

Highlights
-
•
Two novel mutations in PNPLA2 gene have been identified in a neutral lipid storage disease with myopathy (NLSDM) female patient.
-
•
The mutations are located in exon 5 of PNPLA2 and abrogate lipase function.
-
•
The patient showed late onset skeletal muscle myopathy and mild cardiac impairment.
-
•
Clinical cardiac phenotype is milder in NLSDM female patients, beyond genetics.
Keywords: Neutral lipid storage disease with myopathy, Cardiomyopathy, PNPLA2, Triglyceride lipase, Lipid metabolism, Lipid droplets
Abstract
Neutral lipid storage disease with myopathy (NLSDM) presents with skeletal muscle myopathy and severe dilated cardiomyopathy in nearly 40% of cases. NLSDM is caused by mutations in the PNPLA2 gene, which encodes the adipose triglyceride lipase (ATGL). Here we report clinical and genetic findings of a patient carrying two novel PNPLA2 mutations (c.696+4A>G and c.553_565delGTCCCCCTTCTCG). She presented at age 39 with right upper limb abduction weakness slowly progressing over the years with asymmetric involvement of proximal upper and lower limb muscles. Cardiological evaluation through ECG and heart echo scan was normal until the age 53, when mild left ventricular diastolic dysfunction was detected. Molecular analysis revealed that only one type of PNPLA2 transcript, with exon 5 skipping, was expressed in patient cells. Such aberrant mRNA causes the production of a shorter ATGL protein, lacking part of the catalytic domain. This is an intriguing case, displaying severe PNPLA2 mutations with clinical presentation characterized by slight cardiac impairment and full expression of severe asymmetric myopathy.
1. Introduction
Neutral lipid storage disease with myopathy (NLSDM; MIM 610717) is an autosomal recessive disorder characterized by abnormal accumulation of triacylglycerols (TAGs) in cytoplasmic lipid droplets (LDs) in most tissues, including muscle, heart, liver and peripheral blood. A rapid laboratory diagnosis of NLSDM can be easily performed through the detection of lipid vacuoles in peripheral blood leucocytes, also known as Jordans' anomaly [1], [2]. To our best knowledge, forty-six NLSDM patients have been clinically and genetically reported [3], [4], [5], [6], [7], [8], [9], [10], [11], [12], [13]. Clinical symptoms of NLSDM are characterized by progressive myopathy, cardiomyopathy, hepatomegaly, diabetes, chronic pancreatitis, short stature and by high serum creatine kinase levels [14]. The degree of clinical manifestations appears highly variable: from minimal symptoms to a more severe condition, causing physical disability and premature death due to dilated cardiomyopathy. However, since NLSDM is a rare metabolic condition, the pathophysiology of the disease is largely unclear and phenotype–genotype correlations remain incomplete [15]. NLSDM is caused by mutations in PNPLA2 coding for the adipose triglyceride lipase (ATGL), a member of the patatin-like phospholipase domain-containing proteins [3]. This lipase is a lipid droplet-associated protein that catalyses the first step in the hydrolysis of TAGs, stored within LDs [16]. The human ATGL protein consists of 504 amino acids comprising the patatin domain (amino acids 10–178) with catalytic residues S47 and D166, at the N-terminus, and a hydrophobic lipid binding domain at position 315–360 towards the C-terminus [3], [16].Thirty-five PNPLA2 mutations variably affecting protein function or production have been identified so far in NLSDM patients. Many mutations are expected to generate either null alleles or truncated ATGL proteins with the catalytic domain partially lost, all resulting in dramatic impairment of LD metabolism. The outcome in most patients carrying these mutations has been reported as severe. On the contrary, recent studies showed that missense mutations, resulting in an ATGL protein with residual lipolytic activity, may be associated with slowly progressing myopathy and sparing of myocardial muscle [9], [12], [17]. Here we describe clinical and genetic findings in a woman harbouring two novel mutations in PNPLA2. Although these mutations completely abolish lipase activity, our patient showed slowly progressive skeletal muscle weakness with late presentation, in association with mild cardiac impairment.
