

Near-atomic resolution structure of species-D adenovirus 26 (HAdV-D26) reveals structural conservation among human adenoviruses.
Keywords: Human adenovirus D26, HAdV-D26, Ad26, Cryo-electron microscopy, Structure, Major capsid proteins, Minor capsid proteins, Cement proteins
Abstract
Human adenoviruses (HAdVs) cause acute respiratory, ocular, and gastroenteric diseases and are also frequently used as gene and vaccine delivery vectors. Unlike the archetype human adenovirus C5 (HAdV-C5), human adenovirus D26 (HAdV-D26) belongs to species-D HAdVs, which target different cellular receptors, and is differentially recognized by immune surveillance mechanisms. HAdV-D26 is being championed as a lower seroprevalent vaccine and oncolytic vector in preclinical and human clinical studies. To understand the molecular basis for their distinct biological properties and independently validate the structures of minor proteins, we determined the first structure of species-D HAdV at 3.7 Å resolution by cryo-electron microscopy. All the hexon hypervariable regions (HVRs), including HVR1, have been identified and exhibit a distinct organization compared to those of HAdV-C5. Despite the differences in the arrangement of helices in the coiled-coil structures, protein IX molecules form a continuous hexagonal network on the capsid exterior. In addition to the structurally conserved region (3 to 300) of IIIa, we identified an extra helical domain comprising residues 314 to 390 that further stabilizes the vertex region. Multiple (two to three) copies of the cleaved amino-terminal fragment of protein VI (pVIn) are observed in each hexon cavity, suggesting that there could be ≥480 copies of VI present in HAdV-D26. In addition, a localized asymmetric reconstruction of the vertex region provides new details of the three-pronged “claw hold” of the trimeric fiber and its interactions with the penton base. These observations resolve the previous conflicting assignments of the minor proteins and suggest the likely conservation of their organization across different HAdVs.
INTRODUCTION
There are nearly 60 known human adenovirus (HAdV) serotypes that have been classified into seven species (A to G) (1), determined mainly by the hexon hypervariable regions (HVRs) that constitute most of the virion surface (2). The members of different species cause distinct diseases—for example, respiratory infections by the species-B and species-C HAdVs versus moderate to severe eye infections (conjunctivitis and keratoconjunctivitis) and enteric infections by some of the species-D HAdVs. In addition, AdVs (or Ads) are among the most frequently used vectors in gene, vaccine, and cancer therapies (3–5). Currently, most of the structural information on HAdV virions comes from the species-C AdVs, particularly HAdV-C5. Here, we consider structural characterization of a genetically distant serotype in order to evaluate the extent of similarities and differences between different HAdV species as well as to assist in resolving the conflicting interpretations of the structures and locations of certain minor proteins (6–8).
Human species-D HAdV-D26, originally isolated from stool samples (9, 10), is genetically distant and 34% different at the DNA level from the well-characterized HAdV-C5 virus (11). A study carried out in 1963 in male inmate “volunteers” demonstrated that ocular inoculation of HAdV-D26 results in conjunctivitis that peaks within 4 days and viral infection in the throat peaking at 10 days, followed by shedding of virus in the stool for as long as 40 days after eye infection (10). HAdV-D26 vectors are now in preclinical and human clinical studies as oncolytic and vaccine vectors (12–14). The archetype HAdV-C5 has a long-shafted, flexible fiber protein that binds CAR (Coxsackievirus and adenovirus receptor) and mediates strong oncolytic effects in solid tumors but shows reduced activity in hematologic cancers (15). In contrast, HAdV-D26 has a short-shafted fiber that binds CD46 and sialic acid receptors and kills B cell cancers more efficiently than HAd-C5 but is ineffective against the solid tumors (5). Furthermore, HAdV-D26 is not recognized by vitamin K–dependent coagulation factors [for example, factor X (FX) and factor IX (FIX)], and because of that, it does not get targeted to macrophages (16–21). Hence, the structural characterization of HAdV-D26 is of importance, given its genetic diversity, distinct biology, and recent burgeoning use as a therapeutic.
In general, AdVs are composed of a large icosahedral capsid (~925 Å in diameter), comprising three major capsid proteins (MCPs) and four minor proteins organized with an intricate architecture [reviewed by San Martin (22)]. The MCPs consist of 720 copies of hexon protein that are organized as 240 hexon trimers (termed hexons) on a pseudo T = 25 icosahedral lattice; 60 copies of penton base (PB) subunits organized as pentamers occupy each of the 12 icosahedral vertices along with a trimer of fiber molecules, which noncovalently associate with each PB pentamer and project outward from each vertex (Fig. 1). The highly conserved N-terminal (NT) region (residues 1 to 20) of the fibers interacts with the PB, whereas the distal C-terminal (CT) knob domain is responsible for binding to cellular receptors (23). The length of the fibers varies depending on the number of β spiral repeats present between the N and C termini and is specific for a given AdV serotype. HAdV-D26 has a relatively short fiber with 7 to 8 shaft repeats, compared to ~22 repeats in the case of HAdV-C5. Multiple (240) copies of minor protein IX are located on the capsid exterior that stabilizes group of nine hexons (GON) substructures, which constitute icosahedral facets, whereas the other three minor proteins (IIIa, VI, and VIII) are located on the capsid interior. Several copies of IIIa and VIII (60 and 120, respectively) act as “molecular glue” in mediating interactions between the hexons that fortify the capsid. Protein VI, a multifunctional molecule that is involved in the localization of hexons into the nucleus, acts as a cofactor for adenoviral protease (AVP) and also plays a critical role in the lysis of endosomes and subsequent release of partially disassembled virions into the cytoplasm (24–28). Up to three molecules of protein VI can bind to each hexon, thereby packaging hundreds of copies of VI during the assembly. Of note, precursors of the above minor proteins (pIIIa, pVI, and pVIII) and some of the core proteins (pVII, pμ, and pTP) undergo proteolytic processing by the AVP as part of virion maturation, which is essential for forming infectious AdV virions [reviewed by Mangel and San Martin (29)].
Fig. 1. Overall structure and organization of the major and minor capsid proteins in HAdV-D26.
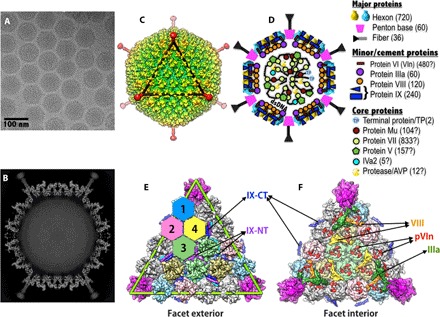
(A) Representative micrograph of frozen-hydrated HAdV-D26 virions (defocus, 1.16 μm) (B) Slice through the center of HAdV-D26 particle showing the cross section of densities of the MCPs and closely associated minor proteins. The shaft repeats and knob domain of the fiber can be seen, and the packaged double-stranded DNA in the middle is disordered. (C) Radially color-coded surface representation of the cryo-EM reconstruction of HAdV-D26, a view down the icosahedral threefold axis. One icosahedral facet is identified by a black triangle. (D) Schematic representation of AdV capsid showing the location and organization of various proteins in the AdV capsid. A list of symbols and the proteins they represent is shown on the right. The copy numbers of the capsid proteins are indicated in parentheses, and “?” indicates the uncertainty in the copy numbers. (E) Exterior view of a triangular facet. The locations of the structurally distinct hexon-1 to hexon-4 are identified by differently colored hexagons, surface representations of the hexons (hexon-1 to hexon-4) are shown in four different colors (light blue, pink, green, and khaki, respectively), and the PB is shown in magenta. The green triangle represents an icosahedral facet and is equivalent to the black triangle in (C). The hexons shown in gray belong to neighboring facets. The locations of the NT triskelions and CT coiled coils (four-helical bundle; 4-HLXB) of protein IX are labeled. (F) Interior view of the facet showing the location of the minor proteins relative to each other and to the MCPs. Surface representations of the proteins IIIa, VIII-U, VIII-V, and pVIn are shown in dark green, orange, yellow, and red, respectively. With the exception of pVIn (red), multiple copies of the same-colored proteins are related to each other by the icosahedral symmetry.
