

SYNOPSIS
Primary hyperparathyroidism (HPT) is a metabolic disease caused by the excessive secretion of parathyroid hormone from one or more neoplastic parathyroid glands. HPT is largely sporadic, however it can be associated with a familial syndrome. The study of such families led to the discovery of tumor suppressor genes whose loss–of-function is now recognized to underlie the development of many sporadic parathyroid tumors. Heritable and acquired oncogenes causing parathyroid neoplasia are also known. Studies of somatic changes in parathyroid tumor DNA and investigation of kindreds with unexplained familial HPT promise to unmask more genes relevant to parathyroid neoplasia.
Keywords: tumor suppressor, oncogene, multiple endocrine neoplasia, MEN1, MEN2A, CDC73, CCND1, RET, CASR, CDKN1B
Introduction
Primary hyperparathyroidism (HPT) is a disorder of mineral metabolism, typically manifesting in hypercalcemia, that results from the excessive secretion of parathyroid hormone from one or more neoplastic parathyroid glands.1 Although HPT is mostly sporadic, familial forms of HPT represent some 2 to 5% of total cases, the majority of which are caused by germline mutation of known HPT-susceptibility genes (Table 1). Investigation of the molecular genetics underlying these rare familial syndromes has yielded significant insight into the pathophysiology of both sporadic and familial parathyroid neoplasms. Signaling involving the G protein-coupled calcium-sensing receptor (CaSR) impacts the hormonal function of parathyroid cells, and the mutation of genes involved in CaSR signaling has also been implicated in familial syndromes of PTH-dependent hypercalcemia. This monograph will review our current knowledge of the genetics of HPT and PTH-dependent hypercalcemia due to benign and malignant parathyroid disease.
Table 1.
Genes implicated in syndromic and sporadic parathyroid tumorigenesis, and related syndromes
| Gene | Protein encoded | Associated hyperparathyroid syndrome: main syndromic manifestations | Features of syndromic parathyroid tumors | Defect in sporadic parathyroid tumors |
|---|---|---|---|---|
| MEN1 | Menin | Multiple endocrine neoplasia type 1 (MEN1): anterior pituitary, parathyroid, enteropancreatic, foregut carcinoid tumors | Multiple, asymmetric tumors typical (> 99% benign) |
Inactivation in ~25–35% of benign tumors; mutation exceedingly rare in cancer |
| CDC73/HRPT2 | Parafibromin | Hyperparathyroidism-jaw tumor syndrome: fibro-osseous jaw, parathyroid, uterine tumors; renal cysts |
Single tumor common (~15% malignant) |
Inactivation in ~70% of cancers; mutation rare in sporadic adenomas |
| CDKN1B | P27(Kip1) | Multiple endocrine neoplasia type 4 (MEN4): anterior pituitary, other involvement varies |
Single to multiple glands (benign in reports to date); can be recurrent | Loss-of-function mutation in ~5% of sporadic adenomas; including germline mutation in sporadic presentation |
| CASR | Calcium-sensing receptor | Familial hypocalciuric hypercalcemia type 1 (FHH1) with heterozygous inactivation; neonatal severe hyperparathyroidism (NSHPT) with homozygous inactivation | FHH1: near-normal size and surgical pathology; altered serum calcium set-point for PTH release NSHPT: Marked enlargement of multiple glands by polyclonal (non-neoplastic) mechanism |
Decreased expression common; mutation exceedingly rare |
| GNA11 | G protein subunit α11 | Familial hypocalciuric hypercalcemia type 2 (FHH2) | ND | ND |
| AP2S1 | adaptor protein-2 sigma subunit | Familial hypocalciuric hypercalcemia type 3 (FHH3): hypercalcemia more severe than in FHH1 | ND | ND |
| RET | c-Ret | Multiple endocrine neoplasia type 2A: medullary thyroid cancer, pheochromocytoma, parathyroid tumors | Single tumor common (> 99% benign) |
Mutation exceedingly rare |
| CCND1/PRAD1 | Cyclin D1 | NA | NA | Overexpression results from DNA rearrangement involving PTH gene |
NA, not applicable
ND, not determined due to lack of relevant published studies
Pathophysiology of Primary Hyperparathyroidism
Maintenance of the serum calcium concentration within a relatively narrow physiologic range is achieved by regulation of parathyroid hormone (PTH) secretion from parathyroid cells in response to changes in the circulating ionized calcium level. The G protein-coupled CaSR located on the surface membrane of the parathyroid chief cells negatively regulates the secretion of PTH.2,3 In clinical medicine, HPT is typically defined by the conjunction of elevated serum ionized calcium with inappropriately elevated PTH.1 Most parathyroid tumors are benign adenomas. Parathyroid carcinoma is a rare cause of HPT seen in less than 1% of cases.
Approximately 2 to 5 % of cases of HPT are associated with familial disease. Study of this small subset has nevertheless provided great insight into the genetic changes and molecular pathways that promote parathyroid neoplasia (Table 1). The most common genetic disorders associated with HPT are MEN1, multiple endocrine neoplasia type 2A (MEN2A), HPT-JT, and familial isolated hyperparathyroidism (FIHP).4–7 Familial hypocalciuric hypercalcemia (FHH) is a related, genetically heterogeneous, largely benign condition of PTH-dependent hypercalcemia often mimicking HPT that does not correct with partial or subtotal parathyroidectomy (PTX).8 These genetic disorders and their relation to the underlying molecular and genetic alterations relevant to parathyroid neoplasia will be discussed in detail below.
Knudson’s Two-Hit Hypothesis and Tumor Suppressor Genes
Some forty-five years ago, Alfred Knudson proposed a model for tumor development based on his epidemiologic analysis of retinoblastoma.9 Familial retinoblastoma is much more rare than sporadic retinoblastoma, yet the former has a much earlier age of onset and is more frequently binocular. According to the “two-hit” hypothesis of neoplasia proposed by Knudson, two events (or “hits”) in an affected cell confer a selective growth advantage and result in its clonal expansion.10
In accordance with clinical and molecular genetic data accrued since his original proposal, Knudson’s concept can be updated. In many hereditary tumor syndromes, an inherited mutation in the germline DNA affecting one allele of a tumor suppressor gene represents the first event or “hit” that is present in all the cells of the affected offspring (Fig. 1). The tendency for bilateral and/or multifocal disease in hereditary tumor syndromes as well as the earlier age of onset are explained by the greater likelihood of any particular cell acquiring a “second hit”, i.e. a somatic mutation in second allele of the same tumor suppressor gene. The “second hit”, that inactivates the remaining wild-type allele, most often results from large subchromosomal or even chromosomal deletion or else DNA rearrangement (Fig. 1). Bi-allelic inactivating mutation of the MEN1 and the CDC73/HRPT2 tumor suppressor genes can frequently be demonstrated in DNA derived from parathyroid tumors, in the context of the familial syndromes MEN1 and HPT-JT, respectively. In the majority of such patients, a germline loss-of-function mutation of the associated tumor suppressor gene can be demonstrated, representing the first “hit” (Fig. 1).
