

Malignant tumors of the central nervous system are the leading cause of cancer death for people under 35 years of age. Glioblastoma multiforme (GBM), the most malignant brain tumor in adults is characterized histologically by pseudopalisading necrosis and abnormal vascular development (Fig. 1). These tumors show infiltration by inflammatory cells and the occurrence of hypoxia (lower than physiological O2 levels) in the pseudopalisading cells surrounding the micronecrotic areas. Microenvironmental hypoxia is not restricted to regions adjacent to the large central necrotic areas of gliomas; in fact, it is present in close proximity to the leading/actively growing edge of GBM, where the tumor is actively expanding radially [1]. The hypoxic cells are active participants in the tumor biology and are refractive to conventional therapies, contributing thus to poor patient survival. Currently, the life expectancy for GBM patients is 1–2 years, stressing the need to better understand the signaling mechanisms that underlie their aggressive growth so that new therapies can be designed.
Fig. 1.
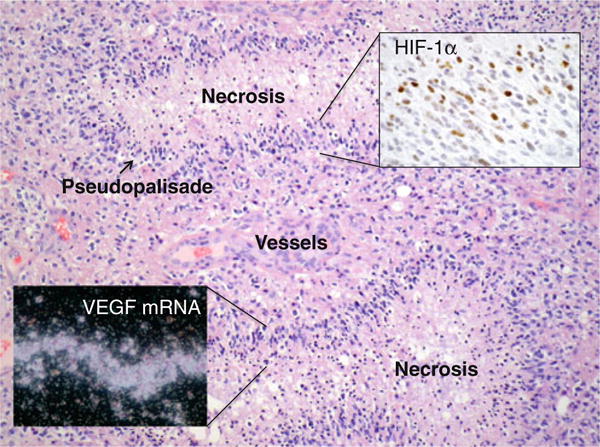
Pathology of human glioblastoma showing regions of pseudopalisading necrosis surrounded by hyperplastic vascular structures. Insets illustrate that pseudopalisading cells express HIF-1α as shown by immunohistochemistry, and VEGF mRNA as shown by in situ hybridization (images courtesy of Daniel J. Brat)
Adaptive changes by which cells respond to hypoxia are mediated largely by the transcription factor hypoxia-inducible factor 1 (HIF-1), which transactivates genes whose products play a role in all hallmarks of cancer and is a major target for anticancer therapy [2]. This is evidenced in GBM by the strong expression of HIF-1α on pseudopalisading cells, which activates the mRNA expression for vascular endothelial growth factor (VEGF), a major inducer of tumor angiogenesis (Fig. 1). HIF-1 is a heterodimeric transcription factor composed of the constitutively expressed HIF-1β and the O2-regulated HIF-1α subunits. The protein levels and activation of the α subunit are tightly controlled by two independent but co-regulated post-translational mechanisms [2]. A family of prolylhydroxylases (PHDs) hydroxylates HIF-1α in the presence of O2. This modification allows binding of the tumor suppressor Von Hippel-Lindau, the substrate recognition unit of an E3 ubiquitin ligase complex, polyubiquitylation, and degradation in the proteasome [3]. Factor-inhibiting HIF (FIH), an asparagine hydroxylase, controls transcriptional activity of HIF-1α by modifying a critical asparagine in the C-terminal activation domain. This modification is also O2-dependent and prevents interaction with the transcriptional co-activators p300/CBP. In the absence of O2, both PHDs and FIH are inactive; HIF accumulates in transcriptionally active form and transactivates hypoxia-inducible genes via binding to the hypoxia-responsive elements (HREs) in their regulatory regions (Fig. 2).
Fig. 2.
