

Summary
Kidney cell death plays a key role in the progression of life-threatening renal diseases, such as acute kidney injury and chronic kidney disease. Injured and dying epithelial and endothelial cells take part in complex communication with the innate immune system, which drives the progression of cell death and the decrease in renal function. To improve our understanding of kidney cell death dynamics and its impact on renal disease, a study approach is needed that facilitates the visualization of renal function and morphology in real time. Intravital multiphoton microscopy of the kidney has been used for more than a decade and made substantial contributions to our understanding of kidney physiology and pathophysiology. It is a unique tool that relates renal structure and function in a time- and spatial-dependent manner. Basic renal function, such as microvascular blood flow regulation and glomerular filtration, can be determined in real time and homeostatic alterations, which are linked inevitably to cell death and can be depicted down to the subcellular level. This review provides an overview of the available techniques to study kidney dysfunction and inflammation in terms of cell death in vivo, and addresses how this novel approach can be used to improve our understanding of cell death dynamics in renal disease.
Keywords: Intravital multiphoton microscopy, acute kidney injury, chronic kidney disease, regulated cell death, renal dysfunction
Kidney cell death plays a key role in the pathology of life-threatening renal diseases, such as acute kidney injury (AKI) and chronic kidney disease (CKD). AKI is a major clinical event, characterized by a rapid decrease in glomerular filtration rate (GFR), and associated with the risk of multiple organ failure owing to the accumulation of metabolic waste.1 The common etiologies of AKI are ischemia-reperfusion injury (IRI), drug-induced renal injury, sepsis, and glomerulonephritis.2 The prevalence of AKI continuously is increasing,3 the mortality is high and AKI often leads to long-term complications such as CKD and end-stage renal disease.4
In chronic kidney disease of various etiologies, such as glomerulosclerosis, cell death and the decrease in renal function are less rapid. CKD often is characterized by the injury and depletion of glomerular epithelial cells.5 Podocytes are postmitotic, highly specialized cells that contribute to the integrity of the glomerular filtration barrier.6 A loss of podocytes leads to the development of proteinuria, which correlates with the decrease in GFR.7 In addition, proteinuria is associated with tubular8 and podocyte9 cell death, owing to albumin overload, which leads to further disease progression. Because podocytes most likely cannot regenerate themselves10 and their regeneration by renal progenitor cells still is discussed controversially,11–13 in particular, cell death of glomerular epithelial cells is dramatic in the course of renal disease.
Cell death classically was categorized into two major types: apoptosis and necrosis. Apoptosis is one of the main cell death mechanisms involved in tubular injury1 and also plays a role in podocyte loss.14 It is defined as a programed process that involves the activation of several caspase proteases and eventually drives the cell into death.15 On the contrary, necrosis commonly was classified as an uncontrolled process, leading to cell membrane rupture and triggering an intense immune response.16 However, intense research over the past decade has shown that necrosis actually may be mediated by several regulated molecular pathways, named necroptosis, ferroptosis, pyroptosis, mitochondria permeability transition, and neutrophil extracellular traps (NET)-induced cell death (NETosis). A detailed report on these new pathways of cell death is beyond the scope of this article but recently was reviewed in detail elsewhere.1,17–19
In AKI and CKD a very dynamic interaction between cell death, inflammation, and epithelial and endothelial dysfunction is involved in disease progression.16,20 To translate basic scientific findings from bench to bedside more efficiently, a novel study approach is needed to capture the dynamic nature of renal pathology and to better understand cell–cell interactions. Therefore, the simultaneous investigation of renal function and morphology in a time-dependent manner is required.
In vivo multiphoton microscopy (MPM) of the living kidney enables simultaneous studies of renal function and morphology, a unique characteristic that is not achieved by any other technique. In vivo imaging of the kidney has been used for more than 10 years and has made substantial contributions to our knowledge of kidney physiology and pathophysiology.21 In contrast to conventional single-photon microscopy, the excitation energy is provided by two or more photons of longer wavelengths, which are absorbed simultaneously. This event requires a high photon density at the focal point, which is achieved by the use of a pulsed laser beam. Several advantages result from this: because light of longer wavelength is less scattered and less powerful, the living tissue is penetrated deeper and is subject to less phototoxicity. In addition, the required photon density for MPM excitation is achievable almost exclusively in the focal point. Therefore, almost no out-of-focus emission is detectable and more sensitive detectors can be used.22
Intravital MPM enables the visualization of kidney structures, such as the glomerular and peritubular vasculature, the proximal tubule (PT), the distal tubule, and the collecting duct (CD). In addition, MPM facilitates a high-powered view on the glomerular filtration barrier and the Bowman’s capsule, including the precise identification and study of parietal cells, podocytes, mesangial cells, endothelial cells (Fig. 1), and the endothelial glycocalyx.23–26 Most importantly, it can be used to determine basic renal function and pathology, such as microvascular blood flow,27 leukocyte rolling and recruitment,28,29 single-nephron glomerular filtration rate (snGFR),27 cell death,30–32 and cell shedding,24 and the assessment of glomerular33–37 and peritubular30 vasculature permeability. Because of the high resolution of this imaging technique, function even can be depicted down to the cell organelle level. MPM has been used successfully to show renin content and release in the living animal,38 to study endocytosis and transcytosis of albumin in proximal39 and glomerular epithelial cells,23 and to visualize mitochondria function including the generation of reactive oxygen species (ROS).28,40 In addition, MPM recently was used to evaluate intracellular Ca2+ changes in vivo using the selective encoding of a green fluorescent protein (GFP)-calmodulin-myosin light chain kinase (M13) fusion protein (GCaMP3), which serves as a calcium indicator in podocytes.41 Figure 1 shows how intravital MPM can be used to visualize basic renal structures and to determine renal function, such as capillary blood flow.
Figure 1.
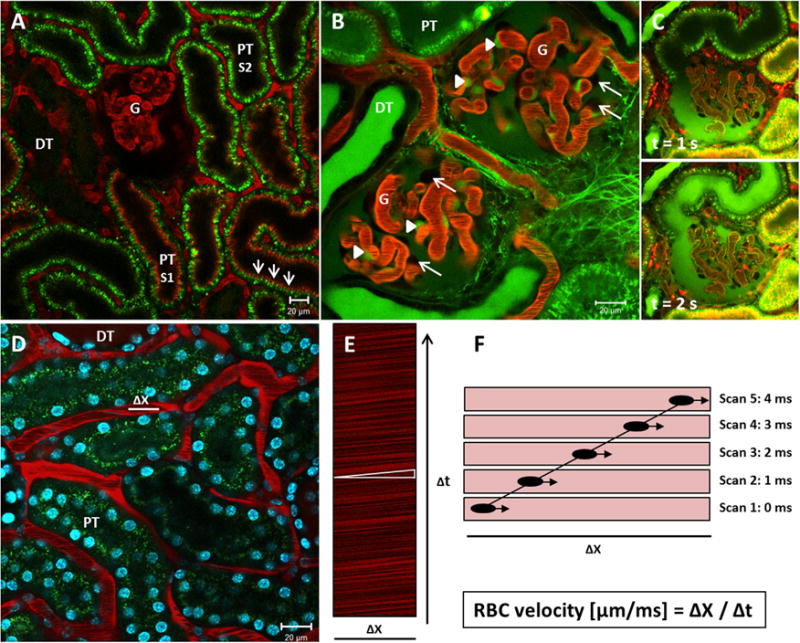
Intravital MPM to study renal function and morphology. (A) Intravital MPM can be used to visualize common renal structures, such as the glomerulus (G), the proximal tubule (PT), and the distal tubule (DT). Images were obtained in a MWF rat. The glomerular and peritubular vasculature is stained red by Alexa 594-albumin, which is barely filtered in the kidneys. Proximal tubules show an intense autofluorescence owing to the high density of NADH-rich mitochondria. S1-PT segments can be distinguished from S2-PT segments by the high uptake of Alexa 594-albumin in the brush border (arrows). Distal tubules show minimal autofluorescence. (B) A continuous infusion of the small-molecular-weight dye, LY, can be applied to show mesangial cells and podocytes. LY is freely filtered into Bowman’s space and therefore stains the urinary space green. Mesangial cells (arrowheads) endocytose LY over time and turn a bright green color. Podocytes (arrows), on the contrary, are excluded from any staining and appear as dark unstained objects at the outer margins of the capillary loops. (C) Intravital MPM can be used to visualize glomerular filtration. Upper and lower panel are images that were extracted from a time series during a bolus injection of LY. Upper panel: The LY is readily filtered into Bowman’s space (green) after the injection, but has not yet reached the urinary pole and the proximal tubule of the same nephron. Lower panel: A second later, the injected LY has streamed down the first segment of the proximal tubule. (D) Image of the renal cortex of a BL6 mouse. Cell nuclei are stained blue by the injection of the cell membrane-permeable dye Hoechst 33342, which binds to DNA. Alexa 594-albumin stains the plasma red, while the RBCs are excluded from the staining and appear as black bands in the lumen while streaming down the capillary. Within a peritubular capillary, a 20-μm longitudinal distance Δx in the central axis is subject to repetitive scans over time Δt. (E) This so-called linescan results in an XT image, in which the movement of the RBCs leaves dark diagonal bands. The slope of these bands (white triangle) is inversely proportional to the velocity of the RBCs. (F) Schematic explanation of the generation of a linescan. The same distance Δx is scanned several times. During the first scan, a RBC is captured in a certain position while streaming down the capillary. During the second scan, which occurs 1 ms later, the same RBC has moved further along the scanned distance. Accordingly, the movement of the RBC can be traced within the following linescans. One diagonal dark band emerges by several scans and indicates the time Δt in which the RBC covered the distance Δx. The velocity of the RBC (μm/ms) is calculated as Δx/Δt.
This review includes the significant contributions of intravital MPM to the investigation of kidney injury, dysfunction, and cell death.
EPITHELIAL DYSFUNCTION
Proximal Tubule Cells in the Center of Necroinflammation
The progression of AKI involves communication between epithelial and endothelial cells with the innate immune system,42 a process defined as necroinflammation.16 Upon necrotic cell death, the rupture of the cell membrane leads to the release of proinflammatory cytosolic content, so-called danger-associated molecular patterns (DAMPs). DAMPs can be recognized by pattern-recognizing receptors (PRRs), which are expressed on cells of the innate immune system. The stimulation of PRRs leads to enhanced cytokine synthesis and local tissue inflammation.16 Similar to immune cells, PT cells participate in an injury-induced inflammatory response. Thus, PT cells express PRRs on their surface, such as the toll-like receptor 4(TLR4), which enable them to sense a proinflammatory environment. Consequently, PT cells are able to respond to endogenous and exogenous stimuli by increasing their ROS and cytokine levels.42 TLR4 expression mediates IRI in several organs, including the kidney, and TLR4-deficient mice show less leukocyte infiltration, tubular damage, and lower serum creatinine levels in response to ischemic renal injury when compared with wild-type mice.43 By promoting increased ROS and cytokine levels and the release of DAMPs upon necrotic cell death, PT cells may communicate with other tubular cells downstream of the nephron, with surrounding endothelial cells and with cells of the innate immune system.16,44
Intravital MPM recently showed the TLR4-mediated uptake of intravenously injected fluorescent endotoxin into S1 segments of the proximal tubule.44 Kalakeche et al44 further investigated the endotoxin-induced oxidative stress within the tubular system. Although S1 proximal tubules were protected from oxidative stress, S2 and S3 segments showed high levels of ROS in response to S1-mediated endotoxin uptake. This suggests a communication between S1 PT cells with downstream portions of the proximal tubule. Furthermore, the TLR1 was expressed exclusively in S2 and S3 segments of the PT, and lipopolysaccharide treatment decreased the TLR1 expression levels in these nephron segments, probably owing to the internalization or the shedding of the receptor. This suggests that S1 proximal tubules are involved mainly in the uptake of endotoxin during sepsis. TLR4-mediated signaling may further generate cytokines, such as tumor necrosis factor α in S1 PT cells to communicate with downstream nephron segments via TLR1 activation.44
In an experimental model of pyelonephritis, GFP-expressing Escherichia coli bacteria were micro-injected into the PT lumen of a superficial nephron.45 The infection then was monitored using intravital MPM. Adhesion of single E coli bacteria on the apical PT wall resulted in a shutdown in blood supply of the adjacent peritubular blood vessels 3 hours after pathogen application. Microdissection followed by pro- and eukaryotic messenger RNA isolation of the affected nephron showed increased cytokine levels. These results suggest a cytokine-mediated communication between proximal tubule and endothelial cells in response to pathogens, which caused direct vasocon-striction in the adjacent capillaries, leaving the affected area isolated and might help to prevent the systemic spread of the infection.45
Mitochondrial Dysfunction and Cell Death
Mitochondria are essential in maintaining cellular energy balance and intracellular Ca2+ signaling. Moreover, mitochondria take part in the generation of ROS.
