

Abstract
The SWI/SNF chromatin remodeling complex is highly conserved from yeast to human and aberrant SWI/SNF complexes contribute to human disease. The Snf5/SMARCB1/INI1 subunit of SWI/SNF is a tumor supressor frequently lost in pediatric rhabdoid cancers. We examined the effects of Snf5 loss on the composition, nucleosome binding, recruitment and remodeling activities of yeast SWI/SNF. The Snf5 subunit is shown by crosslinking-mass spectrometry (CX-MS) and subunit deletion analysis to interact with the ATPase domain of Snf2 and to form a submodule consisting of Snf5, Swp82 and Taf14. Snf5 promotes binding of the Snf2 ATPase domain to nucleosomal DNA and enhances the catalytic and nucleosome remodeling activities of SWI/SNF. Snf5 is also required for SWI/SNF recruitment by acidic transcription factors. RNA-seq analysis suggests both the recruitment and remodeling functions of Snf5 are required in vivo for SWI/SNF regulation of gene expression. Thus, loss of SNF5 alters the structure and function of SWI/SNF.
Keywords: Chromatin remodeling, SWI, SNF, Snf5
eTOC blurb
Mutation of SWI/SNF chromatin remodeling complex subunits contributes to cancer and neurological disorders. Sen et al. report that loss of the Snf5 subunit alters the architecture and function of SWI/SNF in a yeast model system. These findings may reflect changes that occur in pediatric rhabdoid tumors with mutated Snf5.
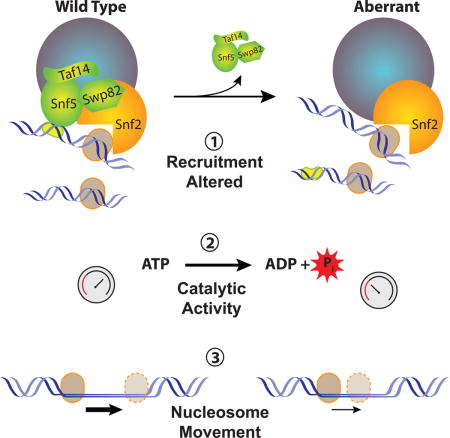
Introduction
The SWI/SNF family of ATP-dependent chromatin remodelers is evolutionarily conserved in eukaryotes from yeast to humans. SWI/SNF complexes have critical roles in controlling stem cell self-renewal, cellular differentiation, and the basic processes of gene expression, DNA replication, repair and recombination (Bartholomew, 2014; Clapier and Cairns, 2009). The key enzymatic activities of SWI/SNF are nucleosome mobilization and disassembly. Alterations in different subunits of SWI/SNF, as uncovered by genomic sequencing, have been linked to cancer and neurological diseases (Kadoch and Crabtree, 2015; Shain and Pollack, 2013; Wilson and Roberts, 2011). In many of these instances loss of a subunit creates an aberrant form of the SWI/SNF complex that is responsible for the abnormality, but the properties of these aberrant complexes remain unknown. In mammals there are potentially over 100 different forms of SWI/SNF that differ in subunit composition, thus making it difficult to understand why loss of a particular subunit through mutation results in the observed phenotype. In the model organism Saccharomyces cerevisiae, there are only two types of SWI/SNF that differ in their catalytic and accessory subunits with a small set of shared subunits. The simplicity of this system makes it easier to understand the molecular defects due to loss of a particular subunit.
The first subunit of SWI/SNF shown to be a tumor suppressor was SNF5 (SMARCB1 / INI1) (Biegel et al., 2002; Sevenet et al., 1999; Versteege et al., 1998). Since then at least six other subunits of SWI/SNF were found in different cancers to be mutated (Helming et al., 2014; Shain and Pollack, 2013). Loss of hSnf5 is a driver mutation in pediatric rhabdoid tumors and the loss of a single copy of SNF5 or Smarcb1 in mice leads to cancer in 100% of the heterozygotes (Roberts et al., 2000; Roberts et al., 2002). In these cases, the loss of Snf5 is not accompanied by genomic instability or rearrangements, but rather is an epigenetic effect that causes changes in gene expression (Hasselblatt et al., 2013; Lawrence et al., 2013; Lee et al., 2012). Also, the loss of Snf5 does not eliminate the entire SWI/SNF complex, but instead forms an aberrant complex containing the catalytic subunit Brg1 that is required for cell viability (Wang et al., 2009). It remains unclear how the loss of Snf5 affects the integrity and underlying enzymatic activity of the SWI/SNF complex due to the difficulty of SWI/SNF purification from mammalian cells. It has been suggested that the complex remains fairly intact and functional since the expression of certain known SWI/SNF target genes are unaffected (Doan et al., 2004). A recent study however has called this into question as the re-expression of Snf5 alters expression of several SWI/SNF target genes and promotes SWI/SNF recruitment to promoters (Kuwahara et al., 2013). Loss of Snf5 has also been shown to affect regulation of the sonic hedgehog and WNT/β-catenin pathways as well as expression of cyclin D1(Kim and Roberts, 2014). Studies with recombinant proteins have suggested that human Snf5 is a vital part of the enzymatic core of SWI/SNF (Phelan et al., 1999), however studies with yeast SWI/SNF find the enzymatic core consists of only the catalytic subunit and two actin related proteins (Yang et al., 2007). Deletion of Snf5 in primary human fibroblasts has similar effects on transcription and chromatin organization as does loss of Brg1 and points to Snf5 being vital for Brg1 activity. There remains a lack of understanding on how loss of Snf5 precisely impacts the enzymatic activity, recruitment and composition of SWI/SNF; which is essential for discerning the role of Snf5 in cancer.
The SWI/SNF complex is highly conserved from yeast to humans, and the human orthologs of Snf2 fully complement the loss of SNF2 in yeast. We have deleted the SNF5 gene of SWI/SNF in yeast to provide important insights into the properties of the aberrant SWI/SNF complex formed in its absence. Our data demonstrate that loss of SNF5 leads to the loss of additional SWI/SNF components and that the activity of the residual SWI/SNF complex is altered relative to the wild type enzyme. These changes in SWI/SNF activity can affect gene expression profiles and signaling pathways that are crucial in cancer development.
Results
A Snf5 submodule composed of Swp82 and Taf14 associates with the SWI/SNF complex via Snf5
The architecture of the 12 subunit yeast SWI/SNF complex was investigated by mapping inter-subunit and inter-domain contacts by crosslinking proximal lysine residues to each other using the homo-bifunctional crosslinker bis-sulfosuccinimdyl suberate (BS3). After crosslinking the free SWI/SNF complex in solution, the protein complex was proteolyzed and crosslinked peptides were fractionated and identified by mass spectrometry. The crosslinks were then mapped onto the linear sequences of the SWI/SNF subunits generating a linkage map for the complex (Fig.s EV1–3). We focused on the interactions involving the Snf5 subunit of SWI/SNF and found that Snf5 crosslinked to 6 of the 12 subunits of SWI/SNF (Fig. 1A). In addition, we deleted SNF5 and determined how SWI/SNF composition was affected by performing Snf6 affinity purification followed by SDS-PAGE and Sypro Ruby® staining. Besides Snf5, Swp82 and Taf14 were the only other SWI/SNF subunits that failed to co-purify with Snf6 (Fig. 1B). The residual complex was shown to be very stable by affinity purification with rigorous washes of 500 mM NaCl and to be a single complex by native gel shift binding assays (Fig. 1C). Consistent with the loss of Swp82 and Taf14 upon deletion of Snf5, the crosslinking-mass spectrometry (CX-MS) data shows that Snf5 extensively crosslinked to both of these subunits. The center of the YEATS domain of Taf14 is crosslinked to the N- and C-terminal halves of Snf5. The Rsc7 homology domain of Swp82 is crosslinked to a region of Snf5 from residues 150 and 174, and another region of Swp82 to the C-terminal end of the Snf5 homology domain. The Snf5 submodule of SWI/SNF consisting of Snf5, Swp82 and Taf14 appeared to be tethered to SWI/SNF through the Snf5 subunit. Deletion of Swp82 did not cause loss of Snf5 or Taf14 from the complex, suggesting Swp82 is not required for the association of Snf5 or Taf14 with SWI/SNF (Fig. 1B). Taf14 is assembled into other chromatin remodelers and transcription factors besides SWI/SNF that do not contain Snf5 or Swp82, and cannot therefore mediate the unique association of the Snf5 module to SWI/SNF(Hampsey, 1998; Peterson et al., 1998; Zhang et al., 2004). Additional deletion analysis has shown that another submodule of SWI/SNF can be formed independent of the Snf5 submodule (data not shown) that consists of Snf2, Snf6, Arp7/9, Rtt102 and Snf11.
Figure 1. Interactions of the Snf5 module in SWI/SNF.
