

Abstract
Many relevant options to improve efficacy and kinetics of sucrose metabolism in Saccharomyces cerevisiae and, thereby, the economics of sucrose-based processes remain to be investigated. An essential first step is to identify all native sucrose-hydrolysing enzymes and sucrose transporters in this yeast, including those that can be activated by suppressor mutations in sucrose-negative strains. A strain in which all known sucrose-transporter genes (MAL11, MAL21, MAL31, MPH2, MPH3) were deleted did not grow on sucrose after 2 months of incubation. In contrast, a strain with deletions in genes encoding sucrose-hydrolysing enzymes (SUC2, MAL12, MAL22, MAL32) still grew on sucrose. Its specific growth rate increased from 0.08 to 0.25 h−1 after sequential batch cultivation. This increase was accompanied by a 3-fold increase of in vitro sucrose-hydrolysis and isomaltase activities, as well as by a 3- to 5-fold upregulation of the isomaltase-encoding genes IMA1 and IMA5. One-step Cas9-mediated deletion of all isomaltase-encoding genes (IMA1-5) completely abolished sucrose hydrolysis. Even after 2 months of incubation, the resulting strain did not grow on sucrose. This sucrose-negative strain can be used as a platform to test metabolic engineering strategies and for fundamental studies into sucrose hydrolysis or transport.
Keywords: disaccharide, isomaltase, laboratory evolution, reverse engineering, multiple gene deletion, real-time PCR
A yeast strain without functional sucrose transport and hydrolysis was constructed and is now ready to be used for metabolic engineering strategies and for fundamental studies into sucrose metabolism.
INTRODUCTION
Sucrose is a cheap substrate for industrial fermentation that is extensively used for ethanol production (Maiorella, Blanch and Wilke 1984; Della-Bianca et al.2013; Gombert and van Maris 2015) and can be used for a wide range of other yeast-based industrial processes (Marques et al.2016; Meadows et al.2016). The main sources of sucrose are sugar cane in the tropics and sugar beet in temperate regions. Extraction of sucrose from feedstocks is inexpensive and no enzymatic pre-treatment is necessary prior to microbial fermentation (Maiorella, Blanch and Wilke 1984; Marques et al.2016). Furthermore, the use of sucrose for industrial biotechnology does not have to compete with food production. For example, in the Brazilian ethanol industry, food and biofuel production can coexist (Mitchell 2008). Therefore, diversifying the range of products that can be produced from sucrose fermentation will have a positive impact on sustainable industrial production of fuels and chemicals (Meadows et al.2016).
Sucrose-based ethanol production employs bakers’ yeast (Saccharomyces cerevisiae), which efficiently consumes sugars and can withstand industrially relevant stresses such as high ethanol and acid concentrations (Della-Bianca et al.2014). To convert sucrose into hexoses, S. cerevisiae secretes invertase (β-fructosidase, sucrose hydrolase; Suc) (Winge and Roberts 1952; Hawthorne 1958). Invertase hydrolyses sucrose into the monomers, glucose and fructose, which subsequently enter the cell through facilitated diffusion via hexose transporters and are metabolised via the classical Embden-Meyerhof-Parnas glycolytic pathway (Lagunas 1993). Alternatively, sucrose can be imported by a proton symporter (Mal11, formerly known as Agt1) and hydrolysed in the cytosol (Stambuk et al.1999). Cytosolically localised invertase and maltases (Malx2) then hydrolyse sucrose intracellularly. In vitro studies have shown that isomaltases can also hydrolyse sucrose, but their contribution to in vivo sucrose metabolism remains to be addressed (Carlson and Botstein 1982; Stambuk et al.1999; Badotti et al.2008; Brown, Murray and Verstrepen 2010; Voordeckers et al.2012; Deng et al.2014). Even closely related yeast strains exhibit significant diversity in the identity and number of representatives from each of the three disaccharide hydrolase-encoding gene families (SUC, MALx2, IMA) (Carlson and Botstein 1983; Brown, Murray and Verstrepen 2010). For example, the genome of the haploid laboratory strain S. cerevisiae CEN.PK113-7D contains five different IMA genes on subtelomeric regions: IMA1 (three copies: CHRIII, CHRVII, CHRXI), IMA2 (CHRXV), IMA3 (CHRIX), IMA4 (CHRX) and IMA5(CHRX) (Teste, François and Parrou 2010; Nijkamp et al.2012), while the related strain CEN.PK102-3A, which was used in this study, does not contain the IMA1 copy at CHRXI (de Kok et al.2011).
Detailed complete knowledge of sucrose transport and hydrolysis is important for both industry and academia. Functional analysis of eukaryotic sucrose transporters and hydrolytic or phosphoroclastic sucrose-cleaving enzymes is often done in S. cerevisiae. An example is the development of a functional screening system for plant sucrose exporters (Zhou, Grof and Patrick 2014). In this system, functional expression of plant sucrose exporters is analysed by monitoring conversion of glucose into sucrose and subsequent sucrose efflux from an S. cerevisiae strain that heterologously expresses sucrose-phosphate synthase (SPS) and sucrose-phosphate phosphatase (SPP) genes. In industry, engineering of sucrose metabolism can contribute to increased product yields on substrate. In a proof-of-principle study with a laboratory strain of S. cerevisiae, Basso et al. (2011) demonstrated an increase in the ethanol yield of S. cerevisiae on sucrose of 11%, when compared to the reference strain, by deleting the secretion signal sequence from the invertase coding gene (SUC2). This modification resulted in a predominant intracellular localisation of invertase activity, thereby creating a necessity for sucrose uptake via proton symport. To avoid intracellular acidification, protons entering the cell via the sucrose-proton symporter have to be expelled by the plasma-membrane H+-ATPase (Pma1). This export mechanism costs one ATP per proton, thus reducing the ATP yield of alcoholic fermentation from 4 to 3 ATP per sucrose molecule. This change in energy coupling was shown to lead to a decrease of the biomass yield and an increase of the ethanol yield of the engineered strain on sucrose.
In metabolic engineering, incomplete knowledge of the targeted pathway can delay strain construction due to the activity of competing pathways or activation of such pathways by suppressor mutations. Although mutants that are unable to grow on sucrose have been reported (Carlson, Osmond and Botstein 1981), it has not yet been investigated whether these strains can regain the ability to grow on sucrose after laboratory evolution (Marques et al.2016), for instance through recruitment of any of the numerous glycoside hydrolases in S. cerevisiae (Yamamoto et al.2004; Naumoff and Naumov 2010; Naumoff 2011). To address this question, S. cerevisiae strains in which either the genes encoding known sucrose transporters or those encoding disaccharide-hydrolysing enzymes were inactivated were subjected to laboratory evolution and mutations underlying the acquired phenotypes were identified by whole-genome resequencing.
MATERIAL AND METHODS
Strains and maintenance
Saccharomyces cerevisiae strains used in this work (Table 1) belong to the CEN.PK family (Entian and Kötter 2007; Nijkamp et al.2012). To prepare stock cultures, yeast strains were grown in 50 mL shake flasks containing 10 mL of either yeast-peptone-dextrose (YPD) medium or, for strains carrying plasmids with auxotrophic marker genes, synthetic medium (SM). YPD contained 10 g L−1 Bacto yeast extract, 20 g L−1 Bacto peptone and 20 g L−1 glucose in demineralised water. SM, supplemented with vitamins, trace elements and 20 g L−1 glucose, was prepared according to Verduyn et al. (1992). Glycerol was added to growing cultures (final concentration 30% v/v) and 1 mL aliquots were stored at –80°C.
Table 1.
