

Abstract
It has been observed that immune cell deterioration occurs in the elderly, as well as a chronic low-grade inflammation called inflammaging. These cellular changes must be driven by numerous changes in gene expression and in fact, both protein-coding and non-coding RNA expression alterations have been observed in peripheral blood mononuclear cells from elder people. In the present work we have studied the expression of small non-coding RNA (microRNA and small nucleolar RNA -snoRNA-) from healthy individuals from 24 to 79 years old. We have observed that the expression of 69 non-coding RNAs (56 microRNAs and 13 snoRNAs) changes progressively with chronological age. According to our results, the age range from 47 to 54 is critical given that it is the period when the expression trend (increasing or decreasing) of age-related small non-coding RNAs is more pronounced. Furthermore, age-related miRNAs regulate genes that are involved in immune, cell cycle and cancer-related processes, which had already been associated to human aging. Therefore, human aging could be studied as a result of progressive molecular changes, and different age ranges should be analysed to cover the whole aging process.
Keywords: microRNA, non-coding RNA, transcriptome, regulation, aging, human, leucocytes
INTRODUCTION
In the last years, aging has become one of the most studied biological processes, due to its increasing incidence in our population and also to its correlation to other highly prevalent diseases, such as cancer or neurodegenerative diseases [1]. It has been estimated that 30 % of the total population in Europe will be older than 65 years in 2050, becoming a huge challenge from the medical and social point of view.
From a biological viewpoint, aging has been defined as a progressive postmaturational decline in physiological capacity, accompanied by an increased susceptibility to disease and an increased mortality risk [2].
It is a complex and multifactorial process in which genetic, lifestyle and environmental factors play a role. The genetic configuration of each individual defines how the organism faces our lifestyle and the interaction with the environment in the aging process [1]. Aging could be defined as the phenotypic expression of cellular senescence in our tissues. Cellular senescence is characterized by cell cycle arrest and it is associated to major changes in gene expression patterns. Moreover, aging reaches most tissues and organs, which is measured as accumulation of senescent cells [3].
A high amount of cells of the immune system acquire senescent features, in the process termed immunosenescence [4]. These age-related changes result in increased susceptibility to viral and bacterial infection, poor vaccination response and higher incidence of agerelated diseases, as stated before. Apart from immune cell deterioration, a chronic low-grade inflammation has also been described in the elderly, the so called inflammaging. Inflammaging results from immune dysfunction and accumulation of senescent cells that promote proinflammatory signals, such as elevated secretion of proinflammatory cytokines and activation of NK-κB transcription factor [5,6].
These cellular and serological changes must be driven by numerous changes in gene expression. Previous works have shown that genomic DNA methylation declines with age [7] and also that there is a change in gene expression at transcriptomic level [8] in peripheral blood cells. Additionally, cellular changes could be led by products of the non-coding part of the genome, non-coding RNAs (ncRNAs).
ncRNAs include a wide variety of RNAs that can be classified according to their size in long ncRNAs (>200 bp) or small ncRNAs (<200 bp). Apart from the well-known ribosomal RNA or transfer RNA, other less characterized ncRNAs are being described to be involved in many cellular processes through the regulation of gene expression. This regulation can be achieved taking part in different processes, such as transcription, mRNA processing or epigenetic control [9].
Among small ncRNA family (sncRNA), microRNAs (miRNA) are the best described class of ncRNAs. They also participate in gene expression regulation, through the binding of target mRNA, triggering either their destabilization or translation inhibition. During the last decade, the implication of miRNAs in virtually all biological processes has been reported, including immune cell development, function [10–12] and immunosenescence [13].
Moreover, several works have studied miRNA expression in peripheral blood cells from donors of different ages, including preterm infants, young, adults, octogenarians and centenarians, in order to find healthy aging miRNA expression patterns [14–18]. Three of these reports, include centenarians in their studies, adding a valuable information about human healthy aging [15–17]. Conversely, Noren Hooten and colleagues compare old subjects (mean age = 64.5 years) with young subjects (mean age = 30 years) [14] while Lai et al compare preterm infants with adults (mean age= 37 years old)[18]. In all these works, the strategy to find age-related miRNAs has been to compare the expression pattern from the oldest subjects to that from youngest subjects.
Small non-coding RNAs include other subtypes apart from miRNAs and, among them, the small nucleolar RNAs (snoRNAs) have been the most widely studied. They perform sequence-specific 2′ -O-methylation and pseudouridylation of ribosomal RNA which takes place in the nucleolus after forming the small nucleolar ribonucleoprotein (snoRNPs) complex [19]. A role for snoRNAs in cancer has been described and they have also been proposed as good candidates for involvement in other diseases [20]. Recently, it has been reported the presence of snoRNA in human plasma samples and identified a subset of 9 snoRNA showing a decreased expression in individuals being older than 66.5 years [21]. This work supports the idea that several types of non-coding RNA may play a role in human aging.
In the present work, we wanted to focus on how blood miRNA expression changes progressively across the life course, from adulthood to the elderly, apart from the classical analysis of comparing elder subjects' expression profile to that of young subjects'. Using a miRNA microarray, we analyzed the expression of 1769 human miRNA and other sncRNAs and we identified 69 sncRNAs that progressively changed with age. Furthermore, this analysis suggests that 50 years could be a turning point in human aging process.
RESULTS
In the present work we wanted to explore how the global expression of sncRNA changes in human aging. To do that, we analysed the expression of sncRNA by microarrays in leucocytes from healthy donors of different ages, ranging from 24 years to 79 years old (Figure 2A). We took two approaches to analyse data. On the one hand, we performed a correlation analysis aiming at identifying progressively changing sncRNA and on the other hand, we applied a more classical differential expression analysis. Finally, we carried out validation experiments both in the same discovery cohort and in an independent cohort of 44 donors (age range: 24-87). This study design, which is depicted in Figure 1, allowed us to study the expression of sncRNA in all the stages of the adulthood and part of the elderly.
Figure 2.

(A) Age distribution of subjects included in the study. (B) Expression of the sncRNA that progresively change with age according to the correlation analysis. y-axis represents the mean z-score values of the sncRNAs that correlate with age. Blue line represents the decreasing sncRNAs and red line represents the increasing sncRNAs. (C) Heatmap that represents the z-score value of the expression of the 69 sncRNAs correlated with age. Columns represent the mean age of the 28 sliding windows and rows are the 69 sncRNAs.
Figure 1.

Study design and analysis workflow followed in the present study.
Correlation and differential expression analysis
The first approach was to assess how the global sncRNA expression changes with chronological age and whether the sncRNA expression was correlated with human aging. The correlation analysis between gene expression and age resulted in 69 sncRNAs (56 miRNA and 13 snoRNA) (Table 1) (Figure 3) that progressively change with age (R>0.75) and have a fold-change greater than 1.5 between the oldest and the youngest sample. Forty-eight sncRNAs show an increasing expression pattern (R>0.75) while 21 have a decreasing expression pattern (R<-0.75) (Figure 2C). The representation of the mean z-score of expression for the decreasing and increasing sncRNAs (Figure 2B), shows that at 50 years old the two lines cross, highlighting that this could be a critical age.
Table 1.
