

A 57-year-old HIV-positive, homosexual man presented with a 1-month history of fatigue, dyspnea, fever, and 10-pound weight loss. Two weeks earlier, he had been admitted to another hospital where an extensive workup failed to demonstrate any occult infection. At that time, he was given an empiric course of intravenous trimethoprim-sulfamethoxazole, ceftriaxone, and azithromycin without any meaningful resolution of his symptoms. On admission to our institution, he appeared frail with a heart rate of 99 BPM, blood pressure 108/62 mmHg, and temperature 99.8°F. Physical examination demonstrated splenomegaly, but no skin or mucosal changes, and no palpable lymph nodes. Complete blood count revealed pancytopenia with WBC 1.0 × 103/μl, and a differential count of 22% neutrophils, 68% lymphocytes, hemoglobin 7.6 g/dl, hematocrit 21.6%, MCV 84 fl, platelet count 26 × 106/μl, and 8% nucleated red blood cells. Peripheral blood smear showed anisopoikilocytosis and macrocytosis (Fig. 1a).
Figure 1.
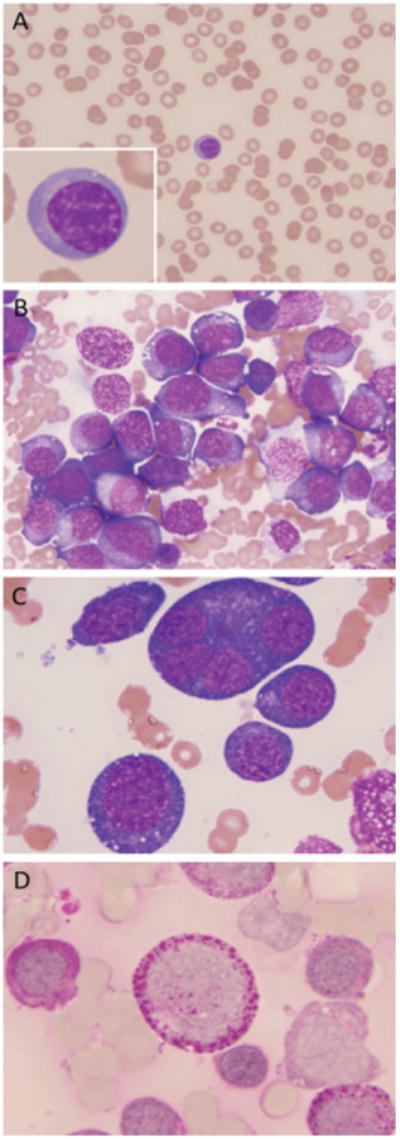
A: Peripheral blood smear with anisopoikilocytosis and one basophilic erythroblast. B: Bone marrow smear shows a cluster of proerythroblasts with dense basophilic cytoplasm and numerous vacuoles. C: High-power view of abnormal, giant multinucleated proerythroblast. D: Proerythroblast (center) with intense cytoplasmic globular “block” PAS staining. [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
Fever and pancytopenia in an HIV-positive patient warrants an immediate investigation to rule out any infection or underlying malignancy. In the setting of HIV infection, the class of offending microorganism depends largely on the degree of immunosuppression. Therefore, an absolute CD4+ T-lymphocyte count as well as HIV-1 viral load should be obtained. Potential infectious pathogens to be considered in our patient include certain viruses (such as cytomegalovirus (CMV), Epstein-Barr virus (EBV), parvovirus B19, human herpesvirus 6 (HHV6), and HIV itself), bacteria (e.g., Mycobacterium tuberculosis, Mycobacterium avium complex), and fungal organisms (e.g., cryptococcus, histoplasmosis, Penicillium marneffei). Importantly, this patient resides in Cape Cod, MA, which is an area endemic for tick-borne diseases such as Ehrlichia and Babesia, and to a lesser extent Tularemia, all of which can present with fevers and pancytopenia. Therefore, Ehrlichia and Babesia should be assiduously searched for in the peripheral blood films. Serology for these tick-borne organisms is less reliable in the acute phase of these diseases as well as in an immunodeficient patient such as ours. The two most important human ehrlichial diseases are human monocytic ehrlichiosis, caused by Ehrlichia chaffeensis that is transmitted by the Lone Star tick, and human granulocytic anaplasmosis, caused by Anaplasma phagocytophilum that is transmitted by the Ixodes scapularis tick. Although the Lone Star tick is not endemic to the Northeast region of the USA, Ixodes scapularis certainly is, and hence anaplasmosis remains an important consideration in our patient. Any history of a tick bite is therefore important, but not necessary in establishing the diagnosis. Ehrlichiae are obligate intracellular bacteria that present as intracytoplasmic inclusions (morulae) seen in leukocytes (neutrophils or monocytes). Erlichiosis results in anemia, leukopenia, thrombocytopenia, and elevated plasma levels of aminotransferases, lactate dehydrogenase, and alkaline phosphatase. Another important cause of fever, pancytopenia, and splenomegaly in an HIV-positive patient is hemophagocytic syndrome. Drug-related bone marrow suppression (e.g., zidovudine) and malignancy such as lymphoproliferative disorders (Hodgkin lymphoma and non-Hodgkin lymphomas) should be ruled out. Less likely, the HIV-positive pancytopenic patient may have a myelodysplastic syndrome and/or acute myelogenous leukemia.