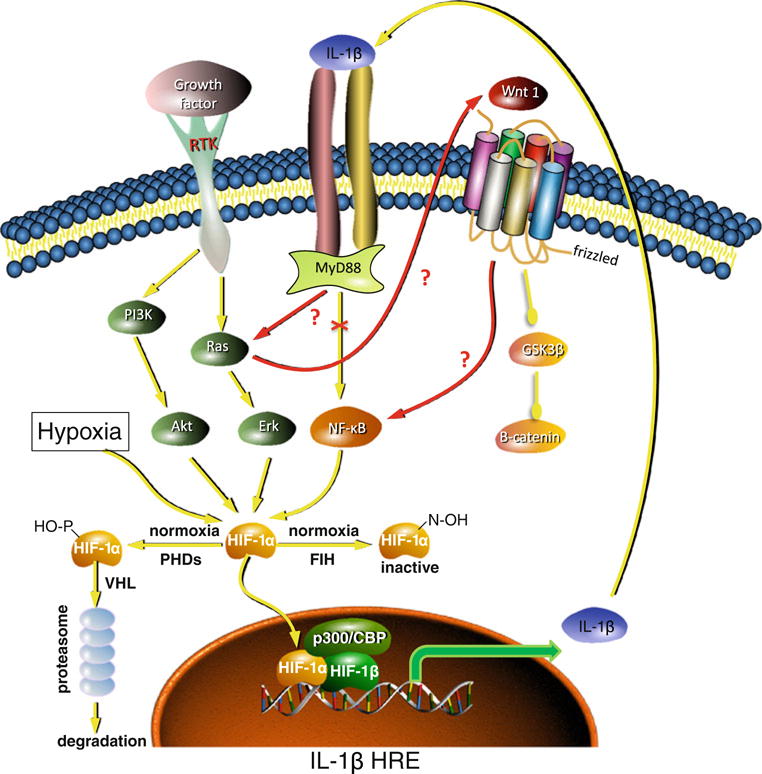
Autocrine loop mediated by IL-1β and HIF in GBM cells, red colored arrows indicate the novel interactions in the IL-1β–MyD88–Ras–Wnt–NF-κB–HIF-1α axis proposed by Sharma et al. [13], yellow colored arrows indicate the previously established interactions
Although the availability of the molecular O2 is the primary physiological regulator of HIF-1 through PHDs and FIH, there is a number of other stimuli (cellular oncogenes, loss of tumor suppressor phosphatase and tensin homolog (PTEN), growth factors, cytokines, vascular hormones, viral proteins) that can lead to induction and activation of HIF under normoxic conditions [4]. In this case, enhanced translation of HIF-1α, mediated via activation of cellular signaling pathways (Ras and PI3-Kinase) overcomes the limiting hydroxylase activity of PHDs/FIH, and HIF-1α accumulates in a non-hydroxylated form. These mechanisms may explain why in human GBM, HIF-1α expression is also found in single tumor cells infiltrating the normal brain, regions which are not known to be hypoxic [5].
Inflammation is an important contributing factor in the development of cancer. This is evidenced in GBM by the presence of immune cell infiltrates and the expression of inflammatory cytokines [6]. HIF-1 plays a pivotal role in linking inflammation and tumorigenesis through its connection with prominent pro-inflammatory mediators such as interleukin (IL)-1β and NF-κB. IL-1β is a pro-inflammatory cytokine with pluripotent activity, including the promotion of angiogenesis, tumor growth, and metastasis, and the IL-1β gene is an HIF-1 target [7]. Conversely, IL-1β can also activate HIF-1α [8], suggesting a potential IL-1β autocrine loop involving HIF-1. Extensive cross talk between HIF and the NF-κB pathway is documented by transcriptional activation of HIF-1α by NF-κB[9] and, by the activation of NF-κB under hypoxia, when the negative regulation of PHD1 on IKKβ is blocked [10]. The interplay between HIF-1 and inflammation, more specifically the role of inflammatory cytokines (IL-1β and tumor necrosis factor (TNF)-α) in the regulation of HIF-1α under normoxia, had been previously reported in GBM [6, 11, 12]. Sharma et al. [13] (this issue) further build upon this prior body of data. They report elevated levels of IL-1β in GBM tumors as compared to adjacent normal tissue, and decided to systematically investigate the IL-1β signaling that leads to HIF-1α expression and activity in malignant glioma cells. Using three different GBM cell lines (T98G, U87MG, and A172) and various genetic and pharmacological approaches, they set out to map the signaling mediators linking IL-1β, activation of the IL-1 receptor complex (IL-1R, MyD88, TRAF6) to HIF-1α. Their initial data confirmed that activation of HIF-1α by IL-1β was preceded by activation of NF-κB. Implication of the NF-κB pathway in the process of IL-1β-mediated HIF-1α activation in GBM provides further support for the role of NF-κB in the regulation of HIF-1α, as observed in other types of cancers [8]. Of note, all three cell lines used in this study have PTEN mutations, so the results have to be interpreted in the context of a preactivated PI3-K pathway, which elevates HIF-1α translation under normoxia. This is important in distinguishing IL-1-mediated signaling in normal vs tumor cells.