Mitochondria are involved in regulated cell death pathways and play a key role in the onset and progression of sepsis-induced,46,47 drug-induced,48,49 and ischemic AKI.49–52 Mitochondria control the intrinsic activation of apoptosis. Upon cell stress, the cytosolic proapoptotic B-cell lymphoma 2 (Bcl-2) family protein Bax translocalizes and inserts into the outer mitochondrial membrane.53 Bax and another activated member of the Bcl-2 family, Bak, oligomerize,54 and thereby induce mitochondrial outer membrane permeabilization. Mitochondrial outer membrane permeabilization leads to the release of mitochondrial pro-death effectors, such as cytochrome c.55 This results in the downstream activation of caspase-3 and caspase-7, which eventually will lead to cell death.56 In human ischemic injury of the kidney, Bax- and Bak-dependent mitochondrial damage seem to be the key mechanism leading to apoptotic cell death, which emphasizes the role of mitochondria in renal injury.57,58
Proximal tubules perform a high level of active transepithelial transport and, consequently, are densely packed with mitochondria to facilitate sufficient adenosine triphosphate (ATP) synthesis. Furthermore, PT cells rely mainly on aerobic ATP generation because their glycolytic capacity is lower compared with other tubular cells.59 For these reasons, PT cells are particularly vulnerable to limitations in oxygen supply.
Several recent MPM studies established dyes and took advantage of endogenous fluorescent markers to determine mitochondrial function in vivo.40,49,60 MPM is a favorable approach to study mitochondria function because it allows the simultaneous assessment of mitochondrial function and structure in many different renal cell types.
Mitochondrial reduced nicotinamide adenine dinucleotide (NADH), the substrate for complex I of the respiratory chain, generates a strong autofluorescence. Because NADH is fluorescent only in its reduced state,61 it can be used as an endogenous fluorophore to evaluate the redox state of the tubular mitochondria.40 In a recent study, a string loop was placed around the renal artery to investigate mitochondrial function before and during a 30-minute ischemic period. Mitochondrial NADH was excited at 720 nm and showed a characteristic basolateral distribution. During ischemia, the NADH fluorescence rapidly increased and was not restored until reperfusion. The investigators concluded that under resting conditions, the proximal tubules are in a relatively oxidized redox state.49 However, during oxygen deprivation, NADH accumulates in PT cells because anaerobic NAD+ regeneration is limited.
A key requirement for normal mitochondrial function is the mitochondrial membrane potential (Δψm), which is generated by the activity of the respiratory chain. In vitro, Δψm is assessed by the use of lipophilic cationic dyes, such as tetramethyl rhodamine methyl ester (TMRM), which load into mitochondria according to Δψm. TMRM also can be used for the intravital MPM investigation of Δψm. After intravenous injection, TMRM quickly loads into tubular and glomerular cells and was used successfully to visualize the effects of ischemia on tubular mitochondria. Within 2 minutes of ischemia, TMRM fluorescence intensity in the proximal tubule decreased and then remained stable for the rest of the 30-minute ischemia period. In contrast, TMRM fluorescence was better maintained in distal tubular mitochondria and decreased more slowly over time. Mitochondria in the collecting duct showed the least depolarization of Δψm during ischemia.49 These results highlight the vulnerability of PT cells to oxygen deprivation, which is consistent with the theory that PT cells depend almost exclusively on aerobic ATP generation.59 Furthermore, the mitochondrial dysfunction during ischemia was accompanied by sustained and severe morphologic impairments. Mitochondrial function and structure are highly associated and electron microscopy images of IRI kidneys showed mitochondria swelling and fragmentation.51 In agreement with these findings, intravital MPM of the reperfused kidney showed the shortening and fragmentation of PT mitochondria in contrast to the normal elongated shape of these cell organelles found in other nephron segments.49 Other dyes that can be used for in vivo determination of Δψm are rhodamine 12349,62 and rhodamine B hexylester: the latter selectively accumulates in vascular mitochondria, but not in mitochondria of tubular cells.30
Reactive oxygen species generated by mitochondria can take part in regulated cell death, such as during apoptosis and ferroptosis. In apoptotic cell death caspase-3 and caspase-7 contribute to ROS generation. More precisely, they cleave a Fe-S-containing subunit of the respiratory chain complex I, which enhances ROS production and amplifies apoptosis by an increased cytochrome c release from the mitochondria.56 Ferroptosis, which contributes to cell death in renal IRI,1 is an iron-dependent pathway of regulated necrosis. It is characterized by depletion of intracellular cysteine, which leads to impaired glutathione (GSH) biosynthesis. GSH is necessary for proper function of the glutathione peroxidase 4, which normally repairs accumulating lipid peroxides. Ferroptosis therefore accumulates lipid ROS species, which are sufficient to cause the death of the cell.56 Furthermore, GSH is a major intracellular antioxidant. It largely determines cellular redox potential and reduces intracellular ROS.63
Monochlorobimane (MCB) is a nonfluorescent dye that becomes fluorescent upon binding to GSH, which allows the determination of intracellular GSH content. The suitability of MCB to study renal GSH levels in vivo recently was investigated. MCB loads from the basolateral side into PTs. Unfortunately, the MCB/ GSH complex quickly is secreted into the tubular lumen, so no steady in vivo labeling in PT cells can be achieved. Nevertheless, the fluorescence signal of the MCB/GSH complex remains stable in endothelial cells over time.49
In ischemia-reperfusion injury, enhanced ROS generation is associated with epithelial and endothelial dysfunction. However, there is a discrepancy regarding whether ROS generation peaks during ischemia or during reperfusion.64 In renal IRI, cellular ROS generation before, during, and after ischemia has not been investigated in real time. We therefore injected the superoxide-sensitive dye dihydroethidium (DHE) into an anaesthetized rat and imaged renal ROS production in vivo. The application of DHE for in vivo imaging of the kidney was established previously.49 When exposed to superoxide, DHE is dehydrated and migrates into the nucleolus, where it binds to nucleic acids and emits a bright red fluorescence. We found a marked increase of nuclear DHE fluorescence in PT cells immediately after the onset of ischemia, which remained increased during reperfusion (Fig. 2). These results are in agreement with experiments performed in cultured cardiomyocytes, in which ischemia generated increased ROS levels within the cells.65 Because oxygen is required for the generation of ROS, it seems paradoxic that hypoxic conditions favor intracellular ROS generation. A possible explanation for this phenomenon might be the role of mitochondria as cell-oxygen sensors. Studies have suggested that hypoxia leads to an enhanced synthesis of ROS by the complex III of the mitochondria respiratory chain. In addition to oxygen, mitochondria-derived ROS are discussed as a second determinant of the hypoxia-induced factor α to adjust the cell to new metabolic conditions. However, this remains controversial.66
Figure 2.
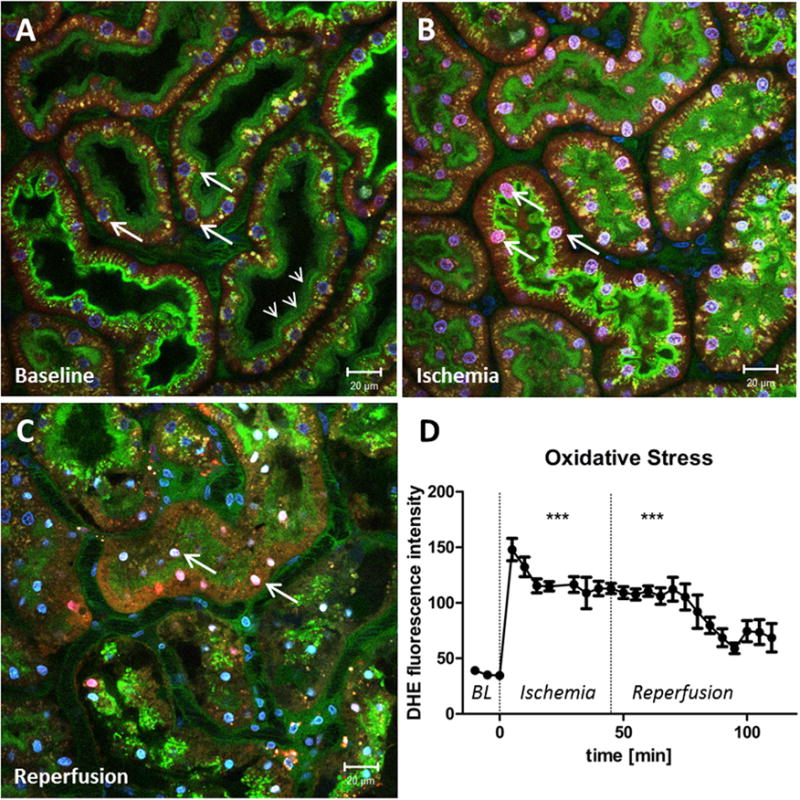
Oxidative stress in proximal tubules is enhanced by ischemia. (A) Renal cortex of a MWF rat during baseline conditions. The peritubular vasculature is labeled green by the injection of fluorescein isothiocyanate albumin. Glomerular-filtered fluorescein isothiocyanate albumin is reabsorbed in the PTs and stains the brush border green (small arrows). Cell nuclei are stained blue by Hoechst 33342 (big arrows). To indicate the generation of reactive oxygen species, DHE was injected into the jugular vein, which is activated to a red dye by superoxide and then translocates to the nucleus. Before ischemia the DHE staining of the PT nuclei is weak and oxidative stress is low. However, DHE intensity of the PT nuclei rapidly increases at the onset of ischemia (arrows in panel B) and remains increased during reperfusion (arrows in panel C). (D) Nuclear fluorescence intensity of DHE in PTs as a function of time before, during, and after ischemia. ***DHE fluorescence is significantly higher during ischemia and reperfusion, when compared with baseline (BL) values (P > .0001).
MitoSOX™ (Thermofischer, Waltham, USA) is a cell-permeable triphenylphoshonium derivate of DHE, which selectively targets mitochondria. When MitoSOX becomes oxidized by superoxide, it shows a red fluorescence. Furthermore, oxidation of MitoSOX occurs only by superoxide, not by other reactive oxygen or nitrogen species.67 A series of studies used MitoSOX in vivo to investigate the role of epithelial superoxide generation in sepsis-induced AKI. Sepsis was induced using the murine cecal ligation and puncture model, and superoxide generation and renal hemodynamics were investigated using intravital video microscopy. MitoSOX fluorescence was generally localized to tubules adjacent to capillaries without blood flow and the administration of the mitochondria-targeted antioxidant Mito-TEMPO (Sigma-Aldrich, St. Louis, USA) partly improved the hemodynamic dysfunction. These results suggest a relationship between tubular ROS generation and hemodynamic dysfunction in sepsis-inducedAKI.46,68
The Role of Ca2+ in Epithelial Dysfunction and Cell Death
Another main determinant of mitochondrial function is the mitochondrial matrix Ca2+ level [Ca2+]m. Increased Ca2+ flux through the mitochondrial Ca2+ uniporter increases [Ca2+]m, which acts as an allosteric activator of the rate-limiting enzymes pyruvate, isocyanate, and α-ketoglutarate dehydrogenase. The activation of these enzymes results in enhanced production of NADH, which increases ATP generation.69 However, if [Ca2+]m levels have an excessively high increase, cell death can be induced via the mitochondrial permeability transition pore (mPTP). mPTP is a nonspecific high-conductance channel in the inner mitochondrial membrane. Opening of the pore leads to unrestricted solute movement, followed by swelling of the mitochondria caused by osmotic water influx. Rupture of the outer mitochondrial membrane as a result of swelling eventually leads to cytochrome c release, which mediates apoptosis.70 mPTP furthermore is involved into necrotic cell death, but the underlying mechanisms are yet not fully understood.1,70 However, mPTP-mediated cell death contributes to renal injury followed by IRI.1 During ischemia, cell acidification contributes to an intra-cellular accumulation of sodium that cannot be eliminated by the Na/K adenosine triphosphatase owing to ATP depletion. As a consequence, the Na+/ Ca2+ exchanger removes sodium at the expense of increasing intracellular Ca2+ levels [Ca2+]i.71 Mitochondrial ROS release further contributes to increasing [Ca2+]i, which is a trigger of mPTP opening, once the Ca2+ enters into the mitochondria.70 In vivo imaging of intracellular Ca2+ alterations would help to better understand the dynamic developments of tubular damage and cell death during renal IRI. However, this has not been performed because of technical difficulties in the application of Ca2+ -sensitive dyes in vivo.21
Strikingly, changes of [Ca2+]i within the living animal recently were visualized, using a genetic approach. Burford et al41 selectively expressed the calcium indicator GCaMP3 in podocytes to study [Ca2+]i changes in podocytes during injury and disease in vivo. In podocytes, Ca2+ homeostasis regulates the remodeling of the actin filaments in the foot processes, which is necessary to maintain the slit diaphragm.72 Rearrangements of the actin cytoskeleton lead to slit diaphragm disruption, foot process effacement, and albuminuria.73 Furthermore, there is evidence that increasing [Ca2+]i is involved in podocyte cell death in albumin-,74 and glucose overload-,75 induced injury. Consistent with this, intravital investigations of podocyte [Ca2+]i in an experimental model of focal segmental glomerulosclerosis (FSGS) showed higher Ca2+ levels within clustered and damaged podocytes compared with unaffected glomerular epithelial cells. Importantly, using this intravital approach, [Ca2+]i and glomerular function can be determined simultaneously. Stimulation of a single podocyte by the use of the laser as a micromanipulator induced an intense increase in [Ca2+]i of the affected cell, which then spread over adjacent podocytes. At the same time, increased leakage of fluorescently labeled albumin was observed, which originated from the injury site and then extended around the adjacent capillaries.41 This study directly visualized and linked changes of podocyte [Ca2+]i to glomerular dysfunction in vivo.