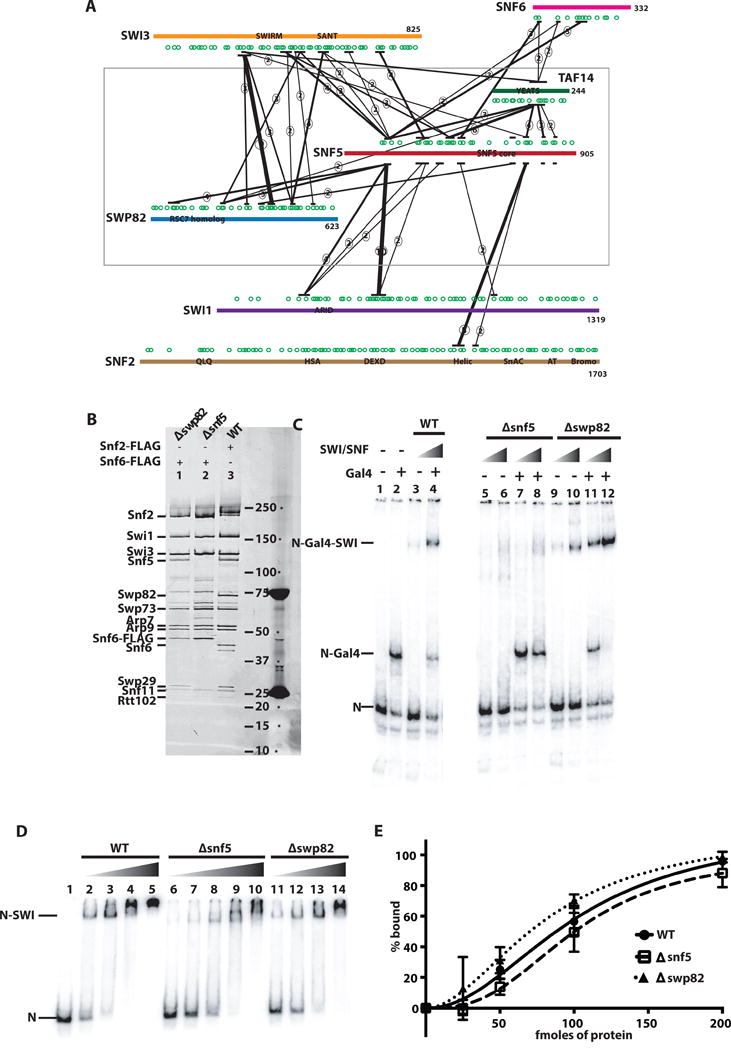
(A) The interactions of Snf5 with other subunits in the SWI/SNF complex were determined by crosslinking with the homo-bifunctional, lysine-specific crosslinker BS3 and subsequent analysis by mass spectrometry. The SWI/SNF subunits shown are Snf2 (brown), Swi1 (purple), Swi3 (orange), Swp82 (blue), Snf6 (magenta) and Taf14 (green). Lysine residues are indicated by green circles and the labeled boxes are the conserved domains. Only those interactions in which 2 or more crosslinks were detected in a region spanning ~30 amino acids are shown and the number of crosslinks detected are indicated by the circled numbers. The thickness of the black line is also indicative of the number of crosslinks detected in that region. (B) SWI/SNF complexes were purified by immuno-affinity chromatography with either Snf5 or Swp82 deleted and analyzed on a 4–20% SDS polyacrylamide gel. Wild type SWI/SNF (lane 3) contains two FLAG tags at the C-terminus of Snf2. Snf6 was tagged with a double FLAG tag at its C terminus in the Δsnf5 (lane 2) and Δswp82 (lane 1) SWI/SNF complexes. (C) Recruitment of wild type and mutant SWI/SNF by the acidic transcription activator Gal4-VP16 was tracked by gel shift analysis. Nucleosomes alone (N), nucleosomes bound by Gal4-VP-16 (N-Gal4) and nucleosomes bound by SWI/SNF with or without Gal4-VP16 (N-Gal4-SWI) migrated at the indicated positions. Gal4-VP16 was added to lanes 2, 4, 7, 8, 11, and 12. Wild type, Δsnf5 and Δswp82 SWI/SNF were add in lanes 3–4, 5–8, and 9–12, respectively. (D) Purified SWI/SNF complexes from wild type (WT) and mutant yeast strains (as indicated) were added to nucleosomes reconstituted with radiolabeled 601 DNA and Xenopus laevis histone octamers and resolved on a 4% native polyacrylamide gels. Free nucleosomes are labeled N and nucleosomes bound by SWI/SNF as N-SWI. (E) The amount of bound nucleosomes versus SWI/SNF was determined from gel shift assays as shown in (B) and plotted. The estimated KD for wild type, Δsnf5 and Δswp82 SWI/SNF were 102 ± 19, 101 ± 15 and 82 ± 21 nM (standard deviation from mean), respectively, and were from three technical replicates.
The Snf5 submodule crosslinked extensively to Swi3 (Fig. 1A), a subunit of SWI/SNF previously shown to be important for scaffolding and assembling the entire SWI/SNF complex (Yang et al., 2007). Most of these crosslinks with Swi3 were localized in or near the SWIRM and SANT domains of Swi3. The N-terminus of Snf5 is also extensively crosslinked to two regions on either side of the ARID domain of Swi1. The ARID domain is found in the ARID1A/1B/2 subunits of mammalian SWI/SNF and these interactions are likely conserved. The Taf14 and Snf5 subunits both crosslink Snf6. Not only is Snf5 required for Swp82 and Taf14 to stably associated with the SWI/SNF complex, homologs of which are not found in mammalian SWI/SNF, but Snf5 is also at the interface with several other well conserved subunits that have homologs in mammalian SWI/SNF such as the ARID1A, ARID1B, ARID2 and BAF155/SMARCC1 subunits.
Snf5 is required for SWI/SNF recruitment by acidic transcription factors
Previous data indicated that the N-terminus of Snf5 and the ARID region of Swi1 are both required for binding of SWI/SNF to acidic activation domains such as VP16 and Gcn4, but deletion of one of these domains alone was insufficient to disrupt binding (Neely et al., 2002; Prochasson et al., 2003). These studies focused primarily on GST pull-down assays to determine recruitment defects and they did not test the ability of the complex missing these domains or the entire Snf5 subunit to be recruited to DNA by these transcription factors. In contrast, we investigated the ability of SWI/SNF complexes lacking the entire Snf5 submodule (Δsnf5), and those lacking only the Swp82 subunit (Δswp82), to bind nucleosomes by gel shift assay. Both complexes were able to bind nucleosomes directly (Fig. 1C, E). SWI/SNF recruitment by the transcription factor containing the DNA binding domain of Gal4 fused to the activation domain of VP16 (Gal4-VP16) was severely affected by loss of Snf5, in contrast to those earlier studies, but not by the loss of Swp82 (Fig. 1D, compare lanes 7–8 to 3–4 and lanes 11–12 to 3–4). These data indicate that Snf5 (and possibly Taf14) is required for SWI/SNF recruitment to chromatin by acidic transcription factors, but not for the complex to directly bind to nucleosomes.
The highly conserved core of Snf5 contacts the surface of the H2A-H2B dimer in nucleosomes
In order to better understand the interactions of the Snf5 subunit when SWI/SNF binds nucleosomes, we mapped the region of Snf5 that contacts the H2A-H2B dimer at residue 109 of H2B using histone photocrosslinking. Nucleosomes are constructed with radiolabeled photocrosslinkers attached to various histone sites and a radiolabel is transferred to the subunit of SWI/SNF that is most proximal to these sites(Dechassa et al., 2008). This approach had previously shown that the Snf5 and Snf2 subunits bind near the H2A-H2B dimer, close to residue 109 of histone H2B and at a lesser extent to residues 19 and 89 of histone H2A and residue 22 of H4, but did not identify the region of Snf5 involved. The photocrosslinked Snf5 subunit from wild type SWI/SNF was gel purified and cleaved at Asn-Gly bonds with hydroxylamine (Fig. 2). Proteolytic conditions were varied to obtain partial and complete digestion (Fig. 2A). Ultimately, the sizes of the radiolabeled proteolytic fragments obtained under these different conditions are diagnostic of the location of the crosslink (Fig. 2C). Appropriate molecular weight size markers were made by expressing different Snf5 constructs representing partial and complete cleavage products using in vitro transcription coupled with translation using cell-free extracts and S-35 labeled methionine (Fig. 2B). We found that under partial cleavage conditions, two proteolytic products of 55 and 61 kDa corresponding to C-terminal fragments of Snf5 are formed due to cleavage at amino acids 358 or 410 (417). Since smaller fragments of 23 or 40 kDa were not detected, the crosslink does not appear to be at either the N- or C-terminal ends of Snf5. More extensive digestion with hydroxylamine generated a 32 kDa fragment that corresponds to the region from amino acid 410 (417) to 699. These data show that the conserved Snf5 homology domain of Snf5 binds to the region of the H2A-H2B near residue 109 of H2B when SWI/SNF stably binds nucleosomes. Yeast Snf5 is a much larger polypeptide than the mammalian Snf5 homolog, and the ~70% of yeast Snf5 that is unique to the yeast complex appears not to make the direct contact with the H2A-H2B acidic pocket. Instead the core homology region of yeast Snf5 that is in common with human SNF5 contacts histones H2A-H2B.
Figure 2. The highly conserved core domain of the Snf5 subunit contacts nucleosomes near the exposed surface of the histones H2A-H2B.