Strains used in this study. The abbreviation malΔ indicates mal11-mal12::loxP mal21-mal22::loxP mal31-32::loxP and mphΔ indicates mph2/3::loxP mph2/3::loxP-hphNT1-loxP.
| Name | Relevant genotype | Parental strain | Origin |
|---|---|---|---|
| CEN.PK113-7D | MATa MAL1x MAL2x MAL3x MAL4x MAL2-8C SUC2 LEU2 URA3 | P. Kötter, Germany | |
| CEN.PK102-3A | MATa MAL1x MAL2x MAL3x MAL4x MAL2-8C SUC2 leu2-112 ura3-52 | P. Kötter, Germany | |
| IMK291 | CEN.PK102-3A MATa leu2-112 ura3-52 MAL2-8C | IMK289 | This study |
| mal11-mal12::loxP mal21-mal22::loxP mal31-32::loxP mph2/3::loxP | |||
| mph2/3::loxP-hphNT1-loxP suc2::loxP-kanMX-loxP | |||
| IMX469 | malΔ mphΔ suc2Δ MAL11-LEU2 | IMK291 | This study |
| IMU048 | malΔ mphΔ suc2Δ MAL11-LEU2 URA3 | IMX469 | This study |
| IMU055 | malΔ mphΔ suc2Δ MAL11-LEU2 MAL12-URA3 | IMX469 | This study |
| IMX470 | malΔ mphΔ suc2Δ LEU2 | IMK291 | This study |
| IMU051 | malΔ mphΔ suc2Δ LEU2 URA3 | IMX470 | This study |
| IMU054 | malΔ mphΔ suc2Δ LEU2 MAL12-URA3 | IMX470 | This study |
| IMZ571 | malΔ mphΔ suc2Δ MAL11-LEU2 cas9-URA3 | IMX469 | This study |
| IMK700 | malΔ mphΔ suc2Δ MAL11-LEU2 cas9-URA3 ima1Δ ima2Δ ima3Δ ima4Δ ima5Δ amdSYM | IMZ571 | This study |
| IMS422 | malΔ mphΔ suc2Δ MAL11-LEU2 URA3 (single colony isolate from evolution, replicate 1) | IMU048 | This study |
| IMS423 | malΔ mphΔ suc2Δ MAL11-LEU2 URA3 (single colony isolate from evolution, replicate 2) | IMU048 | This study |
| IMS424 | malΔ mphΔ suc2Δ MAL11-LEU2 URA3 (single colony isolate from evolution, replicate 3) | IMU048 | This study |
| IMS517 | malΔ mphΔ suc2Δ MAL11-LEU2 | IMS422 | This study |
| IMS518 | malΔ mphΔ suc2Δ MAL11-LEU2 | IMS423 | This study |
| IMS519 | malΔ mphΔ suc2Δ MAL11-LEU2 | IMS424 | This study |
| IMS604 | malΔ mphΔ suc2Δ MAL11-LEU2 cas9-URA3 | IMS517 | This study |
| IMS605 | malΔ mphΔ suc2Δ MAL11-LEU2 cas9-URA3 | IMS518 | This study |
| IMS606 | malΔ mphΔ suc2Δ MAL11-LEU2 cas9-URA3 | IMS519 | This study |
| IMK716 | malΔ mphΔ suc2Δ MAL11-LEU2 cas9-URA3 ima1Δ ima2Δ ima3Δ ima4Δ ima5Δ | IMZ604 | This study |
| IMK717 | malΔ mphΔ suc2Δ MAL11-LEU2 cas9-URA3 ima1Δ ima2Δ ima3Δ ima4Δ ima5Δ | IMZ605 | This study |
| IMK718 | malΔ mphΔ suc2Δ MAL11-LEU2 cas9-URA3 ima1Δ ima2Δ ima3Δ ima4Δ ima5Δ | IMZ606 | This study |
| IMK743 | malΔ mphΔ suc2Δ MAL11-LEU2 ima1Δ ima2Δ ima3Δ ima4Δ ima5Δ amdSYM | IMK700 | This study |
| IMZ620 | malΔ mphΔ suc2Δ MAL11-LEU2 ima1Δ ima2Δ ima3Δ ima4Δ ima5Δ amdSYM IMA1-URA3 | IMK743 | This study |
| IMZ621 | malΔ mphΔ suc2Δ MAL11-LEU2 ima1Δ ima2Δ ima3Δ ima4Δ ima5Δ amdSYM IMA2-URA3 | IMK743 | This study |
| IMZ622 | malΔ mphΔ suc2Δ MAL11-LEU2 ima1Δ ima2Δ ima3Δ ima4Δ ima5Δ amdSYM IMA3,4-URA3 | IMK743 | This study |
| IMZ623 | malΔ mphΔ suc2Δ MAL11-LEU2 ima1Δ ima2Δ ima3Δ ima4Δ ima5Δ amdSYM IMA5-URA3 | IMK743 | This study |
Molecular biology techniques
Diagnostic PCR was performed using DreamTaq DNA Polymerase (Thermo Fisher Scientific, Waltham, MA, USA). PCR amplification for cloning and sequencing purposes was performed with Phusion High-Fidelity DNA Polymerase (Thermo Fisher Scientific). Both enzymes were used according to the manufacturer's instructions. Primers and oligonucleotides were purchased from Sigma-Aldrich (St. Louis, MO, USA). DNA purification from PCR reaction mixtures was done using GenElute PCR Clean-up Kit (Sigma-Aldrich). Separation of DNA fragments was performed in 1% (w/v) agarose gel (Thermo Fisher Scientific) in TAE buffer (40 mM Tris-acetate pH 8.0 and 1 mM EDTA). DNA fragments were purified from gels using the Zymoclean Gel DNA Recovery kit (Zymo Research, Irvine, CA, USA). Plasmid assembly was done with the Gibson Assembly Master Mix (New England Biolabs, Beverly, MA) according to the manufacturer's protocol. Restriction endonucleases (Thermo Fisher Scientific) and T4 DNA ligase (Promega Corporation, Madison, WI, USA) were used according to the manufacturer's instructions. Plasmids were isolated from Escherichia coli using GenElute HP Plasmid Miniprep Kit (Sigma-Aldrich) according to the provided protocol. Yeast genomic DNA was extracted using YeaStar Genomic kit (Zymo Research).
Plasmid construction
Plasmid pUDC156 (Table 2) was assembled by in vivo homologous recombination of a plasmid backbone and an insert fragment containing a yeast expression cassette for the cas9 gene from Streptococcus pyogenes (Kuijpers et al.2013). The plasmid backbone was amplified from MB4917 (Zelle et al.2010) with primers 7833 and 4697 and the cas9-expression cassette was amplified from p414-TEF1p-Cas9-CYC1t (DiCarlo et al.2013) with primers 1768 and 7236 (Table S1, Supporting Information). Both fragments contain 60 bp overlaps with each other and were assembled by co-transformation to S. cerevisiae strain IMX469 resulting in IMZ571. Plasmid pUDR127 (Fig. 1A; Table 2) contains two gRNA cassettes: one for deletion of IMA5 (gRNA-IMA5) and one targeting sequence shared by IMA1, IMA2, IMA3 and IMA4 (gRNA-IMAs). This plasmid was constructed via Gibson assembly of three fragments: two gRNA cassettes overlapping with each other in the 2 μm replicon and a plasmid backbone. The plasmid backbone was obtained via PCR amplification, using pROS11 (Mans et al.2015) as template with primer 6005 (Table S1). The gRNA cassette gRNA-IMA5 was obtained via PCR from pROS11 with primers 8761 and 5975, and cassette gRNA-IMAs was obtained via PCR from pROS11 with primers 8759 and 5974 (Table S1). Plasmid pUDI084 was made by removal of the MAL11 coding sequence from pUDI035 (de Kok et al.2011) using SpeI and NheI restriction sites and subsequent recircularisation of the plasmid using T4 DNA ligase (Table 2). Similarly, pUDE260 was constructed by removal of PGMβ from pUDE063 (de Kok et al.2011) using PvuII sites and subsequent recircularisation of the plasmid (Table 2). The IMA-reinsertion plasmids (pUDE427 to pUDE430) were made by Gibson assembly of an IMA expression cassette and a plasmid backbone. The latter was obtained via PCR with p426-GPD (Mumberg, Müller and Funk 1995) as a template, using primer pairs 7823 and 5975 and 5974 and 7812, which amplify the backbone in two parts to minimise chances of plasmid recircularisation (Table S1). Templates for the IMA expression cassettes were PCR amplified using genomic DNA from strain IMU048 with primers that are specific to each IMA (primers 8607 and 8611 for IMA1; 8608 and 8612 for IMA2; 8609 and 8613 for IMA3,4; and 8610 and 8614 for IMA5). From these templates, the coding sequence of each IMA could be individually amplified using primer pairs 9302 and 9305 (IMA1), 9303 and 9306 (IMA2 and IMA3,4) and 9304 and 9307 (IMA5). Plasmids were transformed to E. coli for storage and amplification, where necessary after isolation of in vivo assembled plasmids from yeast cultures.
Table 2.
Plasmids used in this study.
| Name | Relevant characteristics | Origin |
|---|---|---|
| pUG6 | loxP-KanMX4-loxP | Gueldener et al. (2002) |
| p426-GPD | 2μm URA3 PTDH3-TCYC1 | Mumberg, Müller and Funk (1995) |
| p414-TEF1p-Cas9-CYC1t | CEN6/ARS4 ampR TRP1 PTEF1-cas9-TCYC1 | DiCarlo et al. (2013) |
| MB4917 | CEN6 ampR URA3 | Zelle et al. (2010) |
| pROS11 | 2μm amdSYM gRNA-CAN1.Y gRNA-ADE2.Y | Mans et al. (2015) |
| pUDC156 | CEN6 URA3 PTEF1-cas9-TCYC1 | This study |
| pUDR127 | 2μm amdSYM gRNA-IMA5 gRNA-IMA1,2,3,4 | This study |
| pUDI035 | Integration plasmid LEU2 PTDH3-MAL11-TCYC1 | de Kok et al. (2011) |
| pUDI084 | Integration plasmid LEU2 empty vector | This study |
| pUDE044 | 2μm URA3 PTDH3-Mal12-TADH1 | de Kok et al. (2011) |
| pUDE260 | 2μm URA3 empty vector | This study |
| pUDE427 | 2μm URA3 PTDH3-IMA1-TCYC1 | This study |
| pUDE428 | 2μm URA3 PTDH3-IMA2-TCYC1 | This study |
| pUDE429 | 2μm URA3 PTDH3-IMA3,4-TCYC1 | This study |
| pUDE430 | 2μm URA3 PTDH3-IMA5-TCYC1 | This study |
Figure 1.