Our patient has had a diagnosis of HIV infection for 10 years with a past history of Pneumocystis jiroveci pneumonia. He was compliant with Atripla (efavirenz/emtricitabine/tenofovir) highly active antiretroviral therapy (HAART) and had an undetectable HIV-1 viral load and a CD4+ T-lymphocyte count of 471 cells/mm3 on admission. He was not aware of a tick or mosquito bite, and also he had not noted any skin rashes. He had no recent travel, sick contacts, animal contacts, or occupational exposures. Laboratory investigations were negative for any parasites. Serological studies for EBV, CMV, parvovirus B19, Lyme disease, Cryptococcus neoformans, Coccidioides imitis, and human monocytic and granulocytic ehrlichia were ordered. A set of eight repeated blood cultures were all negative for fungal and bacterial microorganisms. Blood chemistries demonstrated ALT 29 IU/l (reference range, 0–40 IU/l), alkaline phosphatase 91 IU/l (39–117 IU/l), haptoglobin <20 mg/dl (30–200 mg/dl), total bilirubin 1.4 mg/dl (0–1.5 mg/dl), elevated AST 75 IU/l (0–40 IU/l), normal triglycerides level of 113 mg/dl (0–149 mg/dl), elevated fibrinogen level of 569 mg/dl (150–400 mg/dl), markedly elevated LDH level of 1,105 IU/l (94–250 IU/l), and ferritin greater than 2,000 ng/ml (30–400 ng/ml).
For patients who reside in endemic areas for tick-borne illnesses and who present with fever, pancytopenia, and elevated liver enzymes, the spectrum of infectious etiologies should always be broadened even in the absence of a history of tick bites. Examination of a thick preparation of the peripheral blood for parasites has low sensitivity, and in the absence of a positive result, the diagnosis of human ehrlichiosis cannot be excluded. Our patient was therefore started on empirical therapy of intravenous trimethoprim-sulfamethoxazole, ceftriaxone, and azithromycin. To rule out potential bone marrow suppression by HAART, his antiretroviral regimen was withheld. However, no significant improvement in his blood counts was observed. An extensive imaging workup revealed splenomegaly as the sole abnormality. Elevated levels of LDH, together with a low haptoglobin, are consistent with hemolysis. Intravascular hemolysis can be due to a microangiopathic process, immune-mediated hemolysis, G6PD deficiency, paroxysmal cold hemoglobinuria, and paroxysmal nocturnal hemoglobinuria (PNH). The absence of schistocytes in the peripheral smear makes a microangiopathic hemolytic anemia an unlikely cause of hemolysis in this case. PNH was ruled out by flow cytometry, which showed normal expression of CD55 and CD59 (GPI-anchored proteins). Direct antiglobulin test (DAT) was negative, thus excluding antibody-mediated hemolysis. It is important to consider glucose-6-phosphate dehydrogenase (G6PD) deficiency and hemoglobinopathy in a differential diagnosis of DAT-negative hemolytic anemia. Hemoglobinopathies with unstable hemoglobins as well as G6PD deficiency lead to chronic hemolysis. The clinical manifestations of accelerated red blood cell turn-over, such as jaundice and premature bilirubin gallstone formation, were not seen in our patient. Intravascular hemolysis may occasionally result from osmotic lysis following infusion of hypotonic solutions, which did not occur in our patient.
