA methods can identify interacting modules and differentially coexpressed genes (DCG) during infection, compared with controls. Here, we mined the molecular network relationships of the differential coexpression modules and genes using microarray data from spleens of S. Pullorum-infected and uninfected chickens using WGCNA and DCEA (Figure 1). The results complement traditional DEA and add to our understanding of the regulatory mechanisms that occur during Salmonella infection.
Figure 1.

Workflow of the comprehensive gene coexpression network analysis.
2. Materials and Methods
2.1. Microarray Data Harvesting and Processing
A comprehensive transcriptomics dataset derived from microarray analysis of spleens from chickens challenged with 108 CFU of Salmonella enterica serovar Pullorum or mock-challenged with the same volume of distilled water (controls) was obtained from the Gene Expression Omnibus (GEO) database (https://www.ncbi.nlm.nih.gov/geo/) (accession number: GSE59663). The dataset was generated with the Agilent oligo microarray chips containing 43,663 probe sets. In this study, we first streamlined the dataset by excluding 14,920 probe sets that were either unmappable to any gene IDs or mapped to multiple gene IDs. In the case of multiple probe sets mapped to one identical gene, the probe set, which is most often associated with the highest expression level, was maintained to ensure that only one probe set was left to investigate one gene. If more than one probe set was left after the above steps, their intensities were averaged. Finally, a one-to-one match between 13,538 probe sets and 13,538 genes was achieved.
Three biological replicates (chips) for each time point were available in the challenged group for these datasets. However, at each time point in the control group, only one chip was used to hybridize with the equally mixed mRNA sample containing the three control samples. We averaged the replicates for each time point, except at day 21, with the two replicates included and forming the dataset for the challenged group with 10 samples; this dataset was equivalent to the dataset of the control group. The dataset was quantile normalized by the function of normalizeQuantiles in R package limma [20].
2.2. Construction of Weighted Gene Coexpression Network and Identification of Modules
Weighted gene coexpression network analysis (WGCNA) was used to detect coexpression modules from the dataset of challenged samples [14, 21]. The R function of blockwise modules was implemented with the following parameters: power = 12, minModuleSize = 100, and networkType = “signed.” Microarray data were processed as described below.
The pairwise Pearson's correlation coefficients were calculated for all the genes in the challenged groups, followed by the construction of an adjacency matrix using the power function:
| (1) |
where xi and xj were the ith and jth gene expression traits, respectively, which formed a signed weighted correlation network; and β used default value (i.e., β = 12). The topological overlap measure (TOM) was calculated as follows:
| (2) |
where k.total is the sum of connection strengths for a gene with the other network genes. u is the other network genes.
Afterwards, 1-TOM was calculated as a biological important measure for network interconnectedness. Genes with highly similar coexpression relationships were grouped together by performing hierarchical clustering on the topological overlap. Subsequently, genes were hierarchically clustered using 1-TOM as the distance measure and modules were determined by choosing a height cutoff of 0.995 for the resulting dendrogram. Highly similar modules were identified by clustering and merged together using a dynamic tree-cutting algorithm [14]. Eigengene refers to the first principal component for a given module and could be calculated to draw a module trajectory curve [14].
2.3. Identification of Differentially Coexpressed Modules
Differentially coexpressed modules (DCMs) were identified using gene-set coexpression analysis (GSCA) that adopted the length-normalized Euclidean distance to measure the coexpression difference for the pairwise correlations between infected and control groups [16].
| (3) |
where Pm was the number of gene pairs from the pairwise correlation for all the module genes. rpc and rpi were the correlation coefficients for a gene pair in the control and infected groups, respectively.
The null distribution for distance was constructed by permuting samples across conditions for 10,000 times to yield gene-set specific p values. Modules with p value < 0.01 were considered as significantly differentially coexpressed.
2.4. Identification of Differentially Coexpressed Genes
The differential coexpression analysis (DCEA) was implemented by using R package DCGL, which is a useful tool to identify differentially coexpressed genes (DCGs) and differentially coexpressed links (DCLs) [17–19]. The R function DCe was applied and then the p values were adjusted for a false discovery rate (FDR) using the Benjamini-Hochberg method to reduce a large amount of false positive results [22]. The genes with FDR < 0.001 were selected as DCGs.
2.5. Gene Ontology (GO) and Pathway Enrichment for Coexpression Modules
GO enrichment and KEGG pathway analyses for network modules were performed using Database for Annotation, Visualization, and Integrated Discovery (DAVID, v6.7) program using all chickens genes as the background [23, 24]. The modified Fisher's exact test with an adjustment for multiple tests by Benjamini-Hochberg method was used to identify significantly enriched terms for module genes [22].