Apical Membrane Blebbing and Cell Shedding
To facilitate efficient reabsorption of water, solutes, and albumin, the surface area of the proximal tubule is increased, owing to the presence of numerous microvilli. These microvilli are supported by an energy-consuming actin cytoskeleton.76
MPM has been used in several studies to show the severe effects of IRI on the apical membrane of the proximal tubule.31,77 Ischemia leads to apical microvilli breakdown and actin-cytoskeleton derangement, which finally results in membrane blebbing and a reduction of the apical surface area.76 The maintenance of the actin cytoskeleton depends on ATP, and apical membrane blebbing was observed with simultaneous alterations of Δψm during ischemia of the isolated perfused kidney.78 Along with apical membrane blebbing, DAMPs are released into the tubular lumen, which leads to the onset of necroinflammation by activating PRRs of downstream-located PT cells.16 DAMPs and membrane blebs may accumulate further and form tubular casts (Fig. 3), which restrict tubular flow and might contribute to the rapid decrease in GFR observed in AKI.79
Figure 3.
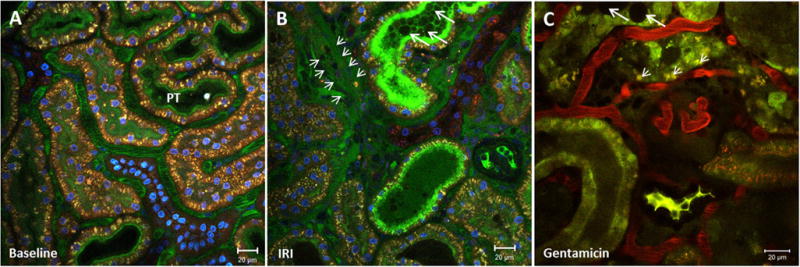
Visualization of apical membrane blebbing and cell shedding. Renal cortex of a MWF rat (A) before and (B) after a 45-minute ischemia period. The peritubular vasculature is stained green by fluorescein isothiocyanate albumin and the nuclei blue by Hoechst 33342. (B) IRI induces necrotic cell death in proximal tubules and leads to apical membrane blebbing, cell shedding, and the release of DAMPs, which accumulate in the tubular lumen and form tubular casts (big arrows). In areas of diminished peritubular blood flow, extravasation and edema formation is visible by the appearance of fluorescein isothiocyanate albumin in the interstitium (small arrows). (C) Renal cortex of a gentamicin-treated rat (100 mg/kg on 5 consecutive days). Proximal tubular dysfunction can be detected by enlarged lysosomes within the PT cells (small arrows). Apical membrane blebbing, shedded cells, and DAMPs form tubular casts (big arrows) in the lumen of some PTs.
Gentamicin-induced AKI (gentamicin-injections on several consecutive days) leads to similar tubular alterations. The first visible abnormalities are enlarged lysosomes, which appear within the first 2 days49 of aminoglycoside treatment. Continuous gentamicin treatment eventually leads to severe brush-border alteration, apical membrane blebbing, and tubular casts as visible in Figure 3.
Apical membrane blebbing inevitably goes along with cell death by necrosis, which can be visualized by intravital MPM. A recent study documented apical membrane blebbing and DAMP release of necrotic cells in vivo.31 Dead cells were visualized by an intravenous injection of propidium iodide (PI). PI is a DNA intercalating fluorescent dye that is impermeable to the intact cell membrane and therefore primarily labels necrotic cell nuclei (Fig. 4). After a 30-minute ischemia period, a time series within the first 60 minutes of reperfusion was performed. The intravital movie showed the PI staining of several PT cell nuclei, indicating the loss of an intact cell membrane. One of the PI-labeled cells swelled and, upon an explosive rupture of the cell membrane, eventually released DAMPs into the tubular lumen. Nuclear PI staining and the obvious membrane rupture of the affected cell suggested necrosis.31
Figure 4.
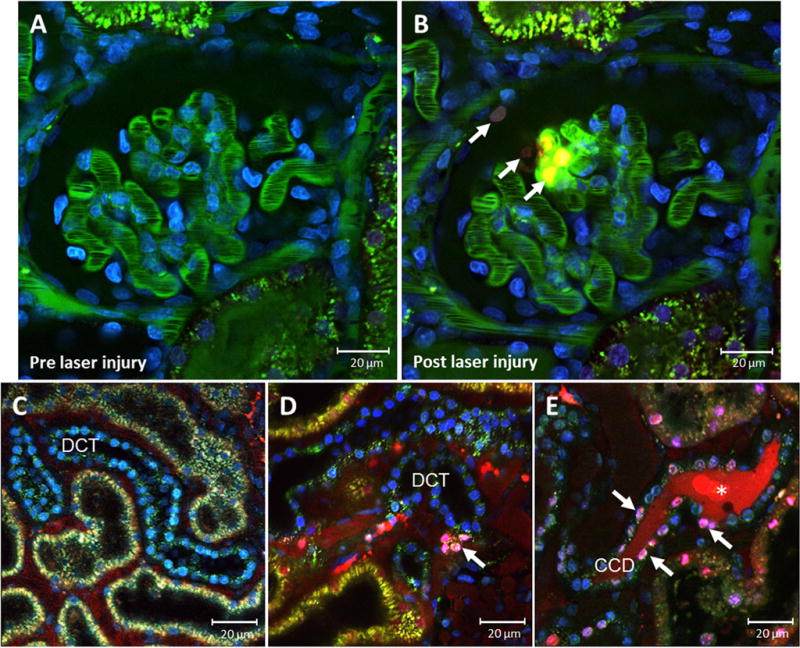
Visualization of necrotic cell death. MWF rat glomerulus before and after laser-induced damage. Glomerular vasculature was stained green by fluorescein isothiocyanate albumin and cell nuclei were stained blue by Hoechst 33342. In addition, PI, a cell membrane–impermeable dye, was injected into the jugular vein to visualize cell necrosis. PI intercalates the DNA of cells with disrupted cell membrane and stains the nuclei red, indicating necrosis. (B) After the laser-induced injury, necrotic cell nuclei are labeled red (arrows) in the glomerulus and in the Bowman’s capsule. Representative in vivo MPM images of cell death in the DCT during (C) control conditions and (D and E) after 3 days of hydrochlorothiazide treatment (6 mg/kg/d via drinking water). MWF rats were injected with Hoechst 33342 (blue), and propidium iodide, which labels dying cells (purple, arrows) in the DCT collecting duct. *Cell debris is shown in the lumen of the cortical collecting duct (CCD).
A recent intravital MPM study showed the dynamic loss of damaged podocytes in an experimental model of FSGS. Puromycin aminonucleoside administration leads to severe alterations of the podocyte actin cytoskeleton and the formation of pseudocysts, which may represent enlargement of the subpodocyte space.24,80 Glomerular epithelial cells can be visualized in vivo by negative staining during a continuous infusion of Lucifer yellow (LY). LY is a small-molecular-weight dye and is filtered freely into Bow-man’s space. During continuous administration, glomerular endothelial and mesangial cells take up the LY over time and turn a bright green color. On the contrary, healthy podocytes are excluded from any staining and appear as dark unstained objects on the outer margins of the glomerular capillary loops24 (Fig. 1). However, intravital MPM showed that damaged podocytes in the puromycin aminonucleoside model lose their cell membrane integrity and small green punctuated regions in the cell were formed as a result of the continuous uptake of LY. Within several minutes these podocytes turned amorphous and the green fluorescence increased further. In the following, intravital MPM showed the rapid detachment of these damaged podocytes, of which most disappeared into the tubular fluid. The disruption of the podocyte cell membrane, which resulted in the accumulation of LY, was consistent with cell necrosis. Most importantly, the loss of podocytes in FSGS correlated with a local disruption of the integrity of the glomerular filtration barrier. Accordingly, the filtration of fluorescently labeled 70-kDa dextran into the Bowman’s space was substantially higher adjacent to damaged and shedded rather than intact podocytes of the same glomerulus.24
Cell Death Visualization Using Intravital MPM
In vivo cell death evaluation was performed mainly in tissues using the terminal deoxynucleotidyl transferase-mediated deoxyuridine triphosphate nick-end labeling (TUNEL) assay. TUNEL staining detects double-strand breaks, which are a hallmark of apoptotic cell death. The TUNEL assay therefore commonly was used to detect apoptotic cell death. Nevertheless, concurrent evidence suggests that endonuclease action also can arise from regulated necrosis, which means that the TUNEL assay might indicate regulated necrosis as well. A distinct differentiation of apoptosis from necrosis therefore requires additional technical efforts, such as immunostaining or Western blot for activated caspase-3.81 Another problem using the TUNEL assay is that it might underestimate tissue apootic cell death owing to the short half-life of apoptosis.82
An elegant way to follow up cell death in renal injury and disease is the intravital approach, which allows the visualization of cell death in a time-dependent manner. Here, we show how propidium iodide can be used to detect kidney cell death in vivo and show the nontraditional effects of a high-dose hydrochlorothiazide treatment. Thiazide diuretics commonly are used for the treatment of hypertension owing to their potent inhibition of NaCl reabsorption in the distal convoluted tubule (DCT) and in the CD.83,84 However, as a nontraditional effect, thiazide treatment also was shown to provoke DCT cell death, thereby shortening the DCT and further contributing to natriuresis.85 This new and unconventional effect of thiazide diuretics, namely to cause cell death of the DCT-CD, is shown in Figure 4. Three days after the thiazide application, intense PI staining was found mainly in the collecting duct, which is excluded from living cells, but labels dying cells.
By the simultaneous use of two different DNA intercalating dyes, intravital MPM can be used further to classify kidney cell death. Kelly et al82 applied the red-emitting PI in addition to Hoechst 33342 (Hoechst), a blue-emitting cell membrane-permeable dye that labels the nuclei of all viable and dying cells. In response to renal IRI, this in vivo approach allowed the differentiation of four different cell types. Viable cells were recognized by normal Hoechst-induced nuclei staining and the lack of PI co-staining. On the contrary, necrotic cells showed bright red PI staining in addition to normal Hoechst-labeled nuclei. Apoptotic cells did not co-label with PI because of an intact cell membrane, but Hoechst staining showed condensed and fragmented cell nuclei, which is characteristic of apoptosis. The last cell type showed the same condensed and fragmented nuclei, but stained positively for PI. These cells were defined as secondary necrotic cells.82 Apoptotic cells normally are eliminated by scavenger cells, such as macrophages. However, if this mechanism is disturbed, apoptotic cells undergo secondary necrosis and autolytic disintegration.86 With this classification the investigators relied on the common assumption that nuclear fragmentation only occurs in apoptotic but not in necrotic cells. However, as mentioned earlier, double-strand breaks and nuclear fragmentation does not seem to be restricted to apoptosis and likely can occur in regulated necrosis as well. Consequently, it cannot entirely be ruled out that the cell type categorized as secondary necrosis also might derive from regulated necrosis.
