

Abstract
We present SS-map, a tool to visualize the secondary structure content of ensembles of proteins. When generating ensembles of intrinsically disordered proteins, we lose the understanding a single native structure gives for folded proteins. It then becomes difficult to visualize the composition of the ensembles or to detect transient helices such as MoRFs. Conformational propensities for single residues also hide the nature of cooperative structures. Here we show how SS-map describes folded and unfolded ensembles of some peptides and gives a new view of the ensembles used to describe intrinsically disordered proteins with residual structure in computational and NMR experiments. This tool is implemented in an open-source python code located at code.google.com/p/ss-map
Keywords: intrinsically disordered proteins, IUP, ensembles, visualization, secondary structure, NMR, polyproline II
Intrinsically disordered proteins (IDPs) exist in solution as ensembles of structures. This raises a challenge to us, humans, as we tend to understand structures by visualizing them,1 and we lack ways to represent ensembles. Ensembles contain structural information, even when IDPs satisfy random-coil statistics.2,3 Some regions of IDPs can adopt secondary structures, at least for a transient time.4 This can be probed with experimental techniques such as NMR, in particular with Residual Dipolar Couplings (RDCs).5-8 Structured regions, termed MoRFs, are key to recognition processes mediated by coupled folding-binding events.9 The interpretation of data derived from NMR is usually done by stating that a certain segment of the protein chain adopts a certain secondary structure in a percentage of the total ensemble, but this conveys information in a difficult way for scientists not familiar with these interpretations. How can the ensembles be represented to better unveil their structure?
When studying protein folding, ensembles coming from computations are represented along the reaction coordinate of native contacts. This shows that for many (small) proteins, folding is a 2-state process. Thus, it is a cooperative event where most of the ensemble at a given temperature is either folded or unfolded. Victor Muñoz has pioneered the study of downhill folders, which fold in a progressive manner.10 How do MoRFs of IDPs behave? Contact order discriminates between 2-state and downhill folders, but it cannot be used in IDPs because it is based on the concept of a well-defined native structure. MoRFs are usually described as the ratio of residues that adopt a certain secondary structure. It is important to differentiate when residues in a fragment independently adopt a conformation in a secondary structure region, from when that fragment contains a true secondary structure, with all the residues adopting that conformation at the same time, even if that structure is only adopted rarely. Indeed, if n residues are in an α-helical region 20% of the time, that does not mean an helix of n residues is present 20% of the time. Whether this happens or not will lead to different experimental results, such as different RDCs, and it would be desirable to visualize the structural differences of these ensembles.
In this communication we present a way to represent the cooperativity or the correlations in secondary structure formation for IDPs, where the use of contact orders or native contacts is impossible. We named our approach SS-map, from Secondary Structure map. We first study 2-folded proteins near its melting temperature to link our SS-map with other visualization techniques used in the protein folding community. Then, we visualize an ensemble of a MoRF from a measles11 and a Sendai5,12 virus nucleoprotein. Finally we reconsider the existence of the polyproline II helix in IDPs.
The SS-map tool is available for download in http://code.google.com/p/ss-map/, under the GNU GPL v3 license. Graphical output from the SS-map is produced with the matplotlib library.13 Details of the simulated ensembles are reported in the supplemental material.
The visualization tool presented in this work extends the calculation of secondary-structure percentage per residue one more dimension: we calculate and show the frequency of having n exactly contiguous residues in a certain secondary structure. For a protein with N+2 residues, this generates a matrix of NxN, where an element (m,n) corresponds to the frequency of having residue m forming a secondary structure element of length exactly n (see for example Fig. 1). Frequencies are normalized, so that if one wants the probability of residue m forming an helix of at least 4 residues, one can get it by summing row m, elements 4 to N.

Figure 1. SS-map representing α-helices for the HPLC-6 peptide at different temperatures [(A): 313K, (B): 320K, (C): 327K)]. Large helices are lost below the melting temperature of 323K and all fragments grow from a central residue. At 320K an ensemble of helices with a wide range of lengths is present but shorter helices are not more abundant than longer ones.
There are different definitions of secondary structure elements. Currently our code can use the definition reported in reference 14, where all the Ramachandran space is assigned to an element; a more restrictive definition as in reference 15; or a user defined rectangular region of the Ramachandran plot. When the ensemble is input as a set of PDB files, SS-map uses the Bio.PDB16 module of Biopython17 to generate dihedral angles. Alternatively, we can use the external code Stride18 to read the secondary structure. Differences in applying these definitions will be discussed below. A schematic workflow with the different possible input and outputs of SS-map is depicted in Figure S1.