2.6. Network Visualization
The complex network bioinformatics software Cytoscape (v3.1.1) was used to visualize the pairwise relationships between genes [25].
3. Results
3.1. Weighted Gene Coexpression Network Analysis
Using blockwiseModules R function (β = 12), a total of 19 modules ranging from 100 to 3,000 genes were recovered for the 13,538 distinct genes in the S. Pullorum-infected group (Table 1). Each module was assigned a unique color, including gray color for the 373 unassigned genes. Genes in the same module shared the same or similar expression patterns that were catalogued by the trajectory curves (Figure 2).
Table 1.
Module preservation and functions.
| Module | Size | Z summary | Function |
|---|---|---|---|
| Lightyellow | 126 | 5.42 | Nucleus (8.40E − 4) |
|
| |||
| Lightgreen | 145 | 0.67 | Jak-STAT signaling pathway (9.73E − 3) |
|
| |||
| Grey60 | 145 | 7.49 | Extracellular matrix (5.10E − 4) |
| Cytoskeleton (7.84E − 3) | |||
|
| |||
| Lightcyan | 155 | 0.52 | Anchored to membrane (4.19E − 3) |
|
| |||
| Midnightblue | 159 | 5.78 | Cytoplasm (1.75E − 4); |
| Organelle membrane (4.67E − 3) | |||
| Endomembrane system (9.27E − 3) | |||
|
| |||
| Cyan | 181 | 8.71 | Cell adhesion molecules (3.24E − 5) |
| Cell adhesion (2.75E − 3) | |||
|
| |||
| Salmon | 248 | 2.14 | Neuroactive ligand-receptor interaction (2.60E − 6) |
|
| |||
| Tan | 287 | 4.86 | Lysosome (4.80E − 3) |
|
| |||
| Greenyellow | 293 | 6.58 | Ligase activity (3.41E − 3) |
|
| |||
| Purple | 298 | 10.93 | Proteasome complex (6.56E − 6) |
| Regulation of cytokine biosynthetic process (3.83E − 3) | |||
| Toll-like receptor signaling pathway (9.52E − 3) | |||
|
| |||
| Magenta | 357 | 1.82 | G-Protein coupled receptor activity (2.88E − 4) |
|
| |||
| Pink | 418 | 13.36 | Cellular macromolecular complex assembly (1.00E − 03) |
|
| |||
| Black | 599 | 1.97 | Postsynaptic membrane (2.35E − 3) |
| Synapse (2.72E − 3) | |||
|
| |||
| Red | 648 | 14.89 | Signal transducer activity (2.07E − 4) |
| Multicellular organism development (2.70E − 4) | |||
|
| |||
| Green | 1056 | 27.63 | Cell cycle phase (9.63E − 13) |
| DNA replication (3.67E − 7) | |||
| Response to DNA damage stimulus (1.45E − 6) | |||
| DNA repair (4.11e − 6) | |||
| Cytoskeleton organization (3.77E − 4) | |||
|
| |||
| Yellow | 1122 | 32.83 | Glucose catabolic process (4.03E − 5) |
| Glycolysis/gluconeogenesis (1.49E − 4) | |||
| Glycolysis (1.79E − 4) | |||
| Glucose metabolic process (3.95E − 4) | |||
|
| |||
| Brown | 1349 | 18.99 | ABC transporters (1.00E − 03) |
|
| |||
| Blue | 2581 | 43.01 | Immune system process (1.31E − 4) |
| Induction of apoptosis (1.98E − 4) | |||
| Antigen processing and presentation (2.06E − 4) | |||
| Lysosome (3.08E − 4) | |||
| Defense response to bacterium (6.06E − 3) | |||
|
| |||
| Turquoise | 2998 | 51.56 | Nervous system development (2.20E − 15) |
| Focal adhesion (2.04E − 9) | |||
| Wnt signaling pathway (7.56E − 9) | |||
| Regulation of actin cytoskeleton (1.64E − 7) | |||
| TGF-beta signaling pathway (4.42E − 7) | |||
Note. The column “Size” gives the gene numbers contained in every module. “Z summary” gives the z score of module preservation. “Function” gives the module functions enriched by DAVID.
Figure 2.

Module expression patterns.