This patient's relatively high CD4+ T-lymphocyte count, together with negative infectious serologies, negative blood cultures, and imaging studies, makes an infectious etiology unlikely; despite a rather typical infection-associated symptomatology. The presence of splenomegaly and nucleated red blood cells in the peripheral blood smear warrants further investigation with primary concern for lymphoma/leukemia, myeloproliferative disorder, or another bone marrow space-occupying process.
During the patient's prior hospitalization, bone marrow biopsy showed a hypercellular marrow with dysplasia in both the erythroid and megakaryocytic line-ages, erythroid hyperplasia, and a left shift in the myeloid series with abnormal localization of immature precursors (ALIP). There was no evidence of any lymphoproliferative disorder, plasma cell neoplasm, granulomatous process, or metastatic tumor. A repeat bone marrow biopsy 3 weeks later demonstrated again a markedly hypercellular bone marrow, comprised almost entirely (80%) of proerythroblasts and basophilic erythroblasts with a marked reduction of polychromatophilic and orthochromatic erythroblasts. Frequent giant proerythroblasts were present, some with vacuolated cytoplasm (Fig. 1B,C). Rare hemophagocytic histiocytes were present. Cytochemical stains revealed PAS “block-positivity” in most of the erythroid precursors (Fig. 1D). The remainder of the bone marrow differential count showed 1% myeloblasts, 5% promyelocytes, 2% myelocytes, 3% metamyelocytes, 4% bands/neutrophils, 2% plasma cells, and 5% lymphocytes. No marrow space fibrosis was noted. Cytogenetic studies were requested on the bone marrow specimen.
The finding of an erythroid dominant, hypercellular marrow with giant proerythroblasts can be a manifestation of severe megaloblastic anemia, erythroleukemia, and occasionally Parvovirus B19 infection, which more often causes red cell aplasia, rather than hyperplasia. The paucity of late erythroblasts in this case indicates that conditions that lead to normal compensatory erythroblastic hyperplasia, such as severe immune and nonimmune hemolysis and blood loss, are unlikely to be the cause of erythroid hyperplasia in our patient.
In patients with megaloblastic anemia, examination of the peripheral blood reveals macrocytic anemia and neutrophils with hypersegmented nuclei. Bone marrow examination is not warranted in these situations, but if performed, may show a predominance of enlarged erythroblasts (megaloblasts), proerythroblasts, and erythroblasts with fine nuclear chromatin. The erythroid series in the marrow also demonstrates multinucleation and nuclear–cytoplasmic dyssynchrony. Maturing granulocytes are abnormal as well, with characteristically enlarged “giant” metamyelocytes and band neutrophils. Megakaryocytes may show hypersegmentation of nuclei. Such findings need to be correlated with decreased levels of vitamin B12 or folate or increased homocysteine and methylmalonic acid in the serum.
Parvovirus B19 infects and replicates within erythroid progenitors producing a characteristic morphologic picture of glassy, eosinophilic, intranuclear inclusions with a clear central halo in these erythroid precursors upon histologic examination of the bone marrow biopsy and aspirate. Infection of erythroid progenitor cells results in temporary cessation of red blood cell production as well as rapid depletion of infected cells by normal cytolytic cell-mediated immunity. As a result, most patients infected with parvovirus B19, including most HIV-infected patients, suffer erythroblastopenia. In a subset of severely immunocompromised HIV-infected patients, however, the cellular immune response is so blunted, that the clearance of infected erythroid precursors is severely limited and infected erythroblasts including giant, multinucleated proerythroblasts accumulate. Identification of parvovirus B19 inclusions in the marrow aspirates and confirmation of infection by in situ hybridization, or immunohistochemical staining and/or PCR of the biopsy are important steps in the assessment of anemia in immunodeficient patients because of the low sensitivity of serology for parvovirus B19 in these patients.
The diagnosis of hemophagocytic syndrome consists of a combination of clinical, laboratory, and histopathological findings. Major criteria include fever, splenomegaly, cytopenias involving two or more cell lines, hypertriglyceridemia, hypofibrinogenemia, and demonstration of hemophagocytosis in the bone marrow, spleen, or lymph nodes without evidence of malignancy. Additional diagnostic criteria include low or absent natural killer cell activity, serum ferritin level >500 μg/l, and soluble CD25 (sIL-2 receptor) ≥2,400 IU/ml [1].