Strategy for deletion of the IMA genes using a shared Cas9 target site. (A) Plasmid pUDR127, that contains two gRNA-cassettes: ‘gRNA-IMAs’ (in red) with a target sequence to cleave all IMA genes except IMA5 and ‘gRNA-IMA5’, in purple. The SNR52 promoter and SUP4 terminator are shown in green and yellow, respectively. Sequences encoding structural RNAs are shown in grey. 2 μm: origin of replication in yeast; pMB1: origin of replication in bacteria; ampR: cassette for ampicillin resistance in E. coli; amdSYM: dominant marker that allows use of acetamide as nitrogen source. (B) Illustration of the shared Cas9 target sequences among the IMA genes. The coding sequence of each IMA is represented by black lines. The red boxes correspond to the shared target sequence. The dark- and light-grey fragments are the homology regions (60 bp) up- and downstream of the target site where the repair fragment (120 bp) can recombine. In view of its sequence divergence from the other IMA genes, an exclusive target sequence, in purple, and repair sites, in dark and light green, were chosen for inactivation of IMA5.
Strain construction
Saccharomyces cerevisiae transformations were carried out according to Gietz and Woods (2002) using 1 μg DNA, unless specified otherwise below. Transformants were selected on agar plates containing SM with 20 g L−1 glucose. The following components were added when necessary: G418 200 mg L−1; uracil 0.15 g L−1; leucine 0.5 g L−1. Cells expressing the amdSYM marker were selected on plates according to Solis-Escalante et al. (2013). Strain IMK289 (de Kok et al.2011) was transformed with a deletion cassette containing loxP-KanMX4-loxP marker. This cassette was amplified from plasmid pUG6 using primers 1482 and 1483 that has homology to the sequence outside the SUC2 open reading frame. The resulting strain was named IMK291. To construct MAL11-expressing strains, vector pUDI035 (Table 2) was linearised with BstEII (restriction site at LEU2 marker) and transformed into IMK291, resulting in strain IMX469 (Table 1). Transformation of pUDE260 and pUDE044 into IMX469 resulted in IMU048 and IMU055, respectively. Linearisation of pUDI084 (Table 2) with BstEII and transformation into IMK291 resulted in IMX470. Transformation of pUDE260 and pUDE044 into IMX470 resulted in IMU051 and IMU054, respectively. IMK700 was made via co-transformation of IMZ571 with 1 μg of plasmid pUDR127 together with 4 μg of each dsDNA repair fragment: one for IMA5 and another one for all the other IMA genes (Fig. 1). Repair fragments were obtained by annealing two complementary PAGE-purified single-stranded oligonucleotides according to Mans et al. (2015): 8592 and 8593 for IMA1-4 and 8763 and 8764 for IMA5 (Table 2). In order to delete IMA genes from strains evolved on sucrose—IMS422, IMS423 and IMS424—pUDE260 was cured from these strains by cultivation on SM plates with 20 g L−1 glucose and 1 g L−1 5΄-fluoroorotic acid (Boeke, La Croute and Fink 1984), resulting in strains IMS517, IMS518 and IMS519, respectively. Transformation of pUDC156 into these three strains yielded strains IMS604, IMS605 and IMS606, respectively. Finally, transformation of pUDR127 and repair fragments for IMA5 and IMA1-4 (as mentioned above) into these three strains yielded strains IMK717, IMK718 and IMK719. For re-insertion of the IMA genes on multicopy plasmids, pUDC156 was cured from IMK700 resulting in IMK743. Then, each IMA overexpression plasmid (named pUDE427-pUDE430, Table 2) was transformed into IMK743 resulting in strains IMZ620-623.
Medium and cultivation
Shake-flask cultures were performed in 500 mL shake flasks containing 100 mL of SM with 20 g L−1 sucrose, in an Innova incubator shaker (New Brunswick Scientific, Edison, NJ, USA) set at 200 rpm and at 30°C under an air atmosphere. For growth rate determinations, cells were inoculated in SM with 20 g L−1 glucose from a frozen stock culture. After reaching stationary phase, the culture was transferred to SM with 20 g L−1 sucrose (initial OD660nm = 0.2) and incubated until exponential growth was observed. Exponentially growing cultures were then transferred to fresh medium (initial OD660nm = 0.2) and samples were taken hourly until stationary phase was reached. Optical density at 660 nm was measured with a Libra S11 spectrophotometer (Biochrom, Cambridge, UK). Specific growth rates were calculated from at least five data points.
Laboratory evolution
Sequential batch cultivation of strain IMU048 was performed in 10 mL SM (Verduyn et al.1992) with 20 g L−1 sucrose, in 50 mL polypropylene tubes (Greiner Bio-One, Frickenhausen, BW) in an Innova incubator shaker (New Brunswick Scientific), at 200 rpm and at 30°C. At the end of each cultivation cycle (OD660nm, 3–4.2), 0.1 mL of culture was transferred to fresh medium to start a next cycle. In total, 30 transfers were carried out in 70 days, which accounts for ∼180 generations. One single-colony isolate from each evolution line was obtained by restreaking three times on non-selective medium (YPD) plates.
Sucrose and glucose determination
Concentrations of sucrose and glucose in culture supernatants were analysed by high-performance liquid chromatography (Agilent 1100 HPLC, Agilent Technologies, Santa Clara, CA), using an Aminex HPX-87H ion exchange column (BioRad, Richmond, CA) coupled to a refractive-index detector and eluted with 0.5 mM H2SO4 at 0.8 mL min−1 and at 40°C. This temperature was used instead of the regularly applied temperature of 60°C to avoid sucrose hydrolysis during analysis.
Enzyme activity assays
For enzyme activity assays, shake-flask cultures on SM with 20 g L−1 sucrose were harvested during exponential growth (OD660nm = 3–4). For strains IMU054, IMU055, IMZ620, IMZ621, IMZ622 and IMZ623, ethanol (20 g L−1) was used as carbon source instead of sucrose. For strains IMK700, IMK716, IMK717 and IMK718, 20 g L−1 ethanol was used as carbon source plus 20 g L−1 sucrose as inducer. Samples were harvested, washed and prepared for sonication according to Postma et al. (1989). Cell extracts were prepared by sonication with 0.7 mm glass beads at 0°C for 2 min at 0.5 min intervals with an MSE sonicator (150 W output; 8 μm peak-to-peak amplitude). Unbroken cells and debris were removed by centrifugation (4°C, 20 min, 47 000 × g). The supernatant was used for enzyme activity assays. For measurement of extracellular enzyme activity, 100 mL of exponentially growing cells (OD660nm = 3–4) was centrifuged (4°C, 10 min, 5000 × g), the supernatant was concentrated up to 200 times with a Vivaspin® 20 filter with a 10 000 MW cut-off (Sartorius Stedim, Aubagne, France) and dialysed overnight against 10 mM potassium-phosphate buffer (pH 7.5) at 4°C. Protein levels in extracellular samples and cell extracts were determined with the Lowry assay (Lowry et al.1951). Sucrose or isomaltose hydrolytic activity was measured at 30°C by monitoring the reduction of NADP+ at 340 nm in a 1 mL reaction mixture containing 50 mM imidazole-HCl (pH 7.0), 1 mM NADP+, 12.5 mM MgCl2, 1 mM ATP, 3.5 units hexokinase, 3.5 units glucose-6-phosphate dehydrogenase and 10–40 μL cell extract. The reaction was started by the addition 100 mM of substrate. An extinction coefficient of 6.3 mM−1 was used for NADPH.