Subsequent analysis using DAVID identified biological features in modules that were potentially associated with the infection by S. Pullorum (Figure 6 and Table 1), such as the Jak-STAT signaling pathway (module lightgreen) [26], the extracellular matrix (ECM) (module grey60) [27], cytoskeleton organization (module green), regulation of the actin cytoskeleton (module blue) [28], G-protein coupled receptor activity (module magenta), Toll-like receptor signaling pathways (module purple), and immune system processes (module blue). ECM genes and cell adhesion genes are significantly enriched in the module grey60 and cyan (FDR = 5.10e − 4 and 2.75e − 3), respectively (Table 1). The grey60 and cyan modules also displayed significant similarity in expression patterns (eigengenes' correlation = 0.76; p = 0.01). These observations were in congruent with those reported earlier by others on the crucial role of host cell ECM proteins and bacterial outer membrane structures in the adhesion and invasion of Salmonella [27].
Figure 6.

(a) Hub genes' network in the infected group. The node colors represent the module colors: the nodes with shape of rectangle are DCGs and the elliptical nodes are non-DCGs. (b) Hub genes' network in the control group. The node colors represent the module colors: the nodes with shape of rectangle are DCGs and the elliptical nodes are non-DCGs.
3.2. Module Stability
To test the reproducibility of the identified modules, we performed a sampling test, in which we randomly selected half of the samples to calculate the new intramodule connectivity. The sampling was repeated 100 times and then the module stability was expressed as the correlation of intramodule connectivity between the original and sampled ones [29]. Most modules displayed good stability; module salmon was the least stable (Figure 3).
Figure 3.
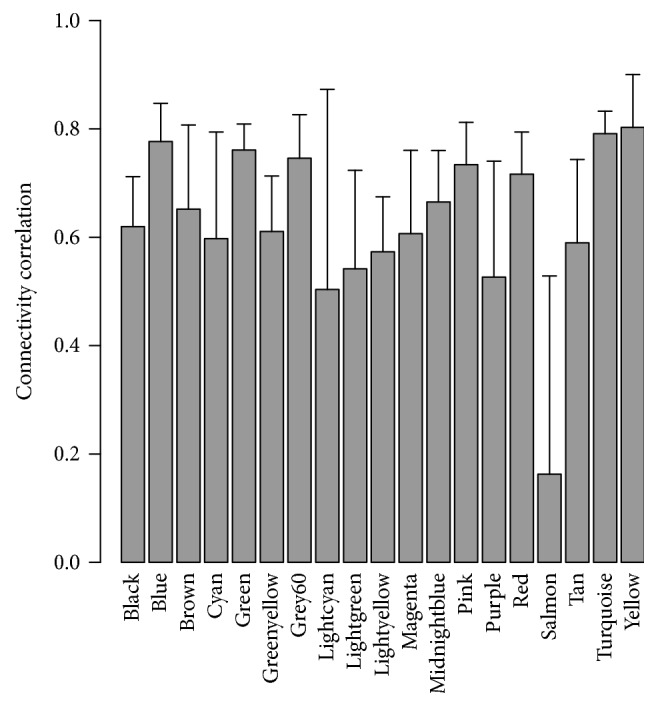
Correlation of intramodule connectivity for each module after 100 samplings.
3.3. Module Preservation Analysis
We investigated whether the S. Pullorum-infected module was preserved in the corresponding controls by testing whether the infection-associated coexpression network can be replicated in the control groups. The preservation scores for all the modules were listed in Table 1, in which Z summary scores <2, between 2 and 10, and >10 indicate no evidence, weak-to-moderate evidence, and strong evidence for module preservation, respectively. Preservation analysis provided strong evidence to support the conservation of modules turquoise, brown, blue, yellow, green, red, pink, and purple, which all contained considerably large numbers of genes, but no evidence to support the preservation of modules lightcyan, lightgreen, magenta, and black associated with the membranes, the Jak-STAT signaling pathway, G-protein coupled receptor activity, and synapses, respectively (Table 1).
3.4. Module Gene Expression and Coexpression Comparison
We compared the module genes' expression and coexpression level between the infected and control groups. The violin plot in Figure 4(a) showed that the gene expression for modules in the infection versus control groups is not significantly different, and the distribution for the expression intensities is similar. Subsequently, we compared the gene coexpression level by calculating the gene connectivity for each module. The module turquoise exhibits the largest connectivities since it includes the largest number of genes (2,998 genes). Modules blue (2,581 genes), yellow (1,122 genes), brown (1,349 genes), and green (1,056 genes), which include a considerable number of genes, display the next highest connectivities. In addition, the coexpression levels are different between modules in the two conditions. The coexpressions are strengthened in the infected state (Figure 4(b)).