In this context, a very recent intravital MPM study introduced a new tool that might be useful to differentiate between secondary necrosis and regulated necrosis in vivo. Hato et al32 first applied Phiphilux G2D2 (Phiphilux), a caspase-3-activated dye in vivo. Phiphilux is well characterized in vitro and is a cell-permeable dye, which emits red fluorescence when cleaved by caspase-3. Importantly, the cleavage by caspase 3 does not interfere with caspase-3 activity,87 which enables an in vivo study of apoptosis. The investigators applied the dye before a 30-minute ischemia period and investigated apoptosis during reperfusion. Within 60 minutes of reperfusion, Phiphilux-positive cells were detected randomly within S2 and S3 segments of the proximal tubule. Co-staining with Hoechst identified condensed nuclei of the Phiphilux-labeled cells. Interestingly, no apoptosis was found in S1 PT cells, which is in agreement with earlier studies that suggested highest caspase-3 activity in S2 and S3 proximal tubules in response to IRI.88 However, it should be mentioned that Phiphilux is cleaved unspecifically within the brush border of S1 proximal tubules by undefined mechanisms. Although this unspecific staining was well distinguishable from the real signal, Phiphilux apparently has to be used with care in vivo.32
Considering the recent development in the research field of regulated necrosis, future applications of Phiphilux together with the established combination of Hoechst and PI may lead to a clearer differentiation of apoptosis and primary and secondary necrosis in vivo.
ENDOTHELIAL DYSFUNCTION
Endothelial cells and microvascular blood flow are local determinants of coagulation, vascular permeability, and inflammation. AKI and CKD both lead to profound peripheral vascular dysfunction, such as alterations in capillary blood flow, coagulopathy, increased vascular permeability with edema, and local tissue inflammation resulting from leukocyte rolling and adhesion.42,89 The presence and extent of vascular dysfunction in renal disease often show a patchy character, with adequately perfused areas in close proximity to areas with diminished blood flow and local tissue inflammation. This finding suggests that the regulation of local tissue perfusion has a considerable impact on the pathology of renal injury and disease.42 Intravital microscopy offers the opportunity to investigate endothelial and microvascular alterations in the living animal during health, injury, and disease.
Microvascular Blood Flow and Endothelial Cell Death
Intravital MPM can be used to determine glomerular and peritubular capillary flow by applying the linescan technique.27 The principal of this measurement takes advantage of the fact that red blood cells (RBCs) appear as dark unstained objects inside the capillaries, while the plasma is labeled with a vasculature dye. As vasculature dyes, large fluorophore-conjugated molecules are used, which are barely filtered in the kidney, such as high-molecular-weight dextrans or albumin. Within the central axis of a capillary, a distinct longitudinal distance is subject to repetitive scans (so-called linescans). The result indicates the movement of the RBCs over the distinct distance (X) within the capillary over time (T) in an XT image. Within the XT image, the movement of the RBCs leaves dark diagonal bands within the data set and the slope of these bands is related inversely to the RBC velocity. The velocity (μm/ms) of the RBCs can be calculated from this data set as ΔX/ΔT(Fig. 1).
Intravital microscopy showed that capillary blood flow is reduced in sepsis-induced90,91 and ischemia reperfusion—induced92,93 AKI. Patchy areas of diminished blood flow are detectable immediately94,95 after reperfusion and remain hypoperfused within 24 and 48 hours after IRI.92,93 Finally, several days after the ischemic injury a severe reduction of the vascular density manifests96,97 a long-term complication of endothelial injury and cell death, which is known as vascular rarefaction. Vascular rarefaction in AKI might accelerate CKD progression98 and also is observed in CKD of other etiologies99 and in the aging kidney.100 Endothelial injury in AKI and CKD is associated with enhanced pro-apoptotic stimuli, such as increased levels of tumor necrosis factor α and interleukin-1.101,102 Furthermore, the expression of endothelial survival stimuli, such as the vascular endothelial growth factor, are decreased.101,103
Impaired microvascular blood flow might contribute to the development of vascular rarefaction. Thus, diminished laminar shear stress on the capillary endo-thelium enhances endothelial apoptosis.104 Furthermore, vascular pericytes migrate away from the capillaries and differentiate into myofibroblasts, which contributes to microvascular destabilization.101 However, in comparison with tubular epithelial cells, the regenerative capacity of endothelial cells is limited. Bromdesoxyuridin (BrdU) stainings up to 7 days after renal IRI showed no significant proliferation of endothelial cells when compared with sham-operated animals. In contrast, tubular proliferation increased continuously after renal ischemia.97 This highlights the significance of local microvascular hypoperfusion and endothelial cell death in the progression of kidney injury and disease.
Intravital MPM is a powerful tool to detect the time-dependent correlations of vascular remodeling and dysfunction. In a recent intravital MPM study a constitutive Tie2-Cre mouse, which expressed YFP in endothelial cells and activated pericytes, was subject to renal ischemia. In the following, the microvascular dysfunction was investigated. The investigators found that Tie2-positive cells migrate from the peritubular capillaries into the interstitium. This could be a result of endothelial mesenchymal transition, a phenotype switch of endothelial cells into fibroblasts, or indicate capillary destabilization caused by migration of activated pericytes. However, the highest level of Tie2-positive cell migration was detected in areas of diminished capillary flow, which directly linked the impairment of local tissue perfusion with vascular remodeling.97
Microvascular Permeability Dysfunction and Edema Formation
Vascular permeability can be detected by the injection of fluorophores, which are conjugated to either high-molecular-weight dextrans or albumin.30 During physiological conditions, plasma proteins, such as albumin, are retained by capillaries. However, in renal disease, such as AKI and CKD, glomerular and peritubular vascular permeability is enhanced, which leads to proteinuria and the formation of edema.25,89,95
Intravital MPM studies have shown that areas of diminished microvascular blood flow are particularly subject to increased vascular permeability and edema formation.95,97 As shown in Figure 3, in the renal cortex, the tubules and peritubular capillaries are located close to each other. The vasculature was labeled using fluorescein isothiocyanate albumin, which was retained within the peritubular vasculature. Next, renal ischemia was applied for 45 minutes. During the 60-minute reperfusion period, extravasation within areas of diminished microvascular blood flow was visible, and fluorescein isothiocyanate albumin appeared in the interstitium (Fig. 3).
Such alterations in microvascular blood flow and permeability are caused by local inflammation, endothelial cell swelling, and variations of the endothelial actin cytoskeleton and of the endothelial cell junctions.42,95 Furthermore, several intravital MPM studies have suggested that coagulopathy plays a major role in microvascular dysfunction during AKI.90,92 Physiological shear stress of the flowing blood on the endothelial surface promotes endothelial cell survival and the release of vasodilators, such as nitric oxide, and substances that inhibit hemostasis.105 Thrombomodulin, which is expressed physiologically by endothelial cells, is a cofactor in the generation of activated protein C (APC). APC is an endogenous anticoagulant and shows reduced serum levels in septic patients.106 In addition, decreased capillary shear stress further promotes the release of vasoconstrictors and favors platelet aggregation and endothelial apoptosis.105 Overall, endothelial dysfunction and cell death result in enhanced microvascular coagulation and decreased local tissue perfusion. Consistent with this hypothesis, the application of soluble thrombomodulin or APC in animal studies of septic90 and ischemic92 AKI showed beneficial effects on microvascular blood flow, peritubular capillary integrity, and local inflammation.
Another important determinant of vascular permeability is the endothelial glycocalyx. The endothelial glycocalyx is a 150- to 500-nm gel-like apical surface layer and common to all blood vessels. It mainly consists of glycosaminoglycans, such as heparin sulfate, hyaluronan, and chondroitin sulfate and heteropolysaccharides. The endothelial glycocalyx acts as a vascular permeability barrier and as an endothelial sensor of hemodynamic shear stress.107 Alterations of the glycocalyx composition are associated with glomerular albuminuria in diabetes,108 FSGS,25 and ischemic-109 and sepsis-induced110 AKI. Furthermore the glycocalyx also contributes to the integrity of non-glomerular vasculature. For example, coronary arterioles showed an increased vascular permeability of albumin in response to glycocalyx disrupture by heparinase treatment.111
A key mechanism of glycocalyx disruption in vivo is increased oxidative stress, which leads to the depolymerization of glycosaminoglycans.107,112 Furthermore, irregular blood flow and shear stress alterations enhance endothelial ROS generation113 and the release of endothelial cell-derived proteases, which cleave glycocalyx components.109 The cleaved components of the glycocalyx then are subject to shedding and excretion.114 Furthermore, extracellular matrix metalloproteinases, which are synthetized by endothelial cells, contribute to glycocalyx shedding.107 In ischemic115 and septic116 AKI, the expression and activation of matrix metalloproteinases is enhanced. Consistent with this finding, Sutton et al115 found decreased vascular permeability and local inflammation after ischemic injury when they investigated the effects of an matrix metal-loproteinase inhibitor using intravital MPM.
A recent intravital MPM study visualized the endothelial glycocalyx in vivo and showed a direct correlation between the structural impairment of the glycocalyx and vascular permeability dysfunction. The investigations were performed in old Munich Wistar Froemter (MWF) rats, which develop spontaneous hypertension, proteiunuria, and FSGS. Salmon et al25 labeled the glycocalyx in vivo by fluorescently labeled wheat germ agglutinin lectin, which binds to N-acetylglucosamine, a component of heparin sulfate and hyaluronan glycosaminoglycans. Because of the high resolution of MPM, the thickness of the glomerular and peritubular endothelial glycocalyx can be determined. In comparison with young and healthy MWF rats, the thickness of the endothelial glycocalyx was reduced in glomerular and mesenteric vessels of old MWF rats. In addition, there was a 50% reduction of the overall coverage of the vasculature with glycocalyx. Importantly, the loss of blood vessel coverage by the endothelial glycocalyx correlated with an increase in glomerular albumin filtration. Similarly, the vascular permeability of mesenteric arteries with impaired endothelial glycocalyx was increased.25 The results of this study highlight the importance of the endothelial glycocalyx for the vascular integrity in vivo and showed how intravital MPM can be used to study vascular dysfunction in other disease etiologies, such as AKI.
Leukocyte Rolling, Adhesion, and Inflammation
Renal injury and disease are associated with increased ROS, heat shock protein, and cytokine signaling, which can lead to the activation of endothelial cells.42 PT cell signaling upon stress and injury especially plays a key role in endothelial cell activation. In septic-induced AKI, proximal tubular stress and ROS release correlates with microvascular blood flow alterations, local inflammation, and coagulation.45,68,91,117 Once activated, endothelial cells show enhanced expression of adhesion molecules, such as CD 54 and P-selectin, which induce leukocyte adhesion.118–120 Furthermore, the disruption of the endothelial glycocalyx leads to modifications of heparin sulfate proteoglycans, which enable the binding of monocyte chemoattractant protein-1 and the enhanced recruitment of leukocytes.121,122 Moreover, increased vascular permeability enhances leukocyte tissue infiltration.95,123
Leukocyte recruitment and local tissue inflammation play a key role in the progression of AKI and CKD.16,124 Cells of the innate and the adaptive immune system are involved in a bidirectional causality between kidney injury and inflammation. Injured tubular epithelial cells release cytokines and ROS and thereby activate endothelial and immune cells. Enhanced cytokine and ROS release in leukocytes in turn serve as a positive feedback pathway, which leads to enhanced inflammation, cell injury, and cell death.16,102 The specific contribution of distinct leukocyte subpopulations in this inflammatory and cytotoxic response recently was reviewed in detail.123,124
Leukocyte recruitment, rolling, and adhesion can be visualized using intravital MPM. Several studies used this technique and contributed to a better understanding of leukocyte recruitment in sepsis-45,90 and ischemia-92,115,125 induced AKI and glomerulonephritis.28,120
Similar to RBCs, leukocytes are excluded from staining when a vasculature dye is injected. However, unlike RBCs, their velocity within the microvasculature is slower, and in disease models they can be identified as round black objects rolling on the endothelial capillary walls.29 In addition, several techniques to label leukocytes in vivo have been established. Leukocytes are labeled most commonly by the intravenous application of the DNA-intercalating dye Hoechst. Because RBCs have no cell nuclei, only white blood cells are stained by Hoechst within the plasma.90,92 Leukocytes also were shown to endocytose LY when continuously infused.24 Furthermore, acridine orange and rhodamine 6G are used, which accumulate in leukocytes after intravenous injection.120,126 However, this approach presents limitations as a result of parenchymal tissue accumulation and phototoxic-induced hemodynamic alterations.127,128 A promising new dye, which apparently avoids these disadvantages, recently was tested for intravital imaging of leukocytes in the liver.127 Carboxyfluorescein diacetate succinimidyl ester does not compromise leukocyte circulation and recruitment,129 and in vivo staining of leukocytes showed a high imaging quality of leukocytes.127 However, carboxyfluorescein diacetate succinimidyl ester has not been used for in vivo imaging of leukocytes in the kidney yet.