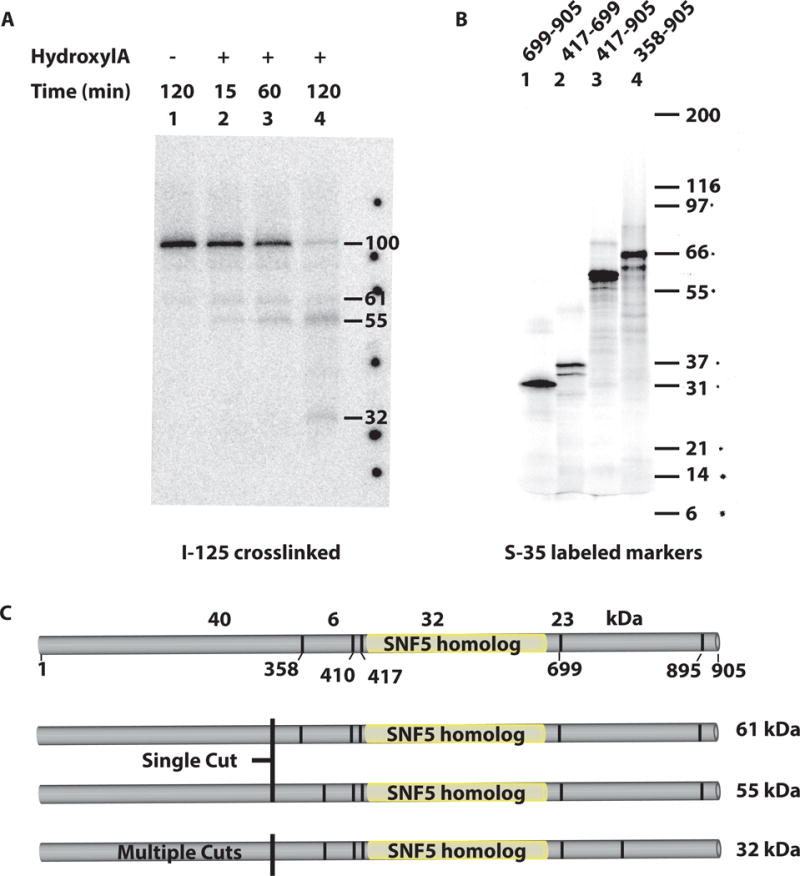
(A) SWI/SNF was recruited to nucleosomes by Gal4-VP16 in presence of competitor DNA to ensure specific SWI/SNF interaction with nucleosomes. After photocrosslinking the labeled Snf5 was isolated and cleaved at Asn-Gly positions with hydroxylamine. The cleaved products were resolved by SDS-PAGE and visualized by phosphorimaging to determine the crosslinked sites using Snf5 marker polypeptides described in section. (B) Four different regions of Snf5 were prepared by in vitro coupled transcription-translation in presence of S-35 methionine. The numbers on the top show the regions of Snf5. (C) The domain organization of Snf5 and the locations of the Asn-Gly cleavage sites are shown. The expected hydroxylamine partial and complete cleavage products are shown.
Snf5 module promotes binding of the ATPase domain of Snf2 to nucleosomal DNA
Next, we analyzed by photocrosslinking the interactions of the Δsnf5 complex with the histone octamer face that is not bound by DNA. We used histone crosslinking to determine whether the interactions between Snf2 and the surface of the H2A-H2B dimer were diminished in the absence of Snf5. SWI/SNF interactions were probed at five sites across the histone octamer face not bound by DNA including H2B109 that was previously identified as a critical site for Snf5 interaction (Fig. 3A). There was no reduction in Snf2 crosslinking at any of the five sites when Snf5 was deleted (Fig. 3B). The Swi1 subunit was modestly detected proximal to these sites only when Snf5 was absent. Our data are consistent with neither Snf5 nor the Snf5 submodule being required for the stable binding of Snf2 to this surface of the nucleosome. The overall affinity of the Δsnf5 complex for nucleosomes was thus not adversely affected compared to wild type SWI/SNF, probably because the contacts with the histone octamer are retained and likely dominate in determining the affinity of SWI/SNF for nucleosomes (Fig. 2C, E).
Figure 3. The catalytic subunit does not make stable contact with nucleosomal DNA in the absence of the Snf5 module, but does with the histone octamer surface.
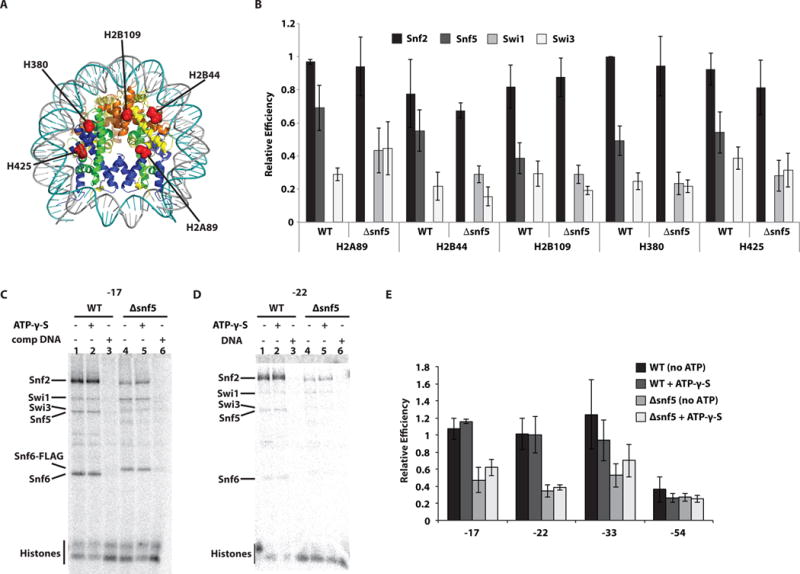
(A) The interactions of wild type (WT) and Δsnf5 SWI/SNF with nucleosomes were mapped by histone photocrosslinking with photoreactive radio-iodinated N-((2-pyridyldithio)ethyl)-4-azidosalicylamide (PEAS) attached to specific histone positions as indicated by histone type and residue position. (B) The relative crosslinking efficiency of Snf2, Snf5, Swi3 and in some instances Swi1 was plotted for each of the five histone positions and for WT and Δsnf5 SWI/SNF. Signals were normalized relative to crosslinking in WT SWI/SNF to residue 80 of histone H3 (H380). (C–D) Purified WT SWI/SNF complex was bound to nucleosomes reconstituted with radiolabeled DNA probes containing photoreactive groups at the positions indicated (“0” corresponding to the dyad). Reactions contained either wild type (lanes 1–3) or Δsnf5 (lanes 4–6) complexes. In some instances 150 μM γ-thio-ATP (lanes 2 and 5) or 13 μg/ml competitor DNA (lanes 3 and 6) were added. (E) The efficiency of Snf2 crosslinking to DNA was plotted for four different positions within nucleosomal DNA and were normalized to nt-17. These experiments were done in triplicates and the standard deviation (SD) is shown.
We next determined how loss of Snf5 changes the interactions of SWI/SNF with nucleosomal DNA by site-specific DNA photocrosslinking. In this method, a phosphorothioate is incorporated into DNA positions near where the ATPase domain of Snf2 binds and are coupled to an aryl azide (Dechassa et al., 2012; Zofall et al., 2006). We track the interactions of Snf2 and its ATPase domain by crosslinking SWI/SNF at these sites. In the wild type SWI/SNF complex, Snf2 crosslinks to DNA facing in and away from the octamer 17 and 22 nt from the dyad axis, respectively (Fig. 3C–D). Under comparable conditions with full binding of the Δsnf5 complex to nucleosomes, Snf2 crosslinking to nucleosomal DNA was reduced 2–3 fold and shows a reduction of Snf2 binding to nucleosomal DNA in the absence of Snf5 (Fig. 3E). Interestingly, the impaired crosslinking of Snf2 in the Δsnf5 complex persisted in the presence a non-hydrolyzable analog of ATP (γ-thio ATP) suggesting that this defect was irreconcilable even when ATP bound to the complex. At a position farther away from where the ATPase domain binds, 54 nt from the dyad axis, there is no difference in the interactions of Snf2 with DNA in the presence or absence of Snf5 (Fig. 3E).
Snf5 contacts and regulates the ATPase domain of Snf2
We suspected that the reduced binding of the ATPase domain to nucleosomal DNA would reduce the ATPase and remodeling activities of SWI/SNF. To better understand how loss of the Snf5 subunit might destabilize the interactions of the ATPase domain with nucleosomal DNA we first re-examined the protein-protein crosslinking data described earlier. These experiments showed that a region of Snf5 close to the Snf5 homology domain crosslinks to the helicase region of the ATPase domain of Snf2 (Fig. 1A). We reinvestigated the crosslinking data by focusing on the subunits of SWI/SNF that crosslink to the Snf2 catalytic subunit and are likely to directly interact. As expected, we found that the Arp7/Arp9/Rtt102 module crosslinks to the HSA domain as shown previously (Fig. 4 and references (Schubert et al., 2013; Szerlong et al., 2008)). The regions of these three proteins that crosslinked to HSA correlated well with those regions of Arp7/Arp9/Rtt102 expected to interact with HSA based on its crystal structure (Schubert et al., 2013). We also identified other subunits not previously known to interact with the HSA domain. The N-terminus of Snf6 and a region spanning residues 1051 to 1064 of Swi1 were proximal to the HSA domain and crosslinked to HSA. The Snf6 and Swi1 subunits were likely not observed previously because they assemble into SWI/SNF independent of any interactions with the HSA domain. In addition a region near the SWIRM domain of Swi3 crosslinked near the AT hook of Snf2, and a region of Swi1 near the ARID domain crosslinked to the SnAC domain of Snf2. These interactions likely also occur in mammalian SWI/SNF given that these regions are conserved between the yeast and mammalian SWI/SNF complexes.