Figure 4.

(a) Violin plot showing the gene expression differences between modules in the infected and control groups. (b) Violin plot showing the gene coexpression connectivity differences between modules in the infected and control groups. .i represents the module in the infected group, and .c represents the module in the control group.
3.5. Identification of Differentially Coexpressed Modules
Gene-set coexpression analysis (GSCA) revealed that 14 of the 19 modules were significantly differentially coexpressed (p < 0.01 by bootstrap sampling test) (Table 2). Among them, modules black (z = 1.97), magenta (z = 1.82), salmon (z = 2.14), and lightcyan (z = 0.52) were significantly differentially coexpressed. These observations were in agreement with the module preservation analysis, in which significantly differentially coexpressed modules (DCM) were only weakly preserved in the control group.
Table 2.
Differentially coexpressed modules enriched with differentially coexpressed genes (DCGs).
| Module | Size | GSCA.p | Hub and DCGs |
|---|---|---|---|
| Black | 599 | 0 | GJA8, LOC421988, MYOCD |
| Magenta | 357 | 0 | C10orf83, PPME1, NRTN, TMEM167B, ABCA5, C10orf58 |
| Salmon | 248 | 0 | CPA5, PRSS2, ARHGEF19, INS, CELA2A, TCERG1L, LOC396296, |
| LOC771434, KCNC2, XKR9 | |||
| Lightcyan | 155 | 5.55E − 16 | LOC769741, TRPA1, C10orf96, CASKIN2, GMCSF, cor6 |
| Brown | 1349 | 2.11E − 09 | — |
| Cyan | 181 | 1.77E − 08 | RAB36, LOC415324, UPK3B, CWH43 |
| Lightgreen | 145 | 4.05E − 07 | CIP1, ZC3H12D, TNS3, SOCS3, LOC415844 |
| Pink | 418 | 4.20E − 07 | ABCG8 |
| Lightyellow | 126 | 2.10E − 05 | DEPDC1, CDCA2 |
| Tan | 287 | 2.23E − 04 | TP53INP1, CNRIP1, NEURL, SEPP1 |
| Midnightblue | 159 | 1.16E − 02 | LOC416257 |
| Greenyellow | 293 | 2.04E − 01 | INVS, ARSH |
| Grey60 | 145 | 2.10E − 01 | — |
| Purple | 298 | 2.51E − 01 | WDR5, NIPA2 |
| Red | 648 | 8.66E − 01 | — |
| Blue | 2581 | 1 | — |
| Green | 1056 | 1 | — |
| Turquoise | 2998 | 1 | — |
| Yellow | 1122 | 1 | — |
Note. The column “Size” gives the gene numbers contained in every module. “GSCA.p” gives the p value calculated by GSCA for modules between the two different conditions and the p value less than 0.05 indicates that the module is significantly differentially coexpressed. “Hub and DCGs” shows the genes which are DCGs in the top ten hub genes.
3.6. Identification of Differentially Coexpressed Genes
A total of 2,856 differentially coexpressed genes (DCG) were selected with a false discovery rate (FDR) of less than 0.001 using the DCe method in the DCGL package. And a total of 284,213 differentially coexpressed links (DCLs) were same signed, 82,619 were differently signed, and 272,491 were switched links.
Furthermore, we mapped the DCGs for each module and found that the DCMs enrich the DCGs. For example, a total of 152 DCGs appeared in the module magenta with Z summary of 1.82 (p = 0), 231 DCGs in module black with Z summary of 1.97 (p = 0), and 147 DCGs in module salmon with Z summary of 2.14 (p = 0). In network biology, a hub gene is a good representative of a module. We identified the hub genes for all of the modules. Table 2 gives the gene names which are not only hub genes but also DCGs in each module.
4. Discussion
We constructed a gene network for the S. Pullorum-infected chickens using weighted gene coexpression network analysis (WGCNA) from the data of time-series microarray. This module detection strategy utilizes the biological variability inherent in the prospective cohort study to reveal the modular organization and function of transcriptional systems. The time series expression profiles allow the study of the transcriptional regulation of these gene coexpression networks during infection. A network-based analysis provides a systems-level understanding of the relationships between members of a network by focusing on genome-wide gene modules rather than individual genes [30]. Differential expression analysis (DEA) aims to identify genes that are expressed significantly higher or lower in one group compared with another. By contrast, WGCNA is not biased toward genes with significant changes in expression. Moreover, the dimensionality of microarray data in the present study was reduced from 13,538 genes to 19 modules, which significantly increased the ability to identify concordant changes in the expression of multiple genes.