For the distinct staining of certain white blood cell (WBC) subtypes, leukocytes can be isolated, purified, and labeled ex vivo using acridine orange. The tagged leukocytes then can be re-injected into an animal for in vivo monitoring.115,126 Furthermore, PKH-26 preferentially stains phagocytic cells, such as neutrophils,130 and fluorescently labeled dextrans accumulate in lysosomes of macrophages.29
Another technique to visualize distinct WBC subtypes is the intravenous injection of leukocyte-specific fluorescent antibodies.28 However, a drawback of this technique is the risk of functional alterations of the leukocytes caused by the interaction of the antibody with its target receptor. On the contrary, a very elegant but expensive approach is the genetic expression of fluorescent proteins under the control of cell-specific promotors.29
A recent study combined in vivo antibody staining and a genetic approach to investigate neutrophil and monocyte behavior in the healthy glomerulus and during glomerulonephritis.28 GFP-expressing monocytes and in vivo Gr1 antibody—stained neutrophils were monitored within the glomerulus using intravital MPM. An unexpected finding of this study was that monocytes and neutrophils also patrol the healthy glomerulus. Thus, GBM antibody—induced inflammation did not increase the total number of recruited leukocytes. However, the leukocyte retention in the glomerulus was enhanced. These results were obtained in healthy, young BL6 mice. To increase the amount of superficial glomeruli for further investigation of leukocyte interaction, some of the experiments were performed in hydronephrotic kidneys (12 weeks of unilateral uretic obstruction [UUO]). In UUO animals, similar results for monocyte and neutrophil behavior were obtained before and after GBM antibody administration. By using the UUO model, the investigators further found a profound role of leukocyte ROS generation in glomerular inflammation. The ROS content of neutrophils in wild-type and Nox2-deficient mice was visualized by the application of DHE. Although the number of adherent neutrophils within the glomerulus remained unaffected in both genotypes, neutrophil ROS production and GBM antibody-induced albuminuria were eliminated in the Nox2-deficient mice. These results showed that the ROS generation in neutrophils is under the control of the reduced nicotinamide adenine dinucleotide phosphate oxidase. Furthermore, the data suggested a major role of neutrophil-mediated ROS production in the disruption of the glomerular filtration barrier in inflammatory disease.28
Because of the short duration of leukocyte retention in the glomerulus during physiological conditions, the histologic detection of WBCs in healthy glomeruli is limited. A histologic section only provides a snapshot of the leukocyte interaction, while intravital MPM allows the monitoring of WBCs in a time-dependent manner. Devi et al28 combined this intravital approach with elegant new genetic tools and gained new insights in the role of neutrophil signaling and the disruption of the glomerular filtration barrier. However, it has to be considered that the data were generated in part using the UUO model, which has an inflammatory character by itself.131
FUTURE DIRECTIONS
Many investigations of AKI and CKD-related tissue injury and cell death are based on autopsies. However, when it comes to AKI, a profound discrepancy between minimal histologic alterations and severe renal dysfunction was detected a long time ago.132 The underlying pathology of AKI includes complex communication of injured epithelial and endothelial cells with the immune system. These interactions drive cell death, microvascular dysfunction, and a rapid decrease in GFR.16,102 Sterile inflammation furthermore also plays a profound role in the progression of CKD.20 For a better understanding of this dynamic pathology, the simultaneous investigation of renal morphology, function, and cell viability is desirable.
Intravital MPM now has been used for more than a decade and has contributed tremendously to our understanding of renal physiology.21 It is a unique approach, which relates structure and function at a cellular and subcellular level with high optical resolution. As outlined in this review, intravital MPM can be used to study epithelial and endothelial dysfunction during kidney injury and disease. Homeostatic alterations, which are highly related to regulated cell death, can be detected readily at a subcellular level. For example, intravital MPM was used successfully to study changes in intracellular calcium41 and ROS68 levels and mitochondrial dysfunction.49 Importantly, this intravital approach allows the investigation of the spatial- and time-dependent relationship between these intracellular alterations and the event of cell death. Furthermore, recent advantages in intravital microscopy enabled serial imaging of the same kidney structures, such as the glomerulus, over several days.26 MPM can be used to study basic renal function, such as microvascular blood flow and permeability, snGFR, and proximal tubular reabsorption.27 Most relevant to the topic of this issue, serial MPM also allows investigators to study the dynamics of cell death, and the effects of kidney cell death on renal function in a time-dependent manner.
In this context, intravital MPM recently showed the shedding of a necrotic podocyte into the Bowman’s space in a model of FSGS.24 Within minutes the integrity of the podocyte cell membrane was lost, before the amorphous cell then rapidly detached from the glomerular basal membrane. This dynamic event resulted in the local increase of glomerular vascular permeability. However, this was restricted to the area of visceral damage, and the filtration of macromolecules adjacent to intact podocytes of the same glomerulus was normal. This emphasizes the significance of podocytes for the integrity of the glomerular filtration barrier. Furthermore, these data point out how intravital MPM can be used to show the dynamics of renal cell death and to trace the resulting functional alterations in the kidney. Therefore, future intravital investigations of cell death and kidney function will help to improve our knowledge of cell death, and its significance in renal disease.
Cell death visualization using intravital MPM is performed most commonly using Hoechst and PI. Hoechst indicates apoptosis by the visualization of nuclear condensation whereas PI labels the nuclei of necrotic cells, with disrupted cell membrane.82 One of the earliest events during apoptosis is the translocation of membrane phosphatidylserine from the inner side of the plasma membrane to the cell surface.133 Annexin V, a 36-kDa protein, binds phosphatidylserine in a Ca2+-dependent manner with high affinity. Annexin V successfully detects apoptosis in vitro and indicates the apoptotic events before nuclear condensation.134 Conjugated to near-infrared fluorophores it also was used for in vivo cell death studies using fluorescence microscopy.135,136 However, the in vivo application of Annexin V is limited owing to pharmacokinetic problems and a low signal/ noise ratio.136 Several small-molecule probes, such as NST-732137 and fluorescent-conjugated zinc (II) dipicolylamine,136 were developed to overcome these problems. NST-732 and zinc (II) dipicolylamine also target phosphatidylserines and show a better signal/noise ratio in vivo. These probes could be valuable in future intravital MPM studies to detect cell death at an early time point. However, it should be mentioned that Annexin V, NST-732, and zinc (II) dipicolylamine also stain necrotic cells. Once the cell membrane is ruptured, these dyes can penetrate then ecrotic cell and then bind to intra-cellular phosphatidylserines.135–137 Thus, they do not allow a distinct differentiation between apoptotic and necrotic cells.
In the past decade, necrosis has received considerable attention in cell death research. Unlike the former assumption that necrosis is an uncontrolled process, there is evidence that necrosis follows regulated pathways.18 However, little is known about the relevance of regulated necrosis in renal disease.1 Therefore, the development of new molecular probes to target specific subtypes of regulated cell death in vivo would be desirable. The caspase-3-activated dye, Phiphilux, recently was established for intravital MPM detection of apoptosis in the kidney. Despite its unspecific activation in S1 PT cells, it indicates apoptosis reliably in other nephron segments and might serve as a promising tool to distinguish apoptosis from regulated necrosis in future studies.32
Necrosis plays a key role in the progression of renal injury by a mechanism called necroinflammation.16 Upon cell membrane rupture, necrotic cells release proinflammatory substances, which can be detected by pattern recognition receptors on tubular epithelial cells and by immune cells. This leads to an inflammatory response of the activated cells, which further enhances inflammation and cell death.16,102 Intense immunologic research increased our understanding of the contribution of leukocyte subtypes to renal inflammation and disease. However, many of these studies were performed in vitro, whereas the detection of leukocyte recruitment often was performed ex vivo in kidney biopsy specimens.138 Although histologic stainings of tissue recruitment of leukocytes only provide a snapshot of their interactions, intravital MPM allows the monitoring of leukocyte rolling and adhesion in real time.28
Devi et al28 recently showed the power of this approach in a model of glomerulonephritis. They used a genetic approach to identify monocytes and neutrophils by the expression of GFP and imaged their actions within the inflamed glomerulus over time. This study showed that instead of an increase of recruited leukocytes in number, rather the retention time of the available leukocytes was enhanced.28
Over the past decades, genetically modified animal models became available and enabled an elegant way to target specific cells in vivo with fluorescent proteins.139 Several studies already combined intravital MPM and genetic approaches for cell fate tracing26,97 and functional measurements.28,41 However, because of technical difficulties, new genetic approaches are available mainly in the mouse.140 In contrast to the MWF rats, a common rat strain used for MPM studies, the glomeruli in most mouse strains are located much deeper within the renal cortex, which aggravates intravital imaging.141 Therefore, Devi et al28 used hydronephrotic kidneys to study the glomerulus. However, a recent study showed that imaging of glomeruli is possible in certain mouse strains141 and several studies that examined glomeruli in the mouse were published.26,41,142 Furthermore, the excitation with light of longer wavelength can further enhance the penetration depth in renal tissue62 and promote the use of genetically modified mouse models for in vivo imaging of the glomerulus.
Given the new finding that necrosis can occur as a regulated process, a new opportunity emerges to interfere with necrosis therapeutically. Several inhibitors of regulated necrosis have been developed and studies have tested their protective effects in renal injury.143–145 In AKI and CKD, nephron loss is compensated for by the hyperfiltration of the remaining nephrons.146 However, an increase in snGFR and workload may exceed the capacity of the nephron, and the peritubular blood oxygen delivery does not deliver sufficient oxygen and metabolic substrates anymore. The result is ischemia and hypoxia. Enhanced ROS generation further drives cellular injury and contributes to a vicious cycle, leading to progressive nephron loss and eventually to end-stage renal disease.146
To improve the outcome of kidney disease, nephron loss must be minimized. Future treatment of patients with inhibitors of regulated cell death might prevent nephron loss. Because kidney cell injury and cell death is linked inevitably to renal dysfunction and structural alterations, MPM is a unique tool to study the full potential of therapeutic interference with regulated cell death in vivo. New intravital serial imaging and genetic labeling strategies will further enhance our knowledge of kidney cell death dynamics in the future.
Footnotes
Financial disclosure and conflict of interest statements: none.