2. Case presentation
The proband is a 54-year-old woman, presenting at age 39 with right upper limb proximal weakness, slowly progressing over the years. Her previous medical history had been unremarkable. At age 47 she noticed muscle weakness in lower limbs. Creatine kinase was 579 U/L (normal range < 190 U/L). Urine organic acids, plasma carnitine and acyl-carnitine profiles were normal. Electromyography performed elsewhere showed a mixed pattern with predominant neurogenic signs and fibrillations in upper limb muscles; nerve conduction studies were normal. The patient, when first admitted in our outpatient clinic at age 49, reported moderate disability due to upper limb muscle weakness. No bulbar symptoms, muscle cramps or pain was reported. Neurological examination showed marked right upper limb weakness with abduction limited to 30 degrees and flexion to 45 degrees, whereas only mild weakness against resistance was observed on the left side. In addition, severe elbow flexion and moderate elbow and finger extension weakness were noticed on the right side. No axial or lower limb weakness was found (Fig. 1a and b). Mild hypertrophy of calves was observed. Tendon retractions or scapular winging was absent. Cranial nerves were normal. Pyramidal or cerebellar signs were absent; deep tendon reflexes were reduced in lower limbs, while triceps and biceps reflexes were inelicitable; superficial and deep sensibility were normal. Muscle MRI, performed at age 49, showed predominant posterior thigh and leg compartment involvement (Fig. 1c and d), as already reported [7].
Fig. 1.
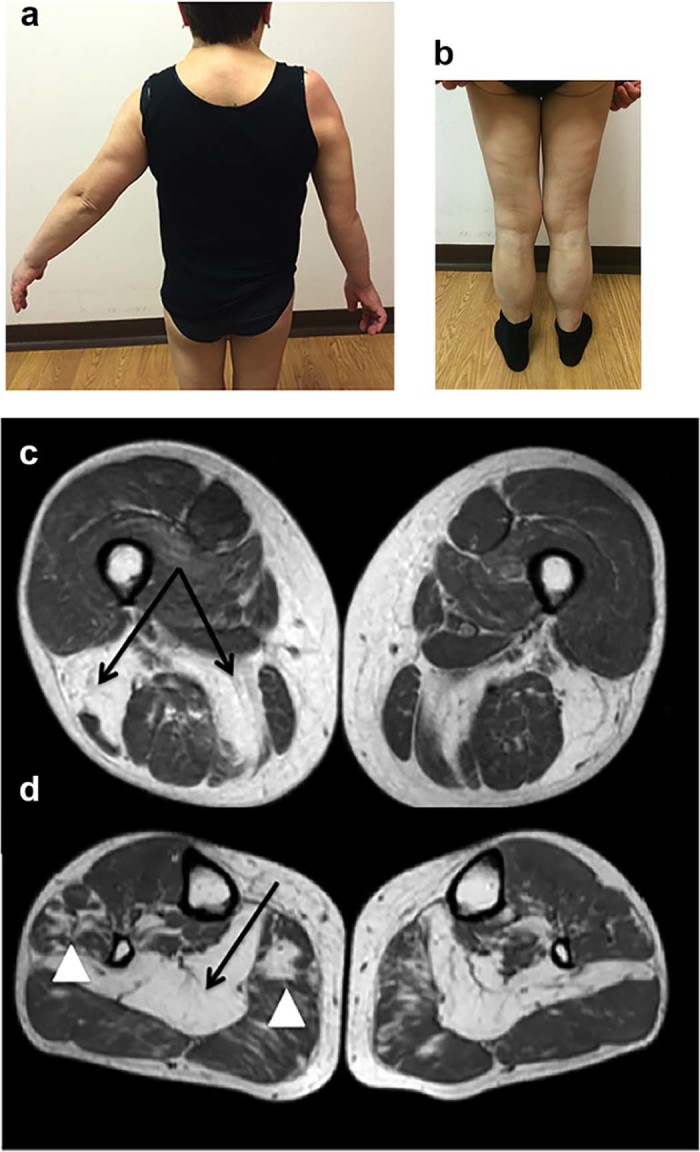
NLSDM patient's clinical and imaging features. (a) Upper limb abduction weakness, predominant on the right side and (b) calves hypertrophy. Thigh (c) and leg (d) muscle MRI: marked T1w changes with almost complete fatty infiltration of biceps femoris and semimembranosus (c) and soleus (d); lower severity involvement has been observed in medial gastrocnemius and peroneal longus (arrowheads) (d).