The information that SS-map presents requires an image for each of the ensembles. This information can be compressed in 2 ways to represent several ensembles in 1 image. The raw-average gives the widely used probability of a certain residue being in the selected conformation, as Figure S2 shows. The column-average gives new and complementary information: the percentage of fragments of a given length. This information can then be combined for different ensembles, for example, at different temperatures, such as in Figures 2 and 3C.
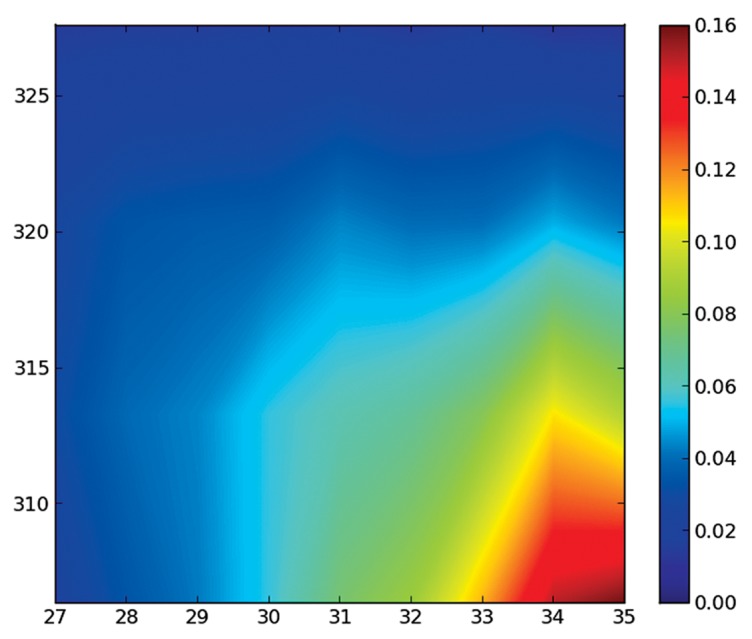
Figure 2. Temperature dependence of the presence of secondary structure elements at different temperatures. The x-axis represents the length of a helix element, and the y-axis the temperature. It shows how long helices are present only at low temperatures, and that helices do not get shorter, they just become much scarcer at higher temperatures (only the region of long helices is plotted, as the remaining region is essentially zero).

Figure 3. SS-map showing β-strands for the GB1p β-hairpin below the folding temperature [(A): 319K and above (B): 327K]. The temperature dependence of the SS-map shows that at all temperatures the most frequent strand has 4 residues (C).
We first present a study of the unfolding of the peptide HPLC-6, which forms an α-helix and has a melting temperature of 323K when simulated with the Profasi force field.19 The percentage of α-helix conformation for each peptide gradually decreases with temperature. This is more prominent at the N- and the C-terminus (Fig. S2; Fig. S3). The SS-map shows that at 313K a long helix spanning most of the residues is the most abundant structure (see Fig. 1). At 320K, this long helix is lost and fragments of different sizes are almost equally present, but in all cases, these fragments grow from the central residue 19. A representation of secondary structure per residue (Fig. S2) suggests that helices get shorter with temperature. This is not true: Long α-helix segments are not less frequent than shorter ones. At 320K, all fragments are rare, and the cumulative percentage of helices larger than 20 residues represents only a 21%. This number, at 313K is of 71%. At 327K, although the overall percentage of α-helix is still 45% (Fig. S2), there is no helix as such, only residues that adopt this conformation independently, without any cooperativity. This information cannot be reflected with the visualizations traditionally used, such as Figure S2, but it is relevant to interpret the results of circular dicroism that revealed a non-negligible percentage of α-helix even at 343K:20 our interpretation is that it was only due to isolated residues in α-helix, and not to true helical segments.
The information of a range of ensembles at different temperatures can be compressed as previously explained. Figure 2 shows that the long helix spanning 34 or 35 residues is lost between 310 and 315K, and then the ensemble is composed of helices of several different lengths. An essentially unfolded ensemble at the melting temperature agrees with recent similar findings for the more complex Protein A.21
We now focus on a structure that forms a β-hairpin, i.e., 2 β-sheets connected by a turn. We have taken a mutated from of the GB1p peptide (GB1m2)22 also studied with the Profasi force field.19 The simulated melting temperature for this peptide is very similar to the previous α-helix, 324K. The SS-map shows two β-strands and an empty 4-residue central region, which corresponds to the β-turn (Fig. 3). Even above the transition temperature, the strands of the hairpin remain the most populated structures, in contrast to the α-helix. The SS-map shows that the unfolded state of this β-hairpin—ensembles above the folding temperature—has different structural characteristics than the unfolded state of the α-helix (Fig. 1; Fig. 2). The temperature profile of the SS-map in Figure 3 also contrasts with the one for the α-helix.