Figure 4. Snf5 contacts the ATPase domain of the catalytic subunit.
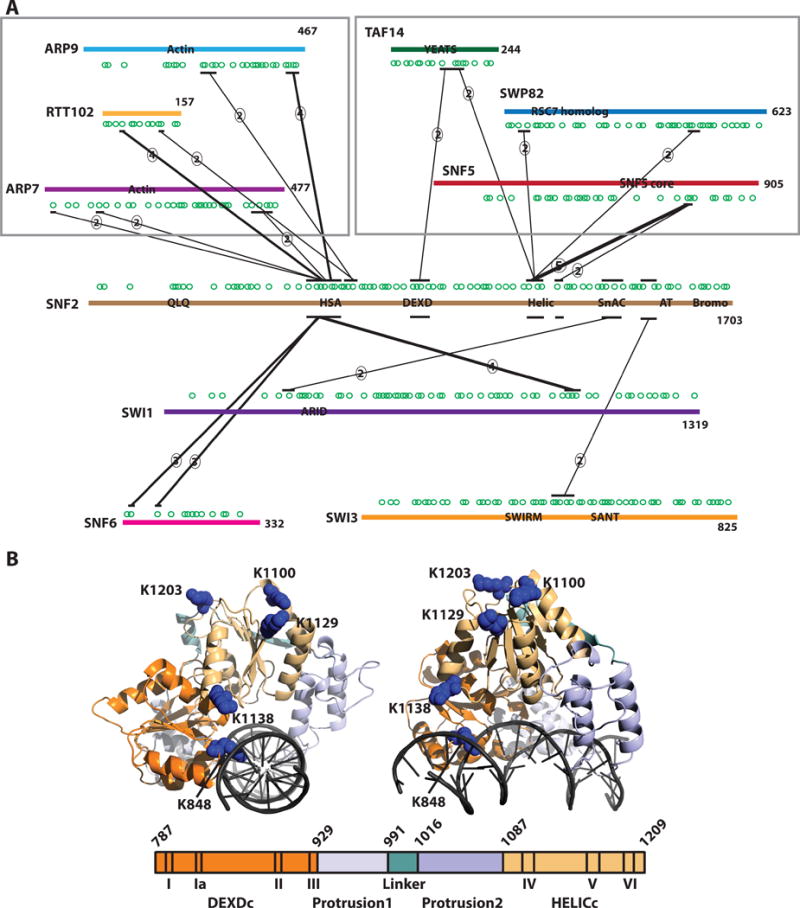
(A) The interactions of Snf2 with other SWI/SNF subunits were examined in the same manner as described for Snf5 in Figure 3. Besides the known interactions of the Arp7, Arp9 and Rtt102 module there also are interactions of the Snf5, Swp82 and Taf14 module with Snf5. Only those crosslinks in which 2 or more crosslinks were detected in a region spanning ~30 amino acids are shown. The total number of crosslinks detected is indicated by the circled number. (B) The ATPase domain of Snf2 bound to DNA was modeled based on the sequence similarity with the ATPase domain of RAD54 (Dechassa et al., 2012). The lysine residues crosslinked to the Snf5 subunit are indicated as blue space filling residues and are primarily in the HELIC lobe of the ATPase domain. Domains are color coded as indicated in the schematic at the bottom of the figure.
The Snf5 submodule crosslinks primarily to the ATPase domain of Snf2, specifically to the helicase domain and to a lesser extent to the DEXD box domain (Fig. 4A and B). The most frequent crosslinks are between the region of Snf5 flanking the Snf5 homology domain and the helicase domain. The only other subunits that crosslinked to the ATPase domain were components of the Snf5 submodule, suggesting that the Snf5 submodule might have a unique role in regulating the activity of the ATPase domain through direct interactions. Based on the location of the Snf5 crosslinks on the surface of the ATPase domain, Snf5 doesn’t appear to regulate the ATPase domain by blocking access to the groove where DNA binds (Fig. 4B). Additionally, the other subunits in the Snf5 module, Swp82 and Taf14 also contacted the ATPase domain further supporting that this module functions cooperatively to modulate the activity of SWI/SNF.
The catalytic efficiencies of wild type and Δsnf5 complexes were determined by measuring the rate of ATP hydrolysis at different ATP concentrations and 5-fold molar excess of mononucleosomes or nucleosomal arrays over SWI/SNF to ensure saturating conditions. Under these conditions, the Δsnf5 complex was not as efficient at hydrolyzing ATP as the wild type SWI/SNF, consistent with the reduced binding of the helicase domain with nucleosomal DNA as previously observed (Fig. 5A–B compared to Fig. 5C–E). The Michaelis-Menten kinetic analysis revealed that the affinity for ATP was unchanged, but the kcat or catalytic efficiency was reduced 2 to 3 fold depending on the substrate (Table EV3). The primary interactions of the Snf5 subunit observed by protein crosslinking and mass spectrometry with the helicase domain of the Snf2 subunit are consistent with Snf5 regulating either the ATPase or DNA translocase activities of SWI/SNF rather than an ability to bind ATP. The reduction in activity was not due to any differences in nucleosome affinity as saturating amounts of nucleosomes were used. Reduced ATPase activity was also detected with free DNA and the Δsnf5 complex, thus showing that the deficiency is not nucleosome specific, even though the effect is less pronounced with DNA than with nucleosomes (Fig. 5C and Table EV3).
Figure 5. Snf5 regulates the catalytic activity of the ATPase domain of SWI/SNF.
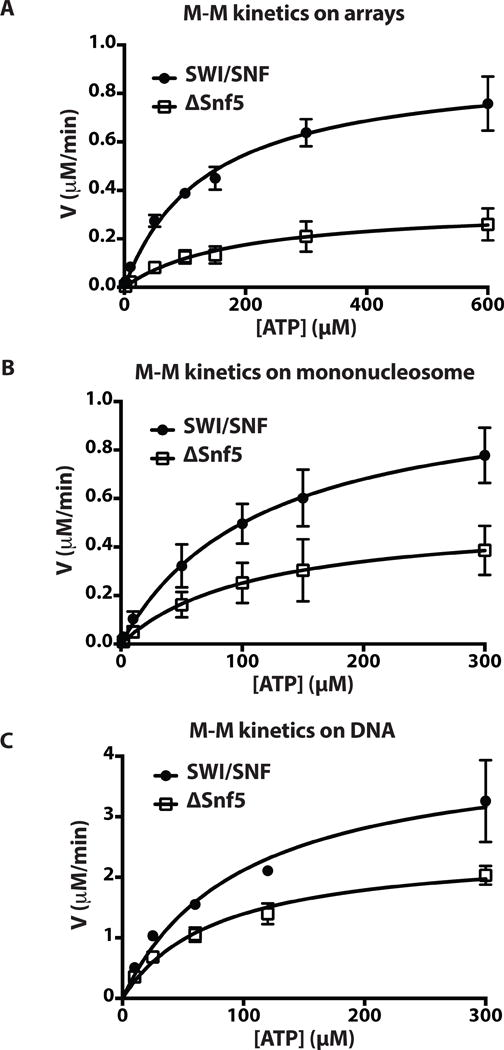
The rate of ATP hydrolysis (V) was determined at different concentrations of ATP and plotted as a function of ATP concentration. KM and kcat values were obtained by fitting data to the Michaelis-Menten equation using GraphPad (PRISM Version 6.0b). Concentrations of substrates and enzymes were (A) 8 nM nucleosomal array (12 repeats of 200bp), 1.6 nM enzyme (WT/Δsnf5) and 2–600 μM ATP; (B) 8 nM 29N59 nucleosome, 1.6 nM enzyme, and 2–300 μM ATP; and (C) 50 nM 100 bp DNA, 10 nM enzyme, and 10–300 μM ATP. Error bars and reported values are mean ± SE from three technical replicates.
Next, we examined the effects of loss of Snf5 on SWI/SNF mobilization of nucleosomes mobilization by mapping DNA movement through the nucleosomes. Here, movement was tracked using a photoactive reporter attached to the histone octamer 54 bp from the dyad axis and DNA is cleaved after crosslinking the histone to DNA. Snapshots are taken at different times to view DNA as it moves past this point in the octamer. Nucleosomes were complelely bound in the remodeling assays and and ATP (4.4 μM) was limiting in order to examine one round of remodeling. The rate at which nucleosomes were moved from their starting position, as evidenced by reduction of DNA cleavage at the reference point 0, was 3 times slower for the Δsnf5 complex than the wild type complex (Fig. 5A–C and Fig. EV5A). By measuring the rate of ATP hydrolysis under these remodeling conditions, we found there was no significant difference in the rate of ATP hydrolysis between the wild type and Δsnf5 complexes, and the defect in remodeling was not coupled to ATP hydrolysis (Fig. 6D). Also without Snf5 SWI/SNF appears to be less processive as can be seen by the slower rate at which longer movements appear in the Δsnf5 complex (Fig. 6A–B and Fig. EV5B–C). Together these data are consistent with Snf5 helping to couple ATP hydrolysis to DNA movement.