References
- 1.Linkrermann A, Chen G, Dong G, et al. Regulated cell death in AKI. J Am Soc Nephrol. 2014;25:2689–701. doi: 10.1681/ASN.2014030262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rahman M, Shad F, Smith MC. Acute kidney injury: a guide to diagnosis and management. Am Fam Physician. 2012;86:631–9. [PubMed] [Google Scholar]
- 3.Hsu RK, McCulloch CE, Dudley RA, Lo LJ, Hsu CY. Temporal changes in incidence of dialysis-requiring AKI. J Am Soc Nephrol. 2013;24:37–42. doi: 10.1681/ASN.2012080800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hsu CY. Yes, AKI truly leads to CKD. J Am Soc Nephrol. 2012;23:967–9. doi: 10.1681/ASN.2012030222. [DOI] [PubMed] [Google Scholar]
- 5.Reidy K, Kaskel FJ. Pathophysiology of focal segmental glomerulosclerosis. Pediatr Nephrol. 2007;22:350–4. doi: 10.1007/s00467-006-0357-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tryggvason K. Unraveling the mechanisms of glomerular ultrafiltration: nephrin, a key component of the slit diaphragm. J Am Soc Nephrol. 1999;10:2440–5. doi: 10.1681/ASN.V10112440. [DOI] [PubMed] [Google Scholar]
- 7.Remuzzi G, Perico N, Macia M, Ruggenenti P. The role of renin-angiotensin-aldosterone system in the progression of chronic kidney disease. Kidney Int Suppl. 2005;99:S57–65. doi: 10.1111/j.1523-1755.2005.09911.x. [DOI] [PubMed] [Google Scholar]
- 8.Abbate M, Zoja C, Corna D, et al. In progressive nephropathies, overload of tubular cells with filtered proteins translates glomerular permeability dysfunction into cellular signals of interstitial inflammation. J Am Soc Nephrol. 1998;9:1213–24. doi: 10.1681/ASN.V971213. [DOI] [PubMed] [Google Scholar]
- 9.Okamura K, Dummer P, Kopp J, et al. Endocytosis of albumin by podocytes elicits an inflammatory response and induces apoptotic cell death. PLoS One. 2013;8:e54817. doi: 10.1371/journal.pone.0054817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lasagni L, Lazzeri E, Shankland SJ, Anders HJ, Romagnani P. Podocyte mitosis-a catastrophe. Curr Mol Med. 2013;13:13–23. doi: 10.2174/1566524011307010013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shankland SJ, Anders HJ, Romagnani P. Glomerular parietal epithelial cells in kidney physiology, pathology, and repair. Curr Opin Nephrol Hypertens. 2013;22:302–9. doi: 10.1097/MNH.0b013e32835fefd4. [DOI] [PubMed] [Google Scholar]
- 12.Berger K, Schulte K, Boor P, et al. The regenerative potential of parietal epithelial cells in adult mice. J Am Soc Nephrol. 2014;25:693–705. doi: 10.1681/ASN.2013050481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wanner N, Hartleben B, Herbach N, et al. Unraveling the role of podocyte turnover in glomerular aging and injury. J Am Soc Nephrol. 2014;25:707–16. doi: 10.1681/ASN.2013050452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tharaux PL, Huber TB. How many ways can a podocyte die? Semin Nephrol. 2012;32:394–404. doi: 10.1016/j.semnephrol.2012.06.011. [DOI] [PubMed] [Google Scholar]
- 15.Singh N. Apoptosis in health and disease and modulation of apoptosis for therapy: an overview. Indian J Clin Biochem. 2007;22:6–16. doi: 10.1007/BF02913307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mulay SR, Linkermann A, Anders HJ. Necroinflammation in kidney disease. J Am Soc Nephrol. 2016;27:27–39. doi: 10.1681/ASN.2015040405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Linkermann A, Green DR. Necroptosis. N Engl J Med. 2014;370:455–65. doi: 10.1056/NEJMra1310050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vanden Berghe T, Linkermann A, Jouan-Lanhouet S, Walc-zak H, Vandenabeele P. Regulated necrosis: the expanding network of non-apoptotic cell death pathways. Nat Rev Mol Cell Biol. 2014;15:135–47. doi: 10.1038/nrm3737. [DOI] [PubMed] [Google Scholar]
- 19.Dixon SJ, Lemberg KM, Lamprecht MR, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149:1060–72. doi: 10.1016/j.cell.2012.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dalla Vestra M, Mussap M, Gallina P, et al. Acute-phase markers of inflammation and glomerular structure in patients with type 2 diabetes. J Am Soc Nephrol. 2005;16(Suppl 1):S78–82. doi: 10.1681/asn.2004110961. [DOI] [PubMed] [Google Scholar]
- 21.Peti-Peterdi J, Burford JL, Hackl MJ. The first decade of using multiphoton microscopy for high-power kidney imaging. Am J Physiol Renal Physiol. 2012;302:F227–33. doi: 10.1152/ajprenal.00561.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Diaspro A, Bianchini P, Vicidomini G, et al. Multi-photon excitation microscopy. Biomed Eng Online. 2006;5:36. doi: 10.1186/1475-925X-5-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schiessl IM, Hammer A, Kattler V, et al. Intravital imaging reveals angiotensin ii-induced transcytosis of albumin by podocytes. J Am Soc Nephrol. 2016;27:731–44. doi: 10.1681/ASN.2014111125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Peti-Peterdi J, Sipos A. A high-powered view of the filtration barrier. J Am Soc Nephrol. 2010;21:1835–41. doi: 10.1681/ASN.2010040378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Salmon AH, Ferguson JK, Burford JL, et al. Loss of the endothelial glycocalyx links albuminuria and vascular dysfunction. J Am Soc Nephrol. 2012;23:1339–50. doi: 10.1681/ASN.2012010017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hackl MJ, Burford JL, Villanueva K, et al. Tracking the fate of glomerular epithelial cells in vivo using serial multiphoton imaging in new mouse models with fluorescent lineage tags. Nat Med. 2013;19:1661–6. doi: 10.1038/nm.3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kang JJ, Toma I, Sipos A, McCulloch F, Peti-Peterdi J. Quantitative imaging of basic functions in renal (patho) physiology. Am J Physiol Renal Physiol. 2006;291:F495–502. doi: 10.1152/ajprenal.00521.2005. [DOI] [PubMed] [Google Scholar]
- 28.Devi S, Li A, Westhorpe CL, et al. Multiphoton imaging reveals a new leukocyte recruitment paradigm in the glomerulus. Nat Med. 2013;19:107–12. doi: 10.1038/nm.3024. [DOI] [PubMed] [Google Scholar]
- 29.Atkinson SJ. Functional intravital imaging of leukocytes in animal models of renal injury. Nephron Physiol. 2006;103:86–90. doi: 10.1159/000092009. [DOI] [PubMed] [Google Scholar]
- 30.Dunn KW, Sutton TA, Sandoval RM. Live-animal imaging of renal function by multiphoton microscopy. Curr Protoc Cytom. 2012:9. doi: 10.1002/0471142956.cy1209s62. Chapter 14, Unit 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Linkermann A, Hackl MJ, Kunzendorf U, et al. Necroptosis in immunity and ischemia-reperfusion injury. Am J Transplant. 2013;13:2797–804. doi: 10.1111/ajt.12448. [DOI] [PubMed] [Google Scholar]
- 32.Hato T, Sandoval R, Dagher PC. The caspase 3 sensor Phiphilux G2D2 is activated non-specifically in S1 renal proximal tubules. IntraVital. 2015;4:2. doi: 10.1080/21659087.2015.1067352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tanner GA. Glomerular sieving coefficient of serum albumin in the rat: a two-photon microscopy study. Am J Physiol Renal Physiol. 2009;296:F1258–65. doi: 10.1152/ajprenal.90638.2008. [DOI] [PubMed] [Google Scholar]
- 34.Peti-Peterdi J. Independent two-photon measurements of albumin GSC give low values. Am J Physiol Renal Physiol. 2009;296:F1255–7. doi: 10.1152/ajprenal.00144.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schiessl IM, Castrop H. Angiotensin II AT2 receptor activation attenuates AT1 receptor-induced increases in the glomerular filtration of albumin: a multiphoton microscopy study. Am J Physiol Renal Physiol. 2013;305:F1189–200. doi: 10.1152/ajprenal.00377.2013. [DOI] [PubMed] [Google Scholar]
- 36.Schiessl IM, Kattler V, Castrop H. In vivo visualization of the antialbuminuric effects of the angiotensin-converting enzyme inhibitor enalapril. J Pharmacol Exp Ther. 2015;353:299–306. doi: 10.1124/jpet.114.222125. [DOI] [PubMed] [Google Scholar]
- 37.Russo LM, Sandoval RM, McKee M, et al. The normal kidney filters nephrotic levels of albumin retrieved by proximal tubule cells: retrieval is disrupted in nephrotic states. Kidney Int. 2007;71:504–13. doi: 10.1038/sj.ki.5002041. [DOI] [PubMed] [Google Scholar]
- 38.Toma I, Kang JJ, Peti-Peterdi J. Imaging renin content and release in the living kidney. Nephron Physiol. 2006;103:71–4. doi: 10.1159/000090622. [DOI] [PubMed] [Google Scholar]
- 39.Dickson LE, Wagner MC, Sandoval RM, Molitoris BA. The proximal tubule and albuminuria: really! J Am Soc Nephrol. 2014;25:443–53. doi: 10.1681/ASN.2013090950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hall AM, Unwin RJ, Parker N, Duchen MR. Multiphoton imaging reveals differences in mitochondrial function between nephron segments. J Am Soc Nephrol. 2009;20:1293–302. doi: 10.1681/ASN.2008070759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Burford JL, Villanueva K, Lam L, et al. Intravital imaging of podocyte calcium in glomerular injury and disease. J Clin Invest. 2014;124:2050–8. doi: 10.1172/JCI71702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Molitoris BA. Therapeutic translation in acute kidney injury: the epithelial/endothelial axis. J Clin Invest. 2014;124:2355–63. doi: 10.1172/JCI72269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wu H, Chen G, Wyburn KR, et al. TLR4 activation mediates kidney ischemia/reperfusion injury. J Clin Invest. 2007;117:2847–59. doi: 10.1172/JCI31008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kalakeche R, Hato T, Rhodes G, et al. Endotoxin uptake by S1 proximal tubular segment causes oxidative stress in the downstream S2 segment. J Am Soc Nephrol. 2011;22:1505–16. doi: 10.1681/ASN.2011020203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mansson LE, Melican K, Boekel J, et al. Real-time studies of the progression of bacterial infections and immediate tissue responses in live animals. Cell Microbiol. 2007;9:413–24. doi: 10.1111/j.1462-5822.2006.00799.x. [DOI] [PubMed] [Google Scholar]
- 46.Patil NK, Parajuli N, MacMillan-Crow LA, Mayeux PR. Inactivation of renal mitochondrial respiratory complexes and manganese superoxide dismutase during sepsis: mitochondria-targeted antioxidant mitigates injury. Am J Physiol Renal Physiol. 2014;306:F734–43. doi: 10.1152/ajprenal.00643.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Galley HF. Oxidative stress and mitochondrial dysfunction in sepsis. Br J Anaesth. 2011;107:57–64. doi: 10.1093/bja/aer093. [DOI] [PubMed] [Google Scholar]
- 48.Simmons CF, Jr, Bogusky RT, Humes HD. Inhibitory effects of gentamicin on renal mitochondrial oxidative phosphorylation. J Pharmacol Exp Ther. 1980;214:709–15. [PubMed] [Google Scholar]
- 49.Hall AM, Rhodes GJ, Sandoval RM, Corridon PR, Molitoris BA. In vivo multiphoton imaging of mitochondrial structure and function during acute kidney injury. Kidney Int. 2013;83:72–83. doi: 10.1038/ki.2012.328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Burke TJ, Wilson DR, Levi M, et al. Role of mitochondria in ischemic acute renal failure. Clin Exp Dial Apheresis. 1983;7:49–61. doi: 10.3109/08860228309076039. [DOI] [PubMed] [Google Scholar]
- 51.Brooks C, Wei Q, Cho SG, Dong Z. Regulation of mitochondrial dynamics in acute kidney injury in cell culture and rodent models. J Clin Invest. 2009;119:1275–85. doi: 10.1172/JCI37829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Plotnikov EY, Kazachenko AV, Vyssokikh MY, et al. The role of mitochondria in oxidative and nitrosative stress during ischemia/reperfusion in the rat kidney. Kidney Int. 2007;72:1493–502. doi: 10.1038/sj.ki.5002568. [DOI] [PubMed] [Google Scholar]