To further decipher the molecular players in this signaling axis, Sharma et al. turned their attention to Ras as prior studies had shown: (1) frequent activation of Ras without mutations in GBMs; (2) downregulation of HIF-1α by Ras inhibitors in GBM, and (3) the capacity of Ras to enhance NF-κB transcriptional activity (see references in Sharma et al. [13]; this issue). They showed that IL-1β treatment of glioma cells elevated Ras activity, and that it occurred in a MyD88-dependent fashion. Moreover, dominant negative Ras (DN-RasN17) abolished IL-1β-induced increase of NF-κB and HIF-1 activity, suggesting Ras dependence. These data suggest that Ras is an intermediate or necessary co-activator in the IL-1-mediated signaling cascade regulating HIF-1 activity through NF-κB. IL-1β treatment further activated the Ras effectors PI3-Kinase and ERK, which are well-established regulators of HIF-1α expression and activation, respectively. Interestingly, simultaneous inhibition of AKT and ERK significantly decreased IL-1β mediated HIF-1α activation, whereas the same treatment had no effect on IL-1β induced NF-κB activity. The authors propose that this provides evidence that in GBM cells two pathways downstream of Ras (AKT/ERK and NF-κB) can independently transduce the IL-1β signal to HIF-1α.
Sharma et al. further interrogated whether Wnt signaling plays a role in IL-1β-mediated activation of HIF-1α. To this end, they found increased Wnt-1 expression in IL-1β-treated glioma cells but, intriguingly, this increase was not accompanied by inhibition of GSK3β or activation of β-catenin, the hallmarks of canonical Wnt pathway activation. siRNA against Wnt-1 reduced IL-1β-mediated activation of HIF-1α as well as NF-κB, leading them to suggest a putative role for Wnt upstream of NF-κB. Further, DN-RasN17 abrogated induction of Wnt-1 expression by IL-1β, leading the authors to propose that induction of Wnt-1 is dependent upon Ras. As for NF-κB, Wnt-1 synergized with AKT and ERK to regulate IL-1β-induced HIF-1α. Together, the authors make the interesting proposition that IL-1β-induced Wnt-1 contributes to regulation of HIF-1α activity through a novel pathway that does not engage the downstream effectors of the canonical Wnt pathway.
The clearly provocative contribution of this report is to place Ras and Wnt-1 in the IL-1β signaling pathway leading to induction of HIF-1α under normoxia in GBM and placing IL-1β–MyD88–Ras–Wnt–NF-κB–HIF-1α in a linear signaling pathway (see Fig. 8e in Sharma et al. [13]). A side branch emanating from Ras also transduces some of the IL-1β signal to HIF-1α via more “classical” AKT/ERK activation. This novel signaling axis may define a framework for translation of inflammatory signaling (IL-1β) into oncogenic signaling, represented by Ras–Wnt–NF–κB–HIF-1α. Importantly, since the IL-1β gene carries an HRE, this signaling can auto-amplify through a feedback activation of IL-1β by HIF-1α, resulting in an autocrine loop.