List of age-related small non-coding RNAs identified by correlation analysis ordered by Pearson correlation coefficient (Pearson R). Fold-change between the last and first sliding window and the expression trend are also shown.
| Probeset Name | Type | Pearson R | FC (last/first) | Trend with age |
|---|---|---|---|---|
| U38B_st | CDBox | −0.947 | 0.364 | decreasing |
| hsa-miR-125b_st | miRNA | −0.941 | 0.569 | decreasing |
| hsa-miR-146a_st | miRNA | −0.917 | 0.561 | decreasing |
| hsa-miR-19b_st | miRNA | −0.913 | 0.530 | decreasing |
| hsa-miR-194_st | miRNA | −0.906 | 0.656 | decreasing |
| hsa-miR-29a_st | miRNA | −0.902 | 0.655 | decreasing |
| hsa-miR-130a_st | miRNA | −0.901 | 0.628 | decreasing |
| hsa-let-7g_st | miRNA | −0.899 | 0.434 | decreasing |
| hsa-let-7a_st | miRNA | −0.897 | 0.578 | decreasing |
| hsa-miR-146b-5p_st | miRNA | −0.896 | 0.457 | decreasing |
| hsa-miR-25_st | miRNA | −0.890 | 0.574 | decreasing |
| U38B_x_st | CDBox | −0.884 | 0.457 | decreasing |
| hsa-miR-15a_st | miRNA | −0.874 | 0.465 | decreasing |
| hsa-miR-126_st | miRNA | −0.871 | 0.554 | decreasing |
| hsa-miR-363_st | miRNA | −0.865 | 0.607 | decreasing |
| hsa-let-7i_st | miRNA | −0.853 | 0.498 | decreasing |
| hsa-miR-629_st | miRNA | −0.847 | 0.612 | decreasing |
| hsa-miR-584_st | miRNA | −0.841 | 0.592 | decreasing |
| hsa-miR-30b_st | miRNA | −0.839 | 0.476 | decreasing |
| ACA40_x_st | HAcaBox | −0.800 | 0.282 | decreasing |
| ACA8_x_st | HAcaBox | −0.781 | 0.644 | decreasing |
| hsa-miR-1271_st | miRNA | 0.768 | 1.497 | increasing |
| U48_st | CDBox | 0.771 | 1.601 | increasing |
| ENSG00000212523_x_st | snoRNA | 0.776 | 1.670 | increasing |
| hsa-miR-500-star_st | miRNA | 0.811 | 1.616 | increasing |
| U46_st | CDBox | 0.820 | 1.510 | increasing |
| U25_st | CDBox | 0.826 | 1.642 | increasing |
| U43_x_st | CDBox | 0.831 | 1.539 | increasing |
| U43_st | CDBox | 0.838 | 1.557 | increasing |
| U91_s_st | scaRna | 0.840 | 1.884 | increasing |
| hsa-miR-1280_st | miRNA | 0.848 | 1.659 | increasing |
| ACA16_st | HAcaBox | 0.853 | 1.498 | increasing |
| hsa-miR-339-5p_st | miRNA | 0.856 | 1.561 | increasing |
| HBII-180A_x_st | CDBox | 0.858 | 1.616 | increasing |
| hsa-miR-663_st | miRNA | 0.865 | 1.500 | increasing |
| hsa-miR-1308_st | miRNA | 0.869 | 1.604 | increasing |
| hsa-miR-25-star_st | miRNA | 0.878 | 1.658 | increasing |
| hsa-miR-1275_st | miRNA | 0.883 | 1.855 | increasing |
| hsa-miR-149-star_st | miRNA | 0.884 | 1.503 | increasing |
| hsa-miR-1228-star_st | miRNA | 0.888 | 1.685 | increasing |
| hsa-miR-766_st | miRNA | 0.894 | 1.730 | increasing |
| hsa-miR-197_st | miRNA | 0.894 | 1.657 | increasing |
| hsa-miR-345_st | miRNA | 0.897 | 1.683 | increasing |
| hsa-miR-574-5p_st | miRNA | 0.898 | 1.585 | increasing |
| hsa-miR-505-star_st | miRNA | 0.907 | 1.535 | increasing |
| hsa-miR-423-3p_st | miRNA | 0.909 | 1.656 | increasing |
| hsa-miR-150-star_st | miRNA | 0.913 | 1.716 | increasing |
| hsa-miR-432_st | miRNA | 0.921 | 2.327 | increasing |
| hsa-miR-1281_st | miRNA | 0.921 | 2.778 | increasing |
| hsa-miR-877_st | miRNA | 0.923 | 1.889 | increasing |
| hsa-miR-486-3p_st | miRNA | 0.925 | 1.678 | increasing |
| hsa-miR-491-5p_st | miRNA | 0.928 | 1.929 | increasing |
| hsa-miR-23a-star_st | miRNA | 0.929 | 2.524 | increasing |
| hsa-miR-339-3p_st | miRNA | 0.929 | 1.520 | increasing |
| hsa-miR-502-3p_st | miRNA | 0.931 | 1.759 | increasing |
| hsa-miR-1307_st | miRNA | 0.938 | 1.836 | increasing |
| hsa-miR-328_st | miRNA | 0.944 | 1.883 | increasing |
| hsa-miR-500_st | miRNA | 0.946 | 2.460 | increasing |
| hsa-miR-744_st | miRNA | 0.949 | 1.845 | increasing |
| hsa-miR-193a-5p_st | miRNA | 0.952 | 2.166 | increasing |
| hsa-miR-574-3p_st | miRNA | 0.954 | 1.811 | increasing |
| hsa-miR-330-3p_st | miRNA | 0.954 | 1.953 | increasing |
| hsa-miR-941_st | miRNA | 0.954 | 2.395 | increasing |
| hsa-miR-425-star_st | miRNA | 0.963 | 1.653 | increasing |
| hsa-miR-423-5p_st | miRNA | 0.963 | 1.998 | increasing |
| hsa-miR-1301_st | miRNA | 0.969 | 1.813 | increasing |
| hsa-miR-378_st | miRNA | 0.975 | 1.537 | increasing |
| hsa-miR-501-5p_st | miRNA | 0.978 | 2.688 | increasing |
| hsa-miR-93-star_st | miRNA | 0.982 | 1.646 | increasing |
Figure 3.
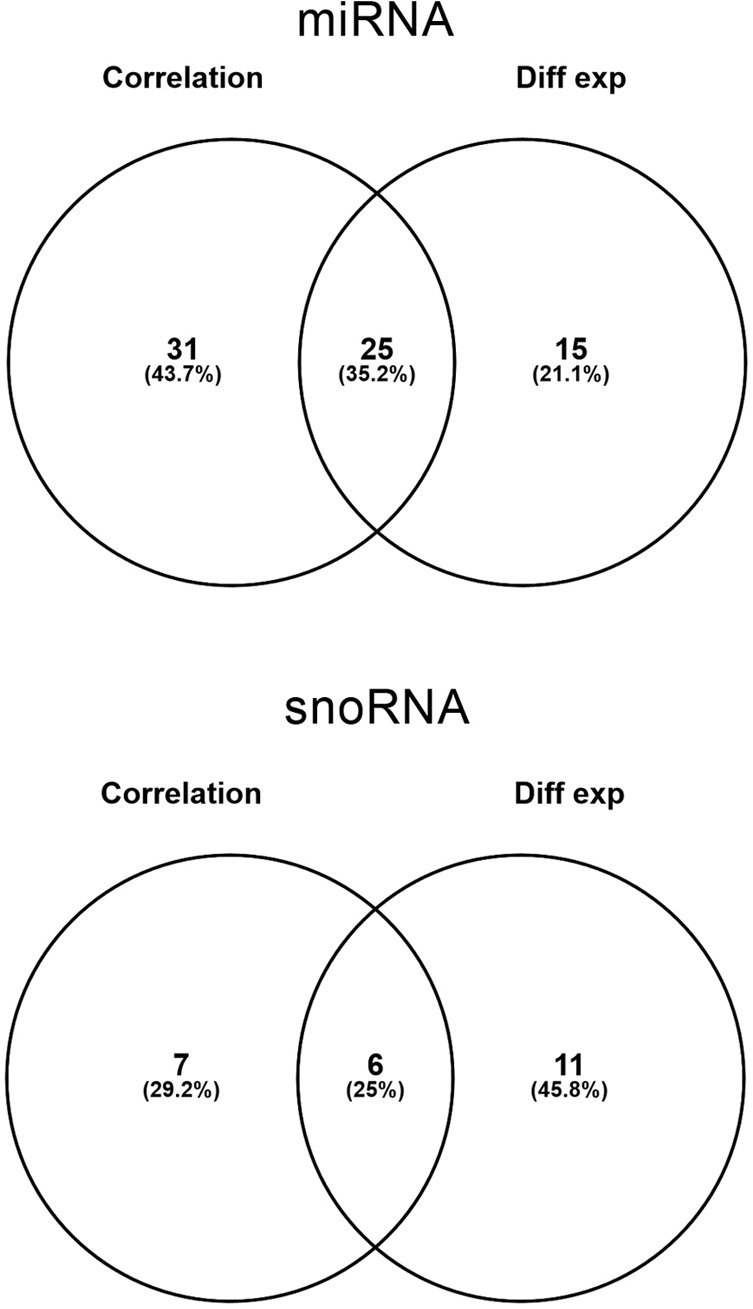
Number of miRNA (top) and snoRNA (bottom) identified by correlation and differential expression analysis and the overlapping probes between two analyses.