A quadriceps muscle biopsy, performed at the same age, revealed myogenic features with vacuoles mainly distributed in hypotrophic type I fibres; staining with Oil Red O showed lipid accumulation (Fig. 2d). No degeneration or regeneration was observed (Fig. 2c). Electron microscopy confirmed the excessive accumulation of lipid droplets without signs of mitochondrial alteration (Fig. 2e and f). Jordans' anomaly was found in the patient leucocytes at age 51 (Fig. 2a). Cultured skin fibroblasts, obtained from a patient dermal biopsy, also revealed an abnormal accumulation of neutral lipids into LDs (Fig. 2b).
Fig. 2.

Histochemical characterization of NLSDM patient. (a) Detection of Jordans' bodies in granulocytes, stained with May-Grünland Giemsa (MGG). (b) Phase contrast image of cultured fibroblasts from the patient reveals increase of lipid droplet storage inside the cells. Consecutive cryosections of the patient muscle biopsy, stained with Gomori trichrome (c) and Oil-Red-O (d), show microvacuoles and abnormal accumulation of lipids. (e,f) Electron microscopy reveals massive line-up of lipid droplets without signs of mitochondrial alteration.
The neurological follow-up showed moderate worsening of the condition and development of moderate muscle weakness of lower limbs, mainly in proximal muscles. In addition, neck flexor muscles were also impaired. The patient remained fully ambulant during the follow-up period.
Cardiological evaluation through ECG and heart echo scan were normal until the age 53, when mild left ventricular diastolic dysfunction was detected, without any progression over the following 12 months. At age 54, heart MRI and standard spirometry were normal while a cardiopulmonary exercise test showed an exercise limitation (peak VO2 = 66% predicted value). All together the cardiopulmonary exercise test data were suggestive of peripheral muscle deconditioning with normal cardiac function.
Family history was negative for neuromuscular disorders. Parents were not consanguineous. In our patient, molecular analysis of PNPLA2 detected the two following novel heterozygous mutations: c.696+4A>G (allele 1) and c.553_565delGTCCCCCTTCTCG (allele 2) (Fig. 3a). The first mutation, inherited from the father, is localized in intron 5 and predicts in frame skipping of exon 5. The aberrant mRNA loses part of the sequence coding for the catalytic site of ATGL protein (Fig. 3b and c). The deletion extends from Arg163 to Leu232, including the Asp166 residue, which is part of the catalytic dyad (Fig. 3e). Hence, the c.696+4A>G mutation, disrupting the ATGL catalytic site, causes total loss of its enzymatic function, as previously shown by functional studies [9], [12], [17]. The c.553_565delGTCCCCCTTCTCG mutation is localized inside exon 5; extensive RT-PCR analysis showed no PNPLA2 mRNA production from this mutant allele (Fig. 3c). This variant was carried also by the 81-year-old mother, who, at age 77, was normal on neurological examination and had a normal muscle biopsy. Both PNPLA2 mutations were not observed in >200 control alleles and were submitted to GenBank (Accession numbers: KU139128 for c.696+4A>G and KU139129 for c.553_565delGTCCCCCTTCTCG).
Fig. 3.