Figure 1. Two-hit loss of function mutation of the CDC73/HRPT2 tumor suppressor in parathyroid neoplasia.
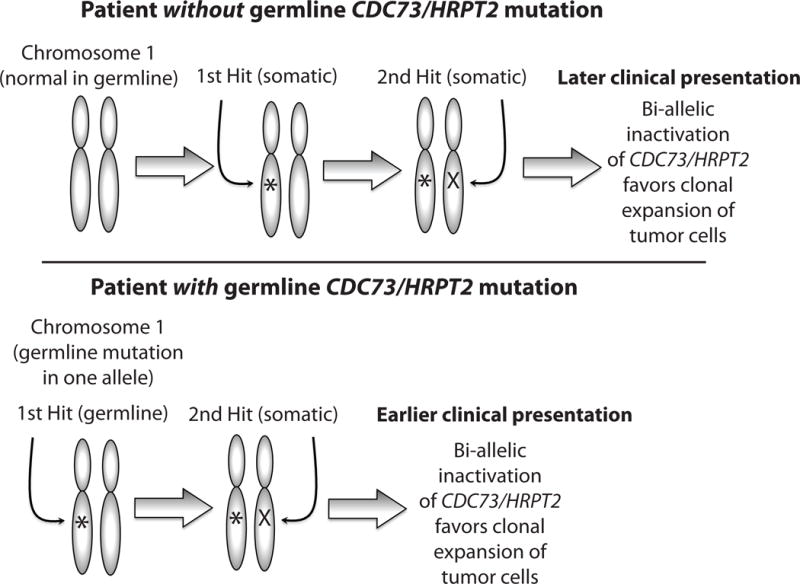
Benign or malignant parathyroid tumors causing hyperparathyroidism (HPT) can result from two-hit loss of function of the CDC73/HRPT2 tumor suppressor gene according to the Knudson hypothesis (see text). The human CDC73/HRPT2 gene is on chromosome 1 at location 1q25. Upper: In a patient without germline CDC73/HRPT2 mutation, both alleles of CDC73/HRPT2 are initially normal in all parathyroid cells. Subsequent step-wise acquired or somatic inactivation of both alleles in the same parathyroid cell results in clonal expansion that may lead to a sporadic parathyroid tumor. Lower: In a patient with germline CDC73/HRPT2 mutation in one allele, an acquired or somatic DNA mutation at the CDC73/HRPT2 locus of the remaining allele in a parathyroid cell results clonal expansion of that cell that may lead to a benign or malignant parathyroid tumor. Patients with germline CDC73/HRPT2 mutation who develop parathyroid adenomas or cancer can present sporadically (if lacking or unaware of relevant family medical history), or belong to a kindred with familial isolated HPT or the hyperparathyroidism-jaw tumor syndrome (HPT-JT). The presence of the first germline mutation at birth in all parathyroid tissue accelerates the acquisition of two hits within a single parathyroid cell and accounts for the earlier age of disease presentation typical of familial forms of HPT such as HPT-JT.
Clinical Features and Genetics of Multiple Endocrine Neoplasia type 1
MEN1 is the most common familial cause of primary hyperparathyroidism.11 MEN1 is characterized by a tendency to develop tumors in the anterior pituitary, parathyroid glands, and enteropancreatic endocrine cells, although tumors in several other endocrine organs and in non-endocrine tissues such as skin and smooth muscle can also be associated with the syndrome.5,12 Some 10% of MEN1 is sporadic.
Familial MEN1 is inherited in an autosomal dominant fashion. Germline inactivation of one allele of the MEN1 gene on chromosome 11q13 confers the tumor susceptibility.13 More than 400 distinct germline mutations in MEN1 have been described in patients and kindreds with MEN1. The vast majority of parathyroid and other syndromic tumors from MEN1 patients can be shown to harbor a somatic mutation or deletion of the second wild-type MEN1 allele.5,14
Molecular genetic analysis of sporadic parathyroid adenomas using conventional DNA sequencing methods identified somatic MEN1 mutation involving at least one allele with a frequency of 3–35%.15–19 The frequency ranged from 26–37% in studies that looked at LOH at 11q13 in sporadic parathyroid adenomas. Studies of sporadic benign parathyroid tumor DNA employing whole exome sequencing (WES) methodology identified somatic MEN1 mutation in some 35% of tumors, similar to results using conventional DNA sequencing methods.20,21 Since HPT is most penetrant feature of MEN1 and is usually its initial manifestation, bona fide MEN1 kindreds may rarely be misassigned a provisional diagnosis of FIHP if only younger mutation carriers are evaluated at the time of initial family ascertainment.
The association of MEN1 mutation with parathyroid carcinoma is rare. At least two cases of parathyroid cancer have been reported in patients with MEN1, one with associated parathyroid adenoma, and the other with bilateral parathyroid carcinoma.22,23
Clinical Features and Genetics of the Hyperparathyroidism-Jaw Tumor Syndrome
HPT-JT is a rare autosomal dominant familial cancer syndrome of variable and incomplete penetrance manifested by HPT, cemento-ossifying tumors of the maxilla and mandible, and less commonly uterine tumors and/or renal cysts.24–26 HPT is the most penetrant feature and usually the presenting manifestation. In contrast to MEN1, parathyroid carcinoma is frequent in HPT-JT, affecting some 20% or more of those with HPT.24–27 Approximately 10% of obligate or genetically confirmed carriers have no clinical manifestations.
A loss-of-function mutation of CDC73/HRPT2 can be demonstrated in the germline of a majority of HPT-JT kindreds.28 Most such CDC73/HRPT2 mutations are predicted to inactivate gene function via frameshift or nonsense mutation.29 Patients with germline deletion of the CDC73/HRPT2 gene have also been reported.30–32 The CDC73/HRPT2 gene encodes the protein parafibromin.28 Parafibromin is a presumed tumor suppressor protein because germline-inactivating mutation of the CDC73/HRPT2 gene predisposes to the full or partial expression of HPT-JT. Somatic mutation of CDC73/HRPT2 is uncommon in sporadic parathyroid adenomas.33 In contrast, mutations of CDC73/HRPT2 are frequently seen in apparently sporadic cases of parathyroid carcinoma.34–36 Interestingly, exome sequence analysis of tumor DNA from parathyroid carcinoma revealed preferential amplification of the mutant CDC73/HRPT2 allele.37 Some 25% of patients with apparently sporadic parathyroid cancer may harbor germline CDC73/HRPT2 mutations, suggesting that such cases may represent undiagnosed or incompletely penetrant HPT-JT.36,38 Besides HPT-JT kindreds, a subset of patients with the FIHP phenotype also harbor germline mutation of CDC73/HRPT2, indicating this familial disorder may represent an incomplete expression of HPT-JT.