At the same time, it should be noted that the study findings do not exclude alternative explanations and certainly provoke a number of intriguing questions. The authors infer from their data the existence of a linear signaling axis, but their findings could equally be compatible with a cross talk between different established pathways (Fig. 2). The proposed IL-1β–MyD88–Ras–Wnt–NF-κB–HIF-1α axis comprises effectors that are frequently deregulated in other types of cancer: Is inclusion of Ras and Wnt in this axis cancer specific? If it is specific, what defines when Ras and Wnt are engaged? The authors do not address how an extracellular growth factor (Wnt 1) acts as an intermediary between Ras and NF-κB and the involvement of frizzled was not examined. How is Wnt-1 activation by IL-1β different from other Wnt pathway agonists and why is the signal not passed down the Wnt/frizzled/GSK3β/β-catenin pathway? Do the authors envision an intracellular role for this molecule or a cross talk between frizzled and IL-1 receptor to facilitate IL-1β signaling, given that the downstream effectors GSK3β and β-catenin are unaffected? Does this NF-κB-activating pathway also lead to amplification of the inflammatory response through activation of other cytokines (TNF-α, IL-6, IL-8)? How does IL-1β affect expression of endogenous hypoxia-inducible genes in vitro and in vivo? So far, the modulation of HIF activity by IL-1β was exclusively measured with a luciferase construct driven by multimerized HREs, designed for highly sensitive monitoring of HIF activity. How does the efficiency of IL-1β-stimulated HIF activation compare with other, well-established activators of HIF such as hypoxia? In which areas of the tumors does it take place? When is this autocrine IL-1β loop initiated in astrocytoma progression? Is it dependent upon initial tumor infiltration by inflammatory cells, which secrete paracrine pro-inflammatory cytokines? While the study provides a model for upregulation of IL-1β at the transcriptional level in GBM, the Cancer Genome Atlas database (http://cancergenome.nih.gov/) does not evidence increased IL-1β mRNA expression in GBM. Clearly, this study is stimulating in that it raises many interesting questions to further validate and understand the new findings.
Though the relationship between inflammation and cancer is widely accepted, the major novelty of this study may be in suggesting that in cancer cells, the normoxic activation of HIF-1α by a key pro-inflammatory cytokine is rewired to become Ras-dependent. This offers a mechanism on how inflammation could be an initiating force behind activation of HIF in the normoxic areas of tumors, particularly in GBM of the mesenchymal subtype, which show activated Ras and immune cell infiltrates. Such pro-inflammatory signaling could further exacerbate pro-HIF signaling already present in PTEN or PI3-Kinase mutant tumor cells, along the leading/actively growing edge of gliomas and in the hypoxic perinecrotic regions [1, 5]. By dissecting the molecular mechanisms underlying HIF-1α accumulation in an inflammatory microenvironment, this study provides important clues as to how complex interactions of several signaling pathways work in tandem to regulate HIF-1α activity. By identifying a series of key regulators and outlining a putative sequence of events, the authors provide a working model on how IL-1β signals to HIF-1α. It will be important to determine whether activation of HIF signaling by IL-1β is sufficient to trigger the many pro-tumorigenic cues controlled by HIF, an aspect which was not yet addressed. Clearly, if the potential HIF-1/IL-1β autocrine loop in GBM is confirmed to be of significance in patient-derived samples, it could have important therapeutic implications. The proposed IL-1β–MyD88–Ras–Wnt–NF-κB–HIF-1α axis identifies a number of intervention points that could disrupt the autocrine signaling and thus reduce both inflammation and the hypoxic phenotype, mediated by IL-1β and HIF-1, respectively.
In conclusion, Sharma et al. are to be congratulated for providing evidence for a potential cancer specific signaling axis wiring IL-1β and HIF through Ras and Wnt, the outcome of which is to fuel an IL-1 autocrine loop, which may drive pro-inflammatory and oncogenic signals via sustained production of NF-κB and HIF transcription factor targets. Further studies are required to validate these new findings and address the importance of this mechanism to tumor growth in patients.
Contributor Information
Stefan Kaluz, Winship Cancer Institute, Emory University, Atlanta, GA, USA.
Erwin G. Van Meir, Winship Cancer Institute, Emory University, Atlanta, GA, USA Department of Hematology and Medical Oncology, School of Medicine, Emory University, Atlanta, GA, USA; Laboratory of Molecular Neuro-Oncology, Department of Neurosurgery School of Medicine and Winship, Cancer Institute, Emory University, Atlanta, GA, USA.