Figure 6. Loss of the Snf5 subunit adversely affects the intrinsic nucleosome mobilizing activity of SWI/SNF.
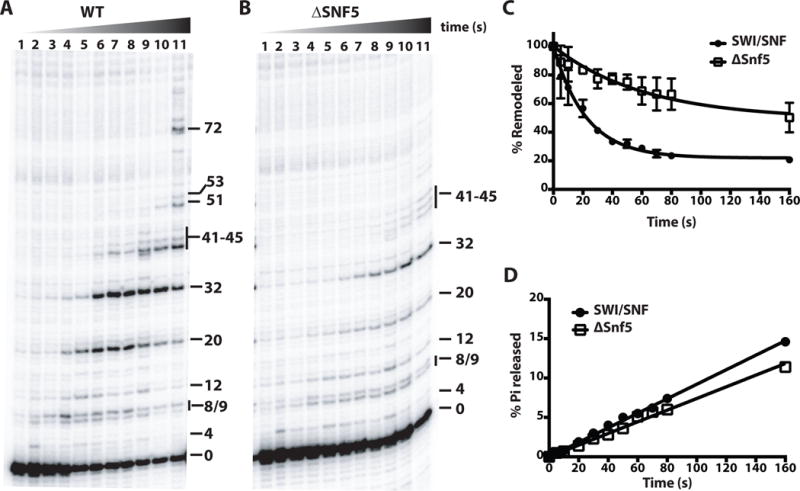
(A–B) Nucleosomes modified at residue 53 of histone H2B were used for monitoring movement of DNA on the octamer surface. DNA cleavage products were resolved on a denaturing 6% polyacrylamide gel and visualized by phosphorimaging. Numbers on the right side of the gel image refer to number of nucleotides (nt) moved from the starting cleavage position (0). Nucleosomes were remodeled with WT (A), and Δsnf5 SWI/SNF (B) for 0, 5, 10, 20, 30, 40, 50, 60, 70, 80 and 160 s using 4.4 μM ATP. (C) The amount of DNA cleaved at starting position (0) was plotted versus time for WT SWI/SNF and Δsnf5 SWI/SNF. Rate constants (k) obtained by fitting data to single exponential function were 0.044± 0.004 s−1 (WT) and 0.016 ± 0.005 s−1 (Δsnf5). Bars and reported values are the mean ± SE from two replicates. (D) ATPase assays were performed in the same conditions as (A and B) and the released radioactive phosphate (Pi) was separated from non-hydrolyzed ATP by thin layer chromatography (TLC). The percentage Pi released is plotted as a function of time. ATP hydrolysis rates obtained were 0.9 μM s−1 (WT) and 0.7 μM s−1 (Δsnf5).
The in vivo role of Snf5 in regulation of gene expression
We examined changes in gene expression at the whole-genome level by RNA-seq analysis with deletion of SNF2, SNF5 or SWP82. The RNA-seq data was analyzed by filtering for only the significant changes in expression observed from the wild type to mutant strains that were 2 fold or greater, p value of <0.05 and were reproducible in biological replicates (Fig. 7 and EV6). The similarities are most striking between wild type and Δswp82 and between Δsnf2 and Δsnf5. These data suggest in most instances, except for a small subset of genes, Swp82 is not important for regulation by SWI/SNF, and similarly Snf5 is important for the majority of SWI/SNF regulated genes. The similarity between Δsnf2 and Δsnf5 could be due to the absence of the complex being equivalent to no SWI/SNF recruitment or the reduction in SWI/SNF activity caused by loss of Snf5. It is therefore noteworthy that there are differences between Δsnf2 and Δsnf5, which suggests the absence of SWI/SNF or its catalytic subunit is not completely equivalent to that when Snf5 is absent.
Figure 7. In vivo targets affected by loss of Snf5 both overlap and are distinct from that of Snf2.
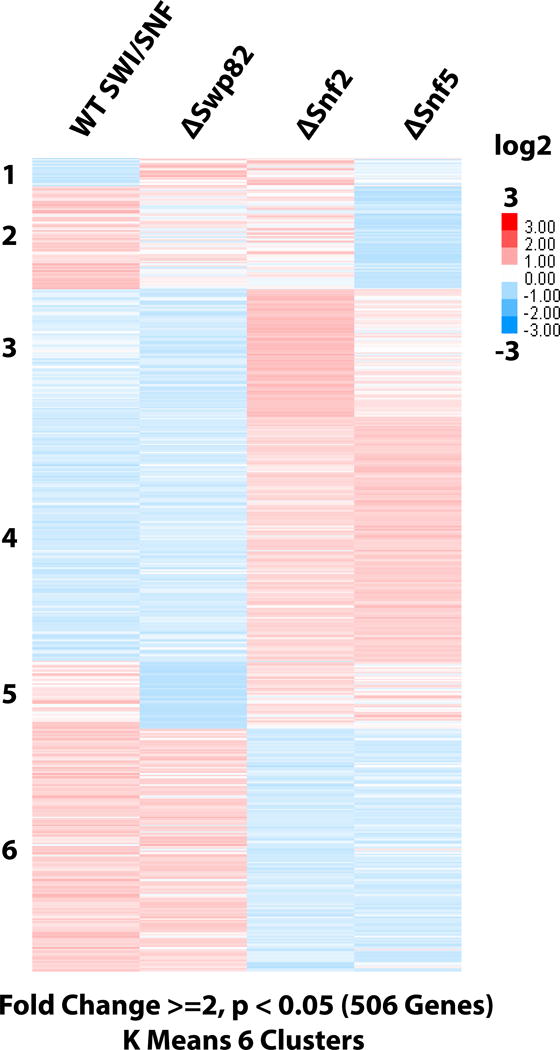
(A) RNA-seq heat map depicts those genes with fold change >= 2 in expression (506) and satisfy the parameters of a p < 0.05. Genes were grouped into 6 clusters using k-Means clustering. A total of 5,935 genes were analyzed.
Differences between the Δsnf2 and Δsnf5 strains might be expected if without Snf5 SWI/SNF is still recruited to a subset of SWI/SNF targets, for example SWI/SNF recruitment via histone acetylation at select promoters (Chatterjee et al., 2011; Hassan et al., 2002; Mitra et al., 2006). Recent work from Jerry Workman and colleagues has shown that loss of Snf5 alters but does not eliminate recruitment of the remaining SWI/SNF complex formed in its absence (manuscript currently under review). The aberrant complex formed and recruited in the absence of Snf5 might function like a dominant negative mutation by blocking sites from other chromatin remodelers while still being non-functional, or by altering chromatin nonetheless to a lesser extent due to its reduced remodeling activity. The genes in cluster 3 appeared to be repressed by SWI/SNF and were activated when Snf2 is absent (Fig.7). The effects of loss of Snf5 on the genes in cluster 3 were not as activating as the loss of Snf2 possibly because the aberrant Δsnf5 recruited to these sites still remodels chromatin, but to a lesser extent. Cluster 2 of Figure 7 shows the loss of Snf5 to be more repressive than the loss of Snf2 where wild type SWI/SNF appears to be activating transcription. In this case the defective SWI/SNF complex could still be recruited and block binding of other remodelers that would otherwise compensate for the loss of SWI/SNF. These differences suggest that in addition to being defective for transcription factor mediated recruitment there may be cases in which the Δsnf5 complex is properly recruited, but because of the reduced remodeling activity is distinct from that when the complex is completely absent. GO term analysis of the genes with significantly altered expression when Snf5 is deleted shows that loss of Snf5 preferentially down regulates genes involved in DNA integration and recombination, and up regulates genes involved in a variety of metabolic processes (Table EV4 and EV5). These types of changes in gene expression could be consistent with the observed role of Snf5 as a tumor suppressor in rhabdoid tumors.
Discussion
Our results show the modular organization of the SWI/SNF complex and the effects of loss of the Snf5 subunit on the properties of yeast SWI/SNF including subunit composition, recruitment by acidic transcription factors, ATPase and nucleosome remodeling activities and gene expression. The aberrant SWI/SNF complex formed in the absence of Snf5 has several critical differences from the wild type enzyme. First, the subunit composition of SWI/SNF is affected by more than just the loss of Snf5 per se as Snf5 associates with several other subunits and is key in stabilizing the association of the Swp82 and Taf14 subunits with the complex. Although the role of the Swp82 subunit is less clear in the SWI/SNF complex, its homolog Rsc7 has been shown to form a critical module with Rsc3, Rsc30 and Htl1 of the RSC complex, a paralog of SWI/SNF, and loss of Rsc7 causes increased sensitivity to DNA damage and cell stress (Wilson et al., 2006). Taf14 is a protein shared by several chromatin remodeling factors such as NuA3, INO80, TFIID and TFIIF complexes. At the N terminus of Taf14 is the YEATS domain that binds to acetylated or crotonylated lysine 9 of histone H3 (H3K9ac or H3K9cr). Taf14 is important for gene transcription and DNA damage repair (Andrews et al., 2016; Shanle et al., 2015; Zhang et al., 2011). Although homologs of Swp82 and Taf14 are not found in mammalian SWI/SNF, other subunits are likely to be lost in human SWI/SNF due to the extensive bridging activity of Snf5 observed in the yeast SWI/SNF complex.
