

Abstract
Tyrosine kinase inhibitor (TKI) therapy for human cancers is not curative, with relapse due to the continuing presence of tumor cells, referred to as minimal residual disease (MRD) cells. MRD stem or progenitor cells survival in the absence of oncogenic kinase signaling, a phenomenon referred to as intrinsic resistance, depends on diverse growth factors. Here, we report that oncogenic kinase and growth factor signaling converge to induce the expression of the signaling proteins c-Fos and Dusp1. Genetic deletion of c-Fos and Dusp1 suppressed tumor growth in a BCR-ABL-induced mouse model of chronic myeloid leukemia (CML). Pharmacological inhibition of c-Fos, Dusp1 and BCR-ABL eradicated MRD in multiple in vivo models, as well as in primary CML patient xenotransplanted mice. Growth factor signaling also conferred TKI resistance and induced c-FOS and DUSP1 expression in tumor cells modeling other types of kinase-driven leukemias. Our data demonstrate that c-Fos and Dusp1 expression levels determine the threshold of TKI efficacy, such that growth factor-induced expression of c-Fos and Dusp1 confers intrinsic resistance to TKI therapy in a wide-ranging set of leukemias, and may represent a unifying Achilles heel of kinase-driven cancers.
Protein kinases are frequently activated in a variety of human cancers and represent attractive drug targets. In this regard, chronic myeloid leukemia (CML) represents an important paradigm, as the success of imatinib in treating CML patients provided proof of concept for targeted anti-kinase therapy and paved the way for the development of tyrosine kinase inhibitor (TKI) therapy for several solid tumor types1,2. Despite the impressive response to TKI therapy in the clinic, it is not curative because a small population of cancer cells are insensitive to treatment; manifesting as minimal residual disease (MRD)3. The cells responsible for MRD in CML are referred to leukemia-initiating cells (LICs), whereas those responsible for MRD in solid tumors are referred to as cancer stem cells (CSCs). In ~50–60 % of CML patients, continuous drug treatment is needed to prevent MRD cells from reinstating the disease4–6. MRD cells serve as a reservoir of cells that can develop TKI resistance by acquiring mutations or by activating alternative survival mechanisms7–9. Even the most potent kinase inhibitors are ineffective against LICs that are present in MRD3,10.
Oncogene addiction refers to the phenomenon in which transformed cells become exquisitely dependent upon a single mutant protein or signaling pathway for survival and proliferation11. The therapeutic response to TKIs is mediated by oncogene addiction to mutant tyrosine kinase oncoproteins11–13. Multiple theories have attempted to explain how cells become oncogene addicted, and how acute oncoprotein inhibition induces cell death, including signaling-network dysregulation, synthetic lethality14,15, genetic streamlining16,17, and oncogenic shock18,19. However, it is still not understood how MRD cells that do not respond to TKI therapy escape addiction from the driver oncogene. Recent studies have revealed that growth factor signaling mediates resistance to TKI therapy in both leukemia and solid organ tumors20–22, but, it remains to be determined if intrinsic resistance conferred by a diverse set of growth factors utilizes distinct or shared molecular pathways. For instance, IL3,IL6, SCF, FLT3L, and GCSF signaling in CML progenitor cells confer intrinsic resistance to imatinib. Likewise, hepatocyte growth factor (HGF) and neuregulin1 (NRG1) confer intrinsic-resistance to BRAF and EGFR inhibitors in solid tumors20–22.
RESULTS
Induced expression of c-Fos and Dusp1 by growth factor signaling confers TKI resistance
To understand how growth factor signaling induces intrinsic resistance to TKI treatment, we modeled growth factor induced-mitigation of TKI response using the interleukin-3 (IL-3)-dependent BaF3 cell line. We generated BaF3 cells with tetracycline-inducible expression of BCR-ABL (BaF3-LTBA, Fig. 1a) as well as those with constitutive BCR-ABL expression (BaF3-BA9). Imatinib treatment of both BaF3-LTBA and BAF3-BA cells caused cell death, whereas addition of IL-3 conferred resistance to imatinib, even in the case of sustained inhibition of BCR-ABL enzymatic activity (Fig. 1b–d and Supplementary Fig. 1a). Likewise, erythropoietin treatment conferred imatinib resistance in the human BCR-ABL+ cell line, K562 (an erythromyeloblastoid leukemia cell line derived from a blast crisis CML patient, Fig. 1e and Supplementary Fig. 1b). Thus, we are able to recapitulate cytokine/growth factor induced resistance to imatinib in vitro.
Figure 1. Expression of c-Fos, Dusp1 and Zfp36 constitutes a common signature of imatinib resistant cells.
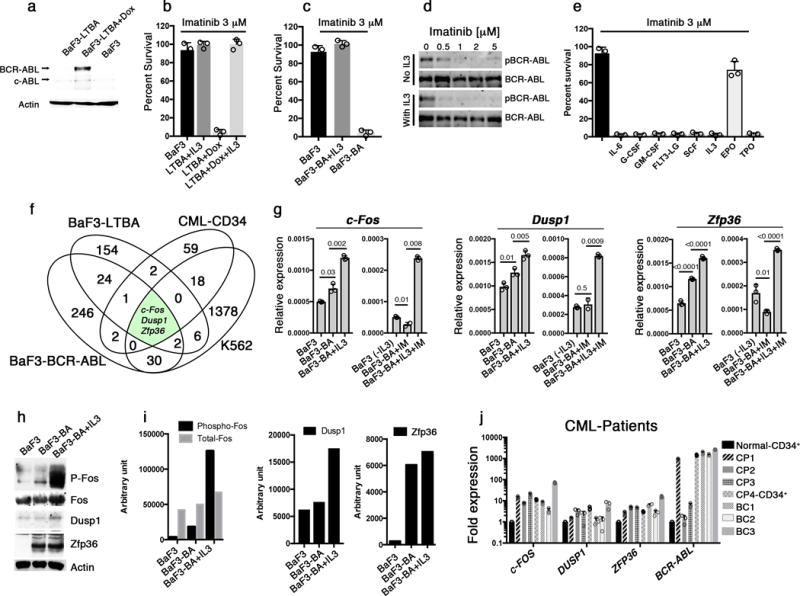
a. Immunoblot analysis of BCR-ABL expression in the indicated cell lines. Doxycycline (Dox) was used to induce BCR-ABL expression in BaF3-LTBA cells.. Actin was used as a loading control and c-Abl represent the endogenous Abl kinase b. Percent survival of BaF3 or BaF3-LBTA (LTBA) cells treated with imatinib (3 μM) without or with IL-3. c. Percent survival of BaF3 or BaF3-BA cells treated with imatinib (3 μM) without or with IL-3. d. Immunoblot analysis of BCR-ABL expression in BaF3-BA cells treated with increasing concentrations of imatinib without or with IL-3. pBCR-ABL, phosphorylated-BCR-ABL e. Percent survival of K562 cells treated with (3 μM) imatinib alone or with the indicated cytokines. f. Venn diagram showing 3 commonly expressed genes among four different experiments described in Supplementary Fig. 1g g. Real-time qPCR analysis of c-Fos, Dusp1 and Zfp36 expression in BaF3 cells either untreated or 1 h after IL-3 was withdrawn from IL-3 treated cells (-IL-3), and in BaF3-BA cells with or without IL-3 treatment or 1 h imatinib (IM) treatment. The data shown are mean ± S.D. from 3 technical replicates of qPCR (P values are shown above the compared bars). (h–i). Immunoblot analysis (h) and densitometric quantification from one representative blot (i) of c-Fos, Dusp1 and Zfp36 expression in BaF3 cells and BaF3-BA cells +/− IL3. j. Real-time qPCR analysis of c-FOS, DUSP1, and ZFP36 expression in primary CML patient peripheral blood mononuclear cells (except for sample CP4, for which CD34+ cells were analyzed) normalized to expression in normal donor CD34+ cells (black bar). Four chronic phase (CP) and three blast crisis (BC) patients were analyzed. Data are shown from two independent qPCR analysis ± S.D. Individual data points are denoted as empty circles in each bar graphs.
We hypothesized that expression of the critical genes mediating TKI resistance would be modulated by BCR-ABL, growth factor signaling and TKI treatment. We therefore compared the expression profiles of BCR-ABL-induced BaF3-LTBA cells with and without IL-3 treatment (192 genes were differentially expressed; Supplementary Fig. 1c), as well as imatinib-treated BAF3-BA cells with and without IL-3 (308 genes were differentially expressed; Supplementary Fig. 1d). Next, we evaluated erythropoietin-modulated gene expression in imatinib-treated K562 cells (1,338 genes were differentially expressed; Supplementary Fig. 1e). Finally, we analyzed existing gene expression profiles from primary CML patient bone-marrow (BM) BCR-ABL+ CD34+ cells collected before and after one week of imatinib treatment23 and identified genes that are differentially expressed in the surviving marrow cells (85 genes were differentially expressed; Supplementary Fig. 1f). When these four data sets were compared, only three differentially expressed genes were common to all comparisons: c-Fos, Dusp1 and Zfp36 (Fig. 1f, and Supplementary Fig. 1g). c-Fos belongs to the family of AP1 (Activator protein 1) transcription factors implicated in the regulation of cell proliferation, survival, apoptosis, transformation and oncogenesis24. Dusp1 (Dual specificity phosphatase-1) is a nuclear protein that provides feedback regulation to MAPK signaling by inactivating MAPKs25, and has been implicated in regulating inflammation, immune regulation and chemoresistance in cancer25,26. Zfp36 is an RNA-binding protein that has been implicated in cancer development, inflammation and immune functions27.
In support of the hypothesis that oncogenic and growth factor signaling modulate Fos, Dusp1 and Zfp36 expression, we found that both BCR-ABL and imatinib induced expression of these genes in BaF3-BA cells (Fig. 1g–i). Likewise, expression analysis of patient samples from chronic and blast phase CML revealed higher (2–10 fold) expression of c-FOS, DUSP1 and ZFP36 compared to normal CD34+ cells (Fig. 1j; the CML patient samples used in this study are described in Supplementary Table 1). Whereas treatment with imatinib alone downregulated the expression of these genes, treatment with imatinib plus growth factor (IL-3) resulted in 4–5 fold higher expression of these genes as compared to BaF3-BA+IM (Fig 1g). In the absence of IL-3, ectopic expression of either Fos or Dusp1 led to modest resistance to imatinib, whereas ectopic expression of Zfp36 did not have an effect (Supplementary Fig. 2a and b). However, similarly to IL-3, ectopic expression of c-Fos, Dusp1 and Zfp36 together in BaF3-BA cells impaired the inhibitory effect of imatinib on cell survival (Supplementary Fig. 2b). Conversely, depletion of c-Fos, Dusp1 and Zfp36 (either each alone or in combination) by shRNA-mediated knockdown reduced BCR-ABL-dependent proliferation and survival in BaF3-BA cells, whereas parental BaF3 cells were not affected (Supplementary Fig. 2c–f). These results suggest that c-Fos, Dusp1, and Zfp36 are functional mediators of growth factor-induced imatinib resistance. Furthermore, knockdown of c-Fos and Dusp1 alone or together sensitized BaF3-BA cells to imatinib, even in the presence of IL-3 (Supplementary Fig. 2g). However, depletion of Zfp36 sensitized parental BaF3 and BaF3-BA cells equally to imatinib, suggesting that, unlike c-Fos and Dusp1, Zfp36 is not differentially required by BCR-ABL-expressing cells. Therefore, we focused subsequent analyses on c-Fos and Dusp1.