The module expression analysis showed that module salmon was the least abundant but exhibited the largest variation in gene expression causing the instability in module construction. Gene expression was the most stable within module midnightblue (Figure 4(a)). The expression distribution for module genes in different conditions (infected versus control) was the same. The coexpression level was further compared. The coexpression comparison showed significant changes for different conditions. We investigated the key modules and genes resulting in the differences. The GSCA analysis identified ten significantly differentially coexpressed modules (DCMs), which are in accordance with the module preservation results that are significant in coexpression differences with little evidence for preservation. Regulatory relationships among genes can be parsed as the pairwise correlations between gene expression levels, so the changes in coexpression patterns between two conditions may indicate dysfunctional regulatory systems in disease [31]. Module lightgreen associated with the Jak-STAT signaling pathway and lightcyan associated with membrane anchoring functions were the two weakest preserved modules (Table 1 and Figure 5). Thus, these modules may be associated with S. Pullorum infection in chickens.
Figure 5.

Violin plot of gene coexpression connectivity for significantly differentially coexpressed modules.
Furthermore, we investigated the driven genes leading to the coexpression difference. The differential coexpression analysis (DCEA) method was applied, and 2,856 differentially coexpressed genes (DCGs) were identified. Compared to the differential expression analysis (DEA), it was found that the overlapping of DCGs with the 234 DEGs (t-test p value less than 0.01) was significant (hypergeometric test p = 1.07e − 07), indicating that differential expression and differential coexpression are somewhat related to each other, which is consistent with a previous report [17]. However, there are many Salmonella infection-related genes identified by the DCEA method. The top one DCG identified is WASF1, which is an important gene in the Salmonella infection pathway and was not identified as a DEG (expression fold change: 1.08; t-test p value of 0.34). The protein encoded by WASF1, a member of the Wiskott-Aldrich syndrome protein family, plays a critical role downstream of Rac, which is a Rho family of small GTPases, in regulating the actin cytoskeleton required for membrane ruffling. This gene associates with an actin nucleation core Arp2/3 complex while enhancing actin polymerization in vitro [32]. Another gene, CDC42 (fold change = 1.02; p = 0.6), a member of the Rho subfamily of actin-organizing small GTP-binding proteins, interacts with WASF1 and is essential for S. Typhimurium entry into host cells [33, 34]. CDC42 was not selected as DCG with a false discovery rate (FDR) of 1.55E − 3, but it interacts with genes PAK7 (fold change = 1.95; p = 0.64) [35, 36], CDC42EP3 (fold change = 1.05; p = 0.81) [37, 38], PAK1 (fold change = 0.97; p = 0.67) [39], PARD6B (fold change = 0.76; p = 0.05) [40, 41], PARD6A (fold change = 1.75; p = 0.90) [29, 40], and IQGAP2 (fold change = 1.58; p = 0.12) [42], which are all identified as DCGs. Carow and Rottenberg reported that gene SOCS3, which was also identified as a DCG (fold change = 1.54; p = 0.15), is a major regulator of infection and inflammation and controls immune homeostasis in physiological and pathological conditions such as infection and autoimmunity [43]. SOCS3 is a hub gene in the module lightgreen associated with the Jak-STAT signaling pathway, an important pathway for Salmonella infection [44]. It is well known that the Jak-STAT pathway can regulate cell growth, apoptosis, immunity, and inflammatory responses and because of its significance in the immune response, the Jak-STAT pathway is often exploited by pathogens [45]. In our study, we found that the Jak-STAT pathway genes were significantly enriched in the module lightgreen which is not detectable in controls. So we think that SOCS3 and the other Jak-STAT pathway genes may together regulate the activity of the organism in infection, which leads this module to be differentially coexpressed.
The above results showed some specific subnetworks for infection, in spite of a common network existing whether in the control or infected group. We constructed two coexpression networks from the top ten hub genes' expression profiles for each module from the two different conditions. As shown in Figures 6(a) and 6(b), common core networks, including the most preserved modules between infected and control groups, were present. However, some closely interacted subnetworks seen during infection disappeared in the control. These infection-specific subnetworks included genes that are members of the Jak-STAT signaling pathway (module lightgreen); others associated with membrane anchoring (module lightcyan), neuroactive ligand-receptor interaction (module salmon), and lysosomal processing (module tan), which suggested that these subnetworks dysregulated the systems during infection. Although only one dataset was used here, due to the lack of published related microarray datasets, these present results advance our understanding of the cell biology and immunoregulatory pathways involved in Salmonella infection in the chicken host.