Clinical Features and Genetics of Multiple Endocrine Neoplasia Type 4
Multiple Endocrine Neoplasia Type 4 (MEN4, sometimes called MENX) is a syndrome originally described by Pellegata et al in a multi-generational family with MEN1-like features, including a proband with acromegaly and HPT but lacking MEN1 mutation.39 Several members of this kindred, including the proband, were shown to harbor a germline heterozygous nonsense mutation in CDKN1B, encoding the cyclin dependent-kinase inhibitor p27(Kip1).39 Investigation of this locus followed from the genetic analysis of rats with the MenX phenotype, a recessively inherited condition characterized by the development of bilateral pheochromocytomas, paragangliomas, parathyroid adenomas and thyroid C cell hyperplasia.39,40 Only a single member of the MEN4/MENX kindred described by Pellegata et al manifested HPT (the proband).39
Since the original report by Pellegata and co-workers,39 several groups have investigated a role for mutation of CDKN1B in parathyroid neoplasia. Apart from the demonstration of HPT linked to CDKN1B mutation in monozygotic twins,41 none of the studies of CDKN1B mutation-positive kindreds expressing MEN1-like tumors but lacking MEN1 germline mutation, and thus characterized by the MEN4 designation, has identified kindreds that include more than one genetically unique member with HPT proven to segregate with germline CDKN1B mutation.39,41–46 At least one study of patients with features of genetic predisposition to HPT failed to identify any cases with germline CDKN1B mutation.18 On the other hand, non-familial presentation of primary HPT due to parathyroid adenomas in association with somatic and germline mutation of CDKN1B has been documented.47
The characterization of CDKN1B as a low-penetrance susceptibility gene for the development of primary parathyroid tumors is supported by good evidence.47,48 As such, CDKN1B remains a reasonable hypothetical candidate for which germline mutation may provide an etiology for some cases of familial HPT. More research will be required, however, to justify including germline inactivating mutation of CDKN1B, associated with MEN4, in the differential diagnosis of familial HPT. At a minimum such justification would seem to require identification and characterization of at least one family that includes more than one genetically unique member with HPT linked to pathologic mutation of CDKN1B in the germline.
Clinical Features and Genetics of Familial Isolated Hyperparathyroidism
FIHP is a genetically heterogeneous, non-syndromic, clinically defined diagnosis of exclusion in kindreds with two or more cases of HPT but lacking the specific features of MEN1, MEN2A, HPT-JT or FHH. While germline mutation of MEN1, CDC73/HRPT2 or CASR may account for a minority of kindreds with the FIHP phenotype upon initial ascertainment,7,49–51 the majority of FIHP kindreds lack mutations in these known HPT-susceptibility genes.7,49 A distinct genetic etiology resulting in the FIHP phenotype has not yet been defined, although a genome-wide screen of seven FIHP families has identified a 1.7 Mb region of suggestive linkage on the short arm of chromosome 2, at location 2p14-p13.3.52
Clinical Features and Genetics of Familial Hypocalciuric Hypercalcemia
FHH is a genetically heterogeneous, clinically benign condition of PTH-dependent hypercalcemia often mimicking HPT (Table 1).8 FHH cases almost always remain hypercalcemic following partial or subtotal PTX. FHH is a transmitted in an autosomal dominant fashion and usually causes mild HPT with relative hypocalciuria; hypercalcemia in FHH is highly penetrant at all ages, even in the perinatal period.8 Most cases of FHH are type 1 (FHH1) and result from heterozygous loss of function mutation of the CASR gene on chromosome 3 that encodes the CaSR.2,3,53–56 The homozygous or compound heterozygous inheritance of two inactive CASR alleles typically results in the phenotype of neonatal severe hyperparathyroidism (NSHPT).54–56 The non-neoplastic nature of the abnormal parathyroids associated with germline CASR loss-of-function mutation was underscored by a recent molecular genetic analysis of enlarged parathyroid glands from a patient with NSHPT that demonstrated generalized polyclonal hyperplasia, rather than the monoclonality that would be expected in bona fide parathyroid tumors.57
Somatic inactivation of CASR has not been found in sporadic parathyroid tumors studied to date,58,59 even though significant loss of CASR expression, not due to allelic loss, has been documented in parathyroid adenomas and very likely contributes to their altered calcium set point for PTH release.60
Type 2 FHH (FHH2) due to germline loss-of-function mutation of GNA11, encoding the G protein α11 subunit61,62, and type 3 FHH (FHH3) due to germline loss-of-function mutation in AP2S1, encoding the adaptor protein-2 sigma subunit involved in clathrin-mediated endocytosis,63–66 have been recently described. Somatic mutation of neither GNA11 nor AP2S1 has been reported in sporadic parathyroid tumors.
Oncogenes and Proto-Oncogenes
Oncogenes derive from naturally occurring genes, referred to as proto-oncogenes, which positively regulate cell division and/or cell growth.67 Oncogenes represent mutationally activated or overexpressed forms of proto-oncogenes that can induce tumor formation, often in a tissue-specific fashion. Proto-oncogenes frequently encode proteins that are involved in signal transduction, particularly in pathways mediating mitogenic signals. Among the etiologies of currently recognized familial cancer syndromes, germline mutational activation of proto-oncogenes is quite rare compared to the germline inactivation of tumor suppressor genes. This is presumably because of the disruptive effect constitutive proliferative signalling created by the activation of most proto-oncogenes would likely have on embryonic and fetal development.