Deletion of c-Fos and Dusp1 abrogates intrinsic TKI resistance
To determine the role of c-Fos and Dusp1 in BCR-ABL-induced leukemogenesis, we determined the effects of deletion of either Dusp1−/− 28 or c-Fosfl/fl 29 (ROSACreERT2c-Fosfl/fl), or both genes (ROSACreERT2c-Fosfl/flDusp1−/−). We used hematopoietic Kit+ cells transduced with BCR-ABL-Ires-YFP retroviruses for in vitro CFU assays and for generating an in vivo CML model (Fig. 2a). Genetic deletion of c-Fos or Dusp1 significantly reduced (by 50%) the number of CFUs generated by BCR-ABL expressing Kit+ cells, whereas CFU generation by control cells (Kit+ cells transduced with a MSCV-Ires-YFP virus) was not affected (Fig. 2b and c). Deletion of Fos and Dusp1 alone sensitized BCR-ABL expressing Kit+ cells to imatinib treatment (~80% reduction compared to 50% reduction in wild-type control). Strikingly, deletion of both c-Fos and Dusp1 suppressed the number of CFUs generated by BCR-ABL expressing cells (~90%), and treatment with imatinib completely eradicated BCR-ABL-positive colonies (Fig. 2d and e). For in vivo analysis, mice were transplanted with 40,000 Kit+ bone-marrow cells expressing BCR-ABL and YFP. Recipient mice developed fatal leukemia with a disease latency of 2–3 weeks30. Imatinib treatment did not result in a significant reduction in leukemic burden compared to vehicle and all mice died within 3–4 weeks (Fig. 2 f and i). In contrast, deletion of Dusp1 delayed BCR-ABL-induced leukemia by one week and deletion of c-Fos led to a disease latency of 7–8 weeks (Fig. 2f–g). Notably, imatinib treatment of mice transplanted with Fos deficient cells for one month led to a significant reduction in leukemic burden (to 0.5–4%) and ~50% of mice survived, and treatment discontinuation did not result in disease relapse (Fig. 2g and j), suggesting the elimination of TKI-resistant MRD. In control experiments in which c-Fos was not deleted (non-tamoxifen treated ROSACreERT2c-Fosfl/fl donor cells), the recipients showed similar disease latency as wild-type (WT) mice in both imatinib treated and untreated groups (Supplementary Fig. 3a and c).
Figure 2. Genetic deletion of c-Fos and Dusp1 increases the response of BCR-ABL-induced leukemia to imatinib.
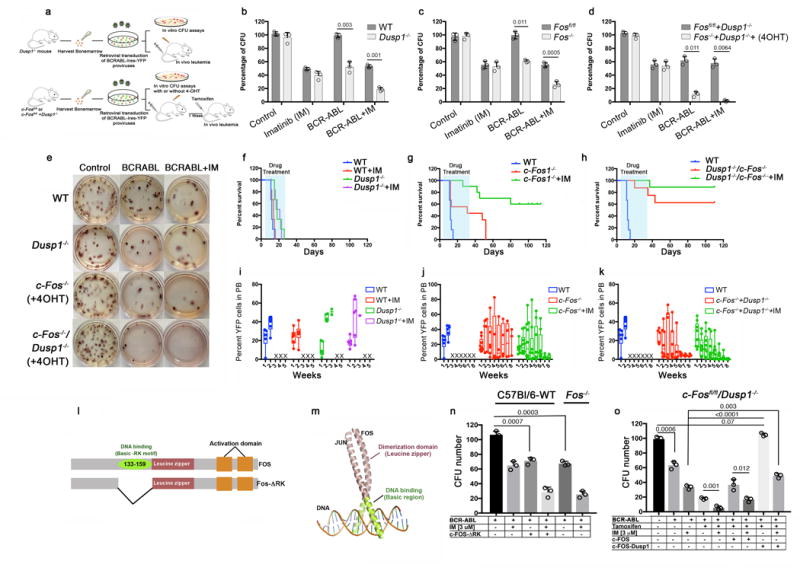
a. Experimental design of in vitro and in vivo experiments to analyze BCR-ABL disease using Dusp1−/−, ROSACreERT2c-Fosfl/fl and ROSACreERT2c-Fosfl/flDusp1−/− mice. Kit+ cells from mouse bone marrow were transduced with BCR-ABL-IRES-YFP retrovirus. Five thousand GFP+ cells were plated for in vitro CFU assays, and 40,000 YFP+ cells were transplanted to monitor in vivo leukemia development in lethally irradiated C57BL/6 mice. b–d. Percent CFUs from Kit+ cells expressing BCR-ABL in the absence of Dusp1 (b), c-Fos (c), and both c-Fos and Dusp1 (d). The data show the mean colony number ± S.D (n = 3; P values are shown above the compared bars). e. Representative photographs of BCR-ABL positive colonies described above (b–d) f–h. Survival curves of mice transplanted with BCR-ABL-YFP transduced Kit+ cells from wild type (WT), Dusp1−/− mice (f) ROSACreERT2c-Fosfl/fl mice (g), and ROSACreERT2c-Fosfl/flDusp1−/− mice (h). The mice were untreated or treated with imatinib. The data shown are from two independent transplantation experiments (n = 6 mice per group); Log Ranked mantel-Cox test, f p < 0.0001 between WT and Dusp1−/− with or without imatinib; g n=12 Log Ranked mantel-Cox test p < 0.0001 between WT and Fos−/− with or without imatinib, h n=12 Log Ranked mantel-Cox test p < 0.0001 between WT and Fos−/− Dusp1−/− with or without Imatinib). In g and h, the transplant recipients were treated with three doses of 2 mg/kg tamoxifen injection to delete c-Fos. i–k. Leukemic burden in mice transplanted with Dusp1−/− (i), c-Fos−/− (j) and c-Fos−/− Dusp1−/− (k) Kit+ cells, measured by the percentage of YFP+ cells in peripheral blood. Dead mice are represented with an X. i. Primary structure of c-Fos and its dominant negative version, c-Fos-ΔRK, which lacks the DNA binding domain consisting of a Basic-RK motif (amino acid residues 133–159). m. Tertiary structure of Fos and Jun bound to AP1-site on DNA, illustrating the homo/heterodimer assembly of Fos with Jun n. Percent CFUs from wild-type (WT) BCR-ABL-YFP+ Kit+ cells expressing dominant negative c-Fos-ΔRK +/− imatinib, compared to Fos−/− BCR-ABL-YFP+ Kit+ cells +/− imatinib. o. Percent CFUs from c-Fos−/−Dusp1−/− Kit+ cells with retroviral-vector-mediated rescue of Fos and Dusp1 expression. Data shown are from two independent experiments ± S.D. (n=3, P values are indicated above the compared bars). Individual data points are denoted as empty circles in each bar graphs.
Deletion of both c-Fos and Dusp1 had a greater effect than deletion of either gene alone, rescuing~60% of the mice from leukemic death with significantly reducing leukemic burden (Fig. 2h,k). Control ROSACreERT2c-Fosfl/flDusp1−/−, not treated with tamoxifen, showed modestly prolonged survival compared to WT (Supplementary Fig. 3b and d), which correlated with lower c-Fos mRNA expression (Supplementary Fig. 3e). Deletion of both c-Fos and Dusp1 combined with a five-week course of imatinib treatment eradicated all leukemic cells from peripheral blood and bone marrow, and discontinuation of treatment did not result in disease relapse, as assessed by YFP+ cell-burden in blood and bone marrow at the end of the experiment, 4 months after tumor cell transplantation (Fig. 2h,k). Thus, genetic deletion of c-Fos and Dusp1 sensitizes BCR-ABL expressing cells (but not wild type cells) to imatinib treatment and eliminates TKI-resistant MRD. Moreover, deletion of c-Fos and Dusp1 in ROSACreERT2c-Fosfl/flDusp1−/− mice by tamoxifen injection did not show any apparent hematopoietic defects, and c-Fos and Dusp1 deficient cells were maintained in the PB and bone marrow (Supplementary Fig. 3f), supporting the concept that these genes are required for BCR-ABL-induced transformation and leukemia development but are dispensable for normal hematopoiesis.
To test whether c-Fos is required for the maintenance of disease, we deleted c-Fos after the onset of disease (three weeks after tumor cell transplantation). At two weeks after transplantation, both wild-type and ROSACreERT2c-Fosfl/flDusp1−/− mice showed an increase in granulocytes (~70–80% compared to 32 % in WT) at the expense of B cells (Supplementary Data Fig 3g). As shown above, deletion of both c-Fos and Dusp1 at week three rescued ~60% of the mice from leukemia (Supplementary Fig 3h). Imatinib treatment rescued c-Fos and Dusp1 deleted mice from leukemia and led to rapid clearance of leukemic cells. Whereas non-imatinib treated mice lacking Fos and Dusp1 showed gradual depletion of leukemic cells, while vector-only control cells lacking Fos and Dusp1 showed stable engraftment (Supplementary Fig 3h,i). Together, these data suggest that genetic loss of both c-Fos and Dusp1 sensitizes leukemic cells to TKI in CML and confers synthetic lethality to BCR-ABL-transformed cells, as leukemic cells are gradually depleted even without TKI treatment.
To rule out potential non-specific effects of c-Fos or Dusp1 deletion on BCR-ABL-induced leukemia, we performed two additional experiments. First, we expressed a dominant negative c-Fos (lacking the DNA binding domain31, Fig. 2l,m) together with BCR-ABL in wild-type (WT-C57Bl/6) bone marrow cells. Expression of dominant negative c-Fos had effects that were similar to those from c-Fos deletion; >50% reduction in the number of BCR-ABL dependent CFUs (Fig. 2n). Second, expression of c-Fos-wild type (WT) partially rescued the phenotype, while expression of both Fos and Dusp1 using a mono-cistronic vector (P2A peptide cleavage) restored CFU numbers to normal levels (Fig. 2o).
Inhibition of c-Fos, Dusp1 and BCR-ABL by small molecule inhibitors cures mice of CML
To test the potential of targeting c-Fos, Dusp1 for therapy, we performed in vitro and in vivo experiments using small molecule inhibitors of c-Fos and Dusp1 (Fig. 3a). CFU analysis of BCR-ABL-LSK cells (bone marrow derived Lin−Sca1+Kit+ represents hematopoietic stem and progenitor cells) treated with c-Fos and Dusp1 inhibitors recapitulated the genetic data. Specifically, combination of a Dusp1 and Dusp6 inhibitor ((E)-2-Benzylidene-3-(cyclohexylamino)-2,3-dihydro-1H-inden-1-one; BCI32), a Fos inhibitor (curcumin33) and a BCR-ABL inhibitor (imatinib) completely suppressed CFU formation (Imatinib+curcumin+BCI ; Supplementary Fig. 4a,b). To address the possibility of off-target effects of curcumin, we tested two other chemically distinct compounds targeting c-Fos, nordihydroguaiaretic acid34 (NDGA) and difluorinated-curcumin35 (DFC). Both NDGA and DFC (Imatinib+NDGA+BCI and Imatinib+DFC+BCI) were effective in suppressing BCR-ABL-dependent CFU formation (Fig. 3b and Supplementary Fig. 4c), suggesting that c-Fos is the relevant target of these compounds. Next, we examined the in vivo efficacy of these compounds using a retroviral bone marrow transduction transplantation leukemogenesis model. A one-month course of treatment with individual compounds with imatinib (BCI, curcumin, and NDGA) or without imatinib (BCI and curcumin), did not effectively treat leukemogenesis; however, treatment with Imatinib+curcumin+BCI, and Imatinib+NDGA+BCI rescued 50 and 60% of the recipient mice, respectively (Supplementary Fig. 4d). In contrast, a one-month course of treatment with Imatinib+DFC+BCI rescued ~90 percent of mice, and we were unable to detect MRD by flow cytometry (Fig. 3c,d).
Figure 3. Chemical inhibition of c-Fos, Dusp1 and BCR-ABL eradicates minimal MRD in mice. a.