Currently, there exist two different models for the structural organization of minor proteins in HAdVs. In the first model (MODEL-1), the protein IX molecules are involved in forming the distinctive triskelions and coiled coils on the HAdV surface, whereas the remaining minor proteins IIIa (underneath the vertex), VI, and VIII are localized to the capsid interior. However, the locations of VI molecules are not clearly identified. MODEL-1 is based on moderate- to high-resolution cryo-electron microscopy (cryo-EM) studies on wild-type and mutant HAdVs and secondary structure considerations (6, 7, 30). In the second model (MODEL-2), the proteins IX and IIIa are located on the capsid exterior and are implicated in forming triskelions and coiled coils augmented with extended polypeptides, respectively, whereas the proteins VI, V (underneath the vertex), and VIII are localized to the interior. MODEL-2 was put forth by x-ray crystallographic studies (8, 31), quasi-symmetry considerations, and some early biochemical (32, 33) as well as low-resolution cryo-EM studies (34, 35). In addition, results from the recent hydrogen-deuterium exchange mass spectrometry studies (36) also played a role in validating the pVI segments observed in the x-ray maps. Although both models were derived on the basis of the studies of HAdV-C5–based vectors, the crystallography studies used a pseudotype vector, termed Ad5F35, composed of HAdV-C5 capsid fitted with a short fiber from HAdV-B35 for the purposes of growing crystals (37). Notably, the number of copies of protein IX in the pseudotype vector Ad5F35 appears to be significantly higher (676 ± 122 copies) compared to the conventional number (240 copies) (38).
One of the motivations for solving the HAdV-D26 structure is to validate the two different models. The structural details of HAdV-D26 reported here support the structures and organization of the minor proteins in MODEL-1 compared to MODEL-2. It is possible that some of the distinct densities observed between the peripentonal hexons (PPHs) in the x-ray maps might actually belong to the extra copies of IX, which were not considered during the interpretation of the x-ray maps (8). In view of these new structural findings, despite the differences in the composition of minor proteins in the Ad5F35 vector and visualization of certain unique densities, MODEL-2 proposed by the x-ray crystallographic studies (8) needs to be reevaluated.
RESULTS AND DISCUSSION
Structure determination of HAdV-D26
We determined the structure of HAdV-D26 using single-particle cryo-EM (see Materials and Methods and fig. S1). The overall resolution of the HAdV-D26 cryo-EM density map is 3.7 Å. However, the local resolution estimations using ResMap (39) suggest that the resolution in the capsid region of HAdV-D26 is closer to 3.0 Å (fig. S2), whereas the resolution limits of the HVRs are estimated to be between 4 and 6 Å. We docked available crystal structures of hexon and PB along with the cryo-EM structures of the minor proteins as rigid bodies into the cryo-EM map and extracted corresponding EM densities into an appropriate pseudocell (see Materials and Methods). The extracted densities were transformed into amplitudes and phases and were subjected to automated model building using the CCP4 program BUCCANEER (40), which resulted in the accurate assignment of 30 to 60% of the amino acids for most individual proteins. These partial models were then used for manual model building using the graphics program O (41). Reciprocal space refinement of the model within an icosahedral asymmetric unit (IAU) was carried out using the program CNS (see Materials and Methods) (42). Although the short fiber is visible in its entirety, the resolution and occupancy of the threefold symmetric fiber have been reduced by the application of fivefold symmetry during the icosahedral particle reconstruction. To obtain further structural details of the fiber in situ and its interactions with the PB, we generated a localized asymmetric reconstruction (LAR) of the vertex region composed of fiber, PB, and PPHs (see Materials and Methods). The LAR of the vertex region revealed a likely three-pronged “claw hold” involving the N termini of the trimeric fiber with elbow-shaped hooks (Fig. 2, D to F).
Fig. 2. Structures of MCPs in HAdV-D26.
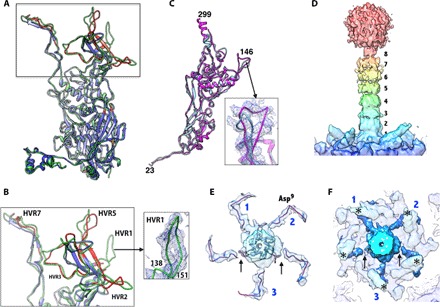
(A) Superposition of the hexon subunits from HAdV-D26 in green and HAdV-C5 [Protein Data Bank (PDB) ID: 3IYN] in blue. The regions of the HAdV-C5 hexon, which differ from those of the HAdV-D26 by RMSDs >2.0 Å, are shown in red, and most of these differences occur in the HVRs. (B) Closeup view of the HVRs (some of them are labeled). The inset shows the EM density for HVR1. (C) Superposition of the PB subunits of HAdV-D26 (magenta) and HAdV-C5 (light blue; PDB ID: 3IYN). The inset shows a closeup view of the variable β3-β4 loop with a six–amino acid insertion in the density. (D) Color-coded surface representation of the fiber density derived from the LAR that highlights the fiber shaft repeats with the receptor binding knob domain located at the top and the PB at the bottom. The eight fiber shaft repeats are labeled. (E) Cutaway top view of the extracted fiber density highlighting the elbow-shaped structures displayed by the fiber NT tails (FNTs). The three fiber tails of the “fiber claw” that are more strongly connected to the central fiber density are labeled as 1, 2, and 3. Arrows identify the locations of the breaks in the density of the remaining two FNTs at this contour level. The backbone trace of the FNT model (residues 2 to 20) derived from the high-resolution HAdV-D26 structure is shown inside the elbow-shaped densities (see also fig. S7). The Asp9 residue is located at the elbow bend. (F) Same view as in (E), showing the elbow-shaped FNTs (dark blue) hooking the base of the protrusion that contains the RGD loop, identified by asterisks (*). Numbers 1, 2, and 3 identify the three FNTs of the fiber claw, and the arrows indicate the break in the density of the remaining two FNTs.
Structure of HAdV-D26
The amino acid sequence identity of various corresponding capsid proteins in HAdV-D26 and HAdV-C5 varies between 47 and 77% (table S1). Despite the sequence differences and the fact that they come from different species (C and D), the overall structure and organization of the HAdV-D26 capsid are mostly similar to those of HAdV-C5 (Fig. 1).