In a second approach, we applied a classical differential expression analysis, comparing the expression of sncRNA between elder and young donors. To set up the age at which we should consider the segregation of two groups, we looked at the correlation analysis. According to those results, 50 years seems to be critical, so we set this age as the cut-off between the two groups. Therefore, we had 21 young samples (age ≤ 50) and 17 elder samples (age >50).
Differential expression analysis identified 39 overexpressed and 18 underexpressed sncRNAs in the elder group compared to the young group (57 sncRNAs in total) (limma test, FDR corrected p<0.05)(Table 2). Among those, there are 40 miRNA and 17 snoRNA.
Table 2.
List of differentially expressed small non-coding RNAs between elder donors and young donors resulted from Limma test. sncRNAs are ordered by increasing fold-change.
| Probeset | Type | adj.P.Val | FC (elder/young) | Change with age |
|---|---|---|---|---|
| hsa-miR-199a-3p_st | miRNA | 0.016454 | 0.331 | DOWN |
| hsa-miR-628-3p_st | miRNA | 0.015405 | 0.385 | DOWN |
| hsa-miR-30e-star_st | miRNA | 0.032863 | 0.393 | DOWN |
| hsa-let-7i_st | miRNA | 0.004641 | 0.455 | DOWN |
| hsa-miR-146b-5p_st | miRNA | 0.040513 | 0.466 | DOWN |
| hsa-let-7g_st | miRNA | 0.015405 | 0.468 | DOWN |
| hsa-miR-584_st | miRNA | 0.014044 | 0.524 | DOWN |
| hsa-miR-25_st | miRNA | 0.006466 | 0.547 | DOWN |
| hsa-let-7a_st | miRNA | 0.001976 | 0.554 | DOWN |
| hsa-miR-126_st | miRNA | 0.032863 | 0.555 | DOWN |
| hsa-miR-629_st | miRNA | 0.015405 | 0.562 | DOWN |
| hsa-miR-98_st | miRNA | 0.032863 | 0.582 | DOWN |
| hsa-miR-26b-star_st | miRNA | 0.040513 | 0.583 | DOWN |
| hsa-miR-146a_st | miRNA | 0.032603 | 0.591 | DOWN |
| hsa-miR-421_st | miRNA | 0.048753 | 0.623 | DOWN |
| hsa-let-7c_st | miRNA | 0.015405 | 0.674 | DOWN |
| hsa-miR-26a_st | miRNA | 0.032863 | 0.743 | DOWN |
| hsa-let-7d_st | miRNA | 0.032690 | 0.769 | DOWN |
| HBII-85-20_x_st | snoRNA | 0.042958 | 1.300 | UP |
| 14qII-1_x_st | snoRNA | 0.032863 | 1.330 | UP |
| ENSG00000200307_st | snoRNA | 0.048753 | 1.355 | UP |
| ACA36_x_st | snoRNA | 0.035892 | 1.419 | UP |
| hsa-miR-93-star_st | miRNA | 0.020493 | 1.425 | UP |
| ENSG00000200377_st | snoRNA | 0.001828 | 1.438 | UP |
| U84_st | snoRNA | 0.044495 | 1.465 | UP |
| hsa-miR-425-star_st | miRNA | 0.016454 | 1.484 | UP |
| hsa-miR-345_st | miRNA | 0.032863 | 1.539 | UP |
| HBII-180C_st | snoRNA | 0.048152 | 1.539 | UP |
| hsa-miR-1228-star_st | miRNA | 0.040755 | 1.542 | UP |
| U53_st | snoRNA | 0.032863 | 1.590 | UP |
| U43_x_st | snoRNA | 0.031213 | 1.591 | UP |
| U43_st | snoRNA | 0.031213 | 1.605 | UP |
| ENSG00000212523_x_st | snoRNA | 0.031213 | 1.618 | UP |
| hsa-miR-423-3p_st | miRNA | 0.017498 | 1.622 | UP |
| HBII-289_st | snoRNA | 0.025374 | 1.646 | UP |
| hsa-miR-339-5p_st | miRNA | 0.006466 | 1.657 | UP |
| U52_st | snoRNA | 0.016454 | 1.657 | UP |
| HBII-180A_x_st | snoRNA | 0.039280 | 1.667 | UP |
| U83_st | snoRNA | 0.046181 | 1.692 | UP |
| hsa-miR-744_st | miRNA | 0.017561 | 1.694 | UP |
| hsa-miR-330-3p_st | miRNA | 0.025221 | 1.736 | UP |
| hsa-miR-1307_st | miRNA | 0.015405 | 1.790 | UP |
| hsa-miR-941_st | miRNA | 0.040513 | 1.880 | UP |
| hsa-miR-944_st | miRNA | 0.009982 | 1.880 | UP |
| hsa-miR-491-5p_st | miRNA | 0.020920 | 1.884 | UP |
| U25_st | snoRNA | 0.005814 | 1.889 | UP |
| hsa-miR-23a-star_st | miRNA | 0.028898 | 1.915 | UP |
| hsa-miR-501-5p_st | miRNA | 0.004144 | 1.951 | UP |
| U91_s_st | snoRNA | 0.006699 | 1.995 | UP |
| hsa-miR-500_st | miRNA | 0.001976 | 2.000 | UP |
| hsa-miR-432_st | miRNA | 0.023427 | 2.030 | UP |
| hsa-miR-1825_st | miRNA | 0.015405 | 2.093 | UP |
| hsa-miR-193b-star_st | miRNA | 0.015405 | 2.386 | UP |
| hsa-miR-1281_st | miRNA | 0.004641 | 2.509 | UP |
| hsa-miR-1228_st | miRNA | 0.000146 | 2.948 | UP |
| hsa-miR-198_st | miRNA | 0.000239 | 5.170 | UP |
| hsa-miR-551b-star_st | miRNA | 0.000443 | 5.398 | UP |
Then, we wanted to compare whether there was any overlap between the sncRNA identified with correlation analysis and differential expression analysis and we found that 25% of the miRNA and 6% of the snoRNA were common between both analyses (Figure 3).