Our patient's serum measurements of vitamin B12, folic acid, and methylmalonic acid were all within normal limits. Serologies for CMV, EBV, HHV6, and parvovirus B19 came back as negative. Immunohistochemistry for parvovirus B19 and CMV performed on the bone marrow sections was also negative. Parvovirus B19 quantitative DNA PCR test failed to detect infection. Large multinucleated proerythroblasts were immunoreactive for the erythroid marker glycophorin and were negative for the immature myeloid marker CD34, the megakaryocytic marker vWF, and the B-cell marker PAX5. Cytochemical stains revealed PAS block-positivity in most of the erythroid precursors. Cytogenetic studies on the bone marrow aspirate demonstrated an abnormal hyperdiploid male karyotype with 52-55 chromosomes, with multiple structural and numerical chromosomal abnormalities including +der(1) add(1) (q44), +6, +10, + 11, +14, +der(21) add(21) (p10), +3∼5mar[cp20].
The presence of rare hemophagocytic histiocytes in the bone marrow is most likely related to increased cell turn-over and inefficient hematopoiesis, and in the absence of supporting laboratory values (e.g., elevated triglycerides, hypofibrinogenemia) and a markedly abnormal karyotype, makes the diagnosis of hemophagocytic syndrome unlikely.
Although severe megaloblastic anemia can mimic the morphologic findings present in our patient's bone marrow, his erythrocytes were normocytic. Moreover folate, vitamin B12, and methylmalonic acid levels were all normal. The possibility of recovery from an acute viral insult (such as parvovirus B-19) was also considered; however, PCR studies performed on the patient's blood as well as immunohistochemical stains for parvovirus were all found to be negative.
Although unexpected, marrow erythroblastosis with dysplastic forms, absence of late erythroblasts, and a concomitant paucity of myeloid and megakaryocytic elements are all consistent with pure erythroleukemia (WHO classification), formerly classified as M6b (French-American-British classification scheme). The pattern of staining with PAS, consisting of large cytoplasmic blotches of stains (so-called “block-positivity”) is characteristic of this form of erythroleukemia. Furthermore, the presence of a hyperdiploid karyotype with a number of complex marker chromosomes is equally characteristic of pure erythroleukemia. In contrast, erythromyeloid leukemia (FAB M6a) usually exhibits karyotypic abnormalities similar to those seen in primary or alkylator-induced myelodysplastic syndrome.
During the patient's hospital stay, his condition deteriorated rapidly. He became increasingly hypoxic and repeat chest X-rays and CT scans failed to reveal any underlying cause. The patient's blood urea nitrogen and creatinine increased rapidly, while his LDH levels were >13,000 IU/l together with a marked increase in liver enzymes. All of these laboratory trends indicated rapid multisystem organ failure. The patient's family ultimately requested that he be made comfort measures only. The patient expired shortly thereafter.
Commentary
The vast majority of hematological malignances in HIV positive patients are of lymphoid lineage. The most common HIV-associated lymphomas are Burkitt lymphoma, diffuse large B-cell lymphoma, primary effusion lymphoma, plasmablastic lymphoma, and now, in the era of HAART, Hodgkin lymphoma. The pathogenesis of these lymphoid malignancies is believed to be chronic antigenic stimulation, cytokine dysregulation, as well as coinfection with EBV and/or HHV8 viruses. In contrast acute myeloid leukemia in the setting of HIV infection is extremely rare, with publications on the topic limited to small series and case reports. Previously reported cases are of HIV-related AML have included FAB M0 [2], M1 [3,4], M2 [5–8], M3 [9,10], M4 [2,10–15], and M5 [14–17], with M2 and M4 subtypes being the most frequently found subtypes. Of interest, myeloid sarcoma (granulocytic sarcoma or chloroma) has also been diagnosed in the context of HIV infection [18]. The role of HIV infection in the pathogenesis of myeloid leukemia is poorly understood. Although HIV was isolated from myeloid progenitors as well as bone marrow stromal cells from an HIV-infected patient, no direct leukemogenic effect was elucidated [11]. A potential mechanism of leukemic evolution is reduced immune tumor surveillance, secondary to HIV immunosuppression.