The Snf5 module also serves two important roles in SWI/SNF recruitment by facilitating in recognition of active histone marks (H3K9ac and H3K9cr) and recognition by particular transcription factors. Earlier data found the N-terminus of Snf5 and a portion of Swi1 together are required for SWI/SNF recruitment by particular transcription factors (Ferreira et al., 2009; Prochasson et al., 2003). In these studies loss of the N terminus of Snf5 alone was insufficient and did not have a strong effect on recruitment. Our data suggest that other parts of Snf5 may also contribute to recruitment as the loss of Snf5 alone and not Swp82 blocked recruitment by a transcription factor containing the VP16 acidic activation domain. In the absence of Snf5, SWI/SNF retains its affinity for nucleosomes and the potential for the Δsnf5 complex being recruited by other mechanisms to some of the SWI/SNF normal targets. ChIP-seq data from the laboratory of Jerry Workman shows that the Δsnf5 complex is recruited to some but not all of the SWI/SNF targets, consistent with our results (in this same issue).
There have been discrepancies as to what constitutes the minimal enzymatic core of the SWI/SNF complex. Some have suggested the Snf2 catalytic subunit with the Arp7 and Arp9 subunits can account for 90% of the enzymatic activity of the entire SWI/SNF complex (Yang et al., 2007). In other experiments with mammalian SWI/SNF, reconstitution with recombinant subunits has suggested the minimal core complex includes Snf5, Brg1 (homolog of Snf2) and BAF155/170 (homologs of Swi3) (Phelan et al., 1999). Although the differences between these two studies could be partially reconciled due to differences between the yeast and human SWI/SNF complexes, our results show that Snf5 is a vital part of the enzymatic core of the yeast SWI/SNF complex much like that shown for the human SWI/SNF complex. Our approach is different than those previously, because we did not attempt to show the role of Snf5 by trying to reconstitute the core enzyme with recombinant proteins. Instead we took a three-pronged approach, first showing the effects of loss of Snf5 on the activity of SWI/SNF in terms of ATPase activity and DNA movement inside of nucleosomes. Second, we provide evidence that Snf5 directly contacts the helicase lobe of Snf2 and can therefore directly regulate the activity of Snf2. And third, the loss of enzymatic activity is associated with reduced binding of the ATPase domain of Snf2 to nucleosomal DNA as shown by mapping SWI/SNF interactions with DNA crosslinking. In other experiments with cryo-electron microscopy, SWI/SNF is observed to bind nucleosomes in a pincer manner with one side of the pincer consisting of Snf5 and the ATPase domain of Snf2, consistent with the vital role of Snf5 in the enzymatic activity of SWI/SNF (manuscript in preparation).
We confirm the importance of the Snf5 subunit for the in vivo activity of SWI/SNF and the validity of our biochemical analysis by RNA-seq. We find that loss of Snf5 affects many of the same targets as loss of the Snf2 catalytic subunit, but nonetheless are not identical. These results mirror much of the same effects seen when BRG1 or SNF5 are separately knocked out in primary human fibroblasts and suggests that we may be examining effects in yeast that are similar to that when Snf5 is absent in humans (Tolstorukov et al., 2013). In some instances loss of Snf5 in yeast has a more severe effect than the loss of the complex caused by deletion of SNF2, consistent with the aberrant, less active complex being recruited to these sites and having a more pronounced effect than its mere absence. These effects are suggested to depend on the genomic landscape of the targets as not all targets behave in this manner. In summary, we have shown that the loss of a single subunit such as Snf5 from SWI/SNF can have pleiotropic effects on SWI/SNF, thereby affecting its subunit composition, recruitment to chromatin, and enzymatic activities; making truly an altered form of the complex.
Experimental Procedures
Purification of SWI/SNF complexes
SWI/SNF complexes with FLAG tagged subunits were purified by immunoaffinity chromatography using anti-FLAG M2 agarose beads (Dechassa et al., 2008). The activities of the purified complexes were tested with binding, remodeling and ATPase assays (Sen et al., 2013). Different preparations of the Δsnf5 complex were purified and compared to ensure a lack of variation in enzymatic activity between preparations.
Nucleosome reconstitution and gel shift assays for binding and recruitment by Gal4-VP16
Mononucleosomes were reconstituted with 5.2 μg of PCR-generated 29N59 DNA (29 and 59 bp of flanking DNA on either side of 601), 100 fmol 32P-labeled 29N59 DNA, and 9 μg wild-type Xenopus laevis refolded octamer at 37°C by a rapid salt dilution method.
For standard binding gel shift assays, SWI/SNF (1.6 to 26 nM in two-fold increments) was titrated into binding reactions containing 6.4 nM radiolabeled mononucleosomal templates in 20mM Na HEPES, 3mM MgCl2, 0.08% NP40, 1.7% glycerol, 60mM NaCl and 200uM PMSF at 30°C for 30 minutes. For recruitment assays, 4.3 nM Gal4-VP16 was bound to 6.4 nM radiolabeled mononucleosomes in presence of 25 ng sonicated salmon sperm competitor DNA for 30 minutes at 30°C. SWI/SNF (13 or 26 nM) was added and incubated at 30°C for 30 minutes. The bound products were resolved on a 4% native polyacrylamide gel (acrylamide:bisacrylamide 79:1) in 0.5X TBE at 4°C and visualized by autoradiography.
ATPase assays
ATPase assays were performed under conditions identical to the remodeling time-course assays (see section on Site-directed mapping) with 0.055 μCi [γ-32P]ATP in a 13.5 μl reaction volume. Reactions were terminated by addition of EDTA and SDS to a final concentration of 37.5 mM and 2% respectively. Reactions were spotted onto a polyethyleneimine cellulose plate (J.T. Baker) and developed with 0.5 M LiCl and 0.5 M formic acid.
Michaelis-Menten kinetics
ATPase assays were performed under multiple turnover conditions with excess nucleosomes (mononucleosomes or 200–12 nucleosomal arrays or DNA) compared to SWI/SNF complex (5:1 molar ratio). ATP concentration was varied from 2–600 μM and non-linear regression analysis of the velocity vs. ATP concentration curves was used to estimate Km and kcat.
DNA-photoaffinity labeling
DNA photoaffinity labeling was performed as described under normal binding conditions (Dechassa et al., 2012). Relative crosslinking was estimated by normalizing the Snf2 band intensity at all positions to the intensity at the-17 position.
Histone cross-linking and label transfer
Histone crosslinking and label transfer was performed as described with the crosslinked products resolved on a 4–20% SDS PAGE and visualized by autoradiography (Dechassa et al., 2008). The intensity of the cross-linked Snf2 band across all samples was quantified relative to the intensity at the H380 position in the WT SWI/SNF complex.
RNA-seq analysis
Yeast strains (wild type and mutants) in the BY4741 background were grown to an OD600 of approximately 0.75 – 1.0 in three independent biological replicates. Total RNA was extracted using the hot acid phenol method (Collart and Oliviero, 2001). Samples were phenol/chloroform extracted and ethanol precipitated before resuspension in 50 μl of diethylpyrocarbonate treated water. After checking the quality of RNA using an Agilent bioanalyzer 2100, a library was prepared using Truseq Stranded mRNA LT Sample Preparation kit (Illumina, RS-122-2101/2102). Initial extension steps of cluster generation were performed on cBot (Illumina, San Diego, CA) using TruSeq Rapid Duo cBot Sample Loading Kit (Illumina) followed by additional cluster generation steps and 76 bp paired end sequencing using an Illumina HiSeq 2500 platform on rapid run mode. Fragments were mapped to the yeast reference genome sacCer3, using TopHat V2.0.10 and Bowtie V2.1.0. Differential gene expression analysis was performed using EdgeR 3.8.6. Data was filtered using fold change >=2 and p < 0.05. The expression values were log transformed and normalized (mean of 0 and standard deviation of 1) using Cluster V 3.0 and grouped by k-Means clustering (6 clusters). Heat maps were generated using Java TreeView V 1.1.6r4. Gene Ontology (GO) enrichment analysis was performed using GORILLA (Eden and Navon, 2009).
Site-directed mapping
Histone octamers containing cysteine at residue 53 of H2B were conjugated to p-azido phenacyl bromide (APB) immediately after the octamer refolding process. Site-directed histone-DNA crosslinking was performed as described previously (Kassabov and Bartholomew, 2004). Nucleosomes were bound with saturating amount of SWI/SNF at 30°C. Nucleosome mobilization was performed using 4.4 μM ATP at 25°C and stopped with excess EDTA (15mM). Lane intensities were normalized for loading bias using Microsoft Excel. Normalized band intensities were plotted as a function of time and fitted into single phase exponential equation using GraphPad (PRISM Version 6.0b). Overlays for different time points were made using the ggplot2 package in R.