In silico analysis
After identifying age-related miRNAs, we wanted to see whether those miRNAs had already been associated to aging. Among the 56 miRNAs that progressively change with age (correlation analysis), 18 had already been associated with aging process and interestingly, 11 had the same expression pattern as in the present study (increased or decreased with age). When we move to the 40 differentially expressed miRNAs between extremes of the cohort (older vs young, differential expression analysis) and compare them with the database, we also found 18 overlapping miRNAs, 8 being in the same direction of deregulation (Figure 4 and Table 3).
Figure 4.
Summary of the number of interesting miRNA identified in each analysis step.

Table 3.
List of miRNA and snoRNA identified in our study which have already been associated to aging according to Digital Aging Atlas database and Serna et al (Serna et al. 2012) and show the same trend with age as in the present paper. Corr: correlation analysis. Diff exp: differential expression analysis.
| miRNA/snoRNA | Analysis | DAA Identifier | Change with age | Tissues (DAA or original article) | References |
|---|---|---|---|---|---|
| let-7a | SW & Diff exp | DAA2533 | decreases | Foreskin | Hackl et al. (2010) |
| let-7a | SW & Diff exp | NA1 | decreases | PBMC | Serna et al. (2012) |
| let-7g | SW & Diff exp | DAA2476 | decreases | Foreskin | Hackl et al. (2010) |
| let-7g | SW & Diff exp | NA1 | decreases | PBMC | Serna et al. (2012) |
| let-7i | SW & Diff exp | DAA2561 | decreases | T Cell | Hackl et al. (2010) |
| let-7i | SW & Diff exp | DAA2503 | decreases | Foreskin | Hackl et al. (2010) |
| miR-125b | SW | DAA2517 | decreases | Foreskin | Hackl et al. (2010) |
| miR-126 | SW & Diff exp | DAA2605 | decreases | Blood | ElSharawy et al. (2012) |
| miR-126 | SW & Diff exp | DAA2504 | decreases | Foreskin | Hackl et al. (2010) |
| miR-1281 | SW & Diff exp | NA1 | increases | PBMC | Serna et al. (2012) |
| miR-130a | SW | DAA2471 | decreases | Foreskin | Hackl et al. (2010) |
| miR-130a | SW | DAA2006 | decreases | Blood, Immune System | Noren Hooten et al. (2010) |
| miR-15a | SW | DAA2553 | decreases | T Cell | Hackl et al. (2010) |
| miR-19b | SW | DAA2548 | decreases | T Cell | Hackl et al. (2010) |
| miR-19b | SW | NA1 | decreases | PBMC | Serna et al. (2012) |
| miR-26a | Diff exp | NA1 | decreases | PBMC | Serna et al. (2012) |
| miR-30b | SW | DAA2486 | decreases | Foreskin | Hackl et al. (2010) |
| miR-423-3p | SW & Diff exp | DAA2545 | increases | Foreskin | Hackl et al. (2010) |
| miR-423-3p | SW & Diff exp | NA1 | increases | PBMC | Serna et al. (2012) |
| miR-98 | Diff exp | DAA2576 | decreases | T Cell | Hackl et al. (2010) |
| U43 | SW & Diff exp | NA1 | increases | PBMC | Serna et al. (2012) |
| U52 | Diff exp | NA1 | increases | PBMC | Serna et al. (2012) |
| U91 | SW & Diff exp | NA1 | increases | PBMC | Serna et al. (2012) |
1The article by Serna et al is not included in Digital Aging Atlas.
As stated before, the main function of miRNAs is to regulate gene expression post-transcriptionally and they act binding to their target mRNAs and causing mRNA degradation or translation inhibition. Therefore, to know which functions were affected by the 56 miRNAs that progressively increase or decrease with age, we searched them in miRTarBase database and found 9370 interactions for 54 miRNA with 4082 genes. Among all those interactions, 458 were supported by strong experimental evidences and included 222 unique genes regulated by miRNAs that decrease with age and 26 unique genes regulated by miRNAs that increase with age (Figure 4). These target genes are involved in immune, cell-cycle and cancer related processes, UV- d image response and other biological processes (Table 4).
Table 4.
List of gene sets that significantly overlap with target genes of age-related miRNA identified by correlation analysis. The overlap analysis has been made using Hallmark gene sets in Molecular Signature Database v5.1.
| Gene Set Name | # Genes in Gene Set (K) | Description | # Genes in Overlap (k) | k/K | p-value | FDR q-value |
|---|---|---|---|---|---|---|
| TARGETS OF 17 DECREASING miRNA | ||||||
| G2M_CHECKPOINT | 200 | Genes involved in the G2/M checkpoint, as in progression through the cell division cycle. | 20 | 0.1 | 1.38E-20 | 6.89E-19 |
| E2F_TARGETS | 200 | Genes encoding cell cycle related targets of E2F transcription factors. | 17 | 0.085 | 1.78E-16 | 2.97E-15 |
| P53_PATHWAY | 200 | Genes involved in p53 pathways and networks. | 17 | 0.085 | 1.78E-16 | 2.97E-15 |
| EPITHELIAL_MESENCHYMAL_TRANSITION | 200 | Genes defining epithelial-mesenchymal transition, as in wound healing, fibrosis and metastasis. | 15 | 0.075 | 7.04E-14 | 5.86E-13 |
| IL2_STAT5_SIGNALING | 200 | Genes up-regulated by STAT5 in response to IL2 stimulation. | 15 | 0.075 | 7.04E-14 | 5.86E-13 |
| TNFA_SIGNALING_VIA_NFKB | 200 | Genes regulated by NF-kB in response to TNF [GeneID=7124]. | 15 | 0.075 | 7.04E-14 | 5.86E-13 |
| ALLOGRAFT_REJECTION | 200 | Genes up-regulated during transplant rejection. | 14 | 0.07 | 1.25E-12 | 8.22E-12 |
| APOPTOSIS | 161 | Genes mediating programmed cell death (apoptosis) by activation of caspases. | 13 | 0.0807 | 1.31E-12 | 8.22E-12 |
| UV_RESPONSE_DN | 144 | Genes down-regulated in response to ultraviolet (UV) radiation. | 12 | 0.0833 | 6.72E-12 | 3.73E-11 |
| PI3K_AKT_MTOR_SIGNALING | 105 | Genes up-regulated by activation of the PI3K/AKT/mTOR pathway. | 10 | 0.0952 | 1.04E-10 | 5.22E-10 |
| INFLAMMATORY_RESPONSE | 200 | Genes defining inflammatory response. | 12 | 0.06 | 3.13E-10 | 1.42E-09 |
| KRAS_SIGNALING_UP | 200 | Genes up-regulated by KRAS activation. | 11 | 0.055 | 4.34E-09 | 1.81E-08 |
| APICAL_JUNCTION | 200 | Genes encoding components of apical junction complex. | 10 | 0.05 | 5.47E-08 | 2.10E-07 |
| COAGULATION | 138 | Genes encoding components of blood coagulation system; also up-regulated in platelets. | 8 | 0.058 | 3.91E-07 | 1.40E-06 |
| ADIPOGENESIS | 200 | Genes up-regulated during adipocyte differentiation (adipogenesis). | 9 | 0.045 | 6.21E-07 | 1.72E-06 |
| COMPLEMENT | 200 | Genes encoding components of the complement system, which is part of the innate immune system. | 9 | 0.045 | 6.21E-07 | 1.72E-06 |