- 53.Sawada M, Sun W, Hayes P, et al. Ku70 suppresses the apoptotic translocation of Bax to mitochondria. Nat Cell Biol. 2003;5:320–9. doi: 10.1038/ncb950. [DOI] [PubMed] [Google Scholar]
- 54.Yi X, Yin XM, Dong Z. Inhibition of Bid-induced apoptosis by Bcl-2. tBid insertion, Bax translocation, and Bax/Bak oligomerization suppressed. J Biol Chem. 2003;278:16992–9. doi: 10.1074/jbc.M300039200. [DOI] [PubMed] [Google Scholar]
- 55.Kagan VE, Tyurin VA, Jiang J, et al. Cytochrome c acts as a cardiolipin oxygenase required for release of proapoptotic factors. Nat Chem Biol. 2005;1:223–32. doi: 10.1038/nchembio727. [DOI] [PubMed] [Google Scholar]
- 56.Dixon SJ, Stockwell BR. The role of iron and reactive oxygen species in cell death. Nat Chem Biol. 2014;10:9–17. doi: 10.1038/nchembio.1416. [DOI] [PubMed] [Google Scholar]
- 57.Castaneda MP, Swiatecka-Urban A, Mitsnefes MM, et al. Activation of mitochondrial apoptotic pathways in human renal allografts after ischemia reperfusion injury. Transplantation. 2003;76:50–4. doi: 10.1097/01.TP.0000069835.95442.9F. [DOI] [PubMed] [Google Scholar]
- 58.Wolfs TG, de Vries B, Walter SJ, et al. Apoptotic cell death is initiated during normothermic ischemia in human kidneys. Am J Transplant. 2005;5:68–75. doi: 10.1111/j.1600-6143.2004.00657.x. [DOI] [PubMed] [Google Scholar]
- 59.Bagnasco S, Good D, Balaban R, Burg M. Lactate production in isolated segments of the rat nephron. Am J Physiol. 1985;248:F522–6. doi: 10.1152/ajprenal.1985.248.4.F522. [DOI] [PubMed] [Google Scholar]
- 60.Dunn KW, Sutton TA, Sandoval RM. Live-animal imaging of renal function by multiphoton microscopy. Curr Protoc Cytom. 2007:9. doi: 10.1002/0471142956.cy1209s41. Chapter 12: Unit 12. [DOI] [PubMed] [Google Scholar]
- 61.Chance B, Schoener B, Oshino R, Itshak F, Nakase Y. Oxidation-reduction ratio studies of mitochondria in freeze-trapped samples. NADH and flavoprotein fluorescence signals. J Biol Chem. 1979;254:4764–71. [PubMed] [Google Scholar]
- 62.Schuh CD, Haenni D, Craigie E, et al. Long wavelength multiphoton excitation is advantageous for intravital kidney imaging. Kidney Int. 2015 doi: 10.1038/ki.2015.323. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 63.Armstrong JS, Steinauer KK, Hornung B, et al. Role of glutathione depletion and reactive oxygen species generation in apoptotic signaling in a human B lymphoma cell line. Cell Death Differ. 2002;9:252–63. doi: 10.1038/sj.cdd.4400959. [DOI] [PubMed] [Google Scholar]
- 64.Baudry N, Laemmel E, Vicaut E. In vivo reactive oxygen species production induced by ischemia in muscle arterioles of mice: involvement of xanthine oxidase and mitochondria. Am J Physiol Heart Circ Physiol. 2008;294:H821–8. doi: 10.1152/ajpheart.00378.2007. [DOI] [PubMed] [Google Scholar]
- 65.Vanden Hoek TL, Li C, Shao Z, Schumacker PT, Becker LB. Significant levels of oxidants are generated by isolated cardiomyocytes during ischemia prior to reperfusion. J Mol Cell Cardiol. 1997;29:2571–83. doi: 10.1006/jmcc.1997.0497. [DOI] [PubMed] [Google Scholar]
- 66.Cash TP, Pan Y, Simon MC. Reactive oxygen species and cellular oxygen sensing. Free Radic Biol Med. 2007;43:1219–25. doi: 10.1016/j.freeradbiomed.2007.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Robinson KM, Janes MS, Pehar M, et al. Selective fluorescent imaging of superoxide in vivo using ethidium-based probes. Proc Natl Acad Sci USA. 2006;103:15038–43. doi: 10.1073/pnas.0601945103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wang Z, Holthoff JH, Seely KA, et al. Development of oxidative stress in the peritubular capillary microenvironment mediates sepsis-induced renal microcirculatory failure and acute kidney injury. Am J Pathol. 2012;180:505–16. doi: 10.1016/j.ajpath.2011.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Denton RM, McCormack JG. Ca2+ transport by mammalian mitochondria and its role in hormone action. Am J Physiol. 1985;249:E543–54. doi: 10.1152/ajpendo.1985.249.6.E543. [DOI] [PubMed] [Google Scholar]
- 70.Bhosale G, Sharpe JA, Sundier SY, Duchen MR. Calcium signaling as a mediator of cell energy demand and a trigger to cell death. Ann N Y Acad Sci. 2015;1350:107–16. doi: 10.1111/nyas.12885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Stys PK, Waxman SG, Ransom BR. Ionic mechanisms of anoxic injury in mammalian CNS white matter: role of Na+ channels and Na(+)-Ca2+ exchanger. J Neurosci. 1992;12:430–9. doi: 10.1523/JNEUROSCI.12-02-00430.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Greka A, Mundel P. Calcium regulates podocyte actin dynamics. Semin Nephrol. 2012;32:319–26. doi: 10.1016/j.semnephrol.2012.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Welsh GI, Saleem MA. The podocyte cytoskeleton—key to a functioning glomerulus in health and disease. Nat Rev Nephrol. 2012;8:14–21. doi: 10.1038/nrneph.2011.151. [DOI] [PubMed] [Google Scholar]
- 74.Chen S, He FF, Wang H, et al. Calcium entry via TRPC6 mediates albumin overload-induced endoplasmic reticulum stress and apoptosis in podocytes. Cell Calcium. 2011;50:523–9. doi: 10.1016/j.ceca.2011.08.008. [DOI] [PubMed] [Google Scholar]
- 75.Yang H, Zhao B, Liao C, et al. High glucose-induced apoptosis in cultured podocytes involves TRPC6-dependent calcium entry via the RhoA/ROCK pathway. Biochem Bio-phys Res Commun. 2013;434:394–400. doi: 10.1016/j.bbrc.2013.03.087. [DOI] [PubMed] [Google Scholar]
- 76.Atkinson SJ, Hosford MA, Molitoris BA. Mechanism of actin polymerization in cellular ATP depletion. J Biol Chem. 2004;279:5194–9. doi: 10.1074/jbc.M306973200. [DOI] [PubMed] [Google Scholar]
- 77.Molitoris BA. Actin cytoskeleton in ischemic acute renal failure. Kidney Int. 2004;66:871–83. doi: 10.1111/j.1523-1755.2004.00818.x. [DOI] [PubMed] [Google Scholar]
- 78.Hall AM, Crawford C, Unwin RJ, Duchen MR, Peppiatt-Wildman CM. Multiphoton imaging of the functioning kidney. J Am Soc Nephrol. 2011;22:1297–304. doi: 10.1681/ASN.2010101054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hall AM, Molitoris BA. Dynamic multiphoton microscopy: focusing light on acute kidney injury. Physiology. 2014;29:334–42. doi: 10.1152/physiol.00010.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Oh J, Beckmann J, Bloch J, et al. Stimulation of the calcium-sensing receptor stabilizes the podocyte cytoskeleton, improves cell survival, and reduces toxin-induced glomerulosclerosis. Kidney Int. 2011;80:483–92. doi: 10.1038/ki.2011.105. [DOI] [PubMed] [Google Scholar]
- 81.Weinlich R, Oberst A, Dillon CP, et al. Protective roles for caspase-8 and cFLIP in adult homeostasis. Cell Rep. 2013;5:340–8. doi: 10.1016/j.celrep.2013.08.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kelly KJ, Sandoval RM, Dunn KW, Molitoris BA, Dagher PC. A novel method to determine specificity and sensitivity of the TUNEL reaction in the quantitation of apoptosis. Am J Physiol Cell Physiol. 2003;284:C1309–18. doi: 10.1152/ajpcell.00353.2002. [DOI] [PubMed] [Google Scholar]
- 83.Terada Y, Knepper MA. Thiazide-sensitive NaCl absorption in rat cortical collecting duct. Am J Physiol. 1990;259:F519–F528. doi: 10.1152/ajprenal.1990.259.3.F519. [DOI] [PubMed] [Google Scholar]
- 84.Eladari D, Chambrey R, Peti-Peterdi J. A new look at electrolyte transport in the distal tubule. Ann Rev Physiol. 2012;74:325–49. doi: 10.1146/annurev-physiol-020911-153225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Loffling J, Loffling-Cueni D, Hegyi I, Kaplan MR. Thiazide treatment of rats provokes apoptosis in distal tubule cells. Kidney Int. 1996;50:1180–90. doi: 10.1038/ki.1996.426. [DOI] [PubMed] [Google Scholar]
- 86.Silva MT. Secondary necrosis: the natural outcome of the complete apoptotic program. FEBS Lett. 2010;584:4491–9. doi: 10.1016/j.febslet.2010.10.046. [DOI] [PubMed] [Google Scholar]
- 87.Liu L, Chahroudi A, Silvestri G, et al. Visualization and quantification of T cell-mediated cytotoxicity using cell-permeable fluorogenic caspase substrates. Nat Med. 2002;8:185–9. doi: 10.1038/nm0202-185. [DOI] [PubMed] [Google Scholar]
- 88.Yang B, Jain S, Pawluczyk IZ, et al. Inflammation and caspase activation in long-term renal ischemia/reperfusion injury and immunosuppression in rats. Kidney Int. 2005;68:2050–67. doi: 10.1111/j.1523-1755.2005.00662.x. [DOI] [PubMed] [Google Scholar]
- 89.Martens CR, Edwards DG. Peripheral vascular dysfunction in chronic kidney disease. Cardiol Res Pract. 2011;2011:267257. doi: 10.4061/2011/267257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Gupta A, Rhodes GJ, Berg DT, et al. Activated protein C ameliorates LPS-induced acute kidney injury and downregulates renal INOS and angiotensin 2. Am J Physiol Renal Physiol. 2007;293:F245–54. doi: 10.1152/ajprenal.00477.2006. [DOI] [PubMed] [Google Scholar]
- 91.Holthoff JH, Wang Z, Seely KA, Gokden N, Mayeux PR. Resveratrol improves renal microcirculation, protects the tubular epithelium, and prolongs survival in a mouse model of sepsis-induced acute kidney injury. Kidney Int. 2012;81:370–8. doi: 10.1038/ki.2011.347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Sharfuddin AA, Sandoval RM, Berg DT, et al. Soluble thrombomodulin protects ischemic kidneys. J Am Soc Nephrol. 2009;20:524–34. doi: 10.1681/ASN.2008060593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Imamura R, Isaka Y, Sandoval RM, et al. Intravital two-photon microscopy assessment of renal protection efficacy of siRNA for p53 in experimental rat kidney transplantation models. Cell Transplant. 2010;19:1659–70. doi: 10.3727/096368910X516619. [DOI] [PubMed] [Google Scholar]
- 94.Yamamoto T, Tada T, Brodsky SV, et al. Intravital video-microscopy of peritubular capillaries in renal ischemia. Am J Physiol Renal Physiol. 2002;282:F1150–5. doi: 10.1152/ajprenal.00310.2001. [DOI] [PubMed] [Google Scholar]
- 95.Sutton TA, Mang HE, Campos SB, et al. Injury of the renal microvascular endothelium alters barrier function after ischemia. Am J Physiol Renal Physiol. 2003;285:F191–8. doi: 10.1152/ajprenal.00042.2003. [DOI] [PubMed] [Google Scholar]
- 96.Horbelt M, Lee SY, Mang HE, et al. Acute and chronic microvascular alterations in a mouse model of ischemic acute kidney injury. Am J Physiol Renal Physiol. 2007;293:F688–F695. doi: 10.1152/ajprenal.00452.2006. [DOI] [PubMed] [Google Scholar]
- 97.Basile DP, Friedrich JL, Spahic J, et al. Impaired endothelial proliferation and mesenchymal transition contribute to vascular rarefaction following acute kidney injury. Am J Physiol Renal Physiol. 2011;300:F721–33. doi: 10.1152/ajprenal.00546.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Okusa MD, Chertow GM, Portilla D, Acute Kidney Injury Advisory Group of the American Society of Nephrology The nexus of acute kidney injury, chronic kidney disease, and World Kidney Day 2009. Clin J Am Soc Nephrol. 2009;4:520–2. doi: 10.2215/CJN.06711208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Bohle A, Mackensen-Haen S, Wehrmann M. Significance of postglomerular capillaries in the pathogenesis of chronic renal failure. Kidney Blood Press Res. 1996;19:191–5. doi: 10.1159/000174072. [DOI] [PubMed] [Google Scholar]