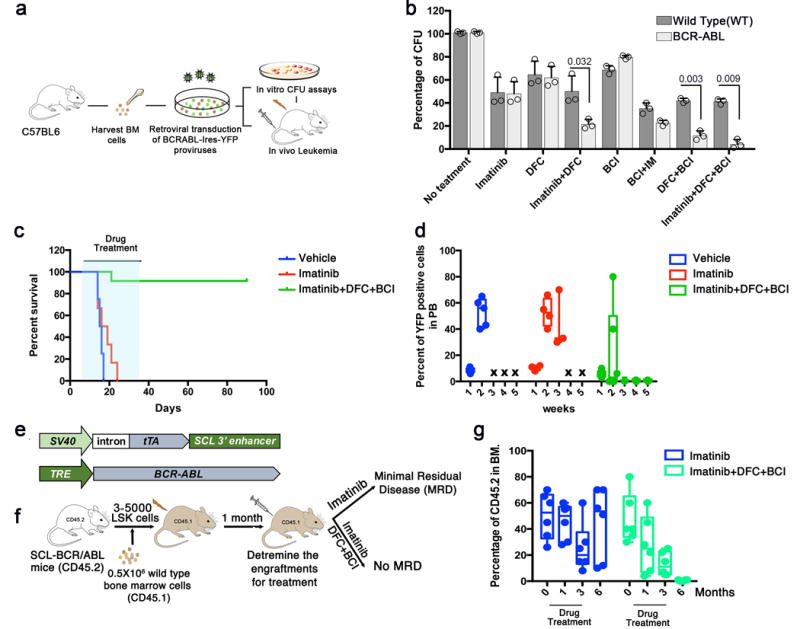
Experimental design for testing the efficacy of small molecule inhibitors of c-Fos (difluorocurumin; DFC) and Dusp 1 ((E)-2-Benzylidene-3-(cyclohexylamino)-2,3-dihydro-1H-inden-1-one; BCI) in vitro and in vivo in CML. b. Percent CFUs from WT and BCR-ABL LSK (Lin−Sca1+Kit+) cells treated with the indicated drugs. The data shown are the mean colony number from two independent experiments ± S.D. (n=3, P values are indicated above the compared bars). c. Survival curve of BCR-ABL-expressing Kit+ cell recipients treated with vehicle (blue), imatinib (red), or a combination of imatinib with DFC and BCI (green). The time period during which the drugs were administered is indicated by light blue shading. Data shown are from one of the two independent transplantation experiments with similar results (n = 5 mice per group; p = 0.0285). d. Percent of YFP+ cells in peripheral blood of mice treated with imatinib or Imatinib+DFC+BCI. e. Schematic structures of the transgenes used in transgenic mice to drive BCR-ABL expression in stem cells. Top, the Scl-3′enhancer drives expression of the tetracycline transactivator-protein (tTA); bottom a tetracycline-responsive promoter (Tet-P) drives BCR-ABL expression. Transgenic mice are fed doxycycline-containing chow; doxycycline withdrawal induces expression of BCR-ABL in hematopoietic stem cells, generating leukemic stem/progenitor cells. f. Experimental design for studying the effects of Dusp1 and c-Fos inhibition in leukemic stem cells. Mice received a competitive transplant of 3,000–5,000 LSK Scl-BCR-ABL cells with 500,000 wild type (WT) total bone marrow cells. Engraftment was evaluated one month after transplantation by flow cytometry (CD45.1 vs. CD45.2); the mice were then treated with imatinib alone or imatinib+DFC+BCI for three months, and the presence of MRD was evaluated at indicated times. g. Percentage of leukemic cells (CD45.2) in bone marrow of BoyJ recipients (CD45.1) at the indicated time points after cell transplantation. Imatinib treatment (blue bars) reduces leukemic burden but not curative treatment with imatinib+DFC+BCI completely eradicated the leukemic cells (green bars). Individual data points are denoted as empty circles in each bar graphs.
Given the lack of MRD in the treated mice, we next wished to test the effectiveness of c-Fos and Dusp1 inhibition in a tumor model in which leukemic stem cells drive leukemogenesis. To this end, we used transgenic mice in which the 3′ enhancer of the SCL gene drives expression of tTA to regulate tet-0-BCR-ABL transgene expression in hematopoietic stem and progenitor cells36 (Fig. 3e). We transplanted 3,000–5,000 bone marrow LSK cells into irradiated BoyJ mice, which developed leukemia within 4–6 weeks. Four weeks after transplantation, drug treatment was started (Fig. 3f). Treatment with imatinib alone for three months resulted in a reduction (~60%) of CML cells in the bone marrow; however, a substantial percentage of BCR-ABL positive cells were present in the bone marrow (Fig. 3g). Moreover, when treatment was discontinued, disease relapse occurred in all mice, replicating the clinical course of TKI therapy in CML (Fig. 3g). In contrast, treatment with Imatinib+DFC+BCI for three months not only eradicated the donor-derived BCR-ABL positive leukemic cells (CD45.2) in the bone marrow, but no relapse occurred when treatment was discontinued (Fig. 3g and Supplementary Fig. 5a–c). Similarly to the retroviral CML model (Fig. 3c), treatment with Imatinib+curcumin+BCI or Imatinib+NDGA+BCI resulted in effective eradication of CML stem progenitor cells, but a few recipient mice in these treatment groups relapsed once treatment was discontinued (Supplementary Fig. 5c). Together, these results suggest that treatment with Imatinib+DFC+BCI is more efficient than the combinations (imatinib+BCI+curcumin or imatinib+BCI+NDGA) at targeting leukemic stem cells that persist in MRD, perhaps due to superior pharmacokinetics of DFC than curcumin35.
Inhibition of c-FOS and DUSP1 sensitizes patient-derived LICs to imatinib
To extend these findings to human cells, we tested the effect of c-FOS and DUSP1 inhibition on the survival of primary CML patient samples (Supplementary Table 1). Mice transplanted with 3,000 CD34+ cells from the primary patient sample CP4 showed robust cell engraftment within two weeks (Fig. 4a,b). Drug treatment was started two weeks after transplantation and continued for a period of 4–6 weeks, using imatinib alone, DFC and BCI, or Imatinib+DFC+BCI. As expected, treatment with Imatinib+DFC+BCI for three weeks resulted in effective inhibition of leukemic cells, whereas treatment with imatinib alone or DFC+BCI was ineffective (Fig. 4b). As previously reported37 leukemic engraftment in this model is not stable, and most mice lost grafts within seven to eight weeks after transplantation (Supplementary Fig. 5d), precluding further investigation of the effect of drug treatment on MRD.
Figure 4. Inhibition of c-Fos, Dusp1 and BCR-ABL selectively eradicates CML cells.
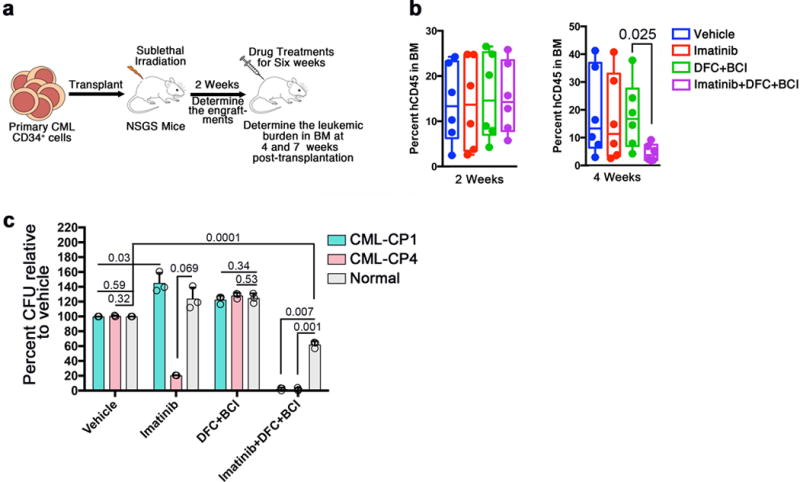
a. Experimental design for the analysis of DUSP1 and c-FOS inhibitor treatment of patient derived CML CD34+ cells. Primary CML cells from CP4 were transplanted into NOD Scid gammaC−/− mice recipients which transgenically express human IL-3, IL-6 and GM-CSF (NSGS); engraftment was assessed after two weeks after transplantation, engrafted mice were treated for six weeks with drug combinations, and leukemic burden was determined at 4 and 7 weeks after transplantation. b. Percent human leukemic cells in the bone marrow of NSGS mice at week 2 (left) and week 4 (right) of treatment. Data shown are from one of two experiments with similar results (n = 6 mice per group. P values are indicated above the compared bars). c. Percent CFU numbers determined by LTC-IC assay for samples from two CML patients and a normal donor treated with vehicle or the indicated drug combinations. Data shown are mean CFU numbers ± S.D (n = 3; P values are indicated above the compared bars). Individual data points are denoted as empty circles in each bar graphs.
Next, to test the efficacy of these drugs on primitive LSCs, we performed LTC-IC (Long Term Culture-Initiating Cells) assays, a stringent in vitro assay for detection of primitive hematopoietic or leukemic stem cells, using mononuclear cells from the CP1 sample, CD34+ cells from the CP4 sample, and CD34+ cells from a healthy donor as a control. As expected, LTC-IC activity was increased in imatinib-treated CP1 cells compared to the vehicle control (Fig 4c), in agreement to previous studies38. However, imatinib treatment of sample CP4 showed a significant decrease in LTC-IC activity compared to the vehicle control (Fig. 4c). Although the basis for the sensitivity of the sensitivity of the CP4 sample to imatinib is unknown—perhaps underlying patient-specific genetic or epigenetic changes confer TKI sensitivity—the difference in the responses between these two samples underscores the heterogeneous nature of LSCs in the context of TKI therapy. Notably, treatment with DFC+BCI did not affect LTC-IC activity in leukemic or normal cells (Fig. 4c). As expected, treatment with DFC+BCI+Imatinib selectively eradicated the LTC-IC activity of leukemic cells (Fig. 4c); however, as opposed to the results with genetic deletion, this drug treatment partially inhibited normal cell growth, perhaps due to off-target toxicity. Taken together, these results provide evidence that a combination of DFC+BCI+imatinib selectivity targets CML stem/progenitor cells but spares normal CD34+ cells.
c-Fos and Dusp1 deficiency alters the AP1 regulated networks
Given that the curcumin and its analogs target many different proteins in addition to c-Fos, we compared the effects of chemical inhibition and genetic loss of c-Fos on gene expression. We performed whole genome RNA-Seq analysis on wild-type Kit+ cells expressing BCR-ABL treated with imatinib, DFC+BCI, DFC+BCI+imatinib and non-treated control. Likewise, BCR-ABL-expressing Kit+ lacking Fos and Dusp1 treated with and without imatinib were subjected to RNA-Seq analysis. We compared gene expression in these samples to that of Kit+ cells transduced with the control vector (pMSCV-Ires-GFP) from wild-type mice. Consistent with the notion that DFC+BCI treatment inhibits c-Fos and Dusp1, we found a striking similarity between DFC+BCI-treated wild-type cells and c-Fos, Dusp1 double knockout BCR-ABL-expressing cells: 146 genes were regulated in common (58 up-regulated and 88 down-regulated) relative to untreated BCR-ABL-expressing cells (Fig. 5a). Further analysis of these differentially expressed genes using Netwalker suggests that inhibition of c-Fos and Dusp1 in the context of BCR-ABL expression leads to both down-regulation of a Fos-Jun-associated gene network and induction of a Jun-JunD-associated gene network (Fig. 5b and c).
Figure 5. Genetic or chemical inhibition of c-Fos and Dusp1 downregulates the Fos-Jun network while activating Jun-JunD target genes.
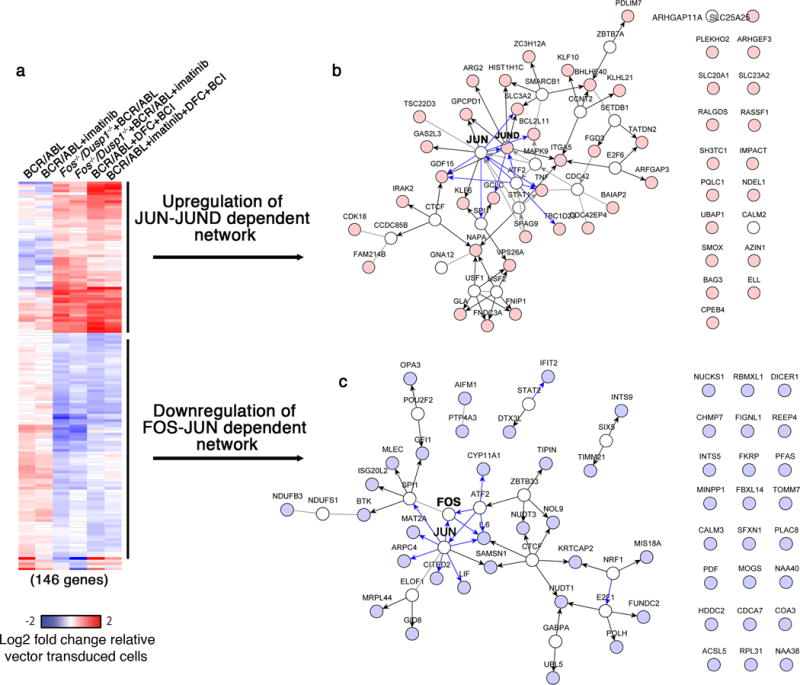
a. Heat map showing commonly modulated genes in BCR-ABL expressing Kit+ cells with c-Fos and Dusp1 deletion and wild type cells treated with Fos (DFC) and Dusp1 (BCI) inhibitors alone or with imatinib. Genetic and chemical inhibition resulted in modulation of 146 genes in common (58 overexpressed and 88 under expressed). b and c. Netwalker analysis shows that overexpressed genes are enriched for genes participating in a Jun-JunD regulated network (b), while downregulated genes are enriched for genes participating in a Fos-Jun regulated network (c).