Peptide mapping
Wild type SWI/SNF was bound to nucleosomes that had been labeled at residue 109 of histone H2B with 125I-PEAS as described previously (Dechassa et al., 2008). After photocrosslinking, the subunits were separated on a 6% SDS-PAGE and the crosslinked Snf5 was visualized by phosphorimaging. Radiolabeled Snf5 was excised from the gel and isolated by electroelution with 50 mM NH4HCO3 and 0.1% SDS (Dechassa et al., 2012). Markers of the proteolytic fragments of Snf5 were made using an in vitro coupled rabbit reticulocyte lysate transcription-translation system from Promega. DNA encoding the different fragments of Snf5 were constructed to contain an upstream T7 promoter and a downstream T7 terminator by PCR amplification of the desired region of Snf5.
Crosslinking and mass spectrometry analysis
SWI/SNF was purified using a Snf2 FLAG-tagged strain as described previously (Kapoor et al., 2015). The complex was further purified over an SP Sepharose column to remove FLAG peptide before crosslinking with BS3 (Thermo Scientific). BS3 was added to 500 μl of purified SWI/SNF (174 fmol/μl) to a final concentration of 2 mM or 5 mM. After 2 hours at RT, the reactions were quenched by addition of 20 μl 1M ammonium bicarbonate, and the proteins were precipitated by addition of TCA to 16.7%. Proteins were resuspended in 100 μl 8 M urea in 2 M ammonium bicarbonate prior to reduction with 10 mM TCEP (tris(2-carboxyethyl)phosphine), and alkylation with 20 mM iodoacetamide. The urea concentration was reduced to 1 M by addition of 100 mM ammonium bicarbonate and trypsin was added at a 20:1 (weight:weight) ratio) for an overnight incubation at 37°C. The resulting peptides were desalted on a C18 column and then fractionated by microcapillary strong cation exchange chromatography (200 μm I.D. X 20 cm). Peptides were loaded onto the column and washed with Buffer A [20% acetonitrile (ACN), 0.1% formic acid (FA)]. Peptides were eluted in 4 steps by increasing the percentage of Buffer B (20% ACN, 1.0 M ammonium formate, pH 3.5): 30% B, 50% B, 70% B and 100%B. A final elution with 10% ACN, 500 mM ammonium acetate was also used. Each fraction was dried and analyzed directly by mass spectrometry.
Peptides were analyzed on a Thermo Scientific Orbitrap Elite with HCD fragmentation and serial MS events that included one FTMS1 event at 30,000 resolution followed by 10 FTMS2 events at 15,000 resolution. Other mass spectrometry settings include: charge state rejection enabled, unassigned reject, charge state 1+, 2+ and 3+ rejected, 4+ and over not rejected; HCD isolation window 3Da, normalize energy 35%; MS mass range: 1500–1000000, use m/z value as masses enabled; FTMS MSn AGC target 5e5, FTMS MSn max ion time 300 ms. An 80 min gradient from 5% ACN, 0.1% FA to 40% ACN, 0.1% FA was used for reverse phase chromatography.
The RAW files were converted to mzXML files. We used the Comet search engine and the Trans-Proteomic Pipline (TPP, http://tools.proteomecenter.org/wiki/index.php?title=Software:TPP) for the identification of unmodified and BS3 mono-modified (monolinks) peptides. For crosslinked peptide searches, we used two different database search algorithms: pLink (Yang et al., 2012) and our in-house designed Nexus with default settings (Knutson et al., 2014) to search a protein database containing the sequences of the 12 SWI/SNF subunits and their reverse decoy sequences. A 5% false discovery rate (FDR) was used for both pLink and Nexus searches. After performing the pLink and Nexus searches, the results were combined and each spectrum was manually evaluated for the quality of the match to each peptide using the COMET/Lorikeet Spectrum Viewer (TPP). Crosslinked peptides were considered confidently identified if at least 4 consecutive b or y ions for each peptide were observed and the majority of the observed ions are accounted for. Twenty intralinked spectra, out of total 3561 spectra (0.56%), corresponding to 20 intralinked peptides, and 151 interlinked spectra, out of total 1781 (8.5%), corresponding to 130 interlinked peptides were removed due to poor spectral support.
Supplementary Material
Highlights.
Transcription defects due to the loss of Snf5 are not the same as the loss of SWI/SNF
Snf5 has a scaffolding role and is required for TAF14 and Swp82 to be in SWI/SNF
Snf5 interacts with the ATPase domain of Snf2 and promotes its binding to DNA
Snf5 is essential for SWI/SNF recruitment by the Gal4-VP16 transcription factor
Acknowledgments
We would like to thank Briana Denehey at MD Anderson Cancer Center for critical reading and feedback on the manuscript, and Soumyadipta Kundu for CX-MS graphics. We want to acknowledge the support provided by the National Institutes of Health R01 GM048413 (BB), P50 GM076547 (JR) and R21 CA175849 (JR). This study also made use of the Science Park NGS Core, supported by CPRIT Core Facility Support Grant RP120348.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Accession numbers
The RNA-seq data has been deposited at GEO and the accession number is GSE85460.
Author contributions
P.S. and B.B. designed the study; P.S., M.L.D., J.P., J.R. and B.B. developed all methods used in the study; P.S., A.H., S.H., J.L., S.B., S. P., M.L.D. and J.P. performed the wet-lab experiments, A.H. and S.H. performed bioinformatics analyses, P.S. and B.B. wrote the manuscript.
Conflict of interest
The authors declare no conflict of interest.
References
- Andrews FH, Shinsky SA, Shanle EK, Bridgers JB, Gest A, Tsun IK, Krajewski K, Shi X, Strahl BD, Kutateladze TG. The Taf14 YEATS domain is a reader of histone crotonylation. Nat Chem Biol. 2016;12:396–398. doi: 10.1038/nchembio.2065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartholomew B. Regulating the chromatin landscape: structural and mechanistic perspectives. Annual review of biochemistry. 2014;83:671–696. doi: 10.1146/annurev-biochem-051810-093157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biegel JA, Kalpana G, Knudsen ES, Packer RJ, Roberts CW, Thiele CJ, Weissman B, Smith M. The role of INI1 and the SWI/SNF complex in the development of rhabdoid tumors: meeting summary from the workshop on childhood atypical teratoid/rhabdoid tumors. Cancer research. 2002;62:323–328. [PubMed] [Google Scholar]
- Chatterjee N, Sinha D, Lemma-Dechassa M, Tan S, Shogren-Knaak MA, Bartholomew B. Histone H3 tail acetylation modulates ATP-dependent remodeling through multiple mechanisms. Nucleic Acids Res. 2011 doi: 10.1093/nar/gkr535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clapier CR, Cairns BR. The biology of chromatin remodeling complexes. Annual review of biochemistry. 2009;78:273–304. doi: 10.1146/annurev.biochem.77.062706.153223. [DOI] [PubMed] [Google Scholar]
- Collart MA, Oliviero S. Preparation of yeast RNA. Curr Protoc Mol Biol. 2001:12. doi: 10.1002/0471142727.mb1312s23. Chapter 13, Unit13. [DOI] [PubMed] [Google Scholar]
- Dechassa ML, Hota SK, Sen P, Chatterjee N, Prasad P, Bartholomew B. Disparity in the DNA translocase domains of SWI/SNF and ISW2. Nucleic Acids Res. 2012 doi: 10.1093/nar/gks007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dechassa ML, Zhang B, Horowitz-Scherer R, Persinger J, Woodcock CL, Peterson CL, Bartholomew B. Architecture of the SWI/SNF-Nucleosome Complex. Mol Cell Biol. 2008 doi: 10.1128/MCB.00693-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doan DN, Veal TM, Yan Z, Wang W, Jones SN, Imbalzano AN. Loss of the INI1 tumor suppressor does not impair the expression of multiple BRG1-dependent genes or the assembly of SWI/SNF enzymes. Oncogene. 2004;23:3462–3473. doi: 10.1038/sj.onc.1207472. [DOI] [PubMed] [Google Scholar]
- Ferreira ME, Prochasson P, Berndt KD, Workman JL, Wright AP. Activator-binding domains of the SWI/SNF chromatin remodeling complex characterized in vitro are required for its recruitment to promoters in vivo. FEBS J. 2009;276:2557–2565. doi: 10.1111/j.1742-4658.2009.06979.x. [DOI] [PubMed] [Google Scholar]
- Hampsey M. Molecular genetics of the RNA polymerase II general transcriptional machinery. Microbiol Mol Biol Rev. 1998;62:465–503. doi: 10.1128/mmbr.62.2.465-503.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassan AH, Prochasson P, Neely KE, Galasinski SC, Chandy M, Carrozza MJ, Workman JL. Function and selectivity of bromodomains in anchoring chromatin-modifying complexes to promoter nucleosomes. Cell. 2002;111:369–379. doi: 10.1016/s0092-8674(02)01005-x. [DOI] [PubMed] [Google Scholar]