Structures of MCPs
Hexon
The overall structure of the HAdV-D26 hexon is also very similar to that of the HAdV-C5 hexon with a root mean square deviation (RMSD) of 1.1 Å for 884 pairs of aligned Cα atoms, which do not include the HVRs (Fig. 2). Significantly, we were able to resolve all the HVRs, including HVR1, which is disordered in the structures of species-C HAdVs (2, 6). Among the HVRs, only HVR1 is shorter in HAdV-D26 by 12 amino acids compared to HAdV-C5, whereas the rest of them are either of the same length or longer by a few residues (table S2). Not surprisingly, the major structural differences between the two hexon structures occur in the HVRs (Fig. 2, A and B, and fig. S3). The differences in the structures and organization of HVRs, particularly HVR7, which contains the putative FX binding site (near residues 423 to 425), correlate well with the biochemical observations that FIX and FX do not bind to HAdV-D26 (16, 17, 20). The binding of FX has been shown to protect HAdV-C5 from immune clearance and target them to the liver, causing liver toxicity (19–21). Apart from the variations in the HVRs, the other significant changes in the hexons occur at the NT and CT regions of the hexons that occupy distinct locations in the AdV capsid (fig. S4), and these are in agreement with the structural changes seen in the HAdV-C5 structure (6). In addition, we observed conformational changes in the V2-α3 helix composed of residues 891 to 896 in hexon-1 and hexon-4 due to interactions with the minor proteins VIII and IIIa (fig. S4 and table S3).
PB and fiber
Although shorter by 50 amino acids, the PB of HAdV-D26 adopts a fiber-bound conformation similar to that of the PBs of HAdV-C2 and HAdV-C5 viruses with an RMSD of 1.3 Å for 446 pairs of Cα atoms (PDB IDs: 1X9T and 3IYN) (Fig. 2C) (6, 43). The main differences between the different PB structures occur in the regions comprising (i) the N terminus, (ii) the Arg-Gly-Asp (RGD)–containing loop, and (iii) the loop (amino acids 139 to 153) connecting the β3 and β4 strands (fig. S5A). The sequence alignments of the PBs show that there is a 12-residue deletion at the N terminus and two deletions in the RGD-containing loop in the PB of HAdV-D26 relative to species-C viruses (fig. S5B). However, the vicinity of the RGD-containing sequence is disordered. A six–amino acid insertion in the loop between the β3 and β4 strands results in associated structural changes (Fig. 2C and fig. S5A). Furthermore, we observed that the conserved NT peptide segment (amino acids 2 to 20) of the fiber molecule is bound at the interface between a pair of PB subunits and is also surface-accessible. This fiber peptide interacts more closely with one of the two PB subunits wrapping around the structural protrusion (amino acids 280 to 330) that contains the RGD loop mainly through hydrophobic and cation-π interactions (fig. S6 and table S4). The observation of five copies of the bound NT fiber peptide, as opposed to three copies, is a result of imposing fivefold symmetry during the icosahedral particle reconstruction. In essence, only three of the five binding sites are occupied (43–45).
It has not been possible to visualize the structure of AdV fibers in situ in any great detail because of their flexibility and the symmetry mismatch of imposing icosahedral fivefold symmetry on the trimeric fiber, which results in the loss of resolution and occupancy during three-dimensional (3D) reconstruction. However, in the case of HAdV-D26, because of its shorter length, we could see the full-length fiber, including the knob domain in the reconstruction, but not in great detail because of the abovementioned symmetry mismatch (Fig. 1, B and C). To acquire more details on the fiber structure in situ, we obtained the LAR of a subvolume of the vertex region comprising fiber, PB, and PPHs without imposing any symmetry and using procedures previously described by Ilca and co-workers (see Materials and Methods) (46). The LAR of the vertex region resulted in a map at ~4.3 Å resolution (Fig. 2, D to F, and fig. S7). However, on the basis of the quality of EM densities, we note that the actual resolution may be lower than what is reported by the program (see Materials and Methods). Despite these efforts, we could not totally eliminate the fivefold symmetry that is implicitly present in the pentameric PB to which the trimeric fiber is attached and is part of the subvolume being reconstructed. However, we were able to better resolve the fiber shaft repeats (Fig. 2D). Excluding the PB and PPHs from the subvolume yielded unintelligible, lower-resolution maps, and using C3 symmetry did not fare better either. This is most likely due to weak fiber density relative to the background of the images. Working with maps obtained from the LAR of the subvolume and by docking the extended high-resolution crystal structure of the AdV-C2 fiber knob and shaft regions (PDB ID: 1QIU) (47), we determined that eight shaft repeats could be accommodated in the HAdV-D26 fiber (Fig. 2D and fig. S7). Furthermore, by subtracting the contribution of the PB and PPHs from the final LAR maps, we located the path of the five FNTs, three of which show continuous density connected to the central shaft of the fiber (identified by the numbers 1, 2, and 3), whereas the remaining two show breaks in the connectivity (Fig. 2, E and F, and fig. S7). Notwithstanding the presence of five FNTs, each tail forms an elbow-shaped hook that may be important for latching onto the PB subunits and is consistent with the high-resolution model of the FNT obtained from the icosahedral reconstruction (fig. S7, E and F). The elbow bends near the Asp9 residue of the FNT and wraps around the loop (amino acids 324 to 330) that forms the base of the disordered RGD (amino acids 309 to 311) loop. The above three tails that are connected to the central fiber density resemble a three-pronged fiber claw, where two FNTs (numbers 1 and 2) occupy adjacent binding sites on the PB, whereas the third FNT binds to the site that is ~144° away from the FNTs numbered 1 and 2 (Fig. 2, E and F, and fig. S7). This particular three-pronged organization of FNTs provides greater stability for the PB-fiber complex and is in agreement with the previous hypotheses (43–45) as well as the arrangement of “prongs” in bird claws seen in nature. Such an arrangement and associated interactions between the trimeric fiber and pentameric PB are likely to be conserved among all the AdVs.
Structures of minor proteins
Protein IX
Of the four structurally distinct molecules of IX, one complete (IX) molecule is ordered in the HAdV-D26 structure and was traced in well-resolved density with defined features for the respective side chains, consistent with the amino acid sequence (Fig. 3A). The NT region (1 to 60) is involved in forming the triskelion structures (fig. S8), whereas the CT region (100 to 134) forms an 11-turn helix that is involved in forming coiled coils (Fig. 3, B to F, and fig. S9). A linker helix (63 to 80) was also built in well-resolved density consistent with a stretch of alanine residues and preceded by SSLDSTA, followed by PSSGSSP flexible sequences that connect the above signature structural elements at the N and C termini of protein IX. The linker region (amino acids 62 to 93) in each of the four independent copies of IX adopts different conformations in forming the coiled-coil structure composed of four helices. Notably, the location of the above IX molecule is not the same as that of the full-length IX molecule reported in the HAdV-C5 cryo-EM structure (6) but is related to it by local threefold symmetry. In addition, we independently traced three other structurally distinct IX molecules, but their linker regions are disordered. Although there is only 47% sequence identity between the IX molecules of HAdV-D26 and HAdV-C5, the overall structure and organization of triskelions and the coiled-coil structures in HAdV-D26 are similar to those of the HAdV-C5 structures (6, 7). The three CT helices originating from one GON facet are arranged in parallel, whereas a fourth helix from a neighboring facet joins them in an antiparallel fashion that results in the formation of a four-helix bundle (4-HLXB) (Fig. 3, C to F). It is likely that the formation of the coiled coils comprising four helices (4-HLXB) is mediated by the hexon subunits during the assembly. There are three such 4-HLXBs, related by icosahedral threefold symmetry, present in each facet (Fig. 3C). However, although the organization of coiled coils appears similar with a left-handed twist, the structural overlay of the 4-HLXBs suggests that the packing of the individual helices and their interactions with the neighboring hexons are different in the two HAdV structures. For example, the antiparallel helix interacts with the FG2 loop (808 to 812) of hexon-4 in the HAdV-D26 structure, whereas the equivalent (antiparallel) helix in HAdV-C5 is displaced by ~10 Å from the former, overlaps with one of the parallel helices of HAdV-D26, and interacts with the V2 barrel of hexon-4 (fig. S9). Thus, the organization of protein IX coiled coils in the HAdV-D26 structure represents a slight variation in the theme. Notably, this exceptional 4-HLXB structure is formed by homopolypeptides at the C termini of protein IX molecules, three of which are oriented in a parallel arrangement and one in an antiparallel manner (Fig. 3, C to F).