The AP-1 transcription factor is a dimeric complex that contains members of the JUN (JUN, JUNB and JUND) and FOS (FOS, FOSB, FRA1 and FRA2) protein families39. The final outcome of AP-1 activity is dependent on AP-1 dimer composition as well as the cellular and genetic context24. Our data show that, in the context of BCR-ABL expression, absence of c-Fos and Dusp1 results in a net AP-1 transcriptional output that is anti-proliferative (reduced expression of Gfi1, Il6, Lif, Cited2 and overexpression of JunD) and pro-apoptotic (overexpression BCL2L11), Fig. 5 b,c. This analysis suggests that acute inhibition of BCR-ABL in c-Fos and Dusp1 inhibited or deficient cells undergo apoptotic shock13,19 due to elevated expression of pro-apoptotic genes.
Dusp6 deficiency confers imatinib resistance
Because BCI inhibits both Dusp6 and Dusp140, to determine whether the therapeutic efficacy of BCI is due to inhibition of Dusp1 or Dusp6, we tested the effects of BCI on MAPK signaling in BaF3 and BaF3-BA cells overexpressing Dusp1 or Dusp6. Although all Dusp family members have the ability to dephosphorylate MAPKs, each Dusp shows a high degree of specificity towards its specific substrates25,26,41,42. For instance, Dusp6 and Dusp9 show a preference for dephosphorylating ERK2 over p38 or JNK43,44, whereas Dusp8, Dusp10 and Dusp16 specifically dephosphorylate JNK and p38 kinases26. Likewise, studies from mice lacking Dusp1 revealed that it preferentially targets p38 and JNK28,45. Overall, DUSP-mediated MAPK regulation is dependent on the cellular, genetic and signaling context25,26,46.
In accord with this complexity, BCI treatment resulted in enhanced phospho-p38 levels in both BaF3 and BaF3-BA cells, and decreased phospho-JNK in BaF3-BA cells; the effects in BaF3-BA cells were observed with or without IL-3 co-treatment (Fig. 6a). Ectopic expression of Dusp1 in BaF3 and BaF3-BA cells reduced phospho-p38 levels, whereas expression of Dusp6 did not modify the levels of phospho-ERK1/2, phospho-p38 or phospho-JNK in either cell type (Fig 6b). Furthermore, overexpression of Dusp1, but not Dusp6, conferred resistance to BCI in BaF3-BA cells (Supplementary Fig 6a). These data support the hypothesis that BCI-mediated inhibition of Dusp1 activates p38 to induce TKI sensitivity and suggests that p38 inhibition would confer resistance to imatinib. Accordingly, we found that inhibition of p38 (using SB202190, a derivative of SB20358047, but not inhibition of JNK (using SP600125, which is 100 fold more selective for inhibition of JNK compared to p3848), conferred imatinib resistance in BaF3-BA cells (Fig. 6c). Notably, co-expression of wild-type Dusp6 with BCR-ABL in WT bone-marrow-derived Kit+ cells resulted in a significant reduction in CFU formation (~55%, P=0.001), but did not affect the cells expressing either vector or Dusp1 (Supplementary Fig 6b). Importantly, over expression of Dusp1 not the Dusp6 conferred resistance to drug (Imatinib+BCI) treatment (Imatinib+BCI), Supplementary Fig 6b. Notably, unlike Dusp1−/− cells, Dusp6−/− cells were resistant to drug (Imatinib+BCI) treatment and displayed normal CFU activity in comparison to untreated wild-type cells (Supplementary Fig 6c). Moreover, ectopic expression of Dusp6 in Dusp6−/− cells abolished drug resistance and restored TKI sensitivity to a normal level (Supplementary Fig. 6c). Previous work has shown that Dusp6 expression may be a requirement for oncogenic transformation in B-ALL but not CML49, and loss of Dusp6 expression in lung cancer confers TKI resistance50. Taken together with these previous results, our data suggest that inhibition of Dusp6 favors cancer cell survival upon TKI treatment, and conversely that inhibition of Dusp1 modulates p38 activity to promote TKI induced cell death in LICs.
Figure 6. Inhibition of Dusp1 activates p38. a.
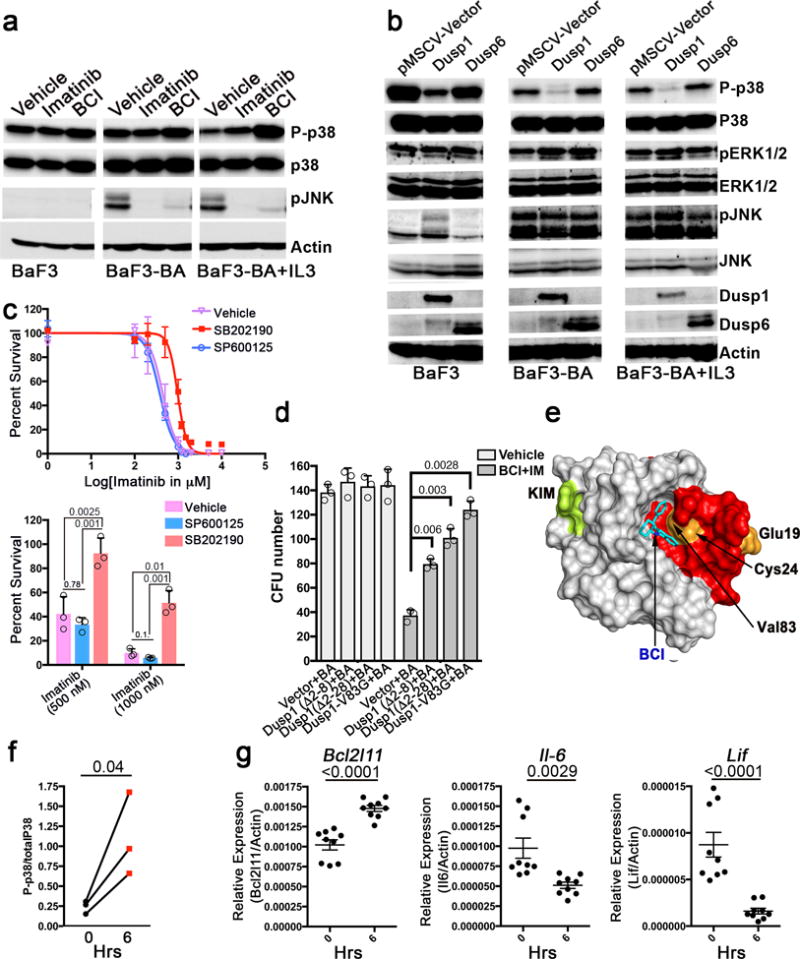
Immunoblot analysis of phospho-p38, total p38, and p-JNK expression in BaF3 and BaF3-BA cells with or without IL-3 showing increased p-P38 levels in BCI treated cells. b. Immunoblot analysis of the indicated proteins in BaF3 and BaF3-BA cells with or without IL-3 expressing Dusp1, Dusp6 and pMSCV (empty vector). c. Top, dose response curve for survival of BaF3-BA cells at increasing imatinib doses, treated with vehicle or the p38 specific inhibitor (SB202190, 500 nM) or a JNK specific inhibitor (SP600125, 500 nM). Bottom, percent cell survival at 500 and 1000 nM imatinib. The data shown are mean values ± S.D. (n = 3; P values are indicated above the compared bars). d. CFU numbers from Kit+ cells co-expressing BCR-ABL (BA) and wild-type or drug-resistant Dusp1 variants. The data shown are mean colony number ± S.D (n = 3; P values are indicated above the compared bars). e. Surface depiction of a structural model of the Dusp1 rhodanese domain, highlighting amino acids affected by BCI resistance mutations, as well as a deletion mutant causing resistance (red). A putative binding pocket for BCI is and KIM (kinase interacting motifs) are indicated The mutations are clustered together in the structure and outline a pocket to which BCI seemingly binds (ΔG= −7.6). f. Ratio of the levels of phospho-p38 to total p38 in peripheral blood cells before and six h after BCI injection into leukemic mice. Data are shown for three mice (n = 3; p =0.04). g. Real-time qPCR analysis showing expression of Bcl2l11, Il6 and Lif in mice before and 6 h after DFC+BCI injection. The data shown are the mean ± S.D from 3 mice in triplicates. (P values are indicated above each comparison). Individual data points are denoted as empty circles in each bar graphs.
Mutations affecting the allosteric domain of Dusp1 confer resistance to BCI
To determine the relevant target of BCI more conclusively, we performed in vitro drug-resistant screening to select for BCI-resistant mutations in Dusp1. BaF3-BA cells transfected with an expression library of randomly-mutagenized Dusp1 construct were used for selection of mutations conferring resistance to different concentrations of BCI (Supplementary Fig 6d). At 1 and 1.5 μM BCI, resistant clones emerged with a frequency of 1,200 and 27 clones per million cells, respectively, whereas selection at 2 μM of BCI did not yield any resistant clones (Supplementary Fig 6e). Twenty-seven clones that emerged in 1.5 μM BCI were randomly selected and further analyzed. Dose-response analysis confirmed their resistance to BCI (Supplementary Fig 6f). Sequencing of the Dusp1 gene in these clones identified four different substitution mutations (E19R, C24G, V83G and E112R) and two deletion mutations at the N-terminus (Δ2-8 and Δ2-28; Supplementary Fig 6g). Because most resistant clones carried two or more mutations, we generated clones with individual mutations for further analysis. Expression of these resistant variants in BaF3-BA cells conferred resistance to BCI (Supplementary Fig. 6h). Notably, the Dusp1-V83G variant demonstrated higher resistance than other tested variants, potentially explaining why it was more frequently observed in the screen (Supplementary Fig. 6h). Similarly, expression of these resistant variants in BCR-ABL expressing BM-derived Kit+ cells conferred resistance to BCI treatment, and the Dusp1-V83G variant conferred greater resistance than the other variants tested (Fig. 6d). We mapped these resistant mutations on a homology-based structural model of Dusp1 and found that they clustered at the N-terminus of the allosteric domain (rhodanese domain) rather than at the catalytic domain (Supplementary Fig. 7). Furthermore, an unbiased docking of BCI using a structural model of the Dusp1 rhodanese domain identified the putative site to which BCI binds, with a predicted free energy of ΔG= −7.63 (Fig. 6e). Taken together, these data provide clear evidence that BCI-induced cell death is mediated by inhibition of Dusp1 (rather than of Dusp6).
In vivo inhibition of Fos and Dusp1 targets by DFC and BCI
To model the therapeutic potential of c-Fos and Dusp1 inhibition in vivo, we performed a pharmacodynamic analysis for BCI and DFC. We obtained peripheral blood mononuclear cells 6 hours before and after BCI injection and measured the levels of phospho-p38 and of the c-Fos-target genes Bcl2l11, Il6, and Lif (Fig. 6 f,g). As expected, cells from mice injected with BCI showed an increase (by 4–8 fold) in phospho-p38 levels (Fig. 6f). Moreover, treatment with DFC+BCI induced Bcl2l11expression while dampening expression of Lif and Il6 (Fig. 6g). Notably, elevated serum IL-6 levels have been reported to be required for leukemic disease development51,52, and IL-6-neutralizing antibodies can suppress disease development53. Our data suggest that these markers (phopsho-p38, IL6, and Lif) could potentially help in testing the efficacy of Fos and Dusp1 inhibition and in evaluating disease progression.
Deletion of c-Fos and Dusp1 blocks BCR-ABL-induced B-ALL development
Given that growth factor signaling mediates intrinsic resistance to TKI therapy in both leukemia and solid organ tumors20–22, we reasoned that c-FOS and DUSP1 might play a critical role in other types of kinase-driven leukemias. First, we tested the role of c-Fos and Dusp1 in BCR-ABL-induced B-ALL, which, like BCR-ABL-induced CML, is driven by a diverse spectrum of oncogenic tyrosine kinases and cytokine receptors54; moreover, similarly to BCR-ABL-induced CML, and most patients with BCR-ABL-induced B-ALL relapse under TKI treatment55. To model B-ALL in vivo in mice, we used bone-marrow-derived mononuclear cells (MNCs) from WT and ROSACreERT2c-Fosfl/flDusp1−/− mice transduced with BCR-ABL-Ires-YFP (P190) retroviruses56. Fos was deleted after two weeks of transplantation by tamoxifen injection. Recipients of wild-type bone-marrow cells developed lethal leukemia with a disease latency of 4–5 weeks; in contrast, deletion of both Fos and Dusp1 led to a complete suppression of disease development and eradication of leukemic cells within 3 weeks after tamoxifen injection (Supplementary Fig. 8a–c). These data provide evidence that loss of c-Fos and Dusp1 is synthetic lethal to BCR-ABL-expressing B-ALL cells. Surprisingly, mice that received BCR-ABL -transduced ROSACreERT2c-Fosfl/flDusp1−/− cells but were not treated with tamoxifen did not develop leukemia; leukemic cells disappeared from these mice, although with a delayed latency compared to tamoxifen-treated mice (Supplementary Fig. 8a–c), most likely due to lower c-Fos mRNA expression in these mice compared to WT mice (Supplementary Fig. 3e) Together, these data suggest that, unlike in CML, deletion of c-Fos and Dusp1 is sufficient to completely eradicate BCR-ABL-induced B-ALL.