We now focus on a true IDP that contains fragments of partial secondary structure. These fragments are called MoRFs and correspond to binding regions of the IDPs.9 Partially ordered regions are a challenge for many biophysical techniques,4 but a successful approach is the use of NMR Residual Dipolar Couplings.6-8 Here we will consider 2 proteins: a Measles virus nucleocapsid protein11 and a Sendai virus nucleoprotein,5 both studied by Blackledge and coworkers. In both proteins, the authors used a random-coil model named Flexible Meccano12,23 to generate an ensemble of structures (Fig. 4). Then they added helical fragments—in a statistically robust way—until they achieved a satisfactory fit of the RDCs. A special conformational treatment was given to the N-capping residues of the helices. The N-capping modifications are not implemented in the public version of Flexible Meccano, and therefore our ensembles differ from the ones used by Blackledge and collegues (see the SI for a further discussion of this point). Table 1 describes the composition of both ensembles.

Figure 4. Helical content for the ensembles that reproduces the experimental RDCs of the Sendai virus nucleoprotein5 (A) and the measles virus nucleoprotein11 (B). Both ensembles were generated with Flexible Meccano by mixing ensembles with pre-defined helical content as detained in Table 1. Although 3 helices were used for the Sendai protein and 4 for the measles protein, the resulting ensemble is more continuous and mixed than Table 1 might suggest.
Table 1. Composition of the ensembles generated with Flexible Meccano12,23 to reproduce the RDCs for the Sendai virus nucleoprotein5 and measles virus nucleoprotein,11 based on the data provided therein.
The analysis of the ensemble using SS-map shows that the picture is more complex than it might seem. For example, helix H1 and H2 in the measles virus protein mix together to give an ensemble of helices that have lengths from 5 to 8 residues. Similarly helices H2 and H3 in the Sendai virus protein cannot really be differentiated and extend from the limits stated in Table 1. In our ensembles helices extend both toward the N-terminal and the C-terminal sense symmetrically, due to the lack of the N-capping treatment.
SS-map helps to bring light to these features, but as a visualization tool it does not substitute the work to determine what constitutes a correct ensemble. Here we have exploited the statistically sound analysis of Blackledge and coworkers to optimize the ensemble to fit the experimental data and we have only considered their best results.
The presence of polyproline II (PPII) helices in IDPs has been studied in several works. It has been related to the unexpected temperature behavior of IDPs24 and its content correlates with the net charge of the IDPs15 because PPII helices are the most stable conformations for charged residues.25 We have analyzed the simulated ensembles of 4 IDPs studied by Pappu and colleagues, but here we only report the results for a poly-glutamine of 34 residues (id. 21 in their work15) because the results are similar for the other IDPs. Among all their reported IDPs, this one has the highest PPII content, as expected from its highest charge. Although the total PPII content is 51%, Figure 5 shows that the longest helices present in the ensembles contain only 5 consecutive residues. To avoid being deceived by single-residue propensities, Pappu and coworkers counted only fragments of 3 or more consecutive residues in PPII conformation. SS-map removes the arbitrariness of that number “3”and conveys more information. As opposed to the α-helix in Figure 1, there is no growing helix from any central residue. Thus, long helices of PPII do not cooperatively form in solution, at least in the models used by Pappu and coworkers.15 Considering that electrostatic interactions in water increase with temperature,26 it would be interesting to study how these ensembles change when heated. We leave that for future work.