Molecular characterization of PNPLA2 mutations. (a) Electropherograms of PNPLA2 exon 5 showing a splice-site mutation (c.696+4A>G) in the first allele and a deletion of 13 bp (c.553_565delGTCCCCCTTCTCG) in the second allele of the patient. (b) Electropherogram of RT-PCR products reveals the skipping of exon 5 in the patient. (c) RT-PCR performed with primers encompassing exons 3, 4, 5 and 6 shows absence of wild-type product (551 bp) in the patient and the presence of a single product of 341 bp, resulting from c.696+4A>G mutation. Patient's mother presents a single wild-type allele and father carries the c.696+4A>G mutation in heterozygous status. (d) Western blot analysis of fibroblasts' extracts shows the wild-type product (56 kDa) in control cells and the mutant protein (48 kDa) in NLSDM fibroblasts. GAPDH has been used as loading control for protein normalization (36 kDa). (e) Schematic representation of the PNPLA2 gene, mRNA and protein in the control subject and in the Italian patient. The diagram of normal PNPLA2 is reported on the left. ATGL protein wild type contains a patatin domain (red) with the catalytic dyad (S47 and D166) and a hydrophobic domain (green). PNPLA2 mutations identified in the NLSDM subject determine: in the first allele (c.696+4A>G) the skipping of exon 5 and the production of a mutant protein lacking part of catalytic site (D166 amino acid); in the second allele (c.553_565delGTCCCCCTTCTCG) the lack of mRNA expression and protein production.
To verify whether patient allele 1, showing the in frame skipping of exon 5, was expressed into patient cells, we performed western blotting analysis of ATGL using total protein extracts from patient fibroblasts. As shown in Fig. 3d, a mutated ATGL protein with lower molecular weight was detected in patient fibroblasts in comparison with control fibroblasts.
Informed consent was obtained from the study participants. Patient investigations were conducted in accordance with protocols approved by the institutional review boards of the Carlo Besta Neurological Institute and the Catholic University of the Sacred Heart.
3. Discussion
The main clinical feature of NLSDM is skeletal muscle myopathy, which is present in 100% of patients. Muscle weakness usually presents in early adult life, between 20 and 30 years. A later onset of the muscle phenotype has been observed in our patient, as well as in some previously reported cases [7], [8], [9], [12], [18]. In these patients, mainly PNPLA2 missense mutations, which partially save lipase activity, have been identified. On the contrary, in our patient with typical muscle phenotype characterized by predominant proximal upper limb muscle weakness, two severe mutations that completely abrogate protein function have been detected. The first mutation causes the skipping of exon 5 and the production of a mutated protein that loses part of the catalytic site, thus abrogating ATGL lipase activity; the second mutation determines complete lack of mRNA expression and protein production (Fig. 3e). A homozygous PNPLA2 mutation affecting the invariant G of the donor splice-site of intron 5 (c.696+1G>C) has previously been described in a Japanese male patient [19]. This mutation caused the production of two aberrant mRNAs: one retaining 93 bp of intron 5 and resulting in a new reading frame shift and a stop at position 162 (p.Val233LeufsX162); the other consisting of a PNPLA2 sequence lacking 210 bp due to in frame skipping of exon 5 (p.Arg163_Leu232del), exactly as the allele 1 of our female patient (with c.696+4A>G mutation). Despite the molecular similarity, some important clinical differences emerged between the Japanese and the Italian patients, concerning, in particular, their cardiac involvement. These differences might be due to homozygous versus heterozygous condition or to modifier genes and epigenetic factors possibly involved in such variable phenotypic expression. In the Japanese patient muscle weakness presented earlier than in our patient and was associated with severe heart involvement presenting at age 33 and requiring heart transplantation. On the contrary, our female patient showed only slight cardiac involvement at age 53.
Although the presence of PNPLA2 severe mutations is similar in men and women, cardiac damage was reported in almost 20% of NLSDM female patients (4 out of 20) and in 55% of male patients (15 out of 27) [13], [20]. Indeed, considering as severe the mutations (identified so far in NLSDM patients) that cause lack of ATGL protein production or expression of truncated proteins with catalytic site only partially conserved, we note that they represent 25% of total PNPLA2 mutations in female patients and 29% in male patients. The latter observation suggests that gender modulates clinical cardiac phenotype in NLSDM, also beyond the severity of mutations in PNPLA2. To this regard, it is known that oestrogens regulate the expression of peroxisome proliferator-activated receptor (PPARα,β/δ,γ) family members. PPARs control mitochondrial metabolism and are mainly involved in fatty acid and glucose utilization in heart [21], [22]. Very recently, Higashi et al. reported a distinct cardiac phenotype in two NLSDM siblings carrying the same homozygous PNPLA2 mutation; according to the hypothesis that there is a gender difference on the phenotypic clinical expression in NLSDM, the male died at age of 31 of heart failure, while his sister was still alive (44 years), although presenting hypertrophic cardiomyopathy [20]. Certainly, additional larger clinical studies are warranted to elucidate whether female gender possibly plays a protective role in NLSDM. NLSDM is an ultra-rare disease, its pathophysiology is largely unclear, phenotype–genotype correlations are incomplete, and a cure is still lacking. To this regard, an international registry for NLSDs, recently established (www.tgcv.org/r/home.htlm), may help to collect worldwide clinical and genetic data and develop common therapeutic protocols.