- 100.Kang DH, Anderson S, Kim YG, et al. Impaired angiogenesis in the aging kidney: vascular endothelial growth factor and thrombospondin-1 in renal disease. Am J Kidney Dis. 2001;37:601–11. doi: 10.1053/ajkd.2001.22087. [DOI] [PubMed] [Google Scholar]
- 101.Kida Y, Tchao BN, Yamaguchi I. Peritubular capillary rarefaction: a new therapeutic target in chronic kidney disease. Pediatr Nephrol. 2014;29:333–42. doi: 10.1007/s00467-013-2430-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Bonventre JV, Zuk A. Ischemic acute renal failure: an inflammatory disease? Kidney Int. 2004;66:480–5. doi: 10.1111/j.1523-1755.2004.761_2.x. [DOI] [PubMed] [Google Scholar]
- 103.Leonard EC, Friedrich JL, Basile DP. VEGF-121 preserves renal microvessel structure and ameliorates secondary renal disease following acute kidney injury. Am J Physiol Renal Physiol. 2008;295:F1648–57. doi: 10.1152/ajprenal.00099.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Dimmeler S, Assmus B, Hermann C, Haendeler J, Zeiher AM. Fluid shear stress stimulates phosphorylation of Akt in human endothelial cells: involvement in suppression of apoptosis. Circ Res. 1998;83:334–41. doi: 10.1161/01.res.83.3.334. [DOI] [PubMed] [Google Scholar]
- 105.Paszkowiak JJ, Dardik A. Arterial wall shear stress: observations from the bench to the bedside. Vasc Endovasc Surg. 2003;37:47–57. doi: 10.1177/153857440303700107. [DOI] [PubMed] [Google Scholar]
- 106.Bernard GR, Vincent JL, Laterre PF, et al. Efficacy and safety of recombinant human activated protein C for severe sepsis. N Engl J Med. 2001;344:699–709. doi: 10.1056/NEJM200103083441001. [DOI] [PubMed] [Google Scholar]
- 107.Mitchell MJ, King MR. Physical biology in cancer. 3. The role of cell glycocalyx in vascular transport of circulating tumor cells. Am J Physiol Cell Physiol. 2014;306:C89–97. doi: 10.1152/ajpcell.00285.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Nieuwdorp M, Mooij HL, Kroon J, et al. Endothelial glycocalyx damage coincides with microalbuminuria in type 1 diabetes. Diabetes. 2006;55:1127–32. doi: 10.2337/diabetes.55.04.06.db05-1619. [DOI] [PubMed] [Google Scholar]
- 109.Mulivor AW, Lipowsky HH. Inflammation- and ischemia-induced shedding of venular glycocalyx. Am J Physiol Heart Circ Physiol. 2004;286:H1672–80. doi: 10.1152/ajpheart.00832.2003. [DOI] [PubMed] [Google Scholar]
- 110.Adembri C, Sgambati E, Vitali L, et al. Sepsis induces albuminuria and alterations in the glomerular filtration barrier: a morphofunctional study in the rat. Crit Care. 2011;15:R277. doi: 10.1186/cc10559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Huxley VH, Williams DA. Role of a glycocalyx on coronary arteriole permeability to proteins: evidence from enzyme treatments. Am J Physiol Heart Circ Physiol. 2000;278:H1177–85. doi: 10.1152/ajpheart.2000.278.4.H1177. [DOI] [PubMed] [Google Scholar]
- 112.Singh A, Ramnath RD, Foster RR, et al. Reactive oxygen species modulate the barrier function of the human glomerular endothelial glycocalyx. PLoS One. 2013;8:e55852. doi: 10.1371/journal.pone.0055852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Hsieh HJ, Liu CA, Huang B, Tseng AH, Wang DL. Shear-induced endothelial mechanotransduction: the interplay between reactive oxygen species (ROS) and nitric oxide (NO) and the pathophysiological implications. J Biomed Sci. 2014;21:3. doi: 10.1186/1423-0127-21-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Stefanidis I, Heintz B, Stocker G, et al. Association between heparan sulfate proteoglycan excretion and proteinuria after renal transplantation. J Am Soc Nephrol. 1996;7:2670–6. doi: 10.1681/ASN.V7122670. [DOI] [PubMed] [Google Scholar]
- 115.Sutton TA, Kelly KJ, Mang HE, et al. Minocycline reduces renal microvascular leakage in a rat model of ischemic renal injury. Am J Physiol Renal Physiol. 2005;288:F91–7. doi: 10.1152/ajprenal.00051.2004. [DOI] [PubMed] [Google Scholar]
- 116.Bojic S, Kotur-Stevuljevic J, Kalezic N, et al. Diagnostic value of matrix metalloproteinase-9 and tissue inhibitor of matrix metalloproteinase-1 in sepsis-associated acute kidney injury. Tohoku J Exp Med. 2015;237:103–9. doi: 10.1620/tjem.237.103. [DOI] [PubMed] [Google Scholar]
- 117.Wu L, Tiwari MM, Messer KJ, et al. Peritubular capillary dysfunction and renal tubular epithelial cell stress following lipopolysaccharide administration in mice. Am J Physiol Renal Physiol. 2007;292:F261–8. doi: 10.1152/ajprenal.00263.2006. [DOI] [PubMed] [Google Scholar]
- 118.Kelly KJ, Williams WW, Jr, Colvin RB, et al. Intercellular adhesion molecule-1-deficient mice are protected against ischemic renal injury. J Clin Invest. 1996;97:1056–63. doi: 10.1172/JCI118498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Takada M, Nadeau KC, Shaw GD, Marquette KA, Tilney NL. The cytokine-adhesion molecule cascade in ischemia/reperfusion injury of the rat kidney. Inhibition by a soluble P-selectin ligand. J Clin Invest. 1997;99:2682–90. doi: 10.1172/JCI119457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Kuligowski MP, Kitching AR, Hickey MJ. Leukocyte recruitment to the inflamed glomerulus: a critical role for platelet-derived P-selectin in the absence of rolling. J Immunol. 2006;176:6991–9. doi: 10.4049/jimmunol.176.11.6991. [DOI] [PubMed] [Google Scholar]
- 121.Celie JW, Rutjes NW, Keuning ED, et al. Subendothelial heparan sulfate proteoglycans become major L-selectin and monocyte chemoattractant protein-1 ligands upon renal ischemia/reperfusion. Am J Pathol. 2007;170:1865–78. doi: 10.2353/ajpath.2007.070061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Constantinescu AA, Vink H, Spaan JA. Endothelial cell glycocalyx modulates immobilization of leukocytes at the endothelial surface. Arterioscler Thromb Vasc Biol. 2003;23:1541–7. doi: 10.1161/01.ATV.0000085630.24353.3D. [DOI] [PubMed] [Google Scholar]
- 123.Jang HR, Rabb H. Immune cells in experimental acute kidney injury. Nat Rev Nephrol. 2015;11:88–101. doi: 10.1038/nrneph.2014.180. [DOI] [PubMed] [Google Scholar]
- 124.Zewinger S, Schumann T, Fliser D, Speer T. Innate immunity in CKD-associated vascular diseases. Nephrol Dial Transplant. 2015 doi: 10.1093/ndt/gfv358. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 125.Camirand G, Li Q, Demetris AJ, et al. Multiphoton intravital microscopy of the transplanted mouse kidney. Am J Transplant. 2011;11:2067–74. doi: 10.1111/j.1600-6143.2011.03671.x. [DOI] [PubMed] [Google Scholar]
- 126.Halin C, Mora JR, Sumen C, von Andrian UH. In vivo imaging of lymphocyte trafficking. Ann Rev Cell Dev Biol. 2005;21:581–603. doi: 10.1146/annurev.cellbio.21.122303.133159. [DOI] [PubMed] [Google Scholar]
- 127.Jbeily N, Claus RA, Dahlke K, et al. Comparative suitability of CFDA-SE and rhodamine 6G for in vivo assessment of leukocyte-endothelium interactions. J Biophotonics. 2014;7:369–75. doi: 10.1002/jbio.201300179. [DOI] [PubMed] [Google Scholar]
- 128.Saetzler RK, Jallo J, Lehr HA, et al. Intravital fluorescence microscopy: impact of light-induced phototoxicity on adhesion of fluorescently labeled leukocytes. J Histochem Cytochem. 1997;45:505–13. doi: 10.1177/002215549704500403. [DOI] [PubMed] [Google Scholar]
- 129.Becker HM, Chen M, Hay JB, Cybulsky MI. Tracking of leukocyte recruitment into tissues of mice by in situ labeling of blood cells with the fluorescent dye CFDA SE. J Immunol Methods. 2004;286:69–78. doi: 10.1016/j.jim.2003.11.016. [DOI] [PubMed] [Google Scholar]
- 130.Albertine KH, Gee MH. In vivo labeling of neutrophils using a fluorescent cell linker. J Leukoc Biol. 1996;59:631–8. doi: 10.1002/jlb.59.5.631. [DOI] [PubMed] [Google Scholar]
- 131.Yang HC, Zuo Y, Fogo AB. Models of chronic kidney disease. Drug Discov Today Dis Models. 2010;7:13–9. doi: 10.1016/j.ddmod.2010.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Brun C, Munck O. Lesions of the kidney in acute renal failure following shock. Lancet. 1957;272:603–7. doi: 10.1016/s0140-6736(57)91069-3. [DOI] [PubMed] [Google Scholar]
- 133.Martin SJ, Reutelingsperger CP, McGahon AJ, et al. Early redistribution of plasma membrane phosphatidylserine is a general feature of apoptosis regardless of the initiating stimulus: inhibition by overexpression of Bcl-2 and Abl. J Exp Med. 1995;182:1545–56. doi: 10.1084/jem.182.5.1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Boersma HH, Kietselaer BL, Stolk LM, et al. Past, present, and future of annexin A5: from protein discovery to clinical applications. J Nucl Med. 2005;46:2035–50. [PubMed] [Google Scholar]
- 135.Zou L, Chen HH, Li D, et al. Imaging lymphoid cell death in vivo during polymicrobial sepsis. Crit Care Med. 2015;43:2303–12. doi: 10.1097/CCM.0000000000001254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Smith BA, Gammon ST, Xiao S, et al. In vivo optical imaging of acute cell death using a near-infrared fluorescent zinc-dipicolylamine probe. Mol Pharm. 2011;8:583–90. doi: 10.1021/mp100395u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Aloya R, Shirvan A, Grimberg H, et al. Molecular imaging of cell death in vivo by a novel small molecule probe. Apoptosis. 2006;11:2089–101. doi: 10.1007/s10495-006-0282-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Nangaku M. Hypoxia and tubulointerstitial injury: a final common pathway to end-stage renal failure. Nephron Exp Nephrol. 2004;98:e8–12. doi: 10.1159/000079927. [DOI] [PubMed] [Google Scholar]
- 139.Nguyen D, Xu T. The expanding role of mouse genetics for understanding human biology and disease. Dis Models Mech. 2008;1:56–66. doi: 10.1242/dmm.000232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Mashimo T. Gene targeting technologies in rats: zinc finger nucleases, transcription activator-like effector nucleases, and clustered regularly interspaced short palindromic repeats. Dev Growth Differ. 2014;56:46–52. doi: 10.1111/dgd.12110. [DOI] [PubMed] [Google Scholar]
- 141.Schiessl IM, Bardehle S, Castrop H. Superficial nephrons in BALB/c and C57BL/6 mice facilitate in vivo multiphoton microscopy of the kidney. PLoS One. 2013;8:e52499. doi: 10.1371/journal.pone.0052499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Sipos A, Toma I, Kang JJ, Rosivall L, Peti-Peterdi J. Advances in renal (patho)physiology using multiphoton microscopy. Kidney Int. 2007;72:1188–91. doi: 10.1038/sj.ki.5002461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Linkermann A, Brasen JH, Himmerkus N, et al. Rip1 (receptor-interacting protein kinase 1) mediates necroptosis and contributes to renal ischemia/reperfusion injury. Kidney Int. 2012;81:751–61. doi: 10.1038/ki.2011.450. [DOI] [PubMed] [Google Scholar]
- 144.Tristao VR, Goncalves PF, Dalboni MA, et al. Nec-1 protects against nonapoptotic cell death in cisplatin-induced kidney injury. Ren Fail. 2012;34:373–7. doi: 10.3109/0886022X.2011.647343. [DOI] [PubMed] [Google Scholar]
- 145.Liang X, Chen Y, Zhang L, et al. Necroptosis, a novel form of caspase-independent cell death, contributes to renal epithelial cell damage in an ATP-depleted renal ischemia model. Mol Med Rep. 2014;10:719–24. doi: 10.3892/mmr.2014.2234. [DOI] [PubMed] [Google Scholar]
- 146.Schnaper HW. Remnant nephron physiology and the progression of chronic kidney disease. Pediatr Nephrol. 2014;29:193–202. doi: 10.1007/s00467-013-2494-8. [DOI] [PMC free article] [PubMed] [Google Scholar]