Acknowledgments
The authors thank Professor Guan Zhu from Texas A&M University and Dr. Patricia Wilkins from CDC, USA, for revising the manuscript. And they also thank the researchers from Yangzhou University for submitting the dataset to GEO. This work was supported by Fundamental Research Program of Chinese Academy of Agricultural Sciences (0032015027) and the Innovative Special Project of Agricultural Sci-Tech and by the Special Fund for Agro-Scientific Research in Public Interest (201303044-7) to Professor Cai's team.
Abbreviations
- DAVID:
Database for Annotation, Visualization, and Integrated Discovery
- DCEA:
Differential coexpression analysis
- DCG:
Differentially coexpressed gene
- DCL:
Differentially coexpressed link
- DCM:
Differentially coexpressed module
- DEA:
Differential expression analysis
- ECM:
Extracellular matrix
- FDR:
False discovery rate
- GO:
Gene Ontology
- GSCA:
Gene-set coexpression analysis
- TOM:
Topological overlap measure
- WGCNA:
Weighted gene coexpression network analysis.
Conflicts of Interest
The authors declare that there are no conflicts of interest.
Authors' Contributions
Bao-Hong Liu contributed to the design and conception of this study, conducted computational experiments, performed and interpreted data, and drafted the manuscript. Jian-Ping Cai conceived this project and participated in its design, helped in interpreting the data, and drafted and revised the manuscript. All authors read and approved the final manuscript.
References
- 1.Khan R. T., Chevenon M., Yuki K. E., Malo D. Genetic dissection of the Ity3 locus identifies a role for Ncf2 co-expression modules and suggests Selp as a candidate gene underlying the Ity3.2 locus. Frontiers in Immunology. 2014;5, article 375 doi: 10.3389/fimmu.2014.00375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Buckle G. C., Walker C. L., Black R. E. Typhoid fever and paratyphoid fever: systematic review to estimate global morbidity and mortality for 2010. Journal of Global Health. 2012;2(1) doi: 10.7189/jogh.01.010401.010401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Santos R. L. Pathobiology of Salmonella, intestinal microbiota, and the host innate immune response. Frontiers in Immunology. 2014;5, article 252 doi: 10.3389/fimmu.2014.00252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Guadarrama C., Villaseñor T., Calva E. The subtleties and contrasts of the LeuO regulator in Salmonella Typhi: implications in the immune response. Frontiers in Immunology. 2014;5, article 581 doi: 10.3389/fimmu.2014.00581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Patel S., McCormick B. A. Mucosal inflammatory response to Salmonella typhimurium infection. Frontiers in Immunology. 2014;5, article 311 doi: 10.3389/fimmu.2014.00311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mooney J. P., Butler B. P., Lokken K. L., et al. The mucosal inflammatory response to non-typhoidal Salmonella in the intestine is blunted by IL-10 during concurrent malaria parasite infection. Mucosal Immunology. 2014;7(6):1302–1311. doi: 10.1038/mi.2014.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bellet M. M., Deriu E., Liu J. Z., et al. Circadian clock regulates the host response to Salmonella. Proceedings of the National Academy of Sciences. 2013;110(24):9897–9902. doi: 10.1073/pnas.1120636110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Huang T.-H., Uthe J. J., Bearson S. M. D., et al. Distinct peripheral blood RNA responses to Salmonella in pigs differing in Salmonella shedding levels: intersection of IFNG, TLR and miRNA pathways. PLoS ONE. 2011;6(12) doi: 10.1371/journal.pone.0028768.e28768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chaussé A., Grépinet O., Bottreau E., et al. Susceptibility to Salmonella carrier-state: a possible Th2 response in susceptible chicks. Veterinary Immunology and Immunopathology. 2014;159(1-2):16–28. doi: 10.1016/j.vetimm.2014.03.001. [DOI] [PubMed] [Google Scholar]