| HYPOXIA | 200 | Genes up-regulated in response to low oxygen levels (hypoxia). | 9 | 0.045 | 6.21E-07 | 1.72E-06 |
| INTERFERON_GAMMA_RESPONSE | 200 | Genes up-regulated in response to IFNG [GeneID=3458]. | 9 | 0.045 | 6.21E-07 | 1.72E-06 |
| UV_RESPONSE_UP | 158 | Genes up-regulated in response to ultraviolet (UV) radiation. | 8 | 0.0506 | 1.09E-06 | 2.87E-06 |
| IL6_JAK_STAT3_SIGNALING | 87 | Genes up-regulated by IL6 [GeneID=3569] via STAT3 [GeneID=6774], e.g., during acute phase response. | 6 | 0.069 | 4.22E-06 | 1.05E-05 |
| TARGETS OF 39 INCREASING miRNA | ||||||
| TNFA_SIGNALING_VIA_NFKB | 200 | Genes regulated by NF-kB in response to TNF [GeneID=7124]. | 6 | 0.03 | 1.35E-09 | 6.75E-08 |
| IL2_STAT5_SIGNALING | 200 | Genes up-regulated by STAT5 in response to IL2 stimulation. | 4 | 0.02 | 4.83E-06 | 1.21E-04 |
| UV_RESPONSE_DN | 144 | Genes down-regulated in response to ultraviolet (UV) radiation. | 3 | 0.0208 | 7.43E-05 | 1.24E-03 |
| APOPTOSIS | 161 | Genes mediating programmed cell death (apoptosis) by activation of caspases. | 3 | 0.0186 | 1.03E-04 | 1.29E-03 |
| EPITHELIAL_MESENCHYMAL_TRANSITION | 200 | Genes defining epithelial-mesenchymal transition, as in wound healing, fibrosis and metastasis. | 3 | 0.015 | 1.96E-04 | 1.40E-03 |
| ESTROGEN_RESPONSE_EARLY | 200 | Genes defining early response to estrogen. | 3 | 0.015 | 1.96E-04 | 1.40E-03 |
| G2M_CHECKPOINT | 200 | Genes involved in the G2/M checkpoint, as in progression through the cell division cycle. | 3 | 0.015 | 1.96E-04 | 1.40E-03 |
| IL6_JAK_STAT3_SIGNALING | 87 | Genes up-regulated by IL6 [GeneID=3569] via STAT3 [GeneID=6774], e.g., during acute phase response. | 2 | 0.023 | 1.12E-03 | 6.99E-03 |
| PI3K_AKT_MTOR_SIGNALING | 105 | Genes up-regulated by activation of the PI3K/AKT/mTOR pathway. | 2 | 0.019 | 1.62E-03 | 9.01E-03 |
| COAGULATION | 138 | Genes encoding components of blood coagulation system; also up-regulated in platelets. | 2 | 0.0145 | 2.78E-03 | 1.39E-02 |
| ALLOGRAFT_REJECTION | 200 | Genes up-regulated during transplant rejection. | 2 | 0.01 | 5.72E-03 | 1.68E-02 |
| E2F_TARGETS | 200 | Genes encoding cell cycle related targets of E2F transcription factors. | 2 | 0.01 | 5.72E-03 | 1.68E-02 |
| GLYCOLYSIS | 200 | Genes encoding proteins involved in glycolysis and gluconeogenesis. | 2 | 0.01 | 5.72E-03 | 1.68E-02 |
| HYPOXIA | 200 | Genes up-regulated in response to low oxygen levels (hypoxia). | 2 | 0.01 | 5.72E-03 | 1.68E-02 |
| INFLAMMATORY_RESPONSE | 200 | Genes defining inflammatory response. | 2 | 0.01 | 5.72E-03 | 1.68E-02 |
| MYOGENESIS | 200 | Genes involved in development of skeletal muscle (myogenesis). | 2 | 0.01 | 5.72E-03 | 1.68E-02 |
| P53_PATHWAY | 200 | Genes involved in p53 pathways and networks. | 2 | 0.01 | 5.72E-03 | 1.68E-02 |
As for age-related snoRNA, even if they are not included in Digital Aging Database, there are three (U43, U52 and U91) which had already been related to aging process by other authors (Table 3).
Validation of candidate miRNA
Once we identified a subset of miRNA and snoRNA related to human aging in leucocytes, we validated some candidates in an independent cohort. The selection of the miRNA for validation was made on the basis of fold-change and previous association with aging. There-fore, taking the list of 69 sncRNA that showed to be correlated with aging, we selected those having a fold change higher than 2 (up or downregulated) between the last and the first sliding window, which yielded a list of 15 sncRNA. Finally, we applied an additional filter, selecting those sncRNA that had already been associated to aging according to Digital Aging Atlas. This selection resulted in a list of 5 candidate miRNA: miR-15a, miR-30b, let-7i, let-7g and miR-1281 (Table 5).
Table 5.
Results for the 5 candidated miRNA selected for validation. Relative quantification (RQ) calculated with RT-qPCR data as well as p-value are shown for both the discovery and validation cohort. For the discovery cohort, microarray results are also shown for comparison purposes.
| discovery cohort | validation cohort | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| ProbeSet Name | miRNA | R-SW | last/first (SW) | FC (elder/young) LIMMA | adj.P.Val LIMMA | RQ (Qpcr) | p-value | RQ (Qpcr) | p-value |
| hsa-miR-1281_st | miR-1281 | 0.921 | 2.778 | 2.509 | 4.64E-03 | 1.127 | 6.60E-01 | - | - |
| hsa-let-7i_st | let-7i | −0.853 | 0.498 | 0.455 | 4.64E-03 | 0.566 | 2.86E-02 | 0.226 | 1.53E-07 |
| hsa-miR-30b_st | miR-30b | −0.839 | 0.476 | 0.503 | 1.05E-01 | 0.777 | 1.07E-01 | 0.419 | 7.66E-07 |
| hsa-miR-15a_st | miR-15a | −0.874 | 0.465 | 0.525 | 9.39E-02 | 0.632 | 1.21E-01 | 0.219 | 1.23E-07 |
| hsa-let-7g_st | let-7g | −0.899 | 0.434 | 0.468 | 1.54E-02 | 0.578 | 1.14E-02 | 0.199 | 1.05E-08 |
First, we assessed by RT-qPCR the expression of candidate miRNA in the same discovery cohort of microarray analysis as a technical validation, which showed that the expression of let-7i and let-7g was significantly lower in the group which was over 50 years (RQ=0.566; p-value= 0.0286 and RQ= 0.578; p-value=0.0281, respectively) (Table 5). miR-15a and miR-30b also had a slightly lower expression in this group, but the difference was not statistically significant (RQ=0.632; p-value=0.121 and RQ=0.777; p-value=0.0591, respectively) (Table 5). On the other hand, miR-1281 did not show any difference in its expression between the two groups (RQ=1.127; p-value=0.672) (Table 5). This RT-qPCR analysis confirmed the results observed in microarray experiment, except for miR-1281 (Figure 5). Even if miR-15a and miR-30b did not show a statistically significant change, we decided to include them in further validation experiments. Therefore, we studied the expression of the four downregulated miRNA in an independent cohort and found a statistically significant underexpression in subjects being older than 50 years (Figure 5A and Table 5).
Figure 5.

Results of the validation experiment of candidate miRNA. (A) The expression of candidate miRNA in elder (over 50) and young (under 50) is shown measured by RT-qPCR in the discovery cohort (upper row) and validation cohort (lower row). In the validation cohort miR-1281 was not analysed. (B) The expression of the validated 4 miRNA with age ranges.