Induction of c-Fos and Dusp1 by oncogenic kinases FLT3-ITD and JAK2-V617F
Next, we analyzed whether growth factor signaling can confer resistance to inhibitors of the oncogenic kinases Flt3 and Jak2. As expected, growth factor signaling conferred resistance to the Flt3 inhibitor AC220 and the Jak2 inhibitor ruxolitinib in BaF3-FLT3-ITD and BaF3-Jak2-V617F cells, respectively (Supplementary Fig. 8d,f). Like BCR-ABL, expression of FLT3-ITD and JAK2-V617F induced the expression of c-Fos and Dusp1 (Supplementary Fig. 8e,g). This induction suggests a more general role for c-Fos and Dusp1 in TKI resistance, which will need to be tested further in animal models.
c-Fos and Dusp1 in normal hematopoietic cells
Given that the leukemic stem cells (LSCs) like hematopoietic stem cells (HSCs) are maintained by growth factor signaling which seemingly supports higher Fos and Dusp1 expression, thus conferring TKI resistance. Based on published data57, HSCs had higher levels of c-Fos and Dusp1 expression than their differentiated progeny (Supplementary Fig. 9 a,b). Expression of BCR-ABL in mouse hematopoietic stem cells (LSK cells) which is bonafide LSCs show induced expression of Fos and Dusp1 compared to vector transduced LSK cells (Supplementary Fig. 9c), perhaps due to the convergence of oncogenic and growth factor signaling at these signaling nodes. Likewise, LSCs (CD34+ and CD38− cells) from CML showed higher expression of Fos and Dusp1 compared to normal HSCs (CD34+ and CD38− cells), Supplementary Fig. 9d. Our expression analysis of BaF3-LTBA+IL-3 cells (Supplementary Fig. 1c) expressing higher Fos and Dusp1 correlates with induced expression of pro-survival and anti-apoptotic genes in these cells compared to BaF3-LTBA cells (Supplementary Fig. 9e), which may represent a protective mechanism. Interestingly, expression of these pro-survival and anti-apoptotic genes is dependent upon Fos and Dusp1 function, as shown using Fos and Dusp1 inhibitors (Supplementary Fig. 9e,f). Altogether, these data provide evidence that higher levels of Fos and Dusp1 are required to maintain induced expression of pro-survival and anti-apoptotic genes, which is critical for tumor maintenance and drug response.
DISCUSSION
Initial excitement over small molecule kinase inhibitor (imatinib) targeting BCR-ABL has been tempered by the observation that LSCs from CML patients can survive TKI treatment, perhaps due to incomplete inhibition of kinase activity, as LSCs displayed residual kinase activity under imatinib treatment. However, later studies using next generation BCR-ABL inhibitor (nilotinib), which fully quenched kinase activity in vivo, yet LSCs still survive58. One could conclude from these data that LSCs are not oncogene dependent, and that BCR-ABL does not represent an Achilles heel in LSCs. According to our model (Supplementary Fig. 9g–i), expression of an activated kinase such as BCR-ABL usurps c-Fos and Dusp1 mediated regulation of cell proliferation and survival. TKI treatment downregulates c-Fos and Dusp1 expression leading to apoptosis in the bulk tumor; however, within TKI resistant cells (LSCs and BaF3-BA+IL3) growth factor signaling can rescue c-Fos and Dusp1 expression leading to sustained expression of pro-survival and anti-apoptotic genes and TKI resistance (Supplementary Figs. 5 b, c and 9f). However, previous work on LSCs from CML showing lack of addiction to BCR-ABL prompted many researchers to identify pathways and targets for therapeutic development, to target LSCs. A number of pathways and drug targets have been identified, such as, PI3/AKT, Tgf-beta-Foxo, Hedgehog, and Wnt-β-catenin pathways59. While inhibition of these targets either alone or in combination with kinase inhibitors inhibit the survival of LSCs, but, to some extent, they also have shown to be detrimental to normal HSCs because targeted proteins regulate cell survival and self-renewal pathways in normal HSCs. Therefore, precluding therapeutic inhibition of these targets in clinic for eradicating the MRD and LSCs.
Altogether, our study shows that c-FOS and DUSP1 are activated by both kinase oncoproteins and growth factors, and that their expression levels are uniquely critical for maintaining the oncogene dependence and growth-factor-mediated rescue of MRD in mouse models of TKI-treated leukemia. Moreover, genetic data revealed that normal HSC do not critically require c-Fos and Dusp1 as mice transplanted with BM cells lacking Fos and Dusp1 do not show any difference in transplantation and maintenance of engraftments compared to WT cells (Supplementary Fig 3). We speculate that kinase-driven cancer cells (LSCs) differ from normal cells (HSCs) because they have adapted to chronic kinase signaling, addicting themselves to elevated c-Fos and Dusp1 activity, a growth factor-induced signaling node. The levels of Fos and Dusp1 may dictate the threshold of TKI efficacy, such that lower levels confer sensitivity whereas higher levels drive intrinsic resistance leading to MRD in leukemia, and perhaps in solid organ cancers. Thus, these proteins may represent a unifying Achilles heel of kinase-driven cancers. Our findings provide proof of principle that minimal residual disease can be treated through inhibition of a convergent signaling node that mediates growth factor-dependence in kinase-induced leukemia.
Methods
Mice
All mice were housed in the barrier facility at Cincinnati Children’s Hospital (CCHMC). All experiments were performed under an IACUC-approved protocol of the Cincinnati Children’s Hospital in accordance with accepted national standards and guidelines. To generate conditional c-Fos mice, c-Fosfl/fl mice29 were crossed with ROSACreERT2 mice (Jackson laboratory, Ban Harbor, ME) to generate ROSACreERT2c-Fosfl/fl mice. To create the double knockout mice, ROSACreERT2c-Fosfl/fl mice were bred with Dusp1−/− mice 28 to generate ROSACreERT2c-Fosfl/flDusp1−/− mice. BoyJ mice weer purchasd from mouse core facility at CCHMC. C57Bl/6 mice were purchased from Jackson laboratory. Scl-tTA transgenic mice were obtained from Dr. Claudia Huettner’s lab. Mouse genotypes were confirmed by PCR analysis using gene specific primers28,29. Athymic Ncr nu/nu, NOD.Cg-Prkdcscid IL2rgtm1Wjl/SzJ (NSG) and hSCF, hIL-3, h-GM-CSF transgenic NSG-derived (NSGS) mice were purchased from the CCHMC mouse core. Six-eight week old mice were used in all experiments. Mice with the indicated genotypes were included in the study without any further pre-selection or formal randomization and both male and female mice were used; we used age- and gender-matched mice. The investigators were not blinded to genotype group allocations.
Human specimens
Umbilical cord blood (UCB) cells, normal BM, CML (p210-BCR-ABL+) and blastic phase leukemia specimens were obtained through Institutional Review Board-approved protocols (Institutional Review Board - Federalwide Assurance #00002988 Cincinnati Childrens Hospital Med Ctr.) and donor informed consent from CCHMC and University of Cincinnati. The patient samples are described in Supplementary Table 1.
Plasmids and constructs
BCR-ABL was cloned into the pLVX-puro and pLVX-Tet-On-Puro (Clontech, USA) plasmids to yield pLVBA and pLTBA for constitutive and inducible expression, respectively. The plasmid pEYKBA9 was digested with EcoRI to release the BCR-ABL fragment that was purified and ligated to EcoRI digested pLVX-puro and pLVX-Tet-On-Puro to generate the plasmids pLVBA and pLTBA, respectively. To create the dominant negative c-Fos (c-Fos-ΔRT), the basic region of FOS DNA binding domain (amino acid residues 133–159) was deleted by PCR using primers (c-FOS-DRK-FP and c-FOS-DRK-RP in table 2) by QuikChange lightning multi-site-directed mutagenesis kit (Agilent Technologies). The PCR reaction was carried out using template DNA (pDonor201-FOS obtained from PlasmID at Harvard cat # HsCD00001156). Subsequently these entry vectors were used to develop retroviral expression clones using destination vector (pMSCV-Ires-GFP.GW) by recombination cloning using LR clonase from Invitrogen. Likewise, retroviral expression vectors for Dusp1 and Dusp6 (pMSCV-Dusp1-Myc-Ires-cherry and pMSCV-Dusp6-Ires-GFP) were created by recombination cloning using entry clones (pENTR-Dusp1, pENTR-Dusp6 obtained from PlasmID at Harvard). BCI resistant mutations of Dusp1 were created by site-directed mutagenesis kit as described above using pMSCV-Dusp1-Myc-Ires-cherry as template. All pENTR clones were confirmed by sequencing. Retroviral expression vectors (pMSCV-Fos-P2A-Dusp1) expressing Fos, and Dusp1 as a polycistronic construct was cloned by recombination cloning using plasmids (pENTR-FOS/HA, pENTR-Dusp1/Myc). The retroviral vector pMSCV-BCR-ABL-IRES-YFP, a gift from Tanishtha Reya 30, was used to express BCR-ABL in primary mouse bone marrow cells for colony formation cell assays and for in vivo transplantation and leukemia development.
Chemical reagents and cytokines
Kinase inhibitor Imatinib was purchased from LC laboratories (Woburn, MA). Inhibitors for c-Fos, diflouro-curcumin (DFC) and curcumin, were purchased from LKT laboratories. The Dusp1 inhibitor BCI was synthesized by Chemzon Scientific Inc. (Montreal, Canada). Mouse cytokines (IL3, SCF, IL6 and Flt3L) were purchased from Peprotech, NJ, USA. Human cytokine erythropoietin was purchased from Amgen Inc., CA. NDGA, tamoxifen, and 4-hydroxy tamoxifen were purchased from Sigma-Aldrich. Hydrocortisone was purchased from STEMCELL technologies.
The source, catalog numbers and dilutions of all antibodies used for Western blotting and FACS analysis are described in Supplementary Table 4.
Cell culture
BaF3, K562, and HEK293T cells were obtained from George Daley’s lab. MS5 was a gift from Jose Cancelas. BaF3, and K562 were cultured and maintained in RPMI supplemented with 10% FBS and 100 IU/ml penicillin, 100 μg/ml streptomycin and 2mM L-glutamine. HEK293T were maintained in DMEM supplemented with 10% FBS and 100 IU/ml penicillin, 100 μg/ml streptomycin and 2mM L-glutamine. BaF3 parental cells were grown in RPMI with 10% WEHI conditioned media, used as a source of IL3. BAF3 cells with BCR-ABL were maintained in RPMI without IL-3 supplementation.
Generation of stable cell lines
Cell stably expressing the BCR-ABL, Fos, Dusp1, Dusp6 were generated by transducing with high titer retroviruses as described earlier60. Inducible expression of BCR-ABL in BaF3 cells was achieved by transducing these cells with pLVX-Tet-On-Hygro viruses (Clontech) followed with selection for hygromycin resistance (selected at 600 μg/ml). Finally, BaF3 cells were transduced with pLTBA-puro viruses. Cells were selected for puromycin resistance at (3 μg/ml), generating the inducible expression cell line BaF3-LTBA.
Cell proliferation assay
1×104 cells were seeded in 96 well plates in 100 μl media with or without growth factors (50ng/ml) and appropriate drug concentrations. The cells were incubated for 60 hours. Cell viability was assessed with the WST-1 reagent (Roche) according to the manufacturer’s recommendations and read with a 96 well plate reader at 450 nm. All assays were performed in triplicate and readings were averaged. A dose response analysis to determine IC50 values was performed by sigmoidal curve fitting in GraphPad6, as described previously60.
Apoptosis assay
BaF3-BA and K562 cells constitutively expressing BCR-ABL were grown with or without growth factors to the logarithmic phase. The cells from each group were treated with Imatinib (5 μM) for six hours. The cells were then stained with APC conjugated Annexin V (BD Biosciences) according to the supplier’s instructions. Five μl of propidium iodide was added to each sample after Annexin-V staining and the cells were subjected to FACS analysis (BD Canto II). Single Annexin-V positive cells were considered as early apoptotic cells while PI and Annexin-V double positive cells were considered as late apoptotic or dead cells.