- 10.Szmolka A., Wiener Z., Matulova M. E., Varmuzova K., Rychlik I. Gene expression profiles of chicken embryo fibroblasts in response to Salmonella Enteritidis infection. PLoS ONE. 2015;10(6) doi: 10.1371/journal.pone.0127708.e0127708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.van Noort V., Snel B., Huynen M. A. The yeast coexpression network has a small-world, scale-free architecture and can be explained by a simple model. EMBO Reports. 2004;5(3):280–284. doi: 10.1038/sj.embor.7400090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stuart J. M., Segal E., Koller D., Kim S. K. A gene-coexpression network for global discovery of conserved genetic modules. Science. 2003;302(5643):249–255. doi: 10.1126/science.1087447. [DOI] [PubMed] [Google Scholar]
- 13.Lee H. K., Hsu A. K., Sajdak J., Qin J., Pavlidis P. Coexpresion analysis of human genes across many microarray data sets. Genome Research. 2004;14(6):1085–1094. doi: 10.1101/gr.1910904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang B., Horvath S. A general framework for weighted gene co-expression network analysis. Statistical Applications in Genetics and Molecular Biology. 2005;4, article 17 doi: 10.2202/1544-6115.1128. [DOI] [PubMed] [Google Scholar]
- 15.Kommadath A., Bao H., Arantes A. S., et al. Gene co-expression network analysis identifies porcine genes associated with variation in Salmonella shedding. BMC Genomics. 2014;15(1, article 452) doi: 10.1186/1471-2164-15-452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Choi Y., Kendziorski C. Statistical methods for gene set co-expression analysis. Bioinformatics. 2009;25(21):2780–2786. doi: 10.1093/bioinformatics/btp502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yu H., Liu B.-H., Ye Z.-Q., Li C., Li Y.-X., Li Y.-Y. Link-based quantitative methods to identify differentially coexpressed genes and gene Pairs. BMC Bioinformatics. 2011;12, article 315 doi: 10.1186/1471-2105-12-315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu B.-H., Yu H., Tu K., Li C., Li Y.-X., Li Y.-Y. DCGL: an R package for identifying differentially coexpressed genes and links from gene expression microarray data. Bioinformatics. 2010;26(20):2637–2638. doi: 10.1093/bioinformatics/btq471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yang J., Yu H., Liu B.-H., et al. DCGL v2.0: an R package for unveiling differential regulation from differential co-expression. PLoS ONE. 2013;8(11) doi: 10.1371/journal.pone.0079729.e79729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Smyth G. K. Linear models and empirical Bayes methods for assessing differential expression in microarray experiments. Statistical Applications in Genetics and Molecular Biology. 2004;3, article 3 doi: 10.2202/1544-6115.1027. [DOI] [PubMed] [Google Scholar]
- 21.Langfelder P., Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics. 2008;9, article 559 doi: 10.1186/1471-2105-9-559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Benjamini Y., Drai D., Elmer G., Kafkafi N., Golani I. Controlling the false discovery rate in behavior genetics research. Behavioural Brain Research. 2001;125(1-2):279–284. doi: 10.1016/s0166-4328(01)00297-2. [DOI] [PubMed] [Google Scholar]
- 23.Ashburner M., Ball C. A., Blake J. A., et al. Gene ontology: tool for the unification of biology. Nature Genetics. 2000;25(1):25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Huang D. W., Sherman B. T., Lempicki R. A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nature Protocols. 2009;4(1):44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 25.Shannon P., Markiel A., Ozier O., et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Research. 2003;13(11):2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Uchiya K.-I., Nikai T. Salmonella pathogenicity island 2-dependent expression of suppressor of cytokine signaling 3 in macrophages. Infection and Immunity. 2005;73(9):5587–5594. doi: 10.1128/IAI.73.9.5587-5594.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Berndt A., Müller J., Borsi L., Kosmehl H., Methner U., Berndt A. Reorganisation of the caecal extracellular matrix upon Salmonella infection—relation between bacterial invasiveness and expression of virulence genes. Veterinary Microbiology. 2009;133(1-2):123–137. doi: 10.1016/j.vetmic.2008.06.025. [DOI] [PubMed] [Google Scholar]
- 28.Guiney D. G., Lesnick M. Targeting of the actin cytoskeleton during infection by Salmonella strains. Clinical Immunology. 2005;114(3):248–255. doi: 10.1016/j.clim.2004.07.014. [DOI] [PubMed] [Google Scholar]
- 29.Liu W., Ye H. Co-expression network analysis identifies transcriptional modules in the mouse liver. Molecular Genetics and Genomics. 2014;289(5):847–853. doi: 10.1007/s00438-014-0859-8. [DOI] [PubMed] [Google Scholar]