Fig. 3. Structure and organization of the protein IX molecules in HAdV-D26.
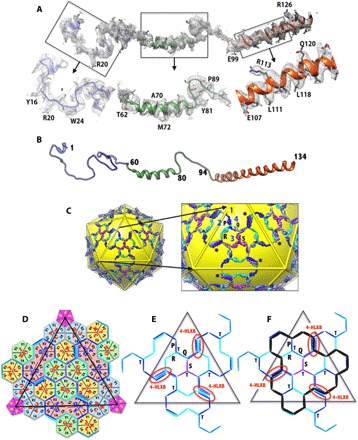
(A) A full-length molecule of IX ordered is shown in the corresponding EM density. The closeup views of different regions of the IX molecule in the EM density are shown below. (B) Ribbon diagram showing the secondary structure of the IX polypeptide with a 5-turn linker α helix in the middle and an 11-turn long α helix at the C terminus. (C) Continuous hexagonal network of IX molecules shown in the background of yellow icosahedron. A closeup view of the protein IX network is shown on the right. The four structurally distinct IX polypeptides (P, Q, R, and S) are shown in different colors (dark blue, light blue, cyan, and purple, respectively), and the same-colored copies are related by the icosahedral symmetry. Numbers 1 to 4 identify the locations of the unique hexons. The full-length molecule (R) ordered is shown in cyan, and the IX molecule (P) that contributes an antiparallel helix is shown in blue. Missing connections between the triskelions and coiled coils in P, Q, and S copies are identified by gray sticks. The triskelions at the icosahedral threefold axes are formed by purple IX molecules (S), whereas those at the local threefold axes are formed by three differently colored IX molecules (P, Q, and R). The four-helical bundle (4-HLXB) structures are formed by the CT helices of four structurally distinct IX-molecules that come from four different triskelions. The CT helices of the three IX molecules (Q, R, and S) are arranged in parallel and come from the same facet, whereas the fourth one (P), which joins in an antiparallel orientation, comes from the neighboring facet; this arrangement is similar to that of HAdV-C5. Asterisks (*) identify some of the 4-HLXBs. (D) Schematic diagram showing the hexagonal network of IX molecules shown as lines/sticks that interlace the hexons, which are represented as hexagons, whereas the PBs are shown as pentagons. The subunits (for example, A, B, and C) that constitute the individual hexons are labeled along with the locations of the V1 and V2 barrels of the individual hexon subunits. The centers of IX triskelions interact with the V2 domains of hexon subunits, whereas the 4-HLXB is located at the interface formed by V1 and V2 domains from two different subunits (K and L) of the hexon-4 capsomer on one side and their local twofold-related counterparts from D and E subunits of hexon-2 that belongs to the adjacent facet. The black triangle represents the boundary of the icosahedral facet. (E) Simplified schematic showing the path of the hexagonal network formed by protein IX molecules within a facet. The labels T and 4-HLXB identify the locations of the triskelions and coiled coils, respectively. The 4-HLXBs are encircled by red ovals. (F) Schematic identifying the locations of the triskelions and 4-HLXB with respect to the GON substructure, whose boundary is identified by the black lines overlaid on top of the protein IX network shown in (E). The 4-HLXBs are identified by red ovals.
The triskelion structures are stabilized by the intertwining polypeptide chains involving the NT residues (1–20) from three IX molecules and held together by backbone hydrogen bonds and nonpolar interactions. The triskelions interact with the B2 and C2 strands of the V2 β barrel of the hexons located at both the local and strict threefold symmetry axes on the capsid exterior (fig. S8). The 4-HLXB structure is held together by distinctive leucine zipper–like interactions between the four CT helices as in the HAdV-C5 structure (6). Remarkably, whereas the NT triskelion structures strictly conform to local threefold symmetry, the CT helices in the 4-HLXB structures do not (fig. S10). This 4-HLXB arrangement is unique to HAdVs because trimeric CT helical bundles are observed on top of the triskelion structures in animal AdVs, in accordance with the local or strict threefold symmetry (48, 49). The 4-HLXB structures in HAdVs, mainly through the interactions of the antiparallel helix, literally connect the protein IX molecules in one facet with those in the neighboring facets, thereby creating a continuous hexagonal protein IX network that stabilizes and straps the GON facets together across the entire particle (Fig. 3, C to F). There are two (icosahedral twofold-related) straps present at each side (icosahedral edge) of the triangular facet, and a total of six such straps link each GON facet with three other neighboring facets (Fig. 3C). We surmise that the above hexagonal protein IX network could stabilize the “wiffle ball–shaped” nucleoprotein complex, which may be formed by the loss of pentons and PPHs during the disassembly of HAdVs. Notably, although it is shown to be dispensable, the loss of above network of interactions in the IX-deleted HAdVs results in lower particle stability and reduced infectivity (50, 51).
Protein VIII
With 77% sequence identity, the structure and location of the obtuse, triangular protein VIII in HAdV-D26 are highly conserved and similar to those of HAdV-C5 (RMSD, 1.1 Å) (Fig. 4, A and B, and fig. S11). The structures of two distinct copies of protein VIII (U and V) are equally well conserved in HAdV-D26 with an RMSD of 0.6 Å. As in the case of the HAdV-C5 structure, the processed NT (1 to 110) and CT fragments (158 to 227) could be traced in well-resolved density with defined features for the respective side chains, whereas the middle fragment (111 to 157) is missing. Each of the obtuse, triangular VIII molecules, spanning nearly 100 Å in length and 30 Å in height, interacts with the bases of four hexon subunits on the capsid interior upon which nearly 90% of its surface area is buried. One of the striking interactions that glue the hexons together on the capsid interior involves the protein VIII molecules. Each VIII molecule disrupts the “native” β strand formation between the CT residues 937 to 942 and 945 to 952 in two different hexons and replaces them by forming antiparallel strands of its own with residues 28 to 36 and 105 to 110, located at either ends of the VIII molecule(s) (fig. S12). These interactions by VIII act as a “molecular clamp” in holding two hexons together—hexon-4(K) and hexon-2(E) by VIII-U and hexon-3′(G) and hexon-3(G) by VIII-V—within the GON structure, respectively. We located three discontinuous, unidentified densities of 5 to 10 amino acids in length, emulating the similar strand formation by the protein VIII with F (hexon-2), I (hexon-3), and J (hexon-4) subunits (fig. S13). On the basis of their relative locations and the chain direction according to antiparallel strand formation, these densities are unlikely to belong to the middle fragment of VIII but may belong to the remains of the scaffolding proteins (52). In addition, the wide end of the obtuse triangle formed by the N and C termini of VIII (1 to 8 and 197 to 227), located at the local threefold symmetry axes along the PPH-PPH and GON-GON interfaces, glues the PPH (by VIII-U) and hexon-2 (by VIII-V) from the neighboring facet (clockwise, as seen from the interior) mainly through hydrophobic interactions (Fig. 1F and fig. S14). Thus, the protein VIII molecules play a critical role in cementing the hexons together within and between the GONs on the capsid interior. The above local threefold junctions located at the GON-GON interfaces are distinct from the threefold junctions found within the GON substructure, which are strengthened by the protein IX triskelion structures on the capsid exterior (fig. S15).