Clinical Features and Genetics of Multiple Endocrine Neoplasia Type 2A
Classical multiple endocrine neoplasia type 2A (MEN2A) is a familial syndrome characterized by medullary thyroid cancer (MTC), pheochromocytoma, and primary HPT. HPT in MEN2A resembles sporadic HPT in its clinical presentation, is usually mild, and is almost always due to benign parathyroid tumors. MEN2A is transmitted in an autosomal dominant fashion. MEN2A is caused by germline gain-of-function mutation in the RET (REarranged during Transfection) proto-oncogene. RET encodes a transmembrane receptor tyrosine kinase which, in conjunction with glial derived neurotrophic factor (GDNF)-family α co-receptors, binds GDNF family ligands.68
Germline gain-of-function mutations of RET are associated with three different endocrine tumor syndromes, all associated with MTC: MEN2A, multiple endocrine neoplasia type 2B (MEN2B), and familial medullary thyroid cancer (FMTC). Parathyroid tumors and HPT are not part of the MEN2B or FMTC disease pattern. Different germline RET mutations can result in the different disease phenotypes. In 95% of patients, MEN2A is associated with germline missense mutations that map to the receptor tyrosine kinase’s extracellular cysteine-rich domain, involving RET codons 609, 611, 618, or 620 of exon 10 or codon 634 of exon 11.69 About 85% of cases of MEN2A result from missense mutation of the cysteine residue present at codon 634.70
The Role of the CCND1 Oncogene in the Pathophysiology of Parathyroid Tumors
The CCND1 (or PRAD1, for parathyroid adenomatosis 1) oncogene was discovered during the molecular genetic analysis of several large sporadic parathyroid adenomas that harbored DNA re-arrangements involving the PTH gene locus on chromosome 11.71–73 The CCND1/PRAD1 oncogene in sporadic parathyroid tumors was identified downstream of a breakpoint caused by the pericentromeric inversion of chromosome 11 DNA.73 The chromosomal inversion positions the 5′ PTH gene regulatory region (normally located at 11p15) just upstream of the CCND1/PRAD1 proto-oncogene resident at 11q13.71–73 The product encoded by the proto-oncogene was subsequently recognized to be a member of the cyclin family based on sequence homology73 and the gene was re-named cyclin D1 (CCND1). It was subsequently shown that transgenic overexpression of CCND1 in the parathyroid tissue of mice causes cell proliferation and recapitulates the metabolic abnormalities typical of HPT in humans.74
CCND1 is overexpressed in some 20 to 40% of sporadic benign parathyroid tumors and in an even higher percentage of parathyroid cancers.75–78 Activating missense mutations in the CCND1 coding region have not been observed in sporadic parathyroid adenomas.79 No somatic chromosomal rearrangements involving CCND1 have been reported in parathyroid carcinoma. Neither germline activating missense mutations nor germline chromosomal translocations/rearrangements involving CCND1 have been identified in any familial form of HPT.
The Role of Other Oncogenes in the Pathophysiology of Parathyroid Tumors
Analysis of eight sporadic parathyroid adenomas by WES and the subsequent targeted sequencing of DNA from an additional 185 parathyroid adenomas demonstrated the Y641N missense mutation in EZH2 (enhancer of zeste 2) in two out of 193 independent parathyroid tumors.20 EZH2 encodes the catalytic subunit of polycomb repressive complex 2 and somatic mutations of EZH2 residue Y641, including Y641N, had previously been reported in certain categories of lymphoma.80 Mutational analysis of a subsequent set of 80 sporadic benign and malignant parathyroid tumors by an independent group did not find any pathogenic EZH2 variants however, suggesting such somatic mutation may be rare.81 In the context of lymphoma, EZH2 is thought to function as a proto-oncogene.80 Since EZH2 Y641N mutation and gene overexpression have been reported in parathyroid tumors, EZH2 may be considered a candidate proto-oncogene in the parathyroid context, also, if further investigation confirms these initial observations. No transgenic mouse models targeting EZH2 mutation or overexpression to parathyroid tissue have yet been reported.
Somatic mutations in the candidate parathyroid proto-oncogene ZFX, a member of the Krüppel associated box domain-containing zinc finger protein transcription factors, were first identified by WES analysis of DNA extracted from 19 parathyroid adenomas and matched germline DNA, with confirmation by direct sequencing of tumor DNA from an additional 111 parathyroid adenomas.82 Several lines of evidence suggest that the somatically acquired mutant ZFX alleles detected in parathyroid tumors may act as oncogenes.83 Development of a transgenic mouse model targeting mutant ZFX protein expression to parathyroid tissue and/or in vitro functional characterization of the mutant ZFX protein will help to clarify the potential significance of ZFX as a parathyroid proto-oncogene.
Future Considerations
It is quite likely that the dysregulation of other genes, besides those discussed above, predispose to parathyroid neoplasia. As previously noted, the susceptibility to parathyroid neoplasia in the majority of FIHP kindreds appears to result from the germline mutation of genes not currently recognized for a role in parathyroid disease: 53 among 76 families initially considered as FIHP in 5 clinical studies that investigated for germline MEN1, CASR and CDC73/HRPT2 gene mutation, or nearly 70%, had no currently recognized syndromic etiology.7,49–51
The existence of currently unidentified parathyroid tumor suppressors and oncogenes is also suggested by the analysis of parathyroid tumors for the loss or gain of specific regions of chromosomal DNA using techniques such as comparative genomic hybridization (CGH). Several investigators have found recurrent loss of chromosomal DNA at the 1p, 6q, 9p, and 13q loci in parathyroid tumors indicating the potential presence there of novel parathyroid tumor suppressor genes.84–87 The possible presence of novel oncogenes at 9q, 16p, 19p, and Xq is suggested by a convergence of results from several laboratories that demonstrate specific chromosomal gain at these loci in benign or malignant parathyroid tumors.84,86–88
WES analysis of benign and malignant parathyroid tumors shows great promise for the identification of somatic and germline gene mutations predisposing to parathyroid neoplasia. The success of this method in the identification of the candidate parathyroid oncogenes EXH220 and ZFX82 has been discussed above. Similarly exome sequence analysis of DNA from parathyroid carcinomas has highlighted the potential significance of recurrent germline and somatic inactivating mutations of PRUNE2 in this context.37 The sensitivity and comprehensive quality of WES and other emerging next generation sequencing modalities will undoubtedly accelerate our understanding of the pathophysiology of familial and sporadic parathyroid neoplasia in the years ahead.
Key Points.
Primary hyperparathyroidism, due to parathyroid tumors, is mostly sporadic.
The molecular genetic investigation of rare syndromic forms of hyperparathyroidism has nevertheless led to significant advances in our understanding of both familial and sporadic parathyroid neoplasia.
Both oncogenes and tumor suppressors have been implicated in the etiology of parathyroid tumors.
The discovery of novel parathyroid tumor susceptibility genes will likely result from the application of next generation sequencing methods to the analysis of sporadic parathyroid tumors and non-syndromic familial cases of hyperparathyroidism.
Acknowledgments
The author thanks his colleagues Drs. Stephen J. Marx, Lee S. Weinstein, Michael T. Collins, Monica C. Skarulis, Sunita K. Agarwal, and Electron Kebebew for their ongoing support and encouragement. The Intramural Research Program of the National Institute of Diabetes and Digestive and Kidney Diseases supported this research.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosure statement: The author has no commercial, financial or personal conflict of interest that has inappropriately influenced the content of this article. The Intramural Research Program of the National Institute of Diabetes and Digestive and Kidney Diseases supported this research.