Real-time quantitative PCR
Exponentially growing cultures (OD660nm = 3–4) in 100 mL SM (Verduyn et al.1992) with 20 g L−1 of sucrose were harvested as described previously (Piper et al.2002). RNA was extracted using the hot-phenol method (Schmitt, Brown and Trumpower 1990), and RNA quality was assayed by electrophoresis using an Agilent BioAnalyzer 2100 (Agilent Technologies). Genomic DNA elimination was performed from 2 μg of total RNA in a 28 μL reaction using the QuantiTec Reverse Transcription kit (Qiagen, Düsseldorf, Germany). Reagents for cDNA synthesis from the same kit were added up to a final volume of 40 μL following the manufacturer's instructions. A portion of 6 μL of cDNA solution diluted 150 times was used in 20 μL qPCR mix that additionally included 10 μL of Rotor-Gene SYBR Green PCR Master Mix (Qiagen) and forward and reverse primers (1 μM each) (Table S1). A Rotor-Gene Q (Qiagen) was used with the following sequence: denaturation at 95°C for 5 min followed by 40 cycles of denaturation at 95°C for 5 s, annealing at 60°C for 10 s and extension at 72°C for 20 s. A melting curve up from 60°C to 95°C was performed to verify primer specificity. PCR efficiency of each primer was determined by a dilution series using a pool of cDNA from different samples. Transcript levels of the ‘housekeeping’ gene UBC6 (Teste et al.2009) were used for data normalisation. Threshold cycles (CT) were exported from Rotor-Gene Q software (version 2.0.2) and analysed using the REST-2009 algorithm (Pfaffl, Horgan and Dempfle 2002). One of the two biological replicates of CEN.PK113-7D was chosen as reference condition (expression ratio = 1).
Sanger and whole-genome sequencing
Sanger sequencing of PCR products of each IMA gene was performed at BaseClear BV (Leiden, The Netherlands). The PCR products were obtained with genomic DNA extracted from strains IMS422, IMS423 and IMS424 and following primers: 8607 and 8611 (IMA1), 8608 and 8612 for (IMA2), 8609 and 86013 for (IMA3,4) and 8610 and 8614 (IMA5) (Table S1). IMA3 and IMA4 were treated as a single gene (IMA3,4) since their coding sequences are identical (Teste, François and Parrou 2010).
DNA for whole genome sequencing was extracted using Qiagen 100/G kit following the manufacturer's protocol (Qiagen, Hilden, Germany). Whole-genome sequencing was performed by Novogene (Beijing, China). A library of 350-bp genomic fragments was created and sequenced paired end (150-bp reads). A minimum data quantity of 4000 MB was generated per strain, representing a minimum 330-fold coverage. The data analysis was performed as described by van den Broek et al. (2015). The sequencing data of the parental strain, IMU048, and of the three evolved isolates, IMS422, IMS423 and IMS424, were deposited at NCBI under the BioProject ID: PRJNA353914.
RESULTS
Experimental design to eliminate non-biological sucrose hydrolysis
Although sucrose is generally stable when dissolved in water, under certain conditions it can be hydrolysed into fructose and glucose, such as for instance by acid catalysis in low pH solutions (Krieble 1935; Wolfenden and Yuan 2008). To investigate possible artefacts created by such non-biological hydrolysis of sucrose, strain IMU051 (malΔ mphΔ suc2Δ), which cannot grow on sucrose, but can grow on hexoses, was incubated in SM shake flasks at pH values between 2 and 7 (Fig. 2). At pH values of 5–7, no growth was observed (Fig. 2A). At pH 3, linear growth was observed (Fig. 2A), which was consistent with the occurrence of sucrose hydrolysis in a sterile culture at this acidic pH (Fig. 2B). While sucrose hydrolysis also occurred at pH 2 (Fig. S2, Supporting Information), no growth was observed at this pH value, since Saccharomyces cerevisiae CEN.PK113-7D cannot grow at this low pH (Della-Bianca et al.2014). To avoid a contribution of non-biological hydrolysis of sucrose to the initiation of growth, all further growth experiments in this study were done in cultures with an initial pH of 6. For those cultures that did show growth, the pH eventually decreases to values below 3. However, since this drop only occurs towards the end of the exponential growth, this did not influence the initiation of growth.
Figure 2.

Non-biological hydrolysis of sucrose caused by medium acidity. (A) Growth of S. cerevisiae strain IMU051 (malΔ mphΔ suc2Δ) in SM with 20 g L−1 sucrose as sole carbon source with initial pH set at different values: pH 2, filled squares; pH 3, filled triangles; pH 4, inverted filled triangles; pH 5, open squares; pH 6, ‘crosses’ and pH 7, ‘plus’. The symbols filled square, open square, cross and plus are overlapping. (B) Sugar concentrations in flasks with SM at an initial pH of 3. Closed symbols: glucose (filled diamonds) and sucrose (filled circles) concentrations in flasks inoculated with IMU051. Open symbols; glucose (open diamonds) and sucrose (open circles) concentrations in flasks without inoculum. At day 0.00 and 1.06: filled circles overlapping with open circles and filled diamonds overlapping with open diamonds. Data were not corrected for evaporation to keep the experimental setup identical to that used for growth studies. The experiment was conducted with two independent replicates, of which one representative replicate is shown. 500 mL shake flasks containing 100 mL SM with sucrose 20 g L−1 were incubated at 30°C and at 200 rpm.
Residual sucrose hydrolysis encoded by unknown genes
To investigate what is needed to completely abolish both sucrose transport and hydrolysis in S. cerevisiae CEN.PK102-3A, a strain was constructed in which the following genes were deleted: SUC2 (encoding invertase); MAL12, MAL22 and MAL32 (encoding maltases); and MAL11, MAL21, MAL31, MPH2, MPH3 (encoding transporters). The resulting strain (IMU051; malΔ mphΔ suc2Δ) did not show any growth over a period of up to 2 months in SM with sucrose as the sole carbon source (Table 3). However, for future metabolic engineering strategies concerning sucrose metabolism, it is important to understand whether this inability to grow is caused by lack of transport or lack of sucrose hydrolysis. To investigate this, three new strains were constructed: one expressing the transporter Mal11 (IMU048; malΔ mphΔ suc2Δ MAL11), one expressing the α-glucosidase Mal12 (IMU054; malΔ mphΔ suc2Δ MAL12) and a control strain expressing both genes (IMU055; malΔ mphΔ suc2Δ MAL11 MAL12). The maltase Mal12 was chosen for intracellular cleavage of sucrose rather than the invertase Suc2, since previous work has shown that extracellular Suc2 activity can be detected even if SUC2 is expressed without the secretion signal sequence (Basso et al.2011).
Table 3.
Maximum specific growth rates of S. cerevisiae strains grown in shake flasks containing SM (initial pH 6.0) with 20 g L−1 sucrose as sole carbon source. For enzyme-activity assays, strains were grown in SM with 20 g L−1 sucrose as sole carbon source. For enzymatic-activity determination, IMU054 and IMU055 were grown in SM with 20 g L−1 ethanol as sole carbon source. Strain IMK700 was grown in SM with 20 g L−1 ethanol as sole carbon source plus 20 g L-1 sucrose as inducer. Averages and mean deviations were obtained from duplicate experiments.
| Strain | Relevant genotype | Growth on sucrose (h−1) | Intracellular sucrose hydrolysis (μmol mg protein−1 min−1) | Extracellular sucrose hydrolysis (μmol mg protein−1 min−1) | Intracellular isomaltose hydrolysis (μmol mg protein−1 min−1) |
|---|---|---|---|---|---|
| CEN.PK113-7D | IMAx MALxx SUC2 | 0.37 ± 0.01 | 0.43 ± 0.01 | 5.40 ± 0.04 | 0.13 ± 0.01 |
| IMU051 | malΔ mphΔ suc2Δ | No growtha | N.D. | N.D. | N.D. |
| IMU054 | malΔ mphΔ suc2Δ MAL12 | No growtha | 2.04 ± 0.07 | N.D. | N.D. |
| IMU055 | malΔ mphΔ suc2Δ MAL11 MAL12 | 0.19 ± 0.01 | 1.67 ± 0.08 | N.D. | N.D. |
| IMU048 | malΔ mphΔ suc2Δ MAL11 | 0.08 ± 0.01 | 0.48 ± 0.03 | B.D. | 1.05 ± 0.04 |
| IMK700 | malΔ mphΔ suc2Δ MAL11 imaΔ cas9 | No growtha | B.D. | N.D. | B.D. |
| IMS422 | IMU048 evolved #1 | 0.25 ± 0.02 | 1.10 ± 0.02 | B.D. | 2.31 ± 0.36 |
| IMS423 | IMU048 evolved #2 | 0.25 ± 0.02 | 1.57 ± 0.08 | B.D. | 1.59 ± 0.13 |
| IMS424 | IMU048 evolved #3 | 0.26 ± 0.02 | 1.10 ± 0.11 | B.D. | 2.16 ± 0.07 |
aIncubation period: 10 days.
B.D.: Below detection limit, i.e. <0.03 μmol mg protein−1 min−1.