Fig. 4. Structures of the minor proteins VIII and IIIa.
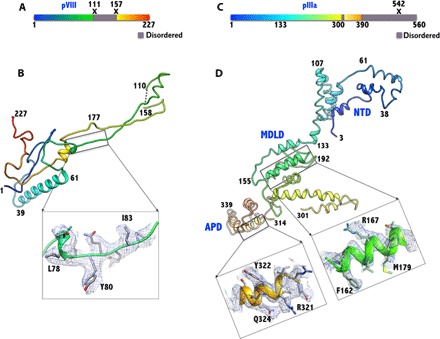
(A) Schematic diagram showing the structurally ordered (colored)/disordered (gray) regions and the locations of proteolytic cleavage sites (X) in the precursor of VIII. (B) The structure of VIII (U) is elongated and has a shape of an obtuse triangle, with a length of ~100 Å and a width (height) of 30 Å. It is composed of two chains, comprising residues 1 to 110 and 158 to 227, which are the result of the proteolytic processing (at residues 111 and 157) by the AVP. The 47-residue middle fragment is disordered. The inset shows the representative electron density in the boxed region. (C) Schematic diagram showing the structurally ordered (colored)/disordered (gray) regions and the location of proteolytic cleavage site (X) in the precursor of IIIa. (D) The structure of the ordered regions of IIIa (amino acids 3 to 390) is mostly composed of α helices and spans 130 Å along the longest dimension and 50 Å wide. The structure of IIIa can be divided into three domains: the NT domain (NTD), the middle domain (MDLD), and the appendage domain (APD). Insets show representative EM densities in the MDLD and the APD. Residues 302 to 313 and 391 to 560 are disordered.
Protein IIIa
The well-ordered region of IIIa (amino acids 3 to 301) in HAdV-D26 is similar to that of HAdV-C5 (RMSD, 1.35 Å; sequence identity, 78%) (Fig. 4, C and D, and fig. S16). In addition, we identified a short helical domain of 77 residues, termed the APD, built into a (weaker) density, contoured at 0.6σ compared to 1.0σ to 2.0σ used for building models in the well-ordered regions of the HAdV-D26 structure. However, the backbone of helices is visible in the reconstructed EM maps and located close to residues 216 to 222 of IIIa (fig. S17). No such density was reported in the HAdV-C5 structure; however, equivalent densities were seen in the temperature-sensitive (ts1) mutant of HAdV-C2 and suggested that they may belong to the precursor segments of IIIa and/or VIII (22, 53). Using the sharpened EM density maps, we assigned this newly identified modular helical domain to residues 314 to 390, but the residues (302 to 313) that connect to the well-ordered domains are disordered (Fig. 4, C and D, and figs. S16 and S17). In addition, the assignment of the APD to IIIa is consistent with the secondary structure predictions comprising six helices, and the APD, which is composed of 77 amino acids, contains more residues than the missing middle fragment of VIII, which comprises only 47 amino acids. Moreover, the secondary structure predictions of the middle fragment of VIII indicate no helices, further bolstering the notion that the APD does not belong to VIII. Aside from the newly identified APD, most of the interactions of IIIa with the PPH and VIII-U are similar to those seen in the HAdV-C5 structure (fig. S18 and table S5). Significantly, a pair of fivefold-related PPHs is held together by strong interactions between the NTDs of the adjacent IIIa molecules bound to the respective PPHs. Residues 30 to 60 of one IIIa interact closely with residues 82 to 140 of the symmetry-related molecule (fig. S18B). Five such interactions hold the five PPHs together in place. These interactions, along with those involving VIII (see below), appear to be critical for cementing the PPHs underneath the vertex region. In addition, the same pair of IIIa molecules (62 to 66 and 103 to 107 of IIIa; 108 to 118 of IIIa-Sym) is involved in a few interactions with the ordered NT residues (23 to 37) of PB (fig. S18C and table S5). However, it appears that a counterclockwise rotation of PB can uncouple these interactions without disrupting the surrounding PPHs. This is in agreement with the observation that penton capsomers can be released without completely dislodging the PPH (54, 55). However, further loosening of the vertex region subsequent to the PB release may result in the eventual release of the PPHs and associated IIIa molecules (26).
The middle domain consisting of residues 132 to 301 primarily interacts with the VIII-U molecule by overlaying on top of the wide end of the obtuse triangle, which together mediate interactions between the PPHs at the opposite end of the above interface stabilized by the NTDs of IIIa molecules (fig. S18, B and D). The newly identified helical domain (APD) (amino acids 314 to 390), located at a distinct local threefold junction, acts as an appendage of the well-ordered parts (3 to 301) of IIIa and particularly stabilizes the interactions between the PPH and GON interfaces (fig. S18D). A similar domain referred to as “molecular stitch” was identified in the HAdV-C2-ts1 mutant structure solved at ~9 Å resolution and was suggested to be responsible for its greater thermostability (22, 53, 56). In this vein, we speculate that the mature form of HAdV-D26 may also be more stable than its counterparts in species-C viruses. The remainder of the IIIa molecule at the C terminus (391 to 560), predicted to be mostly unstructured, is disordered. Furthermore, the arrangement and interactions between the proteins underneath the vertex region bring out a previously unrealized insight into the AdV assembly, that is, that the VIII molecules bind to hexons first, followed by binding of the IIIa molecules, thereby fortifying the interactions between the PPHs and GON substructures. Of note, the association of IIIa and VIII proteins occurs only at one of the two VIII locations, specifically underneath the vertex region. This rules out the possibility that the IIIa and VIII molecules associate together before particle assembly.