References
- 1.Bilezikian JP, Cusano NE, Khan AA, Liu JM, Marcocci C, Bandeira F. Primary hyperparathyroidism. Nat Rev Dis Primers. 2016;2:16033. doi: 10.1038/nrdp.2016.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brown EM, Pollak M, Seidman CE, et al. Calcium-ion-sensing cell-surface receptors. N Engl J Med. 1995;333(4):234–240. doi: 10.1056/NEJM199507273330407. [DOI] [PubMed] [Google Scholar]
- 3.Brown EM. Role of the calcium-sensing receptor in extracellular calcium homeostasis. Best Pract Res Clin Endocrinol Metab. 2013;27(3):333–343. doi: 10.1016/j.beem.2013.02.006. [DOI] [PubMed] [Google Scholar]
- 4.Fraser WD. Hyperparathyroidism. Lancet. 2009;374(9684):145–158. doi: 10.1016/S0140-6736(09)60507-9. [DOI] [PubMed] [Google Scholar]
- 5.Marx SJ. Molecular genetics of multiple endocrine neoplasia types 1 and 2. Nat Rev Cancer. 2005;5(5):367–375. doi: 10.1038/nrc1610. [DOI] [PubMed] [Google Scholar]
- 6.Jackson MART, Hu MI, et al. CDC73-Related Disorders GeneReviews® [Internet] Seattle (WA): University of Washington, Seattle; 2015. pp. 1993–2015. [Google Scholar]
- 7.Simonds WF, James-Newton LA, Agarwal SK, et al. Familial isolated hyperparathyroidism: Clinical and genetic characteristics of thirty-six kindreds. Medicine (Baltimore) 2002;81:1–26. doi: 10.1097/00005792-200201000-00001. [DOI] [PubMed] [Google Scholar]
- 8.Marx SJ, Attie MF, Levine MA, Spiegel AM, Downs RW, Jr, Lasker RD. The hypocalciuric or benign variant of familial hypercalcemia: clinical and biochemical features in fifteen kindreds. Medicine (Baltimore) 1981;60:397–412. doi: 10.1097/00005792-198111000-00002. [DOI] [PubMed] [Google Scholar]
- 9.Knudson AG., Jr Mutation and cancer: statistical study of retinoblastoma. Proc Natl Acad Sci U S A. 1971;68(4):820–823. doi: 10.1073/pnas.68.4.820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Knudson AG. Two genetic hits (more or less) to cancer. Nat Rev Cancer. 2001;1(2):157–162. doi: 10.1038/35101031. [DOI] [PubMed] [Google Scholar]
- 11.Arnold A, Marx SJ. Primer on the Metabolic Bone Diseases and Disorders of Mineral Metabolism. 7. Washington, DC: American Society for Bone and Mineral Research; 2008. Familial Hyperparathyroidism (Including MEN, FHH, and HPT-JT) pp. 361–366. [Google Scholar]
- 12.Schussheim DH, Skarulis MC, Agarwal SK, et al. Multiple endocrine neoplasia type 1: new clinical and basic findings. Trends Endocrinol Metab. 2001;12:173–178. [Google Scholar]
- 13.Chandrasekharappa SC, Guru SC, Manickam P, et al. Positional cloning of the gene for multiple endocrine neoplasia-type 1. Science. 1997;276:404–407. doi: 10.1126/science.276.5311.404. [DOI] [PubMed] [Google Scholar]
- 14.Lemos MC, Thakker RV. Multiple endocrine neoplasia type 1 (MEN1): analysis of 1336 mutations reported in the first decade following identification of the gene. Hum Mutat. 2008;29(1):22–32. doi: 10.1002/humu.20605. [DOI] [PubMed] [Google Scholar]
- 15.Miedlich S, Krohn K, Lamesch P, Muller A, Paschke R. Frequency of somatic MEN1 gene mutations in monoclonal parathyroid tumours of patients with primary hyperparathyroidism. Eur J Endocrinol. 2000;143(1):47–54. doi: 10.1530/eje.0.1430047. [DOI] [PubMed] [Google Scholar]
- 16.Uchino S, Noguchi S, Sato M, et al. Screening of the Men1 gene and discovery of germ-line and somatic mutations in apparently sporadic parathyroid tumors. Cancer Res. 2000;60(19):5553–5557. [PubMed] [Google Scholar]
- 17.Scarpelli D, D’Aloiso L, Arturi F, et al. Novel somatic MEN1 gene alterations in sporadic primary hyperparathyroidism and correlation with clinical characteristics. J Endocrinol Invest. 2004;27(11):1015–1021. doi: 10.1007/BF03345303. [DOI] [PubMed] [Google Scholar]
- 18.Vierimaa O, Villablanca A, Alimov A, et al. Mutation analysis of MEN1, HRPT2, CASR, CDKN1B, and AIP genes in primary hyperparathyroidism patients with features of genetic predisposition. J Endocrinol Invest. 2009;32(6):512–518. doi: 10.1007/BF03346498. [DOI] [PubMed] [Google Scholar]
- 19.Heppner C, Kester MB, Agarwal SK, et al. Somatic mutation of the MEN1 gene in parathyroid tumours. Nature Genet. 1997;16:375–378. doi: 10.1038/ng0897-375. [DOI] [PubMed] [Google Scholar]
- 20.Cromer MK, Starker LF, Choi M, et al. Identification of somatic mutations in parathyroid tumors using whole-exome sequencing. J Clin Endocrinol Metab. 2012;97(9):E1774–1781. doi: 10.1210/jc.2012-1743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Newey PJ, Nesbit MA, Rimmer AJ, et al. Whole-exome sequencing studies of nonhereditary (sporadic) parathyroid adenomas. J Clin Endocrinol Metab. 2012;97(10):E1995–2005. doi: 10.1210/jc.2012-2303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dionisi S, Minisola S, Pepe J, et al. Concurrent parathyroid adenomas and carcinoma in the setting of multiple endocrine neoplasia type 1: presentation as hypercalcemic crisis. Mayo Clin Proc. 2002;77(8):866–869. doi: 10.4065/77.8.866. [DOI] [PubMed] [Google Scholar]
- 23.Shih RY, Fackler S, Maturo S, True MW, Brennan J, Wells D. Parathyroid carcinoma in multiple endocrine neoplasia type 1 with a classic germline mutation. Endocr Pract. 2009;15(6):567–572. doi: 10.4158/EP09045.CRR1. [DOI] [PubMed] [Google Scholar]
- 24.Jackson CE, Norum RA, Boyd SB, et al. Hereditary hyperparathyroidism and multiple ossifying jaw fibromas: a clinically and genetically distinct syndrome. Surgery. 1990;108:1006–1012. [PubMed] [Google Scholar]