- Hasselblatt M, Isken S, Linge A, Eikmeier K, Jeibmann A, Oyen F, Nagel I, Richter J, Bartelheim K, Kordes U, et al. High-resolution genomic analysis suggests the absence of recurrent genomic alterations other than SMARCB1 aberrations in atypical teratoid/rhabdoid tumors. Genes, chromosomes & cancer. 2013;52:185–190. doi: 10.1002/gcc.22018. [DOI] [PubMed] [Google Scholar]
- Helming KC, Wang X, Roberts CW. Vulnerabilities of mutant SWI/SNF complexes in cancer. Cancer Cell. 2014;26:309–317. doi: 10.1016/j.ccr.2014.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadoch C, Crabtree GR. Mammalian SWI/SNF chromatin remodeling complexes and cancer: Mechanistic insights gained from human genomics. Sci Adv. 2015;1:e1500447. doi: 10.1126/sciadv.1500447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapoor P, Bao Y, Xiao J, Luo J, Shen J, Persinger J, Peng G, Ranish J, Bartholomew B, Shen X. Regulation of Mec1 kinase activity by the SWI/SNF chromatin remodeling complex. Genes Dev. 2015;29:591–602. doi: 10.1101/gad.257626.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim KH, Roberts CW. Mechanisms by which SMARCB1 loss drives rhabdoid tumor growth. Cancer genetics. 2014 doi: 10.1016/j.cancergen.2014.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knutson BA, Luo J, Ranish J, Hahn S. Architecture of the Saccharomyces cerevisiae RNA polymerase I Core Factor complex. Nat Struct Mol Biol. 2014;21:810–816. doi: 10.1038/nsmb.2873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuwahara Y, Wei D, Durand J, Weissman BE. SNF5 reexpression in malignant rhabdoid tumors regulates transcription of target genes by recruitment of SWI/SNF complexes and RNAPII to the transcription start site of their promoters. Mol Cancer Res. 2013;11:251–260. doi: 10.1158/1541-7786.MCR-12-0390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence MS, Stojanov P, Polak P, Kryukov GV, Cibulskis K, Sivachenko A, Carter SL, Stewart C, Mermel CH, Roberts SA, et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature. 2013;499:214–218. doi: 10.1038/nature12213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee RS, Stewart C, Carter SL, Ambrogio L, Cibulskis K, Sougnez C, Lawrence MS, Auclair D, Mora J, Golub TR, et al. A remarkably simple genome underlies highly malignant pediatric rhabdoid cancers. The Journal of clinical investigation. 2012;122:2983–2988. doi: 10.1172/JCI64400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitra D, Parnell EJ, Landon JW, Yu Y, Stillman DJ. SWI/SNF binding to the HO promoter requires histone acetylation and stimulates TATA-binding protein recruitment. Mol Cell Biol. 2006;26:4095–4110. doi: 10.1128/MCB.01849-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neely KE, Hassan AH, Brown CE, Howe L, Workman JL. Transcription activator interactions with multiple SWI/SNF subunits. Mol Cell Biol. 2002;22:1615–1625. doi: 10.1128/MCB.22.6.1615-1625.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson CL, Zhao Y, Chait BT. Subunits of the yeast SWI/SNF complex are members of the actin-related protein (ARP) family. J Biol Chem. 1998;273:23641–23644. doi: 10.1074/jbc.273.37.23641. [DOI] [PubMed] [Google Scholar]
- Phelan ML, Sif S, Narlikar GJ, Kingston RE. Reconstitution of a core chromatin remodeling complex from SWI/SNF subunits. Mol Cell. 1999;3:247–253. doi: 10.1016/s1097-2765(00)80315-9. [DOI] [PubMed] [Google Scholar]
- Prochasson P, Neely KE, Hassan AH, Li B, Workman JL. Targeting activity is required for SWI/SNF function in vivo and is accomplished through two partially redundant activator-interaction domains. Mol Cell. 2003;12:983–990. doi: 10.1016/s1097-2765(03)00366-6. [DOI] [PubMed] [Google Scholar]
- Roberts CW, Galusha SA, McMenamin ME, Fletcher CD, Orkin SH. Haploinsufficiency of Snf5 (integrase interactor 1) predisposes to malignant rhabdoid tumors in mice. Proceedings of the National Academy of Sciences of the United States of America. 2000;97:13796–13800. doi: 10.1073/pnas.250492697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts CW, Leroux MM, Fleming MD, Orkin SH. Highly penetrant, rapid tumorigenesis through conditional inversion of the tumor suppressor gene Snf5. Cancer cell. 2002;2:415–425. doi: 10.1016/s1535-6108(02)00185-x. [DOI] [PubMed] [Google Scholar]
- Schubert HL, Wittmeyer J, Kasten MM, Hinata K, Rawling DC, Heroux A, Cairns BR, Hill CP. Structure of an actin-related subcomplex of the SWI/SNF chromatin remodeler. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:3345–3350. doi: 10.1073/pnas.1215379110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen P, Vivas P, Dechassa ML, Mooney AM, Poirier MG, Bartholomew B. The SnAC domain of SWI/SNF is a histone anchor required for remodeling. Mol Cell Biol. 2013;33:360–370. doi: 10.1128/MCB.00922-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sevenet N, Sheridan E, Amram D, Schneider P, Handgretinger R, Delattre O. Constitutional mutations of the hSNF5/INI1 gene predispose to a variety of cancers. Am J Hum Genet. 1999;65:1342–1348. doi: 10.1086/302639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shain AH, Pollack JR. The spectrum of SWI/SNF mutations, ubiquitous in human cancers. PLoS One. 2013;8:e55119. doi: 10.1371/journal.pone.0055119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shanle EK, Andrews FH, Meriesh H, McDaniel SL, Dronamraju R, DiFiore JV, Jha D, Wozniak GG, Bridgers JB, Kerschner JL, et al. Association of Taf14 with acetylated histone H3 directs gene transcription and the DNA damage response. Genes Dev. 2015;29:1795–1800. doi: 10.1101/gad.269977.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szerlong H, Hinata K, Viswanathan R, Erdjument-Bromage H, Tempst P, Cairns BR. The HSA domain binds nuclear actin-related proteins to regulate chromatin-remodeling ATPases. Nat Struct Mol Biol. 2008;15:469–476. doi: 10.1038/nsmb.1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tolstorukov MY, Sansam CG, Lu P, Koellhoffer EC, Helming KC, Alver BH, Tillman EJ, Evans JA, Wilson BG, Park PJ, et al. Swi/Snf chromatin remodeling/tumor suppressor complex establishes nucleosome occupancy at target promoters. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:10165–10170. doi: 10.1073/pnas.1302209110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Versteege I, Sevenet N, Lange J, Rousseau-Merck MF, Ambros P, Handgretinger R, Aurias A, Delattre O. Truncating mutations of hSNF5/INI1 in aggressive paediatric cancer. Nature. 1998;394:203–206. doi: 10.1038/28212. [DOI] [PubMed] [Google Scholar]
- Wang X, Sansam CG, Thom CS, Metzger D, Evans JA, Nguyen PT, Roberts CW. Oncogenesis caused by loss of the SNF5 tumor suppressor is dependent on activity of BRG1, the ATPase of the SWI/SNF chromatin remodeling complex. Cancer Res. 2009;69:8094–8101. doi: 10.1158/0008-5472.CAN-09-0733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson B, Erdjument-Bromage H, Tempst P, Cairns BR. The RSC chromatin remodeling complex bears an essential fungal-specific protein module with broad functional roles. Genetics. 2006;172:795–809. doi: 10.1534/genetics.105.047589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson BG, Roberts CW. SWI/SNF nucleosome remodellers and cancer. Nat Rev Cancer. 2011;11:481–492. doi: 10.1038/nrc3068. [DOI] [PubMed] [Google Scholar]
- Yang B, Wu YJ, Zhu M, Fan SB, Lin J, Zhang K, Li S, Chi H, Li YX, Chen HF, et al. Identification of cross-linked peptides from complex samples. Nat Methods. 2012;9:904–906. doi: 10.1038/nmeth.2099. [DOI] [PubMed] [Google Scholar]
- Yang X, Zaurin R, Beato M, Peterson CL. Swi3p controls SWI/SNF assembly and ATP-dependent H2A-H2B displacement. Nat Struct Mol Biol. 2007;14:540–547. doi: 10.1038/nsmb1238. [DOI] [PubMed] [Google Scholar]
- Zhang H, Richardson DO, Roberts DN, Utley R, Erdjument-Bromage H, Tempst P, Cote J, Cairns BR. The Yaf9 component of the SWR1 and NuA4 complexes is required for proper gene expression, histone H4 acetylation, and Htz1 replacement near telomeres. Mol Cell Biol. 2004;24:9424–9436. doi: 10.1128/MCB.24.21.9424-9436.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Zhang J, Zhang X, Xu C, Tu X. Solution structure of the Taf14 YEATS domain and its roles in cell growth of Saccharomyces cerevisiae. Biochem J. 2011;436:83–90. doi: 10.1042/BJ20110004. [DOI] [PubMed] [Google Scholar]
- Zofall M, Persinger J, Kassabov SR, Bartholomew B. Chromatin remodeling by ISW2 and SWI/SNF requires DNA translocation inside the nucleosome. Nat Struct Mol Biol. 2006;13:339–346. doi: 10.1038/nsmb1071. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.