Protein VI
Protein VI is a multifunctional molecule that is involved in a number of key events in the AdV life cycle. The precursor of VI (pVI) undergoes proteolytic processing at both the N and C termini by the AVP. It has been well established that the cleaved CT (11 amino acids) fragments of VI are released, and some of them remain bound to AVP and act as a cofactor (29, 57). However, the status of the NT fragments was not known until recently (8, 36). In the HAdV-D26 structure, we observed two well-ordered copies of processed N termini of VI (pVIn) bound in the cavity of hexon-1, hexon-2, and hexon-4, whereas there is weak density for three copies of pVIn in hexon-3 (Fig. 5 and see Fig. 1E for hexon numbering). Although hexon-1 to hexon-4 occupy structurally unique positions in the AdV particle, the occupied locations of pVIn may be implicitly averaged while incorporating the threefold symmetric hexons during the assembly. In other words, when a hexon is incorporated, it can be added in one of the three (equivalent) ways that are indistinguishable because of the (local) threefold symmetry. Even after binding to three molecules of pVI, where all the three pVI binding sites are equally occupied, the three ways of incorporating are indistinguishable. However, when only one or two of the three pVI binding sites are occupied, then, the three ways of incorporation are not equivalent. Furthermore, there is no way to constrain all the (60) hexon-1s (partially occupied with pVIs) to be incorporated exactly the same way in lockstep. Each hexon-1 is likely to get incorporated independent of other hexon-1s. This results in implicit averaging of hexon-1s because all hexon-1 positions are considered equivalent, despite the local differences in the occupancy of pVI binding sites and even before performing any icosahedral averaging. The same argument also holds true for hexon-2, hexon-3, and hexon-4. After the imposition of icosahedral symmetry, all the unique positions of hexon-1 to hexon-4 are truly made equivalent, as are the pVIn peptides bound to the respective hexons. However, in the HAdV-D26 structure, two of the three sites show strong density of the pVIn molecules bound to hexon-1, hexon-2, and hexon-4, which indicates that two sites are preferentially occupied over the third site. This, in turn, suggests that, perhaps, there is a certain preference in incorporating hexons in these positions (1, 2, and 4) or loss of pVI from a specific location during the assembly. However, in hexon-3, all the three pVIn sites show equally weak density, which indicates that there is no such preference. Because there is well-resolved density for two copies of pVIn in most of the hexons, it is possible that at least two copies of VI are bound to each hexon, which is in agreement with the biochemical estimation of 1.8 copies of VI per hexon in HAdV-C2 virions (33). This, in turn, suggests that there could be ≥480 copies of VI present in HAdV-D26. Notably, having more VI molecules packaged into AdV virions is an advantage because it significantly increases the chances of endosome lysis, even if only a fraction of them are released from the AdV particles. Alternatively, their release in greater numbers can potentially perforate the endosomal membranes, thus breaking them apart, as has been observed in in vitro studies using liposomes (27).
Fig. 5. Structure and interactions of the cleaved NT fragment of VI (pVIn).
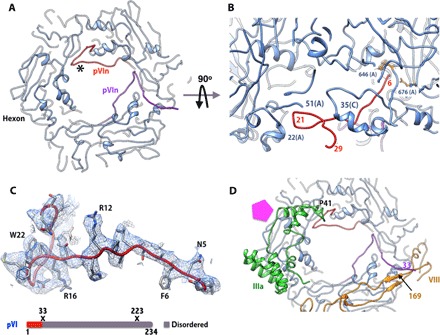
(A) Elongated structure and locations of two copies of pVIn bound to PPH. Only two of the three equivalent binding sites on PPH are occupied. (B) Side view showing the structure and interactions of one of the pVIn molecules. The N terminus of pVIn reaches deep into the hexon cavity near residues 646, 676, and 773, whereas residues 15 to 29 of pVIn interact with residues 48 to 84 of the A subunit and residues 29 to 35 of the C subunit at the entrance of the hexon cavity. (C) Top: Representative EM density for one of the pVIn molecules bound to PPH in (A) (in red; identified by the asterisk). Bottom: Schematic diagram of the structurally ordered (colored)/disordered (gray) regions and the locations of proteolytic cleavage sites (X) in the precursor of VI. (D) Interactions of pVIn with other minor proteins. Residues 25 to 29 of the above pVIn (red) interact with amino acids 42 to 48 of IIIa, whereas the CT residues (amino acids 30 to 33) of the second copy of pVIn (purple) form an antiparallel strand with residues 166 to 169 of VIII (U) (yellow). The pentagon (magenta) identifies the location of the PB.
We traced residues 3 to 33 in two of the pVIn polypeptides bound to hexon-1 and hexon-4 that are located near the VIII molecules (Fig. 5D), whereas residues 3 to 29 were modeled in the rest of the seven ordered pVIn molecules. Interactions between the different copies of pVIn and the hexons are mostly similar. The N terminus of pVIn reaches deep into the hexon cavity and is located near hexon residues Y646, R676, H773, and R872 (Fig. 5B). The well-ordered residues 15 to 29 in all the visible copies closely interact with residues 48 to 54 of one hexon subunit (for example, A) on one side and residues 30 to 37 of the adjacent hexon subunit (for example, C) on the other side at the entrance of the hexon cavity (for example, hexon-1) (Fig. 5B). There are three such equivalent binding sites in each hexon. Some of these interactions are very similar to those observed with a single copy of pVIn seen in the crystal structure of Ad5F35, except that residues 20 to 31 were inadvertently traced into the electron density corresponding to IIIa, perhaps due to the poor quality of the electron density maps resulting from a combined data set acquired from multiple frozen crystals (8). In the HAdV-D26 structure, one of the pVIn molecules (purple) bound to the PPH is ordered until residue 33 and forms an antiparallel strand (involving residues 30 to 33) with the VIII-U molecule (Fig. 5D). Similar interactions are seen between one of the pVIn peptides bound to hexon-4 and VIII-V. On the basis of these close interactions between some of the pVIn peptides and other minor proteins (IIIa and VIII), all of the VI molecules may not be equally available for the cleavage by the AVP in the assembled particle. As previously suggested, the maturation cleavage of VI is necessary to liberate the membrane-lytic (mature) fragment (34 to 239) of VI from the pVIn, which is bound tightly in the hexon cavities (8, 36). However, we could not locate any density corresponding to the mature fragment of VI in the HAdV-D26 structure. The membrane-lytic fragment of VI (amino acids 34 to 223) is likely to reside outside of the hexon cavity on the capsid interior and will be released upon the maturation processing by the AVP. On the basis of these results, the role of protein VI as the capsid cement remains unclear.
CONCLUSIONS
Together, the similarities in the structures and organization of capsid proteins between the genetically distant HAdV-D26 and the archetype HAdV-C5 suggest the overall structural conservation among the HAdVs. In addition, these results support the structure and organization of minor proteins proposed on the basis of the previous cryo-EM studies (6, 7, 30). The differences in hexon HVR5 and HVR7 and their relative proximity to the structurally ordered HVR1 may preclude coagulation factor binding to HAdV-D26, in agreement with experimental observations (16–21). HAdV-D26 contains a short fiber composed of eight shaft repeats, and the elbow structures adopted by the NT tails secure the fiber on the surface trough of the PB via the three-pronged claw hold of the fiber. The subunits in the PB (pentamer) are arranged with a right-handed twist, whereas the elbow-shaped NT tails of the fiber display an opposite (left-handed) twist and latch onto PB like a lid that seals the top of a jar. Among the minor proteins of HAdV-D26, protein VIII is the most similar to that of HAdV-C5 in terms of structure and interactions with hexon bases on the capsid interior. Even with the differences in the local arrangement of CT helices in the coiled-coil structures, the continuous hexagonal lattice formed by the IX polypeptides on the capsid exterior is conserved and straps the GON substructures together in the AdV capsid. Identification of an extra helical domain of IIIa formed by residues 314 to 390 and its interactions with the PPHs and GONs on the capsid interior further stabilize the vertex region. The observation of multiple (two to three) copies of the pVIn bound in the cavity of each hexon suggests that there could be ≥480 copies of VI present in the HAdV-D26 virions. However, the mature fragment of VI is disordered, and its role as the capsid cement remains unclear. Finally, the layered organization of major and minor capsid proteins on the capsid interior suggests a sequence of protein-protein associations during the AdV assembly: Hexons “preloaded” with VI molecules are joined by IX on the exterior and VIII on the interior, perhaps with the assistance of scaffolding proteins, and finally, IIIa binds on top of VIII, further strengthening the vertex region. These results advance the understanding of broader aspects of the structure, assembly, targeting specificities, and cell entry of HAdVs.