In conclusion, we describe a 54-year-old NLSDM female patient showing late onset myopathy in association with slight cardiac involvement, although the identified novel mutations completely abrogate PNPLA2 protein function. Our data expand the allelic spectrum of PNPLA2 mutations, providing further evidence for genetic and clinical NLSDM heterogeneity.
Funding
This work was supported by grant GGP14066 from Telethon Foundation.
Acknowledgements
The authors gratefully acknowledge EuroBioBank and the Telethon Network of Genetic Biobanks (GTB12001F) for providing biological samples.
References
- 1.Jordans G.H. The familial occurrence of fat containing vacuoles in the leukocytes diagnosed in two brothers suffering from dystrophia musculorum progressiva (ERB) Acta Med Scand. 1953;145:419–423. doi: 10.1111/j.0954-6820.1953.tb07038.x. [DOI] [PubMed] [Google Scholar]
- 2.Tavian D., Colombo R. Improved cytochemical method for detecting Jordans' bodies in neutral-lipid storage diseases. J Clin Pathol. 2007;60:956–958. doi: 10.1136/jcp.2006.044917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fischer J., Lefevre C., Morava E. The gene encoding adipose triglyceride lipase (PNPLA2) is mutated in neutral lipid storage disease with myopathy. Nat Genet. 2007;39:28–30. doi: 10.1038/ng1951. [DOI] [PubMed] [Google Scholar]
- 4.Kobayashi K., Inoguchi T., Maeda Y. The lack of the C-terminal domain of adipose triglyceride lipase causes neutral lipid storage disease through impaired interactions with lipid droplets. J Clin Endocrinol Metab. 2008;93:2877–2884. doi: 10.1210/jc.2007-2247. [DOI] [PubMed] [Google Scholar]
- 5.Ohkuma A., Nonaka I., Malicdan M.C. Distal lipid storage myopathy due to PNPLA2 mutation. Neuromuscul Disord. 2008;18:671–674. doi: 10.1016/j.nmd.2008.06.382. [DOI] [PubMed] [Google Scholar]
- 6.Campagna F., Nanni L., Quagliarini F. Novel mutations in the adipose triglyceride lipase gene causing neutral lipid storage disease with myopathy. Biochem Biophys Res Commun. 2008;377:843–846. doi: 10.1016/j.bbrc.2008.10.081. [DOI] [PubMed] [Google Scholar]
- 7.Reilich P., Horvath R., Krause S. The phenotypic spectrum of neutral lipid storage myopathy due to mutations in the PNPLA2 gene. J Neurol. 2011;258:1987–1997. doi: 10.1007/s00415-011-6055-4. [DOI] [PubMed] [Google Scholar]
- 8.Lin P., Li W., Wen B. Novel PNPLA2 gene mutations in Chinese Han patients causing neutral lipid storage disease with myopathy. J Hum Genet. 2012;57:679–681. doi: 10.1038/jhg.2012.84. [DOI] [PubMed] [Google Scholar]
- 9.Tavian D., Missaglia S., Redaelli C. Contribution of novel ATGL missense mutations to the clinical phenotype of NLSD-M: a strikingly low amount of lipase activity may preserve cardiac function. Hum Mol Genet. 2012;21:5318–5328. doi: 10.1093/hmg/dds388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tavian D., Missaglia S., DiMauro S. A late-onset case of neutral lipid storage disease with myopathy, dropped head syndrome, and peripheral nerve involvement. J Genet Syndr Gene Ther. 2013;4:198. [Google Scholar]