A known manifestation of HIV infection in the bone marrow is dyspoietic hematopoiesis termed HIV-myelopathy [19]. HIV-myelopathy, unlike a true myelodysplastic syndrome, is not considered a true stem cell disorder, but rather represents a spectrum of morphological changes secondary to direct HIV effect and HAART. In support of this theory, is the low rate of progression of HIV-myelopathy to acute myelogenous leukemia, unlike cases of true non-HIV-related myelodysplasia [5,20]. In one case report, an HIV-positive patient presented with refractory anemia with excess blasts as a primary diagnosis, which was successfully reversed by HAART [21]. Besides the direct pathogenic effects of HIV on the bone marrow [22], HAART itself may lead to significant dysplastic changes. In animal models, long-term exposure to zidovudine produces myelodysplasia as well as an increase in mitochondrial DNA mutations [22,23]. Cytogenetic studies from previously reported cases of acute myelogenous leukemia in the setting of HIV showed a wide variety of karyotypes including inversion 16, monosomy 7, and t(15;17) with no one persistent abnormality [24].
In the era of HAART, HIV-infected individuals often tolerate chemotherapy as well as their HIV-negative counterparts. Complete hematologic remission has been reported in a series of HIV-infected AML patients. Importantly, survival was highly dependant on the CD4 cell counts [24]. Patients with a CD4count >200 cells/μl had significantly higher median survival time.
Overall, this was a challenging case with many confounding factors, such as the underlying HIV infection and other possible infectious processes. Only after a meticulous clinicopathological investigation with support from the cytogenetic findings, the final diagnosis of acute erythroleukemia was made. The possibility of preexisting myelodysplastic syndrome in this patient with leukemic evolution cannot be excluded. This is the first reported case, to the best of our knowledge, of acute erythroid leukemia (FAB M6b) in a HIV-positive patient.
Acknowledgments
Contract grant sponsor: AIDS Malignancy Consortium/NCI.
Footnotes
Conflict of interest: Nothing to report.
References
- 1.Henter JI, Horne A, Arico M, et al. HLH-2004: Diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2007;48:124–131. doi: 10.1002/pbc.21039. [DOI] [PubMed] [Google Scholar]
- 2.Hentrich M, Rockstroh J, Sandner R, et al. Acute myelogenous leukaemia and myelomonocytic blast crisis following polycythemia vera in HIV positive patients: Report of cases and review of the literature. Ann Oncol. 2000;11:195–200. doi: 10.1023/a:1008304401661. [DOI] [PubMed] [Google Scholar]
- 3.Hengge UR, Schultewolter T, Uppenkamp T. Occurrence and therapy of secondary acute myeloid leukaemia in two HIV-infected patients. AIDS. 1998;12:221–222. [PubMed] [Google Scholar]
- 4.King JA, Nye DM, O'Connor MB, et al. Acute myelogenous leukemia (FAB AML-M1) in the setting of HIV infection and G-CSF therapy: A case report and review of the literature. Ann Hematol. 1998;77:69–73. doi: 10.1007/s002770050415. [DOI] [PubMed] [Google Scholar]
- 5.Napoli VM, Stein SF, Spira TJ, Raskin D. Myelodysplasia progressing to acute myeloblastic leukemia in an HTLV-III virus-positive homosexual man with AIDS-related complex. Am J Clin Pathol. 1986;86:788–791. doi: 10.1093/ajcp/86.6.788. [DOI] [PubMed] [Google Scholar]
- 6.Wijermans PW, ten Kate RW. Successful chemotherapy for acute myeloid leucaemia in HIV-infected patients. Eur J Haematol. 1990;44:136–138. doi: 10.1111/j.1600-0609.1990.tb00365.x. [DOI] [PubMed] [Google Scholar]
- 7.Willumsen L, Ellegaard J, Pedersen B. HIV infection in acute myeloblastic leukemia: A similar case. Am J Clin Pathol. 1987;88:536–537. doi: 10.1093/ajcp/88.4.536a. [DOI] [PubMed] [Google Scholar]