N.D.: Not determined
Combined expression of MAL11 and MAL12 resulted in a maximum specific growth rate of 0.19 h−1 for the control strain IMU055 (Table 3). Even after prolonged incubation of up to 2 months, expression of only the hydrolysing enzyme Mal12 (IMU054) did not enable growth in SM with sucrose as the sole carbon source, indicating that there is no residual sucrose transport activity sufficient to allow growth and that this phenotype is stable. In contrast, the strain that only expressed the transporter Mal11 (IMU048; malΔ mphΔ suc2Δ MAL11) grew at a maximum specific growth rate of 0.08 h−1 in SM with sucrose, after a lag phase of about 4 days (Table 3). All shake-flask cultivations were performed under an air atmosphere. In line with this observation, an activity of sucrose hydrolysis of 0.48 ± 0.03 μmol mg protein−1 min−1 was detected in cell extracts of this strain. Although lower than the activity of 1.67 ± 0.08 μmol mg protein−1 min−1 observed in the reference strain IMU055, this activity was sufficient to sustain growth on sucrose (Table 3).
Laboratory evolution enables sucrose hydrolysis and increased IMA expression
To investigate which genes encode the enzymes responsible for residual sucrose hydrolysis activity in strain IMU048 (malΔ mphΔ suc2Δ MAL11), this strain, which grows slowly in a sucrose-based medium (μ = 0.08 h−1, Table 3), was subjected to sequential batch cultivation in SM with sucrose 2% (w/w) as sole carbon source. After 30 transfers (70 days, corresponding to ∼180 generations), single-colony isolates (IMS422, IMS423 and IMS424) were obtained from three independent evolution experiments. In addition to a 3-fold increase in maximum specific growth rate (from 0.08 to 0.25–0.26 h−1, Table 3), the lag phase of these strains was shortened from 4 days to 1 day.
Enzyme-activity assays were conducted to investigate whether the increased growth rate of the evolved strains on sucrose correlated with sucrose hydrolytic activity. Indeed, intracellular sucrose hydrolysis activities in the evolved strains were 2- to 3-fold higher (1.10–1.57 μmol mg protein−1 min−1) than in the non-evolved parental strain IMU048 (0.48 μmol mg protein−1 min−1). No extracellular sucrose-hydrolysing activity was detected in cultures of these strains. In contrast, in cultures of the reference strain CEN.PK113-7D, extracellular sucrose activity was 5.40 μmol mg protein−1 min−1 while intracellular activity was only 0.43 μmol mg protein−1 min−1 (Table 3). Activity of isomaltases, which are known to also hydrolyse sucrose (Deng et al.2014), was measured to investigate a possible contribution of these enzymes to the observed evolved phenotype. Indeed, isomaltase activity was about 1.5–2 times higher in the evolved strains (1.59–2.31 μmol mg protein−1 min−1) compared to IMU048 (1.05 μmol mg protein−1 min−1) (Table 3). In CEN.PK113-7D, isomaltase activity was only 0.13 μmol mg protein−1 min−1 (Table 3). Since isomaltases cannot hydrolyse maltose (Deng et al.2014), it was checked if the evolved strains could consume this sugar. After 2 months of incubation in SM with maltose, no growth was observed.
Quantitative real-time PCR was performed to investigate if the differences in sucrose and isomaltose hydrolysis observed among reference, unevolved and sucrose-evolved strains could be explained by differences in gene expression. Expression of IMA2, IMA3 and IMA4 was analysed with a single primer pair since the sequences of these genes are highly similar. No difference in expression ratio was observed for the pool consisting of IMA2, IMA3 and IMA4 transcripts among the strains tested (Fig. 3C). In contrast, IMA1 and IMA5 expression was significantly higher in IMU048 (malΔ mphΔ suc2Δ MAL11) than in CEN.PK113-7D. Expression of these genes in the evolved strains was even 3- to 5-fold higher than in the unevolved strain IMU48 (Fig. 3A and B). In all analysed strains, the relative expression level of IMA5 was approximately 5–10 times higher than that of IMA1 (Fig. 3A and B).
Figure 3.
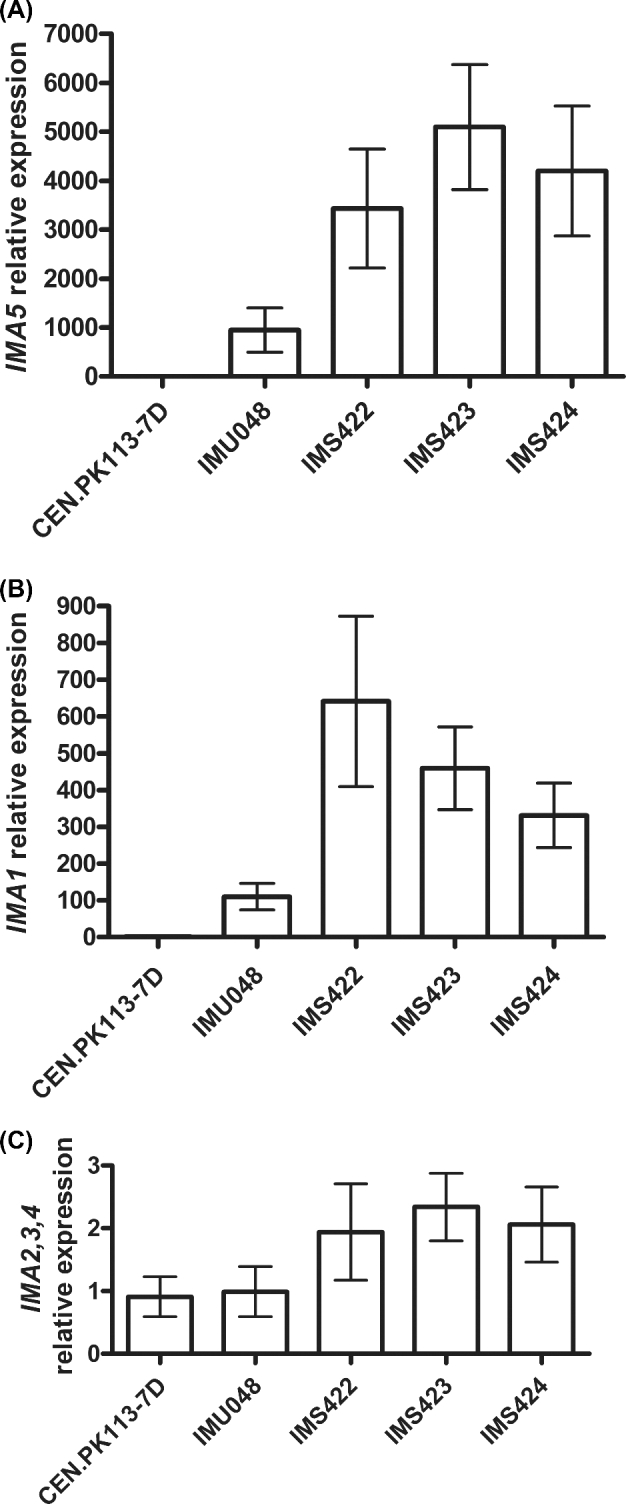
Relative expression of IMA1-5 in S. cerevisiae strains evolved for faster growth on SM with sucrose as the sole carbon source. Relative expression of IMA5(A); IMA1(B); and IMA2, IMA3 and IMA4(C). Expression of IMA2, IMA3 and IMA4 was assayed by a single primer pair since these genes are highly similar in nucleotide sequence. CEN.PK113-7D: reference strain (IMAx MALxx MPHx SUC2); IMU048: unevolved parental strain (genotype: malΔ mphΔ suc2Δ MAL11); and three independently evolved strains (IMS422, IMA423 and IMS424). CEN.PK113-7D was used as calibrator sample (expression set to 1). Averages and standard errors were obtained from independent duplicate biological experiments and three technical replicates. Cells were harvested at mid-exponential phase (OD660nm = 3) from shake-flask cultures on SM with sucrose as sole carbon source.
Deletion of IMA genes is required and sufficient to completely eliminate sucrose hydrolysis
Based on the results shown above, we tested whether deletion of the IMA genes would be sufficient to abolish sucrose consumption in the evolved strains (IMS422, IMS423 and IMS424) and in an unevolved strain (IMZ571; malΔ mphΔ suc2Δ MAL11-LEU2 cas9-URA3) (Table 1). All six copies of IMA genes present in strains IMZ571 and IMS422-424, all derived from strain CEN.PK102-3A (Table 1) (de Kok et al.2011; Nijkamp et al.2012), were deleted in a single transformation step using only two gRNAs cloned in a single plasmid. One of these gRNAs targeted IMA5 while the other simultaneously targeted all other IMA genes. None of the resulting strains (named IMK716, IMK717, IMK718 and IMK700, respectively) grew in SM with sucrose as sole carbon source after 10 days of incubation (Table 4) or after a further 2 months of incubation. Both the sucrose and isomaltose hydrolysis activities in the resulting strains were below the detection limit (Table 4).
Table 4.