RNA isolation and gene expression profiling
Five to six million BaF3 and BaF3-LTBA cells grown with or without doxycycline (500 ng/ml) and IL-3 from the logarithmic phase were collected and resuspended in Qiazol for RNA isolation. Likewise, BaF3-BA cells (with constitutive expression of BCR-ABL) were grown with or without IL-3 and treated with imatinib (3 μM) for 6 hours. Similarly, K562 cell lines were grown with or without erythropoietin (100 U/ml). After 6 hours of imatinib (3 μM) treatment, cells were stained by Annexin V and PI to quantify the levels of apoptotic cell death. The three separate populations of cells (double negative cells (live cells), APC or Annexin V positive cells (early apoptotic cells) and double positive cells (late apoptotic cells)) were sorted in PBS from each of the cell lines. Two million sorted cells from each condition were immediately pelleted and frozen in 700 μl of Qiazol for RNA extraction. Total RNA was quantified and 1 μg of RNA was used for expression profiling on the Mouse ST_1.0 Gene Chip Array (Affymetrix) for BaF3 cells and ExonExprChip.HuGene-1_0-st-v1 (Affymetrix) for K562 cells, at the Cincinnati Children’s Gene Expression Core. The data were collected and .cel files were generated in the MAS5 suite (Affymetrix). The .cel files were imported into GeneSpring-GX 12.6.1(Agilent Technologies) and analyzed using the latest annotation available. All biological replicates were averaged. For the first experiment (experiment 1; conditional expression of BCR-ABL in the presence or absence of IL-3, with the aim of determining changes in gene expression that are modulated by IL-3 in an oncogenic condition), the data was normalized to the median of parental BaF3 cells. After normalization the genes were filtered based on expression, genes with probe intensity value less than 20 percentile in at least one condition were eliminated. For the second experiment (experiment 2; imatinib-induced changes in gene expression in BaF3-BA cells treated with or without IL-3 and the third experiment (experiment 3; imatinib-induced changes in gene expression in K562 cells treated with or without EPO, the data were normalized to the median of all samples followed by filtering based on expression, genes with probe intensity value less than 40 percentile in at least one condition were eliminated. Lists of genes that are differentially expressed were created using filtered genes with fold change analysis between the cells grown with or without the growth factors for all three of the experiments. The fold change cut-off was set to 1.5 for experiment 1 and was set to 2.0 for experiments 2 and 3. A published dataset (GSE12211) that describes gene expression of CML-CD34+ cells during Imatinib therapy23 was similarly analyzed using GeneSpring GX software. The samples were normalized to the median of control sample and filtered based on expression, genes with probe intensity below 50 percentile in at least one condition were eliminated. Gene lists were created containing genes that were differentially expressed by more than 2.0 fold between imatinib-treated and untreated samples. Finally, to identify commonly regulated genes in all four data sets, the data sets were analyzed in GeneSpring GX, and the results are presented as a Venn diagram. The data reported in the manuscript are available in the GEO database (GSE75058).
Real-Time Q-PCR Analysis
Candidate genes picked by microarray analysis were validated by real time Q-PCR. The list of genes analyzed and the primer sequences used to analyze their expression are presented in Supplementary Table 5. Total RNA was isolated as described above. The RNA was first subjected to DNase treatment using DNA free DNase kit (Ambion, Life technologies). Two μg of total RNA was converted to cDNA with Superscript III first strand synthesis kit (Life technologies). Q-PCR reactions were performed with the human gene specific primers (Supplementary Table 5) using the SYBR green method and using a Mastercycler RealPlex2 instrument (Eppendorf). All PCR reactions were performed in triplicate and the real time data was normalized to β-actin expression.
Western Blotting
Four to six million cells were collected and whole cell extracts were prepared using lysis buffer supplemented with a protease inhibitor cocktail (Roche) and phosphatase inhibitor cocktail 2 (Sigma Aldrich), as described previously61. The proteins were resolved on 10% SDS-PAGE gels and transferred to nitrocellulose membranes (Bio-Rad). Membranes were blocked in TBST with 5% non-fat milk and probed with appropriate antibodies as indicated. Densitometry was carried out using ImageJ software.
Colony forming cell assays
Kit+ cells from the BM of WT, Dusp1−/−, ROSACreERT2c-Fosfl/fl, or ROSACreERT2c-Fosfl/fl/Dusp1−/− mice were isolated using the CD117 MicroBead Kit (Miltenyi biotec) according to the manufacturer’s instructions. The cells were incubated overnight in IMDM media supplemented with 10% FBS and a cytokine cocktail with FLT3 (20ng/ml), IL-6(10ng/ml), IL-3(10ng/ml),, and mSCF(50ng/ml),. After 12 hours of stimulation, the cells were transduced with BCR-ABL-IRES-YFP virus using retronectin (Takara). Five thousand YFP positive cells (isolated by FACS) were plated on MethoCult™ GF M3434 (STEMCELL technologies containing Imatinib (3 μM), DFC (0.2 μM) and BCI (0.5 μM) alone or in combinations on 3 replicate plates. Likewise, curcumin (5 μM) or NDGA (5 μM) was used alone or in combination with imatinib (3 μM), and BCI (0.5 μM). Colony numbers were recorded after one week of plating. To delete c-Fos, the cells were plated on MethoCult™ GF M3434 containing 4-hydroxy tamoxifen (1 μg/ml).
BM cell transduction transplantation model of CML
Kit+ cells from the BM of six to eight week old wild type C57BlL/6, Dusp1−/−, ROSACreERT2c-Fosfl/fl, or ROSACreERT2c-Fosfl/fl/Dusp1−/− mice were isolated and transduced with MSCV-BCR-ABL-IRES-YFP as described above. The transduced cells were cultured overnight. The percentage of BCR-ABL positive cells was determined by measuring the level of YFP positive cells after 20 hours of viral transduction using flow cytometry (Fortessa I). Forty thousand YFP positive cells with 0.3 million normal BM cells as carrier were transplanted into each mouse through tail vein injection. After one week of transplantation, engraftment was determined by analyzing the YFP positive cells from the peripheral blood using FACS. Transplanted mice that showed 10–40 % YFP positive cells in the peripheral blood were used for the experiment. Mice that had less than 2 percent YFP positive cells were discarded from the study. To delete the Fosfl/fl allele, tamoxifen (100 mg/kg in corn oil) was i.p. injected into mice one week of transplantation, every days for three consecutive days. After tamoxifen treatment where appropriate the mice were grouped for drug treatments (n=5/group). Mice were monitored for leukemia progression and survival, and the leukemic burden (YFP positive cells) was determined weekly up to 8 weeks in surviving mice. Animal numbers were chosen on the basis of previous experience and published data for transplantation of BCR-ABL positive cells.
BM cell transduction transplantation model of B-ALL
BM cells were harvested from wild-type and ROSACreERT2c-Fosfl/fl/Dusp1−/− mice. Total mononuclear cells (TMNCs) were isolated by gradient centrifugation using Ficoll. Cells were washed and resuspended in 1ml IMDM media + 10% FBS supplemented with SCF (50 ng/ml; Prospec) and IL-7 (20 ng/ml; Peprotech). The cells were transduced with pMSCV–BCRABL(p190)-Ires-GFP virus. After eight hours of transduction, the cells were washed and injected (2×106 cells) into tail veins by i.v. into lethally irradiated C57BL/6 mice. After two weeks of transplantation, the mice were injected with tamoxifen (100 mg/Kg once per day for 3 days [AU: Correct?]) to delete c-Fos. Peripheral blood from the transplanted mice were used to determine the leukemic burden (% GFP-positive cells) and the levels of WBC by complete blood counter (Hemavet, Drew Scientific, Oxford, CT).
BCR-ABL transgenic mouse model
Total BM cells from Scl-tTA transgenic mice 36 were isolated and followed with lineage depletion using lineage antibody cocktail from Milteny biotec according to manufacturer’s instructions. Isolated Lin_ celsl were labeled with anti-Kit and anti-Sca1 antibodies to isolate LSK (Lin− Sca1+ Kit+) cells by FACS. 3,000–5,000 BCR-ABL-LSK cells with 0.3 million helper bone marrow cells from wild-type BoyJ mice were injected via tail vein in to lethally irradiated BoyJ mice. Four weeks post-transplantation, recipient mice were analyzed for CD45.1- and CD45.2-positive cells by FACS analysis to determine leukemic engraftment and chimerism. Mice were grouped into four groups (n=6 per group). Mice were treated with Imatinib (75 mg/kg twice daily) alone and in combination with DC+BCI (both drugs were given at a dose of 10 mg/kg twice daily). Likewise other groups were treated with combinations of Imatinib(75 mg/kg)+curcumin(150 mg/kg) +BCI(10 mg/kg), and Imatinib(75 mg/kg)+NDGA(100 mg/kg) +BCI (10 mg/kg), for 3 months twice a day by i.p. injection. The mice were analyzed for leukemic chimerism by determining the percentage of CD45.2-positive cells once a month for 6 months.
RNAseq analyses of Kit+ cells from wild type and c-Fos and Dusp1 knockout mice
Kit+ cells from the BM of WT and ROSACreERT2c-Fosfl/fl/Dusp1−/− mice were isolated using the CD117 MicroBead Kit (Miltenyi biotec) according to the manufacturer’s instructions. These cells were incubated overnight in IMDM media supplemented with 10% FBS and a cytokine cocktail of FLT3 (50ng/ml), IL-6(10ng/ml), IL-3(10ng/ml),, and mSCF(50ng/ml). After 12 hours of stimulation the cells were transduced with BCR-ABL-IRES-YFP virus using retronectin (Takara). One to two million positive cells (isolated by FACS) from each group was treated for four hours with imatinib alone, DFC+BCI, or imatinib+DFC+BCI (3 μM Imatinib, 0.2 μM DFC and 0.5 μM BCI alone or in combinations. The treatments were done in 2 replicates. To delete FOS 4-hydroxy tamoxifen (1 μg/ml) was added to the media where applicable. Total RNA was isolated, and RNAseq (20 million reads with paired ends) was performed at the DNA sequencing core of Cincinnati Children’s Hospital. The data reported in the manuscript are available in the GEO database (GSE75058). Genes with an absolute log2 change of 1 in BCR-ABL-expressing c-Fos−/−/Dusp1−/− cells (Fos was deleted by adding 4-hydroxy tamoxifen (1 μg/ml) treated with DFC+BCI, compared to BCR-ABL wild type cells (680 genes), were selected. From this list of genes, genes with similar profiles (146) in both c-Fos−/−/Dusp1−/− expressing BCR-ABL and WT cells expressing BCR-ABL that were treated with DFC+BCI were selected. To build a gene network, down- or up-regulated genes were used as seeds to build a coherent network using the GeneConnector functionality in NetWalker suite62.
Patient derived mouse models of CML
3 million CD34+ cells from CML patient CP4 (described in Supplementary Table 1) were transplanted into sub-lethally irradiated 8-week-old NSG mice. After two weeks of transplantation, leukemic engraftment in bone marrow was determined by FACS using mouse and human specific antibodies against CD45. Mice were grouped into four different cohorts (n=6/group) for treatment with vehicle, imatinib (75 mg/kg), DFC+BCI (both at 10 mg/kg) and imatinib (75 mg/kg) +DFC+BCI (both at 10 mg/kg). Drugs were diluted in PBS (vehicle) and administered by intra-peritoneal injection twice daily. Mice were treated for six weeks, and the leukemic burden was determined every two weeks, until week 8 after transplantation.
LTC-IC Assay
The LTC-IC assay was performed according to instructions manual of StemCell Technologies. 5,000 CD34+ cells from patient CP4) or one million total MNCs from patient CP1 were cultured in StemSpan™ SFEM medium containing 50 ng/ml SCF, 5 ng/ml IL-3, 20 ng/ml IL-6, 50 ng/ml Flt3L, and 100 ng/ml GM-CSF for 24 hours in the following conditions: untreated; imatinib (3 μM), DFC (200 nM) + BCI (500 nM), and imatinib (3 μM) + DFC (200 nM) + BCI (500 nM). After 24 hours, the cells were washed in human long-term culture medium (HLTM; MyeloCult H5100 media containing 1 μM hydrocortisone) and were plated on irradiated MS-5 stromal cells. Cultures were maintained for 5 weeks with weekly half-medium changes. Cells were then harvested, counted, and transferred to methylcellulose containing media (MethoCult™ Express, StemCell Technologies) for colony-forming assays. At the end of week five, adherent and non-adherent cells were isolated and plated in methylcellulose, (METHOCULT™ H4434 classic, stem cell technology) for CFU analysis in triplicate. Plates were incubated at 37 °C and colonies were scored two weeks after plating.