- 25.Bradley KJ, Hobbs MR, Buley ID, et al. Uterine tumours are a phenotypic manifestation of the hyperparathyroidism-jaw tumour syndrome. J Intern Med. 2005;257(1):18–26. doi: 10.1111/j.1365-2796.2004.01421.x. [DOI] [PubMed] [Google Scholar]
- 26.Chen JD, Morrison C, Zhang C, Kahnoski K, Carpten JD, Teh BT. Hyperparathyroidism-jaw tumour syndrome. J Intern Med. 2003;253(6):634–642. doi: 10.1046/j.1365-2796.2003.01168.x. [DOI] [PubMed] [Google Scholar]
- 27.Mehta A, Patel D, Rosenberg A, et al. Hyperparathyroidism-jaw tumor syndrome: Results of operative management. Surgery. 2014;156(6):1315–1324. doi: 10.1016/j.surg.2014.08.004. discussion 1324–1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Carpten JD, Robbins CM, Villablanca A, et al. HRPT2, encoding parafibromin, is mutated in hyperparathyroidism-jaw tumor syndrome. Nat Genet. 2002;32(4):676–680. doi: 10.1038/ng1048. [DOI] [PubMed] [Google Scholar]
- 29.Newey PJ, Bowl MR, Thakker RV. Parafibromin–functional insights. J Intern Med. 2009;266(1):84–98. doi: 10.1111/j.1365-2796.2009.02107.x. [DOI] [PubMed] [Google Scholar]
- 30.Domingues R, Tomaz RA, Martins C, Nunes C, Bugalho MJ, Cavaco BM. Identification of the first germline HRPT2 whole-gene deletion in a patient with primary hyperparathyroidism. Clinical endocrinology. 2012;76(1):33–38. doi: 10.1111/j.1365-2265.2011.04184.x. [DOI] [PubMed] [Google Scholar]
- 31.Cascon A, Huarte-Mendicoa CV, Javier Leandro-Garcia L, et al. Detection of the first gross CDC73 germline deletion in an HPT-JT syndrome family. Genes Chromosomes Cancer. 2011;50(11):922–929. doi: 10.1002/gcc.20911. [DOI] [PubMed] [Google Scholar]
- 32.Bricaire L, Odou MF, Cardot-Bauters C, et al. Frequent large germline HRPT2 deletions in a French National cohort of patients with primary hyperparathyroidism. J Clin Endocrinol Metab. 2013;98(2):E403–408. doi: 10.1210/jc.2012-2789. [DOI] [PubMed] [Google Scholar]
- 33.Krebs LJ, Shattuck TM, Arnold A. HRPT2 mutational analysis of typical sporadic parathyroid adenomas. J Clin Endocrinol Metab. 2005;90(9):5015–5017. doi: 10.1210/jc.2005-0717. [DOI] [PubMed] [Google Scholar]
- 34.Howell VM, Haven CJ, Kahnoski K, et al. HRPT2 mutations are associated with malignancy in sporadic parathyroid tumours. J Med Genet. 2003;40(9):657–663. doi: 10.1136/jmg.40.9.657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cetani F, Pardi E, Borsari S, et al. Genetic analyses of the HRPT2 gene in primary hyperparathyroidism: germline and somatic mutations in familial and sporadic parathyroid tumors. The Journal of clinical endocrinology and metabolism. 2004;89(11):5583–5591. doi: 10.1210/jc.2004-0294. [DOI] [PubMed] [Google Scholar]
- 36.Shattuck TM, Valimaki S, Obara T, et al. Somatic and germ-line mutations of the HRPT2 gene in sporadic parathyroid carcinoma. N Engl J Med. 2003;349(18):1722–1729. doi: 10.1056/NEJMoa031237. [DOI] [PubMed] [Google Scholar]
- 37.Yu W, McPherson JR, Stevenson M, et al. Whole-exome sequencing studies of parathyroid carcinomas reveal novel PRUNE2 mutations, distinctive mutational spectra related to APOBEC-catalyzed DNA mutagenesis and mutational enrichment in kinases associated with cell migration and invasion. J Clin Endocrinol Metab. 2015;100(2):E360–364. doi: 10.1210/jc.2014-3238. [DOI] [PubMed] [Google Scholar]
- 38.Cetani F, Pardi E, Borsari S, et al. Genetic analyses of the HRPT2 gene in primary hyperparathyroidism: germline and somatic mutations in familial and sporadic parathyroid tumors. J Clin Endocrinol Metab. 2004;89(11):5583–5591. doi: 10.1210/jc.2004-0294. [DOI] [PubMed] [Google Scholar]
- 39.Pellegata NS, Quintanilla-Martinez L, Siggelkow H, et al. Germ-line mutations in p27Kip1 cause a multiple endocrine neoplasia syndrome in rats and humans. Proc Natl Acad Sci U S A. 2006;103(42):15558–15563. doi: 10.1073/pnas.0603877103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fritz A, Walch A, Piotrowska K, et al. Recessive transmission of a multiple endocrine neoplasia syndrome in the rat. Cancer Res. 2002;62(11):3048–3051. [PubMed] [Google Scholar]
- 41.Agarwal SK, Mateo CM, Marx SJ. Rare germline mutations in cyclin-dependent kinase inhibitor genes in multiple endocrine neoplasia type 1 and related states. J Clin Endocrinol Metab. 2009;94(5):1826–1834. doi: 10.1210/jc.2008-2083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Georgitsi M, Raitila A, Karhu A, et al. Germline CDKN1B/p27Kip1 mutation in multiple endocrine neoplasia. J Clin Endocrinol Metab. 2007;92(8):3321–3325. doi: 10.1210/jc.2006-2843. [DOI] [PubMed] [Google Scholar]
- 43.Molatore S, Marinoni I, Lee M, et al. A novel germline CDKN1B mutation causing multiple endocrine tumors: clinical, genetic and functional characterization. Hum Mutat. 2010;31(11):E1825–1835. doi: 10.1002/humu.21354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Malanga D, De Gisi S, Riccardi M, et al. Functional characterization of a rare germline mutation in the gene encoding the cyclin-dependent kinase inhibitor p27Kip1 (CDKN1B) in a Spanish patient with multiple endocrine neoplasia-like phenotype. Eur J Endocrinol. 2012;166(3):551–560. doi: 10.1530/EJE-11-0929. [DOI] [PubMed] [Google Scholar]
- 45.Occhi G, Regazzo D, Trivellin G, et al. A novel mutation in the upstream open reading frame of the CDKN1B gene causes a MEN4 phenotype. PLoS Genet. 2013;9(3):e1003350. doi: 10.1371/journal.pgen.1003350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tonelli F, Giudici F, Giusti F, et al. A heterozygous frameshift mutation in exon 1 of CDKN1B gene in a patient affected by MEN4 syndrome. Eur J Endocrinol. 2014;171(2):K7–K17. doi: 10.1530/EJE-14-0080. [DOI] [PubMed] [Google Scholar]