References
- 1.Brat DJ, Castellano-Sanchez A, Kaur B, Van Meir EG. Genetic and biologic progression in astrocytomas and their relation to angiogenic dysregulation. Adv Anat Pathol. 2002;9(1):24–36. doi: 10.1097/00125480-200201000-00004. [DOI] [PubMed] [Google Scholar]
- 2.Belozerov VE, Van Meir EG. Hypoxia inducible factor-1: a novel target for cancer therapy. Anticancer Drugs. 2005;16(9):901–909. doi: 10.1097/01.cad.0000180116.85912.69. [DOI] [PubMed] [Google Scholar]
- 3.Ivan M, Kondo K, Yang H, Kim W, Valiando J, Ohh M, et al. HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: implications for O2 sensing. Science. 2001;292(5516):464–468. doi: 10.1126/science.1059817. [DOI] [PubMed] [Google Scholar]
- 4.Semenza GL. Targeting HIF-1 for cancer therapy. Nat Rev Cancer. 2003;3(10):721–732. doi: 10.1038/nrc1187. [DOI] [PubMed] [Google Scholar]
- 5.Zagzag D, Zhong H, Scalzitti J, Laughner E, Simons J, Semenza G. Expression of hypoxia-inducible factor 1 alpha in brain tumors: association with angiogenesis, invasion and progression. Cancer. 2000;88:2606–2618. [PubMed] [Google Scholar]
- 6.Van Meir EG. Cytokines and tumors of the central nervous system. Glia. 1995;15(3):264–288. doi: 10.1002/glia.440150308. [DOI] [PubMed] [Google Scholar]
- 7.Zhang WD, Petrovie JM, Callaghan D, Jones A, Cui H, Howlett C, et al. Evidence that hypoxia-inducible factor-1 (HIF-1) mediates transcriptional activation of interleukin-1 beta (IL-1 beta) in astrocyte cultures. J Neuroimmunol. 2006;174(1–2):63–73. doi: 10.1016/j.jneuroim.2006.01.014. [DOI] [PubMed] [Google Scholar]
- 8.Jung YJ, Isaacs JS, Lee SM, Trepel J, Neckers L. IL-1 beta mediated up-regulation of HIF-1 alpha via an NFkB/COX-2 pathway identifies HIF-1 as a critical link between inflammation and oncogenesis. FASEB J. 2003;17(12):2115–2117. doi: 10.1096/fj.03-0329fje. [DOI] [PubMed] [Google Scholar]
- 9.Rius J, Guma M, Schachtrup C, Akassoglou K, Zinkernagel AS, Nizet V, et al. NF-kappa B links innate immunity to the hypoxic response through transcriptional regulation of HIF-1 alpha. Nature. 2008;453(7196):807–809. doi: 10.1038/nature06905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cummins EP, Berra E, Comerford KM, Ginouves A, Fitzgerald KT, Seeballuck F, et al. Prolyl hydroxylase-1 negatively regulates I kappa B kinase-beta, giving insight into hypoxia-induced NF kappa B activity. Proc Natl Acad Sci USA. 2006;103(48):18154–18159. doi: 10.1073/pnas.0602235103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dey N, Durden DL, Van Meir EG. Cytokine expression and signaling in brain tumors. In: Ransohoff RM, Benveniste EN, editors. Cytokines and the CNS. Taylor & Francis; Boca Raton, London, New York: 2006. [Google Scholar]
- 12.Gauthier T, Hamou M-F, Monod L, Gallay P, Carrel S, de Tribolet N. Expression and release of interleukin-1 by human glioblastoma cells in vitro and in vivo. Acta Neurochir. 1993;121(3–4):199–205. doi: 10.1007/BF01809276. [DOI] [PubMed] [Google Scholar]
- 13.Sharma V, Dixit D, Koul N, Mehta V, Sen E. Ras regulates interleukin-1β-induced HIF-1α transcriptional activity in glioblastoma. J Mol Med. 2010;89(1):1–14. doi: 10.1007/s00109-010-0683-5. [DOI] [PubMed] [Google Scholar]