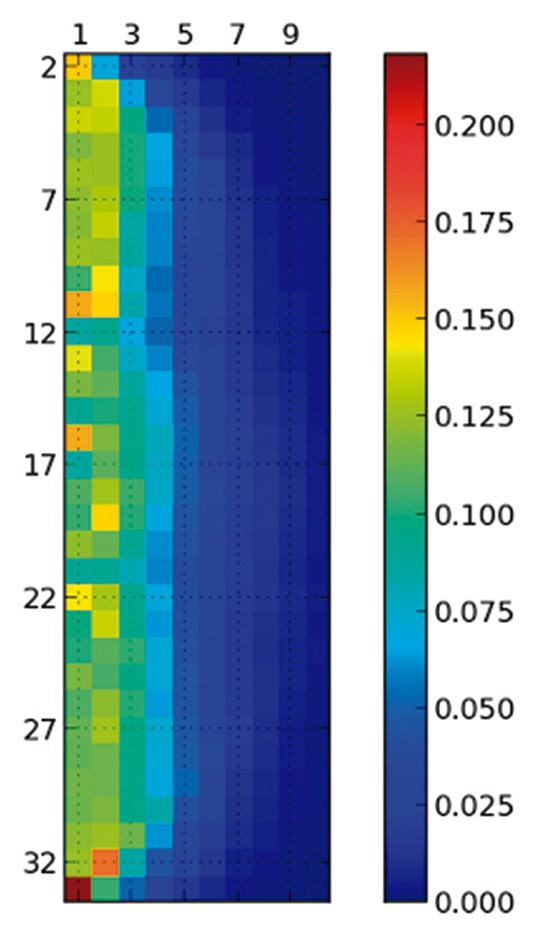
Figure 5. Content of Polyproline II for the 34-residue poly-glutamine, the region defining the polyproline II is the same as in the original study.15 Although the natural propensity of all the residues is to be in PPII with a relevant frequency, the formation of a helix is not a cooperative process and long helices are absent, in contrast to the α-helices of Figure 1.
Although everybody agrees on the qualitative description of α-helices and β-sheets, different groups partition the Ramachandran plot in different regions. For example, Blackledge and coworkers use big rectangular regions so that any point belongs to a given secondary structure.14 Although these regions are larger than what is usually accepted, they allow the classification of all points in the Ramachandran plot. Pappu and colleagues use much more restrictive secondary structure elements,15 closer to more wide-spread definitions such as the one in the Wikipedia.27 In SS-map users can also measure with their own definitions. The effect of these arbitrariness could be more important in IDPs than in folded proteins, due precisely to their higher disorder. Figure 6 shows the ensembles plotted using different criteria. It is interesting that the Stride program never considers a fragments of less than 4 residues to have a secondary structure, to model as closely as possible how crystallographers represent α-helices and β-strands.18 Therefore the results differ in those 1 to 3 residue fragments, but agree almost quantitatively in the rest. The more restrictive definitions used by Pappu and coworkers15 lead to overall lower percentages of secondary structure fragments as expected, but the general picture remains the same (compare Fig. 6 with Fig. 4B). Whether a consensus is necessary or not is something the scientific community has to decide, but our present findings suggest that the structural interpretations do not change significantly with varying definitions.

Figure 6. SS-map for the measles virus nucleoprotein showed if Figure 4 using two different criteria to define the α-helix. The external program Stride (A), which only considers a secondary structure element when it is larger than 4 residues, and the definition used in reference 15 (B), which is approximately circular and much smaller than the region used by Blackledge and coworkers.14
Understanding IDPs with partially folded regions is a challenge to both computation and experiment.4 Conformations cannot be referenced or compared with a native structure and we need new tools to visualize these heterogeneous ensembles. In this work we presented a tool, SS-map, which literally adds a new dimension to the representation of IDPs ensembles. By including the correlation between secondary structure elements in fragments, a more detailed picture emerges. Differences between α-helices, β-strands and PPII regions become more evident. The ensembles used to reproduces RDCs data can also be visualized and compared. SS-map does not optimize or change the ensembles whatsoever, it only extracts information from them and displays it. The results are as realistic as the underlying ensemble is; finding these ensembles remains a challenge.28 Finally, this tool can also be useful to analyze the folding process of small proteins and peptides.29
Glossary
Abbreviations:
- IDP
intrinsically disordered protein
- SS
secondary structure
- RDC
residual dipolar couplling
- PPII
Polyproline II helix
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
Acknowledgments
We would like to thank Rohit Pappu for kindly sharing his data on the study of polyglutamines19 and Martin Blackledge for helpful comments. We acknowledge financial support from the Ministerio de Innovación y Competitividad (CTQ2012-33324) and the Generalitat de Catalunya (2009SGR01472). MSM thanks the Ministerio de Economia y Competitividad for a predoctoral fellowship.