Furthermore, we segregated the cohort in five age ranges to see whether the expression decrease was a gradual process with age, as identified by microarray experiment. We created 5 groups as follows: 24-40 (n=5), 41-60 (n=10), 61-70 (n=7), 71-80 (n=15) and 81-90 (n=7). Then, we represented the expression values of each miRNA (in DCq) for each group and it can be observed that their expression decreases gradually in this ranges, specially in miR-15a, let-7i and let-7g. However, it is true that the group composed of individuals between 80 and 90 years old, have a similar expression to that of 71-80 (Figure 5B).
DISCUSSION
In the present work we have identified a sncRNA signature that shows a progressive expression change with age in peripheral blood leucocytes. This signature involves 69 sncRNAs (56 miRNAs and 13 snoRNAs), 48 of them showing an increasing expression while 21 exhibit a decreasing expression pattern. Our data present that the expression of this sncRNAs subset starts to change around 40 years old and at 47 years, this alteration suffers and acceleration, the main change occurring from 47 to 54 years old. These results highlight that the expression change occurring with age is a progressive process rather than being a sudden event happening in the elderly. Yet, it is true that this progressive change does not follow a uniform pattern, given that we have observed an acceleration between 47 to 54 years.
This expression pattern described for sncRNAs, is similar to that found with mRNA expression analysis previously by our group [8]. Both studies have found a subset of RNAs whose expression changes progressively with chronological age. Yet, the most prominent change described for mRNA starts at a later age than that seen in small RNAs (49 years old in transcriptome and 47 in miRNome) and it is slightly more pronounced. Furthermore, both works determine the critical age in the sixth decade of life, based on the age at which increasing and decreasing mRNA and sncRNA cross (Figure 2B).
The correlation analysis carried out in the present work has been able to identify a subset of 69 probes, the expression of which gradually changes with chronological age. Furthermore, a more classical differential expression analysis has stood out an overlap of 35.2% and 25% of miRNA and snoRNA, respectively, with the correlation analysis, highlighting the validity of the correlation analysis to identify age-related miRNA. It is true, however, that there is an important fraction of sncRNA that have been identified only with one of the analyses. This observation suggests that each of the methods is able to address different questions: the correlation analysis would identify genes showing a gradual expression change correlated with chronological age, and would fail to detect those that suffer a big sudden change in the elderly, whereas differential expression analysis would identify genes presenting the opposite situation.
Among the 71 miRNAs and 24 snoRNAs identified in the present study (with both types of analysis), 46 have previously been reported to change with age by different authors and in different tissues or cell types. Interestingly, 16 sncRNA identified in the present study are in the same direction of change as reported previously (Table 3). Surprisingly, some of those miRNAs had been identified in an in vitro model of foreskin and our research highlights that those miRNAs may have an impact also in the aging of immune system cells. The rest of the miRNAs had also been described in blood cells, reinforcing the role of those miRNAs in senescence and reflecting the validity of our approach. Of note, let-7a, let-7g, let-7i, miR-126, miR-130a and miR-19b have been reported to be decreased and miR-423-3p increased in the elderly by more than one author apart from the present study, supporting their role in healthy aging (Table 3). This observation, points at those miRNAs as candidate molecules to be studied in the future in order to stablish their biological function and understand their implication in human healthy aging.
Moreover, we have confirmed the decreasing expression of miR-15a, miR-30b, let-7i and let-7g with age in and independent cohort by RT-qPCR. As stated before, those miRNA had already been associated to human aging by several authors. Hackl and colleagues, for instance, had also found the downregulation of these miRNA in several cell types and tissues including T cells, which is in accordance with our results [22]. Serna et al, had also related those miRNA to human aging but, they found and overexpression of miR-30b, miR-15a and let-7i in centenarians compared to octogenarians[17]. These data show the opposite trend than what we have observed in our cohort, where those miRNA appear to decrease with age. Nonetheless, they have studied centenarians, octogenarians and young people and they conclude that as for miRNA expression is concerned, centenarians are more similar to young people than to octogenarians. Therefore, the different trend they observed in miR-30b, miR-15a and let-7i expression comparing to our results could be conferred to the fact that the expression of those miRNA in centenarians is very similar to young people.
In order to assess the possible implication of age-related miRNA in human aging, miRNA-target interaction analysis was carried out, which revealed that age associated miRNAs regulate genes involved in immune, cell-cycle and cancer related processes, UV- induced damage response and others. Interestingly, many of the enriched pathways in age-related decreasing miRNA target genes are the same as those found among increasing miRNA targets. Therefore, those biological processes might be regulated either by repressing the expression (through increasing miRNA) of some genes or reducing miRNA-exerted repression (through decreasing miRNA) of some other genes in the same pathways.
Inflammatory pathways had already been associated to aging process [1,23] and our results indicate that some of those pathways are regulated by miRNAs. For instance, NFKB1 appears to be a target of miR-146a-5p, which decreases with chronological age, being a candidate that would explain the increase in this transcription factor activity that have already been described in immunosenescence[6]. The increase of IL-6 and TNFα plasma levels have been described in elder people as well [24–28], and our results point out that IL-6 and TNFα induced signalling pathways via NFkB might be regulated by a subset of miRNAs.
Moreover, the target genes of miRNAs that exhibit a gradual change with chronological age (found in miRTarBase and before filtering by strong evidence of interaction) include several genes whose expression also show a progressive pattern with age, as found in our previous work [8]. Namely, 40 genes (21 downregulated and 19 upregulated) appear to be age-related miRNA targets and show a concordant progressive expression change (increasing miRNA and decreasing target gene, and vice versa) (Figure 4). This observation points out that age-related miRNAs identified in the present work might be responsible for the expression pattern described for a subset of protein-coding genes.
In conclusion, our study identifies a subset of 69 small non-coding RNAs (54 miRNAs and 15 snoRNAs) of which expression in peripheral blood leucocytes changes progressively with chronological age. Furthermore, our data suggest that the age range from 47 to 54 is critical from a molecular point of view, given that sncRNA expression changes start to accelerate. Moreover, these miRNAs regulate several genes that are involved mainly in cancer and immune system-related pathways, which had already been associated to human aging. Therefore aging could be studied as a result of progressive molecular changes, and different age ranges should be analysed to cover the whole aging process.
MATERIALS AND METHODS
Sample donors and sample collection
In order to perform the study, healthy donors were recruited in the Department of Neurology at Donostia University Hospital and the Basque Biobank. The discovery cohort included 38 donors with an age range from 24 to 79 and the validation cohort included 44 donors ranging from 24 to 87 years old. Donors who were over 60 years were examined by a neurologist to ensure their healthy status. Samples from all donors were collected after receiving written informed consent. The study was approved by the hospital's ethics committee.
Peripheral blood (10 ml) was collected by venipuncture after receiving informed consent. Blood samples were collected in tubes containing EDTA as anticoagulant (Vacutainer, Becton Dickinson) and samples were processed and stored at the Basque Biobank.
RNA isolation
Total RNA was isolated from peripheral blood leucocytes with the LeukoLOCK kit (Ambion, ThermoFisher Scientific, 168 Third Avenue, Waltham, MA 02451, USA) using the alternative protocol to capture small RNAs. RNA concentration was measured using a NanoDrop ND-1000 spectrophotometer and RNA integrity was assessed using Agilent 2100 Bioanalyzer with the RNA 6000 Nano Assay Protocol (Agilent Technologies, 5301 Stevens Creek Blvd. Santa Clara, CA 95051, USA). We only included samples with a RNA integrity number higher than 6.
sncRNA expression
Total RNA (500 ng) was labeled using the FlashTag Biotin labeling kit (Genisphere LLC, 2801 Sterling Drive, Hatfield, PA 19440, USA) and hybridized to the GeneChip miRNA 1.0 Array (Affymetrix), which covers 847 human miRNAs and 922 human snoRNAs, following manufacturer's instructions. Briefly, RNA molecules were polyadenylated and a biotin-labeled DNA molecule was attached in a subsequent ligation step. Finally, labeled RNA was hybridized to the array, washed and stained in a GeneChip Fluidics Station 450 and scanned in a GeneChip Scanner 7G (Affymetrix, 3420 Central Expressway, Santa Clara, CA 95051, USA).