MATERIALS AND METHODS
Production of HAdV-D26 virions
Replication-defective HAdV-D26 (RD-HAdV-D26) was constructed and produced as described by Weaver and Barry (13). RD-HAdV-D26 was grown in HEK293 E4pIX (Microbix) cells to provide both E1 and E4 functions to the E1- and E3-deleted HAdV-D26. Final preparations of the virus were produced from 10 Plate CellSTACKS (Corning) and were purified on two rounds of CsCl gradients. The final CsCl viral bands were used for the cryo-EM analysis.
Cryo-EM sample preparation and data collection
The HAdV-D26 (RD-HAdV-D26) samples were dialyzed into glycerol/ethylene glycol–free buffer [40 mM tris (pH 8.1), 300 mM NaCl, and 10 mM CaCl2] and concentrated to 6 mg/ml using Amicon Ultra-4 (Merck Millipore Ltd.) centrifugal filters. Three microliters of sample aliquots was applied twice (separated by blotting in between) (58) to a 1.2/1.3 C-flat grid (Protochips) that had been plasma-cleaned for 6 s at 20 mA using a Gatan Solarus plasma cleaning system. After application of the sample for the second time, the grid was plunge-frozen into liquid ethane using the Gatan Cryoplunge 3 system (CP3) with a blotting time of 3.0 s. Data were acquired on a FEI Titan Krios transmission electron microscope operating at 300 kV and equipped with a Gatan K2 Summit direct detector. A condenser aperture of 70 μm and an objective aperture of 100 μm were used. Coma-free alignment was performed using Leginon software (59). Automated data collection was carried out using Leginon (60) to control both the FEI Titan Krios (used in microprobe mode at a nominal magnification of ×22,500) and the Gatan K2 Summit detector, operated in “counting mode” (pixel size, 1.31 Å) at a dose rate of ~9 counts per physical pixel per second, which corresponds to ~12 electrons per physical pixel per second [when accounting for coincidence loss (61)]. A wait time of 30 s was used after physically moving the stage to each new position and allowing it to settle before acquiring a new movie. Each movie has a total accumulated exposure of 53 e/Å2 fractionated into 38 frames of 200 ms each (yielding movies of 7.6 s). About 2000 micrographs were acquired using a defocus range between 0.8 and 3.0 μm under focus.
Cryo-EM data processing and structure determination
Whole-frame alignment was carried out using the dosefgpu_driftcorr program (61) to account for stage drift and beam-induced motion. We used a frame offset of 7 and a B factor of 1000 pixels2 to align the movie frames (62). Motion-corrected sums from each movie were used for further processing. The image contrast transfer function parameters were estimated for each micrograph using the program ctffind3 (63). The HAdV-D26 particles were automatically picked from the images using FindEM (64), via a template derived from 100 manually picked particles using the Appion pipeline (65). After manual inspection/selection/rejection, a total of 19,590 particles were extracted using a box size of 1200 pixels2 and were binned by 2 (BIN2). An HAdV model, generated using the program pdb2mrc (66) and the PDB files (4CWU or 3IYN, with the minor proteins excluded) and low-pass–filtered to 60 Å, was used as the starting model for HAdV-D26 image reconstruction using the program Frealign v9.11 (67). After four cycles of refinement using the BIN2 data in mode 3, the unbinned stack was introduced for four cycles in mode 1. The resulting maps showed apparent densities corresponding to minor proteins that were not part of the initial model. The resolution estimates for the 75% best correlating particles after the application of a spherical mask with inner and outer radii of 280 and 510 Å, respectively, were 3.69 Å (Part_FSC, 0.143) and 3.82 Å (FSC, 0.143) (fig. S1). In addition, we transferred the images and box (coordinate) files of the selected particles (from Appion) to RELION and extracted the particles with box sizes of 800 and 1024 pixels2. These particles were subjected to 3D classification using a fourfold binned stack with C1 symmetry. After 25 iterations of 3D classification, two best classes containing 12,841 particles were subjected to 3D refinement using the unbinned 800-pixel2 (1.31 Å per pixel) particles using icosahedral (I1) symmetry. However, the memory requirements did not permit the use of boxed particles at 1024 pixels2 in RELION v1.4. The gold standard resolution based on comparing two half-maps was estimated to be 3.72 Å (fig. S1). The local resolution estimations using ResMap (39) suggested that the resolution in the contiguous region of the capsid was closer to 3.0 Å (fig. S2). In the end, the quality of the maps resulting from both programs was very similar. However, because the RELION 3D maps obtained with a box size of 800 pixels3 (1.31 Å per pixel) resulted in the truncation of the fiber knobs, we chose to work with the Frealign 3D maps, which were generated with a box size of 1200 pixels3. Multiple sharpened maps, obtained by applying different negative temperature factors (−100, −150, and −200 Å2 using the program bfactor.exe), along with the unsharpened map, were used for model building.
Localized asymmetric reconstructions
Using the refined virus particle orientations and positions from the converged icosahedral particle reconstruction, we determined the location and orientation of the fiber subparticles comprising PB and PPHs along the fivefold axis and their symmetry-related counterparts, as described by Ilca and co-workers (46) and in the documentation (www.opic.ox.ac.uk/wiki/index.php?title=Localized_reconstruction). We extracted the fiber subparticles in a box of 256 × 256 pixels (1.31 Å per pixel) centered at 475 Å from the center of the particle. This resulted in 154,092 subparticles. We obtained the initial reconstruction and subsequent 3D classifications with the C1 symmetry using RELION (68). We selected four classes (from the six classes) containing 112,602 subparticles, which were used to obtain an unsymmetrized final reconstruction using Frealign (67). The final estimated resolutions were 5.0 and 4.3 Å for the FSC and Part_FSC values, respectively, with an FSC cutoff of 0.143. We also tried masked refinements by defining a mask around the fiber region to exclude the effect of PB and PPHs on LAR and by using C3 symmetry. None of these trials provided better maps than those obtained with C1 symmetry and without the mask (fig. S7).
Model building and refinement
The available models of hexon and PB capsomers of HAdV-C5 (PDB ID: 1P30, 1X9T, and 3IYN) were docked into the EM density map (sharpened with a B factor of −150 Å2) using Chimera. Density corresponding to the individual (unique) capsomers was extracted into an appropriate pseudocell (for example, a = 144 Å, b = 144 Å, c = 160 Å, α = 90°, β = 90°, and γ = 90°) and transformed into amplitudes and phases using the CCP4 and RAVE suite of programs (step 1) (69, 70). The automated model building program BUCCANEER (40, 71), available in the CCP4 suite of programs (69), was used to obtain initial models for HAdV-D26 capsid proteins. The chimera-docked models, amplitudes, and phases of each capsomer and appropriate amino acid sequence files (in the FASTA format) were used as the input to BUCCANNER (step 2). The resulting models from BUCCANEER contained 30 to 60% regions of correctly assigned sequence for a given protein and served as good initial models for manual model building using the graphics program O (step 3) (41). We used differently sharpened maps at various contour levels (0.5σ to 2.0σ) for manual model building. All the unique capsomers (hexon-1 to hexon-4 and monomer of PB) were individually built. In the case of minor proteins, we manually traced the polyalanine backbone for each of the proteins. Then, the above steps 1 to 3 were used to obtain the atomic models for the minor proteins. We relied on the “Lego” commands in O (41) to assign proper backbone conformation particularly in the weakly ordered regions (for example, certain HVRs) and to adjust side-chain rotamer conformations. Once the models for all the proteins (hexons, PB, IIIa, VIs, VIIIs, and IXs) that constitute one IAU were built, the density corresponding to ¼ of a virion, comprising 15-fold icosahedral symmetry, was extracted into a pseudocell (for example, a = 1089.92 Å, b = 544.96 Å, c = 544.96 Å, α = 90°, β = 90°, and γ = 90°) and transformed into amplitudes and phases. The 15-fold redundant icosahedral symmetry present in ¼ of a virion is equivalent to 15-fold “non-crystallographic symmetry” (NCS). The above pseudo-observations were used to refine the model in the IAU, containing 13,378 amino acid residues (105,753 atoms), using 15-fold strict NCS via the program CNS using the maximum likelihood target function using amplitudes (mlf) (42). The final R factor (R free) of the IAU model is 0.3343 (0.3363).