References
- 1.Gan J, Norman C. . 2012 Visualization Challenge. Science 2013; 339:509; http://dx.doi.org/ 10.1126/science.339.6119.509 [DOI] [Google Scholar]
- 2.Fitzkee NC, Rose GD. . Reassessing random-coil statistics in unfolded proteins. Proc Natl Acad Sci U S A 2004; 101:12497 - 502; http://dx.doi.org/ 10.1073/pnas.0404236101; PMID: 15314216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jha AK, Colubri A, Freed KF, Sosnick TR. . Statistical coil model of the unfolded state: resolving the reconciliation problem. Proc Natl Acad Sci U S A 2005; 102:13099 - 104; http://dx.doi.org/ 10.1073/pnas.0506078102; PMID: 16131545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dyson HJ. . Expanding the proteome: disordered and alternatively folded proteins. Q Rev Biophys 2011; 44:467 - 518; http://dx.doi.org/ 10.1017/S0033583511000060; PMID: 21729349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jensen MR, Houben K, Lescop E, Blanchard L, Ruigrok RWH, Blackledge M. . Quantitative conformational analysis of partially folded proteins from residual dipolar couplings: application to the molecular recognition element of Sendai virus nucleoprotein. J Am Chem Soc 2008; 130:8055 - 61; http://dx.doi.org/ 10.1021/ja801332d; PMID: 18507376 [DOI] [PubMed] [Google Scholar]
- 6.Schneider R, Huang JR, Yao M, Communie G, Ozenne V, Mollica L, et al. . Towards a robust description of intrinsic protein disorder using nuclear magnetic resonance spectroscopy. Mol Biosyst 2012; 8:58 - 68; http://dx.doi.org/ 10.1039/c1mb05291h; PMID: 21874206 [DOI] [PubMed] [Google Scholar]
- 7.Jensen MR, Markwick PRL, Meier S, Griesinger C, Zweckstetter M, Grzesiek S, et al. . Quantitative determination of the conformational properties of partially folded and intrinsically disordered proteins using NMR dipolar couplings. Structure 2009; 17:1169 - 85; http://dx.doi.org/ 10.1016/j.str.2009.08.001; PMID: 19748338 [DOI] [PubMed] [Google Scholar]
- 8.Marsh JA, Neale C, Jack FE, Choy W-Y, Lee AY, Crowhurst KA, et al. . Improved structural characterizations of the drkN SH3 domain unfolded state suggest a compact ensemble with native-like and non-native structure. J Mol Biol 2007; 367:1494 - 510; http://dx.doi.org/ 10.1016/j.jmb.2007.01.038; PMID: 17320108 [DOI] [PubMed] [Google Scholar]
- 9.Mohan A, Oldfield CJ, Radivojac P, Vacic V, Cortese MS, Dunker AK, et al. . Analysis of molecular recognition features (MoRFs). J Mol Biol 2006; 362:1043 - 59; http://dx.doi.org/ 10.1016/j.jmb.2006.07.087; PMID: 16935303 [DOI] [PubMed] [Google Scholar]
- 10.Garcia-Mira MM, Sadqi M, Fischer N, Sanchez-Ruiz JM, Muñoz V. . Experimental identification of downhill protein folding. Science 2002; 298:2191 - 5; http://dx.doi.org/ 10.1126/science.1077809; PMID: 12481137 [DOI] [PubMed] [Google Scholar]
- 11.Jensen MR, Communie G, Ribeiro EA Jr., Martinez N, Desfosses A, Salmon L, et al. . Intrinsic disorder in measles virus nucleocapsids. Proc Natl Acad Sci U S A 2011; 108:9839 - 44; http://dx.doi.org/ 10.1073/pnas.1103270108; PMID: 21613569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bernadó P, Blanchard L, Timmins P, Marion D, Ruigrok RWH, Blackledge M. . A structural model for unfolded proteins from residual dipolar couplings and small-angle x-ray scattering. Proc Natl Acad Sci U S A 2005; 102:17002 - 7; http://dx.doi.org/ 10.1073/pnas.0506202102; PMID: 16284250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hunter JD. . Matplotlib: A 2D Graphics Environment. Comput Sci Eng 2007; 9:90 - 5; http://dx.doi.org/ 10.1109/MCSE.2007.55 [DOI] [Google Scholar]
- 14.Nodet G, Salmon L, Ozenne V, Meier S, Jensen MR, Blackledge M. . Quantitative description of backbone conformational sampling of unfolded proteins at amino acid resolution from NMR residual dipolar couplings. J Am Chem Soc 2009; 131:17908 - 18; http://dx.doi.org/ 10.1021/ja9069024; PMID: 19908838 [DOI] [PubMed] [Google Scholar]