- 11.Laforêt P., Ørngreen M., Preisler N., Andersen G., Vissing J. Blocked muscle fat oxidation during exercise in neutral lipid storage disease. Arch Neurol. 2012;69:530–533. doi: 10.1001/archneurol.2011.631. [DOI] [PubMed] [Google Scholar]
- 12.Missaglia S., Tasca E., Angelini C., Moro L., Tavian D. Novel missense mutations in PNPLA2 causing late onset and clinical heterogeneity of neutral lipid storage disease with myopathy in three siblings. Mol Genet Metab. 2015;115:110–117. doi: 10.1016/j.ymgme.2015.05.001. [DOI] [PubMed] [Google Scholar]
- 13.Pasanisi M.B., Missaglia S., Cassandrini D. Severe cardiomyopathy in a young patient with complete deficiency of adipose triglyceride lipase due to a novel mutation in PNPLA2 gene. Int J Cardiol. 2016;207:165–167. doi: 10.1016/j.ijcard.2016.01.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kaneko K., Kuroda H., Izumi R. A novel mutation in PNPLA2 causes neutral lipid storage with myopathy and triglyceride deposit cardiomyovasculopathy: a case report and literature review. Neuromuscl Disord. 2014;24:634–641. doi: 10.1016/j.nmd.2014.04.001. [DOI] [PubMed] [Google Scholar]
- 15.Perrin L., Féasson L., Furby A. PNPLA2 mutation: a paediatric case with early onset but indolent course. Neuromuscl Disord. 2013;23:986–991. doi: 10.1016/j.nmd.2013.08.008. [DOI] [PubMed] [Google Scholar]
- 16.Schweiger M., Lass A., Zimmermann R., Eichmann T.O., Zechner R. Neutral lipid storage disease: genetic disorders caused by mutations in adipose triglyceride lipase/PNPLA2 or CGI-58/ ABHD5. Am J Physiol Endocrinol Metab. 2009;297:289–296. doi: 10.1152/ajpendo.00099.2009. [DOI] [PubMed] [Google Scholar]
- 17.Pennisi E.M., Missaglia S., Dimauro S., Bernardi C., Akman H.O., Tavian D. A myopathy with unusual features caused by PNPLA2 gene mutations. Muscle Nerve. 2015;51:609–613. doi: 10.1002/mus.24477. [DOI] [PubMed] [Google Scholar]
- 18.Ash D.B., Papadimitriou D., Hays A.P., Dimauro S., Hirano M. A novel mutation in PNPLA2 leading to neutral lipid storage disease with myopathy. Arch Neurol. 2012;69:1190–1192. doi: 10.1001/archneurol.2011.2600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hirano K., Tanaka T., Ikeda Y. Genetic mutations in adipose triglyceride lipase and myocardial up-regulation of peroxisome proliferated activated receptor-γ in patients with triglyceride deposit cardiomyovasculopathy. Biochem Biophys Res Commun. 2014;10:574–579. doi: 10.1016/j.bbrc.2013.12.003. [DOI] [PubMed] [Google Scholar]
- 20.Higashi M., Hirano K., Kobayashi K., Ikeda Y., Issiki A., Otsuka T. Distinct cardiac phenotype between two homozygotes born in a village with accumulation of a genetic deficiency of adipose triglyceride lipase. Int J Cardiol. 2015;192:30–32. doi: 10.1016/j.ijcard.2015.05.004. [DOI] [PubMed] [Google Scholar]
- 21.Regitz-Zagrosek V. Therapeutic implications of the gender-specific aspects of cardiovascular disease. Nat Rev Drug Discov. 2006;5:425–438. doi: 10.1038/nrd2032. [DOI] [PubMed] [Google Scholar]
- 22.Finck B.N. The PPAR regulatory system in cardiac physiology and disease. Cardiovasc Res. 2007;73:269–277. doi: 10.1016/j.cardiores.2006.08.023. [DOI] [PubMed] [Google Scholar]