Pharmacodynamic analysis of Dusp1 and c-Fos targets
Phospho-p38 analysis
Three leukemic mice (8–12 weeks old), which had been transplanted with BCR-ABL expressing Kit+ cells, were injected with BCI (10 mg/kg) by intraperitoneal injection 4 weeks post-transplant. Phospho-p38 levels were quantified in peripheral blood MNCs using the Phosflow™ kit (BD Biosciences) according to the supplier’s instructions. In brief, blood was collected from each mouse before and six hours after drug injection. Mononuclear cells were isolated by RBC depletion: RBCs were lysed twice using 4 ml Pharmlyse solution (BD Biosciences) per 100 μl of peripheral blood by mixing and incubation on ice for 5 min. After the second lysis step, cell pellets were washed with 1 ml 2% BSA in PBS followed by fixation using 100 μl of fixation and permeabilization solutions (BD Biosciences) for 20 min at 4 °C in the dark. The pellets were washed with 1 ml of 1X BD Perm/wash buffer. After fixation, the cells were blocked using 300 μl 2% BSA in Perm/wash buffer (BD Biosciences) at room temperature for 20 min. The cells were divided into three equal aliquots, 100 μl each (~1million), and incubated with 1 μl total p-38 antibody, 1 μl phospho-p38 antibody, or 1 μl isotype IgG control, overnight at 4 °C.. The cells were then washed and incubated with secondary antibody (1 μl of Alexa flour-488 conjugated secondary antibody) for 1 h at room temperature. All antibody sources and dilutions are presented in the supplementary table 4. Cells were washed once again and suspended in 200 μl PBS. Data was acquired by FACS on Fortessa instrument, and the data were analyzed by FlowJo software. The mean fluorescent intensity (MFI) of the IgG control was deducted from the MFI of the experimental samples. The MFI values of phospho-p38 were normalized to those of total p38 to determine the phospho-p38 levels after BCI injection.
Quantitative gene expression analysis of target genes
Three leukemic mice (8–12 weeks old), which had been transplanted with BCR-ABL expressing Kit+ cells, were injected with DFC+BCI (10 mg/kg each). Peripheral blood was collected from each mouse before and six hours after drug injection. Mononuclear cells were isolated by RBC depletion, as above. MNCs were pelleted and re-suspended in Qiazol lysis buffer (Qiagen). Total RNA was extracted, followed by cDNA synthesis and Q-PCR analysis of Bcl2l11, Lif, and Il6 using gene specific primers (Supplementary table 5).
Drug preparation
All drugs were prepared as 10 mM stocks in DMSO and stored in −20 °C until use. For in vivo injection, the stocks of Imatinib and BCI were diluted in PBS, whereas the DFC stock was diluted in alkaline PBS containing 15 mM sodium hydroxide. All drugs were injected into mice via i.p injection.
FACS analysis
Peripheral blood (PB) cells were collected from transplanted recipient mice once per week via tail bleeding. 20 μl of blood was lysed using Pharmlyse solution (BD Biosciences) and the remaining mononucleated cells were pelleted by centrifugation. The cell pellets were washed once with cold PBS. The percent leukemic chimerism was determined by quantifying the levels of YFP positive cells (BCR-ABL-Ires-YFP), analyzed by FACS. BM cells from mice transplanted with Scl-ttA-BCR-ABL cells or patient were aspirated from the mouse femurs to determine the levels of CD45.2 and human CD45. These BM cells were blocked with FcR block (BD bioscience) followed by staining with anti-mouse FITC labeled CD45.1 and anti-mouse PE CD45.2. All antibody sources and dilutions are presented in the supplementary table 4. The FACS analysis was performed on an LSRII instrument and the data were analyzed using FACSDIVA software.
For the analysis of human grafts, bone marrow cells were aspirated every two weeks from femurs of the transplanted mice. RBCs were lysed using RBC lysis buffer as above and the total MNCs were pelleted by centrifugation. The pellet was washed once with cold 1X PBS. The cells were blocked with human FcR block and mouse FcR Block (Miltenyi Biotec) followed by staining with anti-human CD45 FITC and anti-mouse CD45 APC Cy7 overnight at 4 °C. All antibody sources and dilutions are presented in the supplementary table 4. The FACS analysis was performed on an LSRII instrument and the data were analyzed using FACSDIVA software.
For differential analysis of peripheral blood (PB) cells, 20 μl of blood was lysed using RBC lysis buffer and the TMNCs were pelleted by centrifugation and washing as described above. The cells were then blocked for 10 minutes at room temperature using mouse FcR blocking reagent (Miltenyi Biotec) followed by staining with the following antibodies for 30 minutes at 4 °C: anti-CD11b (recognizes monocytes), anti-CD3 (recognizes T cells), anti-B220 (recognizes B cells) and anti-Gr1 (recognizes granulocytes) All antibody sources and dilutions are presented in the supplementary table 4. The FACS analysis was performed on an LSRII instrument and the data were analyzed using FloJo software.
Random mutagenesis and screening of Dusp1 mutants
A Gateway entry clone containing mouse Dusp1 cDNA was purchased from the Harvard PalsmiD repository (cat # MmCD00312825). The Dusp1 coding region was transferred into the retroviral gateway vector pMSCV-Ires-GFP.GW60 by recombination cloning. Mutagenesis and resistance screening were performed as described previously9. Mutants isolated in the screen were engineered into the pMSCV-Dusp1-Ires-GFP vector, using the QuikChange II XL Site-Directed Mutagenesis Kit (Agilent). The sequence of each point mutation was confirmed by sequence analysis.
Dusp1 structural modeling and inhibitor docking
Structural models of Dusp1 domains were built by homology-based modeling using SWISS-MODEL software, and crystal structures, as described previously60. A model of the Dusp1 phosphatase domain was built using the crystal structures of Dusp4 (PDB: 3EZZ; Dusp4 has 85% sequence identity with Dusp1 and Dusp6 (PDB: 1MKP; Dusp6 has 48% sequence identity with Dusp1. The structure of the N-terminal rhodanese domain of Dusp1 was built using the crystal structure of Dusp16 (PDB: 2VSW; the N-terminal domain of Dusp16 has 26% sequence identity to Dusp1). Only three structures of a Dusp allosteric domain are available in the database (for Dusp6, Dusp10 and Dusp16), and a sequence analysis did not show sufficient sequence identity of the Dusp1 rhodanese domain with the N-terminal domains of Dusp6 and Dusp10 (less than 17%); we therefore only used the Dusp16 structure (PDB: 2VSW) to build the model.
Blind docking of BCI to the rhodanese domain model of Dusp1 was performed using SwissDock software60. Modules with the most favorable energies were clustered. For each cluster, binding modules with the lowest energy (i.e., the most likely to represent true binding) were selected to validate the model by site-directed mutagenesis, as mutation V83G conferred resistance to BCI. Figures were generated using PyMol software.
Statistical analysis
Unless otherwise specified, results are depicted as the mean ± S.D. Statistical analyses were performed using one tail Student’s t test using GraphPad Prism (v6 GraphPad). Mantel-Cox test was used to perform Kaplan-Meier survival analysis in GraphPad Prism (v6, GraphPad).
Supplementary Material
Editorial summary.
Growth factor signaling in BCR-ABL-expressing chronic myeloid leukemia stem cells confers resistance to imatinib therapy. Growth factor signaling induces the expression of c-Fos and Dusp1 proteins, which prevents the drug induced cell death of leukemic stem cells. Combined inhibition of BCR-ABL, c-Fos and DUSP1 eradicated leukemia in vivo, pointing to a new therapeutic strategy to eradicate the disease for kinase-driven leukemias.
Acknowledgments
The authors are thankful to Prof. Harinder Singh and Yi Zheng for providing critical feedback on this study. We are thankful to George Daley for providing the BaF3-BA cells and Tanishtha Reya for MSCV-BCR-ABL-Ires-YFP constructs. We are thankful to Prof. Martin Carroll for providing the patient samples from the CML blast crisis. This study was supported by grants to M.A. from NCI (1RO1CA155091), Leukemia Research Foundation and V Foundation.
Footnotes
Accession codes
Data Availability Statements
The data that support the findings of this study is available from the corresponding author upon request. The GEO accession number for the microarray data reported in this paper is GSE75058.
Author Contributions
Experiments were conceived and designed by M.K. and M.A. Experiments were performed by M.K., Z.K., A.G., E.H., S.R., Z.S., M.F.B., K.K., T.L., M.X., J.C.M., J.A.C., and H.L.G. Bioinformatics analysis of microarrays and RNA-seq data were performed by M.K. and K.K. The manuscript was written by M.K. and M.A.
Author Information
The authors declare no competing financial interests. Correspondence and requests for materials should be addressed to M.A.