Maximum specific growth rates of S. cerevisiae strains grown in shake flasks containing SM (initial pH 6.0) with 20 g L−1 sucrose as sole carbon source. For enzymatic activity determination, strains were grown in SM with 20 g L−1 ethanol as sole carbon source (sucrose was added besides ethanol to the culture of strains IMK716-18 as an inducer). Averages and mean deviations were obtained from duplicate experiments.
| Strain | Relevant genotype | Growth on sucrose (h−1) | Intracellular sucrose hydrolysis (μmol mg protein−1 min−1) | Intracellular isomaltose hydrolysis (μmol mg protein−1 min−1) |
|---|---|---|---|---|
| IMK716 | IMU048 evolved #1 cas9 imaΔ | No growtha | B.D. | B.D. |
| IMK717 | IMU048 evolved #2 cas9 imaΔ | No growtha | B.D. | B.D. |
| IMK718 | IMU048 evolved #3 cas9 imaΔ | No growtha | B.D. | B.D. |
| IMZ620 | malΔ mphΔ suc2Δ MAL11 imaΔ IMA1 | 0.19 ± 0.01 | 2.38 ± 0.03 | 4.60 ± 0.54 |
| IMZ621 | malΔ mphΔ suc2Δ MAL11 imaΔ IMA2 | 0.19 ± 0.01 | 2.88 ± 0.28 | 1.95 ± 0.03 |
| IMZ622 | malΔ mphΔ suc2Δ MAL11 imaΔ IMA3,4 | 0.17 ± 0.01 | 0.92 ± 0.03 | 0.64 ± 0.14 |
| IMZ623 | malΔ mphΔ suc2Δ MAL11 imaΔ IMA5 | 0.04 ± 0.01 | 0.17 ± 0.03 | 6.11 ± 0.18 |
aIncubation period: 10 days.
B.D.: Below detection limit, i.e. <0.03 μmol mg protein−1 min−1.
IMA1 and MAL23-C copy number increased in evolved strains
Sanger sequencing of the IMA genes including their promoter regions did not reveal any mutations (IMA genes from the evolved strains in comparison to that of the parental). Whole genome sequencing of the parental strain IMU048 revealed that, upon sequential cycle of deletions using loxP sites, the strain became aneuploid for CHRIII and CHRVII. Regarding CHRIII, the parental strain gained an extra copy after deletions using loxP sites, truncating the majority of the left arm. Sequencing of three independently evolved isolates indicated a further increase in copy number of the right arm of CHRIII (Fig. 4). Relative to the left arm of the same chromosome (until approximately position 90 000), the region between positions 90 000 and 150 000, which includes the centromere, showed an approximately 2-fold increase in read depth. The right-arm distal end (beyond position 150 000) even showed a 3-fold higher increase in read depth in all three evolved strains relative to the distal end of the left arm (Fig. 4). Retrotransposons (e.g. YCLWΔ15 and YCRCΔ6) located near positions 90 000 and 150 000 may have contributed to translocation/duplication events at these loci. The duplicated CHRIII region in the parental strain IMU048 and triplicated in the evolved isolates harboured IMA1 and MAL23-C. Additionally, a region from CHRVII located between retrotransposons (from position 70 700 to 82 000) was found duplicated already in the parental strain and, in the evolved strains, this duplication was lost.
Figure 4.
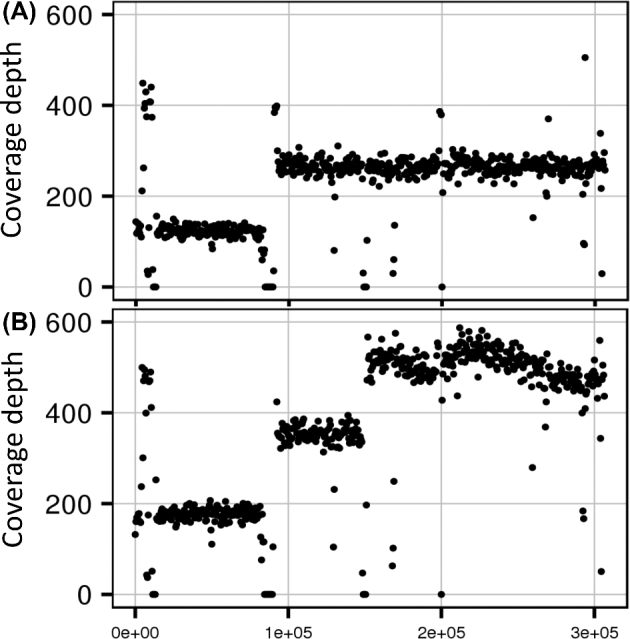
Depth of coverage analysis of chromosome III from unevolved strain IMU048 (A) and the evolved strain IMS422 (B). Sequencing reads from the IMU048 and IMS422 have been mapped onto the CEN.PK113-7D genome sequence (Nijkamp et al.2012). Read count data represent the average coverage of non-overlapping 500 bp window. These data are representative of the other two evolved strains (IMS423 and IMS424).
To investigate the impact of an increased expression of IMA genes during growth on sucrose, they were individually overexpressed in a strain in which all known sucrose-hydrolysing activities were inactivated (IMK743; malΔ mphΔ suc2Δ MAL11 imaΔ) (Table 1). All four constructed strains grew in a sucrose-based medium: strains IMZ620 and IMZ621 (overexpression of IMA1 and IMA2, respectively) both grew at specific growth rate of 0.19 h−1. Accordingly, sucrose and isomaltose hydrolysis activities in these strains were ca. 2 μmol mg protein−1 min−1 (Table 4). Strain IMZ622, which overexpressed IMA3/4, grew at 0.17 h−1, while IMZ623 (IMA5 overexpressed) grew at 0.04 h−1. Sucrose hydrolysis activities in cell extracts of strains IMZ622 and IMZ623 were 0.92 and 0.17 μmol mg protein−1 min−1, respectively (Table 4). No maltose hydrolysis was detected in any of the IMA-overexpressing strains.
DISCUSSION
This study confirms the power of laboratory evolution in identifying suppressor mutations of seemingly essential genes (Liu et al.2015). Prolonged incubation of the malΔ mphΔ suc2Δ MAL11 imaΔ strain for up to 2 months demonstrated that deletion of the IMA genes is both required and sufficient to eliminate growth on sucrose in a strain lacking invertase and maltase activities. Deletion of the disaccharide transporter genes MAL11, MAL21, MAL31, MPH2 and MPH3 similarly resulted in a sucrose-negative phenotype that was stable for over 2 months of incubation in a sucrose-containing medium. The phenotypic stability of the sucrose-negative strains generated in this study makes them a suitable platform for use in diverse applications: (i) screening heterologous disaccharide transporters (e.g. characterisation of plant sucrose transporters in yeast; study maltose/maltotriose transporters relevant for beer brewing (Alves et al.2008)); (ii) screening and characterisation of disaccharide hydrolases and phosphorylases; (iii) metabolic engineering of disaccharide metabolism for improving the production of biofuels and other bio-based chemicals.
Before laboratory evolution for improved growth on sucrose, strain IMU048 (malΔ mphΔ suc2Δ MAL11) already showed a much higher expression of IMA1 and IMA5 compared to CEN.PK113-7D. A main difference between IMU048 and CEN.PK113-7D is the location of sucrose hydrolysis. In the reference strain CEN.PK113-7D, sucrose is predominantly hydrolysed extracellularly (Basso et al.2011), whereas in IMU048 sucrose is imported via Mal11 (proton symporter) and hydrolysed in the cytosol. The presence of intracellular sucrose could possibly activate transcription factors such as the MAL activators MALx3 (Alamäe et al.2003; Weinhandl et al.2014). It is important to clarify that the deletions carried out in this study included maltases and MAL transporters but not the MAL activators (MAL13, MAL23-C and MAL33). MAL23-C (known as MAL2-8c) is constitutively expressed (Gibson et al.1997) but might undergo posttranslational inhibition by chaperones, as has been shown for Mal63, for which the inhibition is relieved when maltose is present (Bali et al.2003; Ran, Bali and Michels 2008). Teste, François and Parrou (2010) showed that the IMA1 and IMA5 promoter regions contain one and three binding sites for MALx3, respectively. The other IMA promoters do not contain binding sites for MALx3 (Teste, François and Parrou 2010). The same authors showed that maltose induces higher expression levels of IMA5 than of IMA1, and has no effect on the expression levels of IMA2, IMA3 and IMA4. The strongly increased transcript levels of IMA1 (110-fold) and IMA5 (950-fold) in IMU048, relative to those in CEN.PK113-7D (Fig. 3A and B), but not of IMA2,3,4 (Fig. 3C), is in line with the hypothesis that sucrose activates the MAL-activators. Extracellular glucose and fructose released in cultures of the strain CEN.PK113-7D by the action of Suc2 might further contribute to the repression of the MAL activators (Hu et al.1995; Horák 2013) in this reference strain.