- 30.Miller J. A., Oldham M. C., Geschwind D. H. A systems level analysis of transcriptional changes in Alzheimer's disease and normal aging. Journal of Neuroscience. 2008;28(6):1410–1420. doi: 10.1523/JNEUROSCI.4098-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhai Y., Franco L. M., Atmar R. L., et al. Host transcriptional response to influenza and other acute respiratory viral infections—a prospective cohort study. PLoS Pathogens. 2015;11(6) doi: 10.1371/journal.ppat.1004869.e1004869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen Z., Borek D., Padrick S. B., et al. Structure and control of the actin regulatory WAVE complex. Nature. 2010;468(7323):533–538. doi: 10.1038/nature09623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen L.-M., Hobbie S., Galán J. E. Requirement of CDC42 for Salmonella-induced cytoskeletal and nuclear responses. Science. 1996;274(5295):2115–2118. doi: 10.1126/science.274.5295.2115. [DOI] [PubMed] [Google Scholar]
- 34.Hallstrom K., McCormick B. A. Salmonella interaction with and passage through the intestinal mucosa: through the lens of the organism. Frontiers in Microbiology. 2011;2:p. 88. doi: 10.3389/fmicb.2011.00088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pandey A., Dan I., Kristiansen T. Z., et al. Cloning and characterization of PAK5, a novel member of mammalian p21-activated kinase-II subfamily that is predominantly expressed in brain. Oncogene. 2002;21(24):3939–3948. doi: 10.1038/sj/onc/1205478. [DOI] [PubMed] [Google Scholar]
- 36.Dan C., Nath N., Liberto M., Minden A. PAK5, a new brain-specific kinase, promotes neurite outgrowth in N1E-115 cells. Molecular and Cellular Biology. 2002;22(2):567–577. doi: 10.1128/MCB.22.2.567-577.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Joberty G., Perlungher R. R., Macara I. G. The Borgs, a new family of Cdc42 and TC10 GTPase-interacting proteins. Molecular and Cellular Biology. 1999;19(10):6585–6597. doi: 10.1128/mcb.19.10.6585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Alberts A. S., Bouquin N., Johnston L. H., Treisman R. Analysis of RhoA-binding proteins reveals an interaction domain conserved in heterotrimeric G protein β subunits and the yeast response regulator protein Skn7. Journal of Biological Chemistry. 1998;273(15):8616–8622. doi: 10.1074/jbc.273.15.8616. [DOI] [PubMed] [Google Scholar]
- 39.Zhang B., Chernoff J., Zheng Y. Interaction of Rac1 with GTPase-activating proteins and putative effectors. A comparison with Cdc42 and RhoA. The Journal of Biological Chemistry. 1998;273(15):8776–8782. doi: 10.1074/jbc.273.15.8776. [DOI] [PubMed] [Google Scholar]
- 40.Joberty G., Petersen C., Gao L., Macara I. G. The cell-polarity protein Par6 links Par3 and atypical protein kinase C to Cdc42. Nature Cell Biology. 2000;2(8):531–539. doi: 10.1038/35019573. [DOI] [PubMed] [Google Scholar]
- 41.Noda Y., Takeya R., Ohno S., Naito S., Ito T., Sumimoto H. Human homologues of the Caenorhabditis elegans cell polarity protein PAR6 as an adaptor that links the small GTPases Rac and Cdc42 to atypical protein kinase C. Genes to Cells. 2001;6(2):107–119. doi: 10.1046/j.1365-2443.2001.00404.x. [DOI] [PubMed] [Google Scholar]
- 42.Brill S., Li S., Lyman C. W., et al. The Ras GTPase-activating-protein-related human protein IQGAP2 harbors a potential actin binding domain and interacts with calmodulin and Rho family GTPases. Molecular and Cellular Biology. 1996;16(9):4869–4878. doi: 10.1128/MCB.16.9.4869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Carow B., Rottenberg M. E. SOCS3, a major regulator of infection and inflammation. Frontiers in Immunology. 2014;5, article 58 doi: 10.3389/fimmu.2014.00058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liu X., Lu R., Xia Y., Wu S., Sun J. Eukaryotic signaling pathways targeted by Salmonella effector protein AvrA in intestinal infection in vivo. BMC Microbiology. 2010;10:p. 326. doi: 10.1186/1471-2180-10-326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yang L., Zhang Y.-J. Antagonizing cytokine-mediated JAK-STAT signaling by porcine reproductive and respiratory syndrome virus. Veterinary Microbiology. 2016 doi: 10.1016/j.vetmic.2016.12.036. [DOI] [PMC free article] [PubMed] [Google Scholar]