- 47.Costa-Guda J, Marinoni I, Molatore S, Pellegata NS, Arnold A. Somatic mutation and germline sequence abnormalities in CDKN1B, encoding p27Kip1, in sporadic parathyroid adenomas. J Clin Endocrinol Metab. 2011;96(4):E701–706. doi: 10.1210/jc.2010-1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Costa-Guda J, Arnold A. Genetic and epigenetic changes in sporadic endocrine tumors: parathyroid tumors. Molecular and cellular endocrinology. 2014;386(1–2):46–54. doi: 10.1016/j.mce.2013.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Simonds WF, Robbins CM, Agarwal SK, Hendy GN, Carpten JD, Marx SJ. Familial isolated hyperparathyroidism is rarely caused by germline mutation in HRPT2, the gene for the hyperparathyroidism-jaw tumor syndrome. J Clin Endocrinol Metab. 2004;89(1):96–102. doi: 10.1210/jc.2003-030675. [DOI] [PubMed] [Google Scholar]
- 50.Warner J, Epstein M, Sweet A, et al. Genetic testing in familial isolated hyperparathyroidism: unexpected results and their implications. J Med Genet. 2004;41(3):155–160. doi: 10.1136/jmg.2003.016725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cetani F, Pardi E, Ambrogini E, et al. Genetic analyses in familial isolated hyperparathyroidism: implication for clinical assessment and surgical management. Clinical endocrinology. 2006;64(2):146–152. doi: 10.1111/j.1365-2265.2006.02438.x. [DOI] [PubMed] [Google Scholar]
- 52.Warner JV, Nyholt DR, Busfield F, et al. Familial isolated hyperparathyroidism is linked to a 1.7 Mb region on chromosome 2p13.3-14. J Med Genet. 2006;43(3):e12. doi: 10.1136/jmg.2005.035766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Brown EM. Familial hypocalciuric hypercalcemia and other disorders with resistance to extracellular calcium. Endocrinol Metab Clin North Am. 2000;29(3):503–522. doi: 10.1016/s0889-8529(05)70148-1. [DOI] [PubMed] [Google Scholar]
- 54.Pollak MR, Brown EM, Chou Y-HW, et al. Mutations in the human Ca2+-sensing receptor gene cause familial hypocalciuric hypercalcemia and neonatal severe hyperparathyroidism. Cell. 1993;75:1297–1303. doi: 10.1016/0092-8674(93)90617-y. [DOI] [PubMed] [Google Scholar]
- 55.Brown EM. Familial hypocalciuric hypercalcemia and other disorders with resistance to extracellular calcium. Endocrinol Metabol Clin North Am. 2000;29:503–522. doi: 10.1016/s0889-8529(05)70148-1. [DOI] [PubMed] [Google Scholar]
- 56.Hendy GN, D’Souza-Li L, Yang B, Canaff L, Cole DE. Mutations of the calcium-sensing receptor (CASR) in familial hypocalciuric hypercalcemia, neonatal severe hyperparathyroidism, and autosomal dominant hypocalcemia. Hum Mutat. 2000;16:281–296. doi: 10.1002/1098-1004(200010)16:4<281::AID-HUMU1>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- 57.Corrado KR, Andrade SC, Bellizzi J, D’Souza-Li L, Arnold A. Polyclonality of Parathyroid Tumors in Neonatal Severe Hyperparathyroidism. J Bone Miner Res. 2015;30(10):1797–1802. doi: 10.1002/jbmr.2516. [DOI] [PubMed] [Google Scholar]
- 58.Hosokawa Y, Pollak MR, Brown EM, Arnold A. Mutational analysis of the extracellular Ca(2+)-sensing receptor gene in human parathyroid tumors. J Clin Endocrinol Metab. 1995;80(11):3107–3110. doi: 10.1210/jcem.80.11.7593409. [DOI] [PubMed] [Google Scholar]
- 59.Cetani F, Pinchera A, Pardi E, et al. No evidence for mutations in the calcium-sensing receptor gene in sporadic parathyroid adenomas. J Bone Miner Res. 1999;14(6):878–882. doi: 10.1359/jbmr.1999.14.6.878. [DOI] [PubMed] [Google Scholar]
- 60.Farnebo F, Enberg U, Grimelius L, et al. Tumor-specific decreased expression of calcium sensing receptor messenger ribonucleic acid in sporadic primary hyperparathyroidism. J Clin Endocrinol Metab. 1997;82(10):3481–3486. doi: 10.1210/jcem.82.10.4300. [DOI] [PubMed] [Google Scholar]
- 61.Nesbit MA, Hannan FM, Howles SA, et al. Mutations affecting G-protein subunit alpha11 in hypercalcemia and hypocalcemia. N Engl J Med. 2013;368(26):2476–2486. doi: 10.1056/NEJMoa1300253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gorvin CM, Cranston T, Hannan FM, et al. A G-protein Subunit-alpha11 Loss-of-Function Mutation, Thr54Met, Causes Familial Hypocalciuric Hypercalcemia Type 2 (FHH2) J Bone Miner Res. 2016;31(6):1200–1206. doi: 10.1002/jbmr.2778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Nesbit MA, Hannan FM, Howles SA, et al. Mutations in AP2S1 cause familial hypocalciuric hypercalcemia type 3. Nat Genet. 2013;45(1):93–97. doi: 10.1038/ng.2492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hendy GN, Canaff L, Newfield RS, et al. Codon Arg15 mutations of the AP2S1 gene: common occurrence in familial hypocalciuric hypercalcemia cases negative for calcium-sensing receptor (CASR) mutations. J Clin Endocrinol Metab. 2014;99(7):E1311–1315. doi: 10.1210/jc.2014-1120. [DOI] [PubMed] [Google Scholar]
- 65.Hannan FM, Howles SA, Rogers A, et al. Adaptor protein-2 sigma subunit mutations causing familial hypocalciuric hypercalcaemia type 3 (FHH3) demonstrate genotype-phenotype correlations, codon bias and dominant-negative effects. Hum Mol Genet. 2015;24(18):5079–5092. doi: 10.1093/hmg/ddv226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Vargas-Poussou R, Mansour-Hendili L, Baron S, et al. Familial Hypocalciuric Hypercalcemia Types 1 and 3 and Primary Hyperparathyroidism: Similarities and Differences. J Clin Endocrinol Metab. 2016;101(5):2185–2195. doi: 10.1210/jc.2015-3442. [DOI] [PubMed] [Google Scholar]