As mentioned above, the complement of genes belonging to the disaccharide hydrolase gene families varies greatly among Saccharomyces cerevisiae strains. With even closely related strains showing a high degree of genetic variation, it is important to have access to the genome sequence of the immediate parental strain that is used as a platform on which a metabolic engineering strategy will be implemented. The importance of strain-dependent gene contents is exemplified by a literature debate on the number of binding sites for MALx3 in the promoters of IMA1 and IMA5. In contrast to Teste, François and Parrou (2010), Pougach et al. (2014) stated that the IMA5 promoter only has a single binding site for MALx3. In our study, Sanger sequencing of the promoter region of IMA5 confirmed the presence of three MALx3 binding sites previously identified by Teste, François and Parrou (2010): the motif CGGN{9}CGG was found at positions –141 and –543 and motif MGCN{9}MGS at position –516 (Sirenko, Ni and Needleman 1995). This confirmation does not exclude the possibility that the data of Pougach et al. (2014) are correct, since they used a different S. cerevisiae strain (KV5000, which originates from BY4741, an S288c-derived strain) whilst Teste, François and Parrou (2010) used the CEN.PK113-7D strain.
After laboratory evolution, expression of both IMA1 and IMA5 increased by 3-fold compared to the unevolved IMU048. Whole-genome sequencing showed that the strain subjected to evolution (IMU048) already contains a duplication of a large part of CHRIII, which starts before the centromere and extends until the right end of the chromosome (Fig. 4). Likewise, CHRVII of IMU048 contains a duplicated region between retrotransposon sites that was lost after the evolution. Therefore, genes located in this CHRVII region might not contribute to sucrose consumption. Although such aneuploidies often go undetected, this mechanism has also been previously identified in a fraction of deletion mutants generated by the Saccharomyces genome deletion consortium (Winzeler et al.1999; Hughes et al.2000). Several cases reported that the extra chromosome harboured a close homologue of the gene deleted. In our study, deletion of MAL31 and MAL32 may have led to the aneuploidy of CHRIII that harbours the MAL2 locus. Additionally both unevolved (IMU048) and evolved strains (IMS422-4) carried structural variant of the CHRIII. Such rearrangement, especially at chromosome 3, has been extensively reported in the literature as result of recombination between retrotransposons located around positions 90 000 and 150 000 (Mieczkowski, Lemoine and Petes 2006; de Kok et al.2012). In all the three evolved strains obtained in this study, a third copy of the mentioned region was found (Fig. 4). This region not only contains IMA1 but also MAL23-C. If, as discussed above, sucrose indeed activates MAL23-C, IMA1 expression level could be increased by increase in copy number of the gene encoding its activator, MAL23-C. While in this study overexpression of IMA1 was shown to result in faster growth on sucrose, IMA5 was shown to be predominantly an isomaltase and overexpression resulted in only very slow growth and low intracellular sucrose hydrolysis activity (Table 4) (Deng et al.2014). It therefore seems likely that the increased expression of IMA5 is collateral to the upregulation of MAL23-C.
In addition to the scientific value of the sucrose-negative strain platform, this study demonstrates how a single CRISPR targeting sequence can be used to simultaneously delete multiple genes in S. cerevisiae. This strategy allows the deletion and/or modification of entire gene families in a single transformation event.
Supplementary Material
Acknowledgments
We thank Stefan de Kok for the construction of strain IMK291 and Harmen M. van Rossum for his advice on CRISPR/Cas9 genome editing.
SUPPLEMENTARY DATA
Supplementary data are available at FEMSYR online.
FUNDING
Wesley Leoricy Marques, Robert Mans, Jack T. Pronk and Antonius J. A. van Maris were supported by the BE-Basic R&D Program, which was granted an FES subsidy from the Dutch Ministry of Economic Affairs, Agriculture and Innovation (EL&I). In addition, Wesley Leoricy Marques was supported by the Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP, São Paulo, Brazil), grant numbers 2012/05548-1 and 2012/16630-0. This work was carried out as part of a Dual Degree PhD project under the agreement between UNICAMP and Delft University of Technology. Rosa Lorizolla Cordeiro was supported by FAPESP, grant number 2014/07962-5.
Conflict of interest. None declared.
REFERENCES
- Alamäe T, Pärn P, Viigand K et al. . Regulation of the Hansenula polymorpha maltase gene promoter in H. polymorpha and Saccharomyces cerevisiae. FEMS Yeast Res 2003;4:165–73 [DOI] [PubMed] [Google Scholar]
- Alves SL, Herberts RA, Hollatz C et al. . Molecular analysis of maltotriose active transport and fermentation by Saccharomyces cerevisiae reveals a determinant role for the AGT1 permease. Appl Environ Microb 2008;74:1494–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badotti F, Dário MG, Alves SL et al. . Switching the mode of sucrose utilization by Saccharomyces cerevisiae. Microb Cell Fact 2008;7:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bali M, Zhang B, Morano KA et al. . The Hsp90 molecular chaperone complex regulates maltose induction and stability of the Saccharomyces MAL gene transcription activator Mal63p. J Biol Chem 2003;278:47441–8 [DOI] [PubMed] [Google Scholar]
- Basso TO, de Kok S, Dario M et al. . Engineering topology and kinetics of sucrose metabolism in Saccharomyces cerevisiae for improved ethanol yield. Metab Eng 2011;13:694–703 [DOI] [PubMed] [Google Scholar]
- Boeke JD, La Croute F, Fink GR. A positive selection for mutants lacking orotidine-5΄-phosphate decarboxylase activity in yeast: 5-fluoro-orotic acid resistance. Mol Gen Genet 1984;197:345–6 [DOI] [PubMed] [Google Scholar]
- Brown CA, Murray AW, Verstrepen KJ. Rapid expansion and functional divergence of subtelomeric gene families in yeasts. Curr Biol 2010;20:895–903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlson M, Botstein D. Two differentially regulated mRNAs with different 5΄ ends encode secreted and intracellular forms of yeast invertase. Cell 1982;28:145–54 [DOI] [PubMed] [Google Scholar]
- Carlson M, Botstein D. Organization of the SUC gene family in Saccharomyces. Mol Cell Biol 1983;3:351–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlson M, Osmond BC, Botstein D. Mutants of yeast defective in sucrose utilization. Genetics 1981;98:25–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Kok S, Nijkamp JF, Oud B et al. . Laboratory evolution of new lactate transporter genes in a jen1Δ; mutant of Saccharomyces cerevisiae and their identification as ADY2 alleles by whole-genome resequencing and transcriptome analysis. FEMS Yeast Res 2012;12:359–74 [DOI] [PubMed] [Google Scholar]
- de Kok S, Yilmaz D, Suir E et al. . Increasing free-energy (ATP) conservation in maltose-grown Saccharomyces cerevisiae by expression of a heterologous maltose phosphorylase. Metab Eng 2011;13:518–26 [DOI] [PubMed] [Google Scholar]
- Della-Bianca BE, Basso TO, Stambuk BU et al. . What do we know about the yeast strains from the Brazilian fuel ethanol industry? Appl Microbiol Biot 2013;97:979–91 [DOI] [PubMed] [Google Scholar]
- Della-Bianca BE, de Hulster E, Pronk JT et al. . Physiology of the fuel ethanol strain Saccharomyces cerevisiae PE-2 at low pH indicates a context-dependent performance relevant for industrial applications. FEMS Yeast Res 2014;14:1196–205 [DOI] [PubMed] [Google Scholar]
- Deng X, Petitjean M, Teste M-A et al. . Similarities and differences in the biochemical and enzymological properties of the four isomaltases from Saccharomyces cerevisiae. FEBS Open Bio 2014;4:200–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiCarlo JE, Norville JE, Mali P et al. . Genome engineering in Saccharomyces cerevisiae using CRISPR-Cas systems. Nucleic Acids Res 2013;41:4336–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Entian KD, Kötter P. 25 yeast genetic strain and plasmid collections. Methods Microbiol 2007;36:629–66 [Google Scholar]
- Gibson AW, Wojciechowicz LA, Danzi SE et al. . Constitutive mutations of the Saccharomyces cerevisiae mal-activator genes MAL23, MAL43, MAL63, and MAL64. Genetics 1997;146:1287–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gietz BRD, Woods RA. Transformation of yeast by lithium acetate/single-stranded carrier DNA/polyethylene glycol method. Methods Enzymol 2002;350:87–96 [DOI] [PubMed] [Google Scholar]