- 15.Mao AH, Crick SL, Vitalis A, Chicoine CL, Pappu RV. . Net charge per residue modulates conformational ensembles of intrinsically disordered proteins. Proc Natl Acad Sci U S A 2010; 107:8183 - 8; http://dx.doi.org/ 10.1073/pnas.0911107107; PMID: 20404210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hamelryck T, Manderick B. . PDB file parser and structure class implemented in Python. Bioinformatics 2003; 19:2308 - 10; http://dx.doi.org/ 10.1093/bioinformatics/btg299; PMID: 14630660 [DOI] [PubMed] [Google Scholar]
- 17.Cock PJA, Antao T, Chang JT, Chapman BA, Cox CJ, Dalke A, et al. . Biopython: freely available Python tools for computational molecular biology and bioinformatics. Bioinformatics 2009; 25:1422 - 3; http://dx.doi.org/ 10.1093/bioinformatics/btp163; PMID: 19304878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Frishman D, Argos P. . Knowledge-based protein secondary structure assignment. Proteins 1995; 23:566 - 79; http://dx.doi.org/ 10.1002/prot.340230412; PMID: 8749853 [DOI] [PubMed] [Google Scholar]
- 19.Irbäck A, Mitternacht S, Mohanty S. . An effective all-atom potential for proteins. PMC Biophys 2009; 2:2; http://dx.doi.org/ 10.1186/1757-5036-2-2; PMID: 19356242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chakrabartty A, Ananthanarayanan VS, Hew CL. . Structure-function relationships in a winter flounder antifreeze polypeptide. I. Stabilization of an α-helical antifreeze polypeptide by charged-group and hydrophobic interactions. J Biol Chem 1989; 264:11307 - 12; PMID: 2738067 [PubMed] [Google Scholar]
- 21.Maisuradze GG, Liwo A, Ołdziej S, Scheraga HA. . Evidence, from simulations, of a single state with residual native structure at the thermal denaturation midpoint of a small globular protein. J Am Chem Soc 2010; 132:9444 - 52; http://dx.doi.org/ 10.1021/ja1031503; PMID: 20568747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fesinmeyer RM, Hudson FM, Andersen NH. . Enhanced hairpin stability through loop design: the case of the protein G B1 domain hairpin. J Am Chem Soc 2004; 126:7238 - 43; http://dx.doi.org/ 10.1021/ja0379520; PMID: 15186161 [DOI] [PubMed] [Google Scholar]
- 23.Ozenne V, Bauer F, Salmon L, Huang J-R, Jensen MR, Segard S, et al. . Flexible-meccano: a tool for the generation of explicit ensemble descriptions of intrinsically disordered proteins and their associated experimental observables. Bioinformatics 2012; 28:1463 - 70; http://dx.doi.org/ 10.1093/bioinformatics/bts172; PMID: 22613562 [DOI] [PubMed] [Google Scholar]
- 24.Kjaergaard M, Nørholm A-B, Hendus-Altenburger R, Pedersen SF, Poulsen FM, Kragelund BB. . Temperature-dependent structural changes in intrinsically disordered proteins: formation of alpha-helices or loss of polyproline II?. Protein Sci 2010; 19:1555 - 64; http://dx.doi.org/ 10.1002/pro.435; PMID: 20556825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Krimm S, Mark JE. . Conformations of polypeptides with ionized side chains of equal length. Proc Natl Acad Sci U S A 1968; 60:1122 - 9; http://dx.doi.org/ 10.1073/pnas.60.4.1122; PMID: 16591670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Thomas AS, Elcock AH. . Molecular simulations suggest protein salt bridges are uniquely suited to life at high temperatures. J Am Chem Soc 2004; 126:2208 - 14; http://dx.doi.org/ 10.1021/ja039159c; PMID: 14971956 [DOI] [PubMed] [Google Scholar]
- 27.Wikipedia. Ramachandran Plot, http://en.wikipedia.org/wiki/Ramachandran_plot
- 28.Fisher CK, Stultz CM. . Constructing ensembles for intrinsically disordered proteins. Curr Opin Struct Biol 2011; 21:426 - 31; http://dx.doi.org/ 10.1016/j.sbi.2011.04.001; PMID: 21530234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Irbäck A, Mohanty S. . PROFASI: A Monte Carlo simulation package for protein folding and aggregation. J Comput Chem 2006; 27:1548 - 55; http://dx.doi.org/ 10.1002/jcc.20452; PMID: 16847934 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.