- 67.Harris TJ, McCormick F. The molecular pathology of cancer. Nat Rev Clin Oncol. 2010;7(5):251–265. doi: 10.1038/nrclinonc.2010.41. [DOI] [PubMed] [Google Scholar]
- 68.Wells SA, Jr, Santoro M. Targeting the RET pathway in thyroid cancer. Clin Cancer Res. 2009;15(23):7119–7123. doi: 10.1158/1078-0432.CCR-08-2742. [DOI] [PubMed] [Google Scholar]
- 69.Frank-Raue K, Raue F. Hereditary Medullary Thyroid Cancer Genotype-Phenotype Correlation. Recent Results Cancer Res. 2015;204:139–156. doi: 10.1007/978-3-319-22542-5_6. [DOI] [PubMed] [Google Scholar]
- 70.Eng C, Clayton D, Schuffenecker I, et al. The relationship between specific RET proto-oncogene mutations and disease phenotype in multiple endocrine neoplasia type 2. International RET mutation consortium analysis. JAMA. 1996;276(19):1575–1579. [PubMed] [Google Scholar]
- 71.Arnold A, Kim HG, Gaz RD, et al. Molecular cloning and chromosomal mapping of DNA rearranged with the parathyroid hormone gene in a parathyroid adenoma. J Clin Invest. 1989;83(6):2034–2040. doi: 10.1172/JCI114114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Rosenberg CL, Kim HG, Shows TB, Kronenberg HM, Arnold A. Rearrangement and overexpression of D11S287E, a candidate oncogene on chromosome 11q13 in benign parathyroid tumors. Oncogene. 1991;6(3):449–453. [PubMed] [Google Scholar]
- 73.Motokura T, Bloom T, Kim HG, et al. A novel cyclin encoded by a bcl1-linked candidate oncogene. Nature. 1991;350(6318):512–515. doi: 10.1038/350512a0. [DOI] [PubMed] [Google Scholar]
- 74.Imanishi Y, Hosokawa Y, Yoshimoto K, et al. Primary hyperparathyroidism caused by parathyroid-targeted overexpression of cyclin D1 in transgenic mice. J Clin Invest. 2001;107(9):1093–1102. doi: 10.1172/JCI10523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hsi ED, Zukerberg LR, Yang WI, Arnold A. Cyclin D1/PRAD1 expression in parathyroid adenomas: an immunohistochemical study. J Clin Endocrinol Metab. 1996;81(5):1736–1739. doi: 10.1210/jcem.81.5.8626826. [DOI] [PubMed] [Google Scholar]
- 76.Hemmer S, Wasenius VM, Haglund C, et al. Deletion of 11q23 and cyclin D1 overexpression are frequent aberrations in parathyroid adenomas. Am J Pathol. 2001;158(4):1355–1362. doi: 10.1016/S0002-9440(10)64086-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Tominaga Y, Tsuzuki T, Uchida K, et al. Expression of PRAD1/cyclin D1, retinoblastoma gene products, and Ki67 in parathyroid hyperplasia caused by chronic renal failure versus primary adenoma. Kidney Int. 1999;55(4):1375–1383. doi: 10.1046/j.1523-1755.1999.00396.x. [DOI] [PubMed] [Google Scholar]
- 78.Vasef MA, Brynes RK, Sturm M, Bromley C, Robinson RA. Expression of cyclin D1 in parathyroid carcinomas, adenomas, and hyperplasias: a paraffin immunohistochemical study. Mod Pathol. 1999;12(4):412–416. [PubMed] [Google Scholar]
- 79.Hosokawa Y, Tu T, Tahara H, Smith AP, Arnold A. Absence of cyclin D1/PRAD1 point mutations in human breast cancers and parathyroid adenomas and identification of a new cyclin D1 gene polymorphism. Cancer Lett. 1995;93(2):165–170. doi: 10.1016/0304-3835(95)03805-7. [DOI] [PubMed] [Google Scholar]
- 80.Yap DB, Chu J, Berg T, et al. Somatic mutations at EZH2 Y641 act dominantly through a mechanism of selectively altered PRC2 catalytic activity, to increase H3K27 trimethylation. Blood. 2011;117(8):2451–2459. doi: 10.1182/blood-2010-11-321208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Sanpaolo E, Miroballo M, Corbetta S, et al. EZH2 and ZFX oncogenes in malignant behaviour of parathyroid neoplasms. Endocrine. 2016 doi: 10.1007/s12020-016-0892-y. [DOI] [PubMed] [Google Scholar]
- 82.Soong CP, Arnold A. Recurrent ZFX mutations in human sporadic parathyroid adenomas. Oncoscience. 2014;1(5):360–366. doi: 10.18632/oncoscience.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Arnold A, Soong CP. New role for ZFX in oncogenesis. Cell Cycle. 2014;13(22):3465–3466. doi: 10.4161/15384101.2014.980693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Palanisamy N, Imanishi Y, Rao PH, Tahara H, Chaganti RS, Arnold A. Novel chromosomal abnormalities identified by comparative genomic hybridization in parathyroid adenomas. J Clin Endocrinol Metab. 1998;83(5):1766–1770. doi: 10.1210/jcem.83.5.4806. [DOI] [PubMed] [Google Scholar]
- 85.Agarwal SK, Schrock E, Kester MB, et al. Comparative genomic hybridization analysis of human parathyroid tumors. Cancer Genet Cytogenet. 1998;106:30–36. doi: 10.1016/s0165-4608(98)00049-1. [DOI] [PubMed] [Google Scholar]
- 86.Farnebo F, Kytölä S, Teh BT, et al. Alternative genetic pathways in parathyroid tumorigenesis. J Clin Endocrinol Metab. 1999;84:3775–3780. doi: 10.1210/jcem.84.10.6057. [DOI] [PubMed] [Google Scholar]
- 87.Kytölä S, Farnebo F, Obara T, et al. Patterns of chromosomal imbalances in parathyroid carcinomas. Am J Pathol. 2000;157:579–586. doi: 10.1016/S0002-9440(10)64568-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Garcia JL, Tardio JC, Gutierrez NC, et al. Chromosomal imbalances identified by comparative genomic hybridization in sporadic parathyroid adenomas. Eur J Endocrinol. 2002;146(2):209–213. doi: 10.1530/eje.0.1460209. [DOI] [PubMed] [Google Scholar]