- Gombert AK, van Maris AJ. Improving conversion yield of fermentable sugars into fuel ethanol in 1st generation yeast-based production processes. Curr Opin Biotechnol 2015;33:81–6 [DOI] [PubMed] [Google Scholar]
- Gueldener U, Heinisch J, Koehler GJ et al. . A second set of loxP marker cassettes for Cre-mediated multiple gene knockouts in budding yeast. Nucleic Acids Res 2002;30:e23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawthorne DC. The genetics of alpha-methyl-glucoside fermentation in saccharomyces. Heredity (Edinb) 1958;12:273–84 [Google Scholar]
- Horák J. Regulations of sugar transporters: insights from yeast. Curr Genet 2013;59:1–31 [DOI] [PubMed] [Google Scholar]
- Hu Z, Nehlin JO, Ronne H et al. . MIG1-dependent and MIG1-independent glucose regulation of MAL gene expression in Saccharomyces cerevisiae. Curr Genet 1995;28:258–66 [DOI] [PubMed] [Google Scholar]
- Hughes TR, Roberts CJ, Dai H et al. . Widespread aneuploidy revealed by DNA microarray expression profiling. Nat Genet 2000;25:333–7 [DOI] [PubMed] [Google Scholar]
- Krieble VK. Activities and the hydrolysis of sucrose with concentrated acids. J Am Chem Soc 1935;57:15–9 [Google Scholar]
- Kuijpers NG, Solis-Escalante D, Bosman L et al. . A versatile, efficient strategy for assembly of multi-fragment expression vectors in Saccharomyces cerevisiae using 60 bp synthetic recombination sequences. Microb Cell Fact 2013;12:47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagunas R. Sugar transport in Saccharomyces cerevisiae. FEMS Microbiol Rev 1993;10:229–42 [DOI] [PubMed] [Google Scholar]
- Liu G, Yong MYJ, Yurieva M et al. . Gene essentiality is a quantitative property linked to cellular evolvability. Cell 2015;163:1388–99 [DOI] [PubMed] [Google Scholar]
- Lowry OH, Rosebrough NJ, Farr AL et al. . Protein measurement with the Folin phenol reagent. J Biol Chem 1951;193:265–75 [PubMed] [Google Scholar]
- Maiorella BL, Blanch HW, Wilke CR. Economic evaluation of alternative ethanol fermentation processes. Biotechnol Bioeng 1984;26:1003–25 [DOI] [PubMed] [Google Scholar]
- Mans R, van Rossum HM, Wijsman M et al. . CRISPR/Cas9: a molecular Swiss army knife for simultaneous introduction of multiple genetic modifications in Saccharomyces cerevisiae. FEMS Yeast Res 2015;15, DOI: 10.1093/femsyr/fov004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marques WL, Raghavendran V, Stambuk BU et al. . Sucrose and Saccharomyces cerevisiae: a relationship most sweet. FEMS Yeast Res 2016:16, DOI: 10.1093/femsyr/fov107 [DOI] [PubMed] [Google Scholar]
- Meadows AL, Hawkins KM, Tsegaye Y et al. . Rewriting yeast central carbon metabolism for industrial isoprenoid production. Nature 2016;537:694–7 [DOI] [PubMed] [Google Scholar]
- Mieczkowski PA, Lemoine FJ, Petes TD. Recombination between retrotransposons as a source of chromosome rearrangements in the yeast Saccharomyces cerevisiae. DNA Repair 2006;5:1010–20 [DOI] [PubMed] [Google Scholar]
- Mitchell DA. Note on Rising Food Prices. The World Bank Development Prospects Group, 2008, http://www.bio-based.eu/foodcrops/media/08-07ANoteonRisingFoodPrices.pdf (date last accessed, 10 August 2016) [Google Scholar]
- Mumberg D, Müller R, Funk M. Yeast vectors for the controlled expression of heterologous proteins in different genetic backgrounds. Gene 1995;156:119–22 [DOI] [PubMed] [Google Scholar]
- Naumoff DG. Hierarchical classification of glycoside hydrolases. Biochemistry 2011;76:622–35 [DOI] [PubMed] [Google Scholar]
- Naumoff DG, Naumov GI. Discovery of a novel family of α-glucosidase IMA genes in yeast Saccharomyces cerevisiae. Dokl Biochem Biophys 2010;432:549–51 [DOI] [PubMed] [Google Scholar]
- Nijkamp JF, van den Broek M, Datema E et al. . De novo sequencing, assembly and analysis of the genome of the laboratory strain Saccharomyces cerevisiae CEN.PK113-7D, a model for modern industrial biotechnology. Microb Cell Fact 2012;11:36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfaffl MW, Horgan GW, Dempfle L. Relative expression software tool (REST) for group-wise comparison and statistical analysis of relative expression results in real-time PCR. Nucleic Acids Res 2002;30:e36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piper MDW, Daran-Lapujade P, Bro C et al. . Reproducibility of oligonucleotide microarray transcriptome analyses. An interlaboratory comparison using chemostat cultures of Saccharomyces cerevisiae. J Biol Chem 2002;277:37001–8 [DOI] [PubMed] [Google Scholar]
- Postma E, Verduyn C, Scheffers WA et al. . Enzymic analysis of the crabtree effect in glucose-limited chemostat cultures of Saccharomyces cerevisiae. Appl Environ Microb 1989;55:468–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pougach K, Voet A, Kondrashov FA et al. . Duplication of a promiscuous transcription factor drives the emergence of a new regulatory network. Nat Commun 2014;5:4868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran F, Bali M, Michels CA. Hsp90/Hsp70 chaperone machine regulation of the saccharomyces MAL-activator as determined in vivo using noninducible and constitutive mutant alleles. Genetics 2008;179:331–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt ME, Brown T, Trumpower BL. A rapid and simple method for preparation of RNA from Saccharomyces cerevisiae. Nucleic Acids Res 1990;18:3091–2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sirenko OI, Ni B, Needleman RB. Purification and binding properties of the Mal63p activator of Saccharomyces cerevisiae. Curr Genet 1995;27:509–16 [DOI] [PubMed] [Google Scholar]
- Solis-Escalante D, Kuijpers NG, Bongaerts N et al. . amdSYM, a new dominant recyclable marker cassette for Saccharomyces cerevisiae. FEMS Yeast Res 2013;13:126–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stambuk BU, da Silva MA, Panek AD et al. . Active alpha-glucoside transport in Saccharomyces cerevisiae. FEMS Microbiol Lett 1999;170:105–10 [DOI] [PubMed] [Google Scholar]
- Teste M-A, Duquenne M, François JM et al. . Validation of reference genes for quantitative expression analysis by real-time RT-PCR in Saccharomyces cerevisiae. BMC Mol Biol 2009;10:99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teste M-A, François JM, Parrou J-L. Characterization of a new multigene family encoding isomaltases in the yeast Saccharomyces cerevisiae, the IMA family. J Biol Chem 2010;285:26815–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van den Broek M, Bolat I, Nijkamp JF et al. . Chromosomal copy number variation in Saccharomyces pastorianus is evidence for extensive genome dynamics in industrial lager brewing strains. Appl Environ Microb 2015;81:6253–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verduyn C, Postma E, Scheffers WA et al. . Effect of benzoic acid on metabolic fluxes in yeasts: a continuous-culture study on the regulation of respiration and alcoholic fermentation. Yeast 1992;8:501–17 [DOI] [PubMed] [Google Scholar]
- Voordeckers K, Brown CA, Vanneste K et al. . Reconstruction of ancestral metabolic enzymes reveals molecular mechanisms underlying evolutionary innovation through gene duplication. PLoS Biol 2012;10:e1001446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinhandl K, Winkler M, Glieder A et al. . Carbon source dependent promoters in yeasts. Microb Cell Fact 2014;13:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winge O, Roberts C. The relation between the polymeric genes for maltose raffinose, and sucrose fermentation in yeasts. Cr Trav Lab Carlsb 1952;25:141–71 [PubMed] [Google Scholar]
- Winzeler EA, Shoemaker DD, Astromoff A et al. . Functional characterization of the S. cerevisiae genome by gene deletion and parallel analysis. Science 1999;285:901–906 [DOI] [PubMed] [Google Scholar]
- Wolfenden R, Yuan Y. Rates of spontaneous cleavage of glucose, fructose, sucrose, and trehalose in water, and the catalytic proficiencies of invertase and trehalase. J Am Chem Soc 2008;130:7548–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto K, Nakayama A, Yamamoto Y et al. . Val216 decides the substrate specificity of alpha-glucosidase in Saccharomyces cerevisiae. Eur J Biochem 2004;271:3414–20 [DOI] [PubMed] [Google Scholar]
- Zelle RM, Trueheart J, Harrison JC et al. . Phosphoenolpyruvate carboxykinase as the sole anaplerotic enzyme in Saccharomyces cerevisiae. Appl Environ Microb 2010;76:5383–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Grof CP, Patrick JW. Proof of concept for a novel functional screening system for plant sucrose effluxers. J Biol Methods 2014;1:1–6 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.