Microarray data analysis
Data normalization and filtering
We first performed raw data analysis, including a detection step (a probe set is detected above background with an associated p-value) resulting in a true/false call and a quantile normalization step using the miRNA QC Tool software v1.0.33.0 (Affymetrix, 3420 Central Expressway, Santa Clara, CA 95051, USA). In a subsequent filtering step, all non-human probesets were removed, resulting in a final list of 1769 probesets for downstream analysis. The data discussed in this publication have been deposited in NCBI's Gene Expression Omnibus [29] and are accessible through GEO Series accession number GSE89042 (www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE89042).
Correlation analysis
The workflow followed in this study for data analysis is shown in figure 4. In order to detect the genes whose expression progressively increase or decrease with age, the Pearson's correlation was calculated between the age and each of the sncRNA expression. To do that, we first performed a sliding window analysis. Briefly, we ordered the samples by age from the youngest to the oldest, made a group of first 10 samples (first window) and calculated the average expression for each probeset in this window. Then, we slid the window one sample to the right and calculated the average expression for each probeset in this second window. We carried out this computation to a total of 28 windows, obtaining at the end a matrix of 28 values for each probeset, being each value the average expression of 10 samples. Z-scores were computed for this matrix and these values were used for calculating the correlation with age. Finally, to identify the most significant sncRNAs, we selected those having a R > 0.75 and with a > 50% expression difference between the first and the last sliding window.
Differential expression analysis
With the aim of identifying any sncRNA that was differentially expressed between elder subjects and young subjects, these two groups were compared using the “limma” package from Bioconductor [30]. According to the correlation analysis, the cut-off to establish the two groups (young and elder) was set at age 50. Differentially expressed sncRNAs were defined as those passing the false discovery rate (FDR) corrected p-value, set at 0.05 (Figure 4). Venn diagrams were created using Venny 2.1 (http://bioinfogp.cnb.csic.es/tools/venny/index.html).
Search for microRNA targets and biological function
In order to get the targets of miRNAs, miRTarBase database was used, which gathers experimentally validated miRNA-target interactions (MTI) [31]. First, using a tool available at miRandola web (http://atlas.dmi.unict.it/mirandola/index.php) [32] we converted the miRNA names from miRBase v11 (in which the microarray used is based) to miRbase v20 to match the nomenclature of miRTarBase. Afterwards, we found the miRNA-target interactions registered in miRTarBase for the miRNAs of interest and removed the interactions that have weak evidence according to the database. Afterwards, we computed overlaps of miRNA target genes with Hallmark gene sets from Molecular Signature Database v5.0 on Gene Set Enrichment analysis website [33], considering only the resulting top twenty gene sets from significant list (FDR < 0.05).
Finally, we wanted to evaluate whether the miRNAs identified in the present study had already been related to aging process in other works. To do that, we compared our age-related miRNAs with those compiled in Digital Aging Atlas [34] and the article by Serna et al [17], which is not included in this database. The snoRNA are not included in this database either.
Validation of candidate miRNA
Validation of candidate miRNA were performed by reverse transcription quantitative PCR (RT-qPCR) using miScript PCR system (Qiagen, QIAGEN Strasse 1 40724 Hilden, Germany). Total RNA (200 ng) was reverse transcribed using miScript II Reverse Transcription kit using the HiSpec buffer and following the manufacturer's protocol in a Veriti thermal cycler (Applied Biosystems, ThermoFisher Scientific, 168 Third Avenue, Waltham, MA 02451, USA). Afterwards, qPCR was carried out with miScript SYBR Green PCR kit, adding 3 ng of cDNA as input, as specified in the protocol. We used the following miScript primer Assays to amplify miRNAs of interest: Hs_let-7g_2, Hs_let-7i_1, Hs_1281_1, Hs_miR-15a_1, Hs_miR-30b_1 and Hs_miR-191_1 as the reference gene. The PCR was carried out in a CFX384 thermal cycler (BioRad, 1000 Alfred Nobel Drive, Hercules, CA 94547, USA).
Raw data were processed in Bio-Rad CFX Manager software and the subsequent analysis to calculate relative expression was carried out in Excel software using 2−DDCT method[35]. Statistical analysis and plots were performed in RStudio software version 1.0.44 running R version 3.3.2. To compare the expression of candidate miRNA between donors who were over 50 years and those who were under 50 years old we applied a Wilcoxon runk sum test.
Acknowledgments
We want to thank all the participants for their collaboration.
Footnotes
CONFLICTS OF INTEREST
The authors state that they do not have any conflict of interest to declare.
FUNDING
This work has been partially supported by Spanish Network of Multiple Sclerosis (REEM), FEDER Funds (grant numbers CP10/00539 and PI13/02277) and by Instituto de Salud Carlos III (grant number PS09/02105). AA and IOQ are supported by a pre-doctoral fellowship from the Department of Education of the Basque Government (PRE_2016_2_0063 and PRE_2016_2_0042 respectively).