- 8.Monfardini S, Vaccher E, Pizzocaro G, et al. Unusual malignant tumours in 49 patients with HIV infection. AIDS. 1989;3:449–452. doi: 10.1097/00002030-198907000-00008. [DOI] [PubMed] [Google Scholar]
- 9.Calvo R, Ribera JM, Battle M, et al. Acute promyelocytic leukemia in a HIV seropositive patient. Leuk Lymphoma. 1997;26:621–624. doi: 10.3109/10428199709050899. [DOI] [PubMed] [Google Scholar]
- 10.Sutton L, Guenel P, Tanguy ML, et al. Acute myeloid leukaemia in human immunodeficiency virus-infected adults: Epidemiology, treatment feasibility and outcome. Br J Haematol. 2001;112:900–908. doi: 10.1046/j.1365-2141.2001.02661.x. [DOI] [PubMed] [Google Scholar]
- 11.Murthy AR, Ho D, Goetz MB. Relationship between acute myelomonoblastic leukemia and infection due to human immunodeficiency virus. Rev Infect Dis. 1991;13:254–256. doi: 10.1093/clinids/13.2.254. [DOI] [PubMed] [Google Scholar]
- 12.Peters BS, Matthews J, Gompels M, et al. Acute myeloblastic leukaemia in AIDS. AIDS. 1990;4:367–368. doi: 10.1097/00002030-199004000-00016. [DOI] [PubMed] [Google Scholar]
- 13.Puppo F, Scudeletti M, Murgia L, et al. Acute myelomonocytic leukaemia in an HIV-infected patient. AIDS. 1992;6:136–137. [PubMed] [Google Scholar]
- 14.al-Bahar S, Pandita R, Dhabhar BN, al-Bahar E. Human immunodeficiency virus (HIV) infection associated with acute myeloblastic leukemia in a low HIV prevalence area. Acta Haematol. 1994;91:52–53. doi: 10.1159/000204248. [DOI] [PubMed] [Google Scholar]
- 15.Costello RT, Sainty D, Heuberger L, et al. Third case of acute monocytic leukemia (M5) occurring in an HIV-seropositive man: A case report. Am J Hematol. 1995;49:356–357. doi: 10.1002/ajh.2830490419. [DOI] [PubMed] [Google Scholar]
- 16.Mansberg R, Rowlings PA, Yip MY, Rozenberg MC. First and second complete remissions in a HIV positive patient following remission induction therapy for acute non-lymphoblastic leukaemia. Aust N Z J Med. 1991;21:55–57. doi: 10.1111/j.1445-5994.1991.tb03004.x. [DOI] [PubMed] [Google Scholar]
- 17.De la Salmoniere P, Janier M, Gilquin J, et al. Chicken pox and acute monocytic leukaemia skin lesions in an HIV-seropositive man. Clin Exp Dermatol. 1994;19:505–506. doi: 10.1111/j.1365-2230.1994.tb01258.x. [DOI] [PubMed] [Google Scholar]
- 18.Rizzo M, Magro G, Castaldo P, Tucci L. Granulocytic sarcoma (chloroma) in HIV patient: A report. Forensic Sci Int. 2004;146(Suppl):S57–S58. doi: 10.1016/j.forsciint.2004.09.018. [DOI] [PubMed] [Google Scholar]
- 19.Thiele J, Zirbes TK, Bertsch HP, et al. AIDS-related bone marrow lesions– myelodysplastic features or predominant inflammatory-reactive changes (HIV-myelopathy)? A comparative morphometric study by immunohistochemistry with special emphasis on apoptosis and PCNA-labeling. Anal Cell Pathol. 1996;11:141–157. [PubMed] [Google Scholar]
- 20.Schneider DR, Picker LJ. Myelodysplasia in the acquired immune deficiency syndrome. Am J Clin Pathol. 1985;84:144–152. doi: 10.1093/ajcp/84.2.144. [DOI] [PubMed] [Google Scholar]
- 21.Modest GA, Cooley TP, Zacks JF. HIV and refractory anemia with excess blasts (RAEB) Am J Hematol. 2002;70:318–319. doi: 10.1002/ajh.10113. [DOI] [PubMed] [Google Scholar]
- 22.Inoue T, Cronkite EP, Hirabayashi Y, et al. Lifetime treatment of mice with azidothymidine (AZT) produces myelodysplasia. Leukemia. 1997;11(Suppl 3):123–127. [PubMed] [Google Scholar]
- 23.Chan SS, Santos JH, Meyer JN, et al. Mitochondrial toxicity in hearts of CD-1 mice following perinatal exposure to AZT, 3TC, or AZT/3TC in combination. Environ Mol Mutagen. 2007;48:190–200. doi: 10.1002/em.20191. [DOI] [PubMed] [Google Scholar]
- 24.Aboulafia DM, Meneses M, Ginsberg S, et al. Acute myeloid leukemia in patients infected with HIV-1. AIDS. 2002;16:865–876. doi: 10.1097/00002030-200204120-00006. [DOI] [PubMed] [Google Scholar]