References
- 1.Daley GQ, Van Etten RA, Baltimore D. Induction of chronic myelogenous leukemia in mice by the P210bcr/abl gene of the Philadelphia chromosome. Science. 1990;247:824–830. doi: 10.1126/science.2406902. [DOI] [PubMed] [Google Scholar]
- 2.Druker BJ, et al. Effects of a selective inhibitor of the Abl tyrosine kinase on the growth of Bcr-Abl positive cells. Nat Med. 1996;2:561–566. doi: 10.1038/nm0596-561. [DOI] [PubMed] [Google Scholar]
- 3.O’Hare T, Zabriskie MS, Eiring AM, Deininger MW. Pushing the limits of targeted therapy in chronic myeloid leukaemia. Nat Rev Cancer. 2012;12:513–526. doi: 10.1038/nrc3317. [DOI] [PubMed] [Google Scholar]
- 4.Rousselot P, et al. Imatinib mesylate discontinuation in patients with chronic myelogenous leukemia in complete molecular remission for more than 2 years. Blood. 2007;109:58–60. doi: 10.1182/blood-2006-03-011239. [DOI] [PubMed] [Google Scholar]
- 5.Mahon FX, et al. Discontinuation of imatinib in patients with chronic myeloid leukaemia who have maintained complete molecular remission for at least 2 years: the prospective, multicentre Stop Imatinib (STIM) trial. Lancet Oncol. 2010;11:1029–1035. doi: 10.1016/S1470-2045(10)70233-3. [DOI] [PubMed] [Google Scholar]
- 6.Ross DM, et al. Safety and efficacy of imatinib cessation for CML patients with stable undetectable minimal residual disease: results from the TWISTER study. Blood. 2013;122:515–522. doi: 10.1182/blood-2013-02-483750. [DOI] [PubMed] [Google Scholar]
- 7.Chu S, et al. Detection of BCR-ABL kinase mutations in CD34+ cells from chronic myelogenous leukemia patients in complete cytogenetic remission on imatinib mesylate treatment. Blood. 2005;105:2093–2098. doi: 10.1182/blood-2004-03-1114. [DOI] [PubMed] [Google Scholar]
- 8.Savona M, Talpaz M. Getting to the stem of chronic myeloid leukaemia. Nat Rev Cancer. 2008;8:341–350. doi: 10.1038/nrc2368. [DOI] [PubMed] [Google Scholar]
- 9.Azam M, Latek RR, Daley GQ. Mechanisms of autoinhibition and STI-571/imatinib resistance revealed by mutagenesis of BCR-ABL. Cell. 2003;112:831–843. doi: 10.1016/s0092-8674(03)00190-9. [DOI] [PubMed] [Google Scholar]
- 10.Krause DS, Van Etten RA. Tyrosine kinases as targets for cancer therapy. N Engl J Med. 2005;353:172–187. doi: 10.1056/NEJMra044389. doi:353/2/172[pii] 10.1056/NEJMra04438. [DOI] [PubMed] [Google Scholar]
- 11.Weinstein IB. Cancer. Addiction to oncogenes–the Achilles heal of cancer. Science. 2002;297:63–64. doi: 10.1126/science.1073096. [DOI] [PubMed] [Google Scholar]
- 12.Sawyers CL. Shifting paradigms: the seeds of oncogene addiction. Nat Med. 2009;15:1158–1161. doi: 10.1038/nm1009-1158. [DOI] [PubMed] [Google Scholar]
- 13.Pagliarini R, Shao W, Sellers WR. Oncogene addiction: pathways of therapeutic response, resistance, and road maps toward a cure. EMBO Rep. 2015;16:280–296. doi: 10.15252/embr.201439949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Reddy A, Kaelin WG., Jr Using cancer genetics to guide the selection of anticancer drug targets. Curr Opin Pharmacol. 2002;2:366–373. doi: 10.1016/s1471-4892(02)00178-9. doi:S1471489202001789 [pii] [DOI] [PubMed] [Google Scholar]
- 15.Kaelin WG., Jr The concept of synthetic lethality in the context of anticancer therapy. Nat Rev Cancer. 2005;5:689–698. doi: 10.1038/nrc1691. doi:nrc1691 [pii] 10.1038/nrc1691. [DOI] [PubMed] [Google Scholar]
- 16.Kamb A. Consequences of nonadaptive alterations in cancer. Mol Biol Cell. 2003;14:2201–2205. doi: 10.1091/mbc.E02-11-0732. E02-11-0732 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mills GB, Lu Y, Kohn EC. Linking molecular therapeutics to molecular diagnostics: inhibition of the FRAP/RAFT/TOR component of the I3K pathway preferentially blocks PTEN mutant cells in vitro and in vivo. Proc Natl Acad Sci U S A. 2001;98:10031–10033. doi: 10.1073/pnas.191379498. 98/18/10031 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sharma SV, Settleman J. Exploiting the balance between life and death: targeted cancer therapy and "oncogenic shock". Biochem Pharmacol. 80:666–673. doi: 10.1016/j.bcp.2010.03.001. doi:S0006-2952(10)00165-6 [pii] 10.1016/j.bcp.2010.03.001. [DOI] [PubMed] [Google Scholar]
- 19.Sharma SV, Settleman J. Oncogene addiction: setting the stage for molecularly targeted cancer therapy. Genes Dev. 2007;21:3214–3231. doi: 10.1101/gad.1609907. [DOI] [PubMed] [Google Scholar]
- 20.Corbin AS, et al. Human chronic myeloid leukemia stem cells are insensitive to imatinib despite inhibition of BCR-ABL activity. J Clin Invest. 2011;121:396–409. doi: 10.1172/JCI35721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Straussman R, et al. Tumour micro-environment elicits innate resistance to RAF inhibitors through HGF secretion. Nature. 2012;487:500–504. doi: 10.1038/nature11183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wilson TR, et al. Widespread potential for growth-factor-driven resistance to anticancer kinase inhibitors. Nature. 2012;487:505–509. doi: 10.1038/nature11249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bruennert D, et al. Early in vivo changes of the transcriptome in Philadelphia chromosome-positive CD34+ cells from patients with chronic myelogenous leukaemia following imatinib therapy. Leukemia. 2009;23:983–985. doi: 10.1038/leu.2008.337. doi:leu2008337 [pii] 10.1038/leu.2008.337. [DOI] [PubMed] [Google Scholar]
- 24.Eferl R, Wagner EF. AP-1: a double-edged sword in tumorigenesis. Nat Rev Cancer. 2003;3:859–868. doi: 10.1038/nrc1209. [DOI] [PubMed] [Google Scholar]
- 25.Lawan A, Shi H, Gatzke F, Bennett AM. Diversity and specificity of the mitogen-activated protein kinase phosphatase-1 functions. Cell Mol Life Sci. 2013;70:223–237. doi: 10.1007/s00018-012-1041-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jeffrey KL, Camps M, Rommel C, Mackay CR. Targeting dual-specificity phosphatases: manipulating MAP kinase signalling and immune responses. Nat Rev Drug Discov. 2007;6:391–403. doi: 10.1038/nrd2289. [DOI] [PubMed] [Google Scholar]
- 27.Brooks SA, Blackshear PJ. Tristetraprolin (TTP): interactions with mRNA and proteins, and current thoughts on mechanisms of action. Biochimica et biophysica acta. 2013;1829:666–679. doi: 10.1016/j.bbagrm.2013.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dorfman K, et al. Disruption of the erp/mkp-1 gene does not affect mouse development: normal MAP kinase activity in ERP/MKP-1-deficient fibroblasts. Oncogene. 1996;13:925–931. [PubMed] [Google Scholar]
- 29.Zhang J, et al. c-fos regulates neuronal excitability and survival. Nat Genet. 2002;30:416–420. doi: 10.1038/ng859. [DOI] [PubMed] [Google Scholar]
- 30.Zhao C, et al. Hedgehog signalling is essential for maintenance of cancer stem cells in myeloid leukaemia. Nature. 2009;458:776–779. doi: 10.1038/nature07737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ransone LJ, Visvader J, Wamsley P, Verma IM. Trans-dominant negative mutants of Fos and Jun. Proc Natl Acad Sci U S A. 1990;87:3806–3810. doi: 10.1073/pnas.87.10.3806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Molina G, et al. Zebrafish chemical screening reveals an inhibitor of Dusp6 that expands cardiac cell lineages. Nat Chem Biol. 2009;5:680–687. doi: 10.1038/nchembio.190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Huang TS, Lee SC, Lin JK. Suppression of c-Jun/AP-1 activation by an inhibitor of tumor promotion in mouse fibroblast cells. Proc Natl Acad Sci U S A. 1991;88:5292–5296. doi: 10.1073/pnas.88.12.5292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Park S, Lee DK, Yang CH. Inhibition of fos-jun-DNA complex formation by dihydroguaiaretic acid and in vitro cytotoxic effects on cancer cells. Cancer Lett. 1998;127:23–28. doi: 10.1016/s0304-3835(97)00543-0. [DOI] [PubMed] [Google Scholar]
- 35.Padhye S, et al. Fluorocurcumins as cyclooxygenase-2 inhibitor: molecular docking, pharmacokinetics and tissue distribution in mice. Pharm Res. 2009;26:2438–2445. doi: 10.1007/s11095-009-9955-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Koschmieder S, et al. Inducible chronic phase of myeloid leukemia with expansion of hematopoietic stem cells in a transgenic model of BCR-ABL leukemogenesis. Blood. 2005;105:324–334. doi: 10.1182/blood-2003-12-4369. [DOI] [PubMed] [Google Scholar]
- 37.Li L, et al. Activation of p53 by SIRT1 inhibition enhances elimination of CML leukemia stem cells in combination with imatinib. Cancer Cell. 2012;21:266–281. doi: 10.1016/j.ccr.2011.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Copland M, et al. BMS-214662 potently induces apoptosis of chronic myeloid leukemia stem and progenitor cells and synergizes with tyrosine kinase inhibitors. Blood. 2008;111:2843–2853. doi: 10.1182/blood-2007-09-112573. [DOI] [PubMed] [Google Scholar]
- 39.Angel P, Karin M. The role of Jun, Fos and the AP-1 complex in cell-proliferation and transformation. Biochim Biophys Acta. 1991;1072:129–157. doi: 10.1016/0304-419x(91)90011-9. [DOI] [PubMed] [Google Scholar]
- 40.Molina G, et al. Zebrafish chemical screening reveals an inhibitor of Dusp6 that expands cardiac cell lineages. Nature Chemical Biology. 2009;5:680–687. doi: 10.1038/nchembio.190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Owens DM, Keyse SM. Differential regulation of MAP kinase signalling by dual-specificity protein phosphatases. Oncogene. 2007;26:3203–3213. doi: 10.1038/sj.onc.1210412. [DOI] [PubMed] [Google Scholar]
- 42.Boutros T, Chevet E, Metrakos P. Mitogen-activated protein (MAP) kinase/MAP kinase phosphatase regulation: roles in cell growth, death, and cancer. Pharmacol Rev. 2008;60:261–310. doi: 10.1124/pr.107.00106. [DOI] [PubMed] [Google Scholar]
- 43.Groom LA, Sneddon AA, Alessi DR, Dowd S, Keyse SM. Differential regulation of the MAP, SAP and RK/p38 kinases by Pyst1, a novel cytosolic dual-specificity phosphatase. EMBO J. 1996;15:3621–3632. [PMC free article] [PubMed] [Google Scholar]
- 44.Fjeld CC, Rice AE, Kim Y, Gee KR, Denu JM. Mechanistic basis for catalytic activation of mitogen-activated protein kinase phosphatase 3 by extracellular signal-regulated kinase. The Journal of biological chemistry. 2000;275:6749–6757. doi: 10.1074/jbc.275.10.6749. [DOI] [PubMed] [Google Scholar]
- 45.Zhao Q, et al. MAP kinase phosphatase 1 controls innate immune responses and suppresses endotoxic shock. J Exp Med. 2006;203:131–140. doi: 10.1084/jem.20051794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hirsch DD, Stork PJ. Mitogen-activated protein kinase phosphatases inactivate stress-activated protein kinase pathways in vivo. The Journal of biological chemistry. 1997;272:4568–4575. doi: 10.1074/jbc.272.7.4568. [DOI] [PubMed] [Google Scholar]
- 47.Young PR, et al. Pyridinyl imidazole inhibitors of p38 mitogen-activated protein kinase bind in the ATP site. The Journal of biological chemistry. 1997;272:12116–12121. doi: 10.1074/jbc.272.18.12116. [DOI] [PubMed] [Google Scholar]
- 48.Bennett BL, et al. SP600125, an anthrapyrazolone inhibitor of Jun N-terminal kinase. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:13681–13686. doi: 10.1073/pnas.251194298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shojaee S, et al. Erk Negative Feedback Control Enables Pre-B Cell Transformation and Represents a Therapeutic Target in Acute Lymphoblastic Leukemia. Cancer Cell. 2015;28:114–128. doi: 10.1016/j.ccell.2015.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hrustanovic G, et al. RAS-MAPK dependence underlies a rational polytherapy strategy in EML4-ALK-positive lung cancer. Nat Med. 2015;21:1038–1047. doi: 10.1038/nm.3930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhang B, et al. Altered microenvironmental regulation of leukemic and normal stem cells in chronic myelogenous leukemia. Cancer Cell. 2012;21:577–592. doi: 10.1016/j.ccr.2012.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Reynaud D, et al. IL-6 controls leukemic multipotent progenitor cell fate and contributes to chronic myelogenous leukemia development. Cancer Cell. 2011;20:661–673. doi: 10.1016/j.ccr.2011.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Welner RS, et al. Treatment of chronic myelogenous leukemia by blocking cytokine alterations found in normal stem and progenitor cells. Cancer Cell. 2015;27:671–681. doi: 10.1016/j.ccell.2015.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Roberts KG, et al. Genetic alterations activating kinase and cytokine receptor signaling in high-risk acute lymphoblastic leukemia. Cancer Cell. 2012;22:153–166. doi: 10.1016/j.ccr.2012.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Druker BJ, et al. Activity of a specific inhibitor of the BCR-ABL tyrosine kinase in the blast crisis of chronic myeloid leukemia and acute lymphoblastic leukemia with the Philadelphia chromosome. N Engl J Med. 2001;344:1038–1042. doi: 10.1056/NEJM200104053441402. [DOI] [PubMed] [Google Scholar]
- 56.Chang KH, et al. Vav3 collaborates wit p190-BCR-ABL in lymphoid progenitor leukemogenesis, proliferation, and survival. Blood. 2012;120:800–811. doi: 10.1182/blood-2011-06-361709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bagger FO, et al. BloodSpot: a database of gene expression profiles and transcriptional programs for healthy and malignant haematopoiesis. Nucleic Acids Res. 2016;44:D917–924. doi: 10.1093/nar/gkv1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jorgensen HG, Allan EK, Jordanides NE, Mountford JC, Holyoake TL. Nilotinib exerts equipotent antiproliferative effects to imatinib and does not induce apoptosis in CD34+ CML cells. Blood. 2007;109:4016–4019. doi: 10.1182/blood-2006-11-057521. [DOI] [PubMed] [Google Scholar]
- 59.Holyoake TL, Vetrie D. The chronic myeloid leukemia stem cell: stemming the tide of persistence. Blood. 2017 doi: 10.1182/blood-2016-09-696013. [DOI] [PubMed] [Google Scholar]
- 60.Kesarwani M, et al. Targeting substrate-site in Jak2 kinase prevents emergence of genetic resistance. Scientific reports. 2015;5:14538. doi: 10.1038/srep14538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Azam M, Seeliger MA, Gray NS, Kuriyan J, Daley GQ. Activation of tyrosine kinases by mutation of the gatekeeper threonine. Nat Struct Mol Biol. 2008;15:1109–1118. doi: 10.1038/nsmb.1486. doi:nsmb.1486 [pii] 10.1038/nsmb.1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Komurov K, Dursun S, Erdin S, Ram PT. NetWalker: a contextual network analysis tool for functional genomics. BMC Genomics. 2012;13:282. doi: 10.1186/1471-2164-13-282. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.