REFERENCES
- 1.López-Otín C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell. 2013;153:1194–217. doi: 10.1016/j.cell.2013.05.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mangerich A, Bürkle A. Pleiotropic cellular functions of PARP1 in longevity and aging: genome maintenance meets inflammation. Oxid Med Cell Longev. 2012;2012:321653. doi: 10.1155/2012/321653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wang C, Jurk D, Maddick M, Nelson G, Martin-Ruiz C, von Zglinicki T. DNA damage response and cellular senescence in tissues of aging mice. Aging Cell. 2009;8:311–23. doi: 10.1111/j.1474-9726.2009.00481.x. [DOI] [PubMed] [Google Scholar]
- 4.Castelo-Branco C, Soveral I. The immune system and aging: a review. Gynecol Endocrinol. 2014;30:16–22. doi: 10.3109/09513590.2013.852531. [DOI] [PubMed] [Google Scholar]
- 5.Franceschi C, Bonafè M, Valensin S, Olivieri F, De Luca M, Ottaviani E, De Benedictis G. Inflamm-aging. An evolutionary perspective on immunosenescence. Ann N Y Acad Sci. 2000;908:244–54. doi: 10.1111/j.1749-6632.2000.tb06651.x. [DOI] [PubMed] [Google Scholar]
- 6.Salminen A, Kaarniranta K. NF-kappaB signaling in the aging process. J Clin Immunol. 2009;29:397–405. doi: 10.1007/s10875-009-9296-6. [DOI] [PubMed] [Google Scholar]
- 7.Bollati V, Schwartz J, Wright R, Litonjua A, Tarantini L, Suh H, Sparrow D, Vokonas P, Baccarelli A. Decline in genomic DNA methylation through aging in a cohort of elderly subjects. Mech Ageing Dev. 2009;130:234–39. doi: 10.1016/j.mad.2008.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Irizar H, Goñi J, Alzualde A, Castillo-Triviño T, Olascoaga J, Lopez de Munain A, Otaegui D. Age gene expression and coexpression progressive signatures in peripheral blood leukocytes. Exp Gerontol. 2015;72:50–56. doi: 10.1016/j.exger.2015.09.003. [DOI] [PubMed] [Google Scholar]
- 9.Angrand PO, Vennin C, Le Bourhis X, Adriaenssens E. The role of long non-coding RNAs in genome formatting and expression. Front Genet. 2015;6:165. doi: 10.3389/fgene.2015.00165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li X, Jin P. Roles of small regulatory RNAs in determining neuronal identity. Nat Rev Neurosci. 2010;11:329–38. doi: 10.1038/nrn2739. [DOI] [PubMed] [Google Scholar]
- 11.Baltimore D, Boldin MP, O'Connell RM, Rao DS, Taganov KD. MicroRNAs: new regulators of immune cell development and function. Nat Immunol. 2008;9:839–45. doi: 10.1038/ni.f.209. [DOI] [PubMed] [Google Scholar]
- 12.Gangaraju VK, Lin H. MicroRNAs: key regulators of stem cells. Nat Rev Mol Cell Biol. 2009;10:116–25. doi: 10.1038/nrm2621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kroesen BJ, Teteloshvili N, Smigielska-Czepiel K, Brouwer E, Boots AM, van den Berg A, Kluiver J. Immuno-miRs: critical regulators of T-cell development, function and ageing. Immunology. 2015;144:1–10. doi: 10.1111/imm.12367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Noren Hooten N, Abdelmohsen K, Gorospe M, Ejiogu N, Zonderman AB, Evans MK. microRNA expression patterns reveal differential expression of target genes with age. PLoS One. 2010;5:e10724. doi: 10.1371/journal.pone.0010724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.ElSharawy A, Keller A, Flachsbart F, Wendschlag A, Jacobs G, Kefer N, Brefort T, Leidinger P, Backes C, Meese E, Schreiber S, Rosenstiel P, Franke A, Nebel A. Genome-wide miRNA signatures of human longevity. Aging Cell. 2012;11:607–16. doi: 10.1111/j.1474-9726.2012.00824.x. [DOI] [PubMed] [Google Scholar]
- 16.Gombar S, Jung HJ, Dong F, Calder B, Atzmon G, Barzilai N, Tian XL, Pothof J, Hoeijmakers JH, Campisi J, Vijg J, Suh Y. Comprehensive microRNA profiling in B-cells of human centenarians by massively parallel sequencing. BMC Genomics. 2012;13:353. doi: 10.1186/1471-2164-13-353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Serna E, Gambini J, Borras C, Abdelaziz KM, Belenguer A, Sanchis P, Avellana JA, Rodriguez-Mañas L, Viña J. Centenarians, but not octogenarians, up-regulate the expression of microRNAs. Sci Rep. 2012;2:961. doi: 10.1038/srep00961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lai CY, Wu YT, Yu SL, Yu YH, Lee SY, Liu CM, Hsieh WS, Hwu HG, Chen PC, Jeng SF, Chen WJ. Modulated expression of human peripheral blood microRNAs from infancy to adulthood and its role in aging. Aging Cell. 2014;13:679–89. doi: 10.1111/acel.12225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Matera AG, Terns RM, Terns MP. Non-coding RNAs: lessons from the small nuclear and small nucleolar RNAs. Nat Rev Mol Cell Biol. 2007;8:209–20. doi: 10.1038/nrm2124. [DOI] [PubMed] [Google Scholar]
- 20.Esteller M. Non-coding RNAs in human disease. Nat Rev Genet. 2011;12:861–74. doi: 10.1038/nrg3074. [DOI] [PubMed] [Google Scholar]
- 21.Freedman JE, Gerstein M, Mick E, Rozowsky J, Levy D, Kitchen R, Das S, Shah R, Danielson K, Beaulieu L, Navarro FC, Wang Y, Galeev TR, et al. Diverse human extracellular RNAs are widely detected in human plasma. Nat Commun. 2016;7:11106. doi: 10.1038/ncomms11106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hackl M, Brunner S, Fortschegger K, Schreiner C, Micutkova L, Mück C, Laschober GT, Lepperdinger G, Sampson N, Berger P, Herndler-Brandstetter D, Wieser M, Kühnel H, et al. miR-17, miR-19b, miR-20a, and miR-106a are down-regulated in human aging. Aging Cell. 2010;9:291–96. doi: 10.1111/j.1474-9726.2010.00549.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ostan R, Bucci L, Capri M, Salvioli S, Scurti M, Pini E, Monti D, Franceschi C. Immunosenescence and immunogenetics of human longevity. Neuro-immunomodulation. 2008;15:224–40. doi: 10.1159/000156466. [DOI] [PubMed] [Google Scholar]
- 24.Alberro A, Sáenz-Cuesta M, Muñoz-Culla M, Mateo-Abad M, Gonzalez E, Carrasco-Garcia E, Araúzo-Bravo MJ, Matheu A, Vergara I, Otaegui D. Inflammaging and Frailty Status Do Not Result in an Increased Extracellular Vesicle Concentration in Circulation. Int J Mol Sci. 2016;17:1–7. doi: 10.3390/ijms17071168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ferrucci L, Corsi A, Lauretani F, Bandinelli S, Bartali B, Taub DD, Guralnik JM, Longo DL. The origins of age-related proinflammatory state. Blood. 2005;105:2294–99. doi: 10.1182/blood-2004-07-2599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Palmeri M, Misiano G, Malaguarnera M, Forte GI, Vaccarino L, Milano S, Scola L, Caruso C, Motta M, Maugeri D, Lio D. Cytokine serum profile in a group of Sicilian nonagenarians. J Immunoassay Immunochem. 2012;33:82–90. doi: 10.1080/15321819.2011.601781. [DOI] [PubMed] [Google Scholar]
- 27.Valiathan R, Ashman M, Asthana D. Effects of Ageing on the Immune System: infants to Elderly. Scand J Immunol. 2016;83:255–66. doi: 10.1111/sji.12413. [DOI] [PubMed] [Google Scholar]
- 28.Minciullo PL, Catalano A, Mandraffino G, Casciaro M, Crucitti A, Maltese G, Morabito N, Lasco A, Gangemi S, Basile G. Inflammaging and Anti-Inflammaging: The Role of Cytokines in Extreme Longevity. Arch Immunol Ther Exp (Warsz) 2016;64:111–26. doi: 10.1007/s00005-015-0377-3. Springer International Publishing. [DOI] [PubMed] [Google Scholar]
- 29.Edgar R, Domrachev M, Lash AE. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002;30:207–10. doi: 10.1093/nar/30.1.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43:e47. doi: 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chou CH, Chang NW, Shrestha S, Hsu SD, Lin YL, Lee WH, Yang CD, Hong HC, Wei TY, Tu SJ, Tsai TR, Ho SY, Jian TY, et al. miRTarBase 2016: updates to the experimentally validated miRNA-target interactions database. Nucleic Acids Res. 2016;44:D239–47. doi: 10.1093/nar/gkv1258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Russo F, Di Bella S, Nigita G, Macca V, Laganà A, Giugno R, Pulvirenti A, Ferro A, Laganà A, Giugno R, Pulvirenti A, Ferro A. miRandola: Extracellular Circulating MicroRNAs Database. PLoS One. 2012;7:1–6. doi: 10.1371/journal.pone.0047786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA. 2005;102:15545–50. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Craig T, Smelick C, Tacutu R, Wuttke D, Wood SH, Stanley H, Janssens G, Savitskaya E, Moskalev A, Arking R, de Magalhães JP. The Digital Ageing Atlas: integrating the diversity of age-related changes into a unified resource. Nucleic Acids Res. 2015;43:D873–78. doi: 10.1093/nar/gku843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) Method. Methods. 2001;25:402–08. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]