Structural analysis and figure generation
The quality of each model was assessed using the programs CNS (42) and PROCHECK (72). Structural analyses (for example, structural superposition) were carried out using Chimera (73) and/or the Superpose program in the CCP4 suite (69). The positions of HVRs in the HAdV-26 hexon were identified by aligning its amino acid sequence with that of the HAdV-C5 hexon and obtaining the ranges of residues in the corresponding HVRs, as defined in previous work (2, 74). Amino acid sequence alignments were carried out using a CLUSTALW server (www.genome.jp/tools/clustalw/) (75), and secondary structure prediction was carried out using the PSIPRED server (http://bioinf.cs.ucl.ac.uk/psipred/) (76). All the figures were generated using Chimera (73), and a few screenshots were obtained using the graphics program O (41).
Supplementary Material
Acknowledgments
We thank B. Carragher and C. Potter, the directors of the National Resource for Automated Molecular Microscopy (NRAMM) facility, for their support and encouragement. V.S.R. thanks J. Speir for his assistance with the data acquisition on Titan Krios and N. Grigorieff for his help with and advice on using the Frealign program. V.S.R. also acknowledges the inspiring discussions with G. Nemerow and J. Johnson. Funding: This work was supported by NIH grants R01AI070771 and R21AI103692 (to V.S.R). The NRAMM facility is funded by NIH grant GM103310 (to B. Carragher and C. Potter). Author contributions: M.A.B. and M.E.B. expressed and purified HAdV-D26 virions. D.V. and M.G.C. prepared the grids and collected cryo-EM images. D.V. and V.S.R. processed the data. V.S.R., D.V., and X.Y. selected the particles, performed the initial data analysis, and obtained cryo-EM image reconstructions. X.Y. and F.J.A. obtained the LARs of the fiber subvolumes and performed the postreconstruction analyses. V.S.R. built and refined the atomic models. V.S.R. and X.Y. interpreted the structure. V.S.R., F.J.A., and M.A.B. wrote the manuscript with input from the coauthors. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors. The cryo-EM map and the atomic model of HAdV-D26 have been deposited in the Electron Microscopy Data Bank and the PDB under accession codes EMD-8471 and 5TX1, respectively.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/3/5/e1602670/DC1
table S1. Number of amino acids and sequence identity between the respective capsid proteins in HAdV-D26 and HAdV-C5.
table S2. Residues forming the HVRs in HAdV-D26 versus HAdV-C5.
table S3. Structural characteristics and interactions of the N and C termini of hexon subunits in HAdV-D26.
table S4. Fiber-PB interactions in HAdV-D26.
table S5. Protein IIIa interactions with the major and minor proteins in HAdV-D26.
table S6. Data collection and refinement statistics of HAdV-D26.
fig. S1. Resolution estimations of the cryo-EM reconstruction of HAdV-D26.
fig. S2. Estimation of local resolution of the HAdV-D26 cryo-EM reconstruction.
fig. S3. Structural similarities and differences between the hexon subunits of HAdV-D26 and HAdV-C5.
fig. S4. Structural similarities and differences between the different hexon structures in HAdV-D26.
fig. S5. Structural superposition and sequence alignment of PBs from HAdV-D26 and HAdV-C5.
fig. S6. Interactions between the conserved N terminus of the fiber and the PB in HAdV-D26.
fig. S7. The structure and analysis of the fiber molecule obtained by LAR.
fig. S8. Interactions between the protein IX triskelions and the hexons.
fig. S9. The structural overlay of the 4-HLXBs of IX from HAdV-D26 and HAdV-C5.
fig. S10. Structural superposition of the protein IX molecules.
fig. S11. Structural conservation in protein VIII molecules from HAdV-D26 and HAdV-C5.
fig. S12. Molecular clamping interactions by the protein VIII.
fig. S13. β strand formation by the unidentified densities.
fig. S14. Gluing interactions of the PPHs by protein VIII.
fig. S15. Structures, locations, and organization of minor proteins relative to the MCPs in HAdV-D26.
fig. S16. Structural similarities of the IIIa proteins from HAdV-D26 and HAdV-C5.
fig. S17. Cryo-EM density and fit of the APD of IIIa in HAdV-D26.
fig. S18. Gluing interactions involving IIIa, PPHs, and PB.
REFERENCES AND NOTES
- 1.Davison A. J., Benkő M., Harrach B., Genetic content and evolution of adenoviruses. J. Gen. Virol. 84, 2895–2908 (2003). [DOI] [PubMed] [Google Scholar]
- 2.Rux J. J., Burnett R. M., Type-specific epitope locations revealed by X-ray crystallographic study of adenovirus type 5 hexon. Mol. Ther. 1, 18–30 (2000). [DOI] [PubMed] [Google Scholar]
- 3.Abbink P., Lemckert A. A. C., Ewald B. A., Lynch D. M., Denholtz M., Smits S., Holterman L., Damen I., Vogels R., Thorner A. R., O’Brien K. L., Carville A., Mansfield K. G., Goudsmit J., Havenga M. J. E., Barouch D. H., Comparative seroprevalence and immunogenicity of six rare serotype recombinant adenovirus vaccine vectors from subgroups B and D. J. Virol. 81, 4654–4663 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bouard D., Alazard-Dany N., Cosset F.-L., Viral vectors: From virology to transgene expression. Br. J. Pharmacol. 157, 153–165 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen C. Y., Senac J. S., Weaver E. A., May S. M., Jelinek D. F., Greipp P., Witzig T., Barry M. A., Species D adenoviruses as oncolytics against B-cell cancers. Clin. Cancer Res. 17, 6712–6722 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liu H., Jin L., Koh S. B. S., Atanasov I., Schein S., Wu L., Zhou Z. H., Atomic structure of human adenovirus by cryo-EM reveals interactions among protein networks. Science 329, 1038–1043 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Saban S. D., Silvestry M., Nemerow G. R., Stewart P. L., Visualization of α-helices in a 6-Ångstrom resolution cryoelectron microscopy structure of adenovirus allows refinement of capsid protein assignments. J. Virol. 80, 12049–12059 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Reddy V. S., Nemerow G. R., Structures and organization of adenovirus cement proteins provide insights into the role of capsid maturation in virus entry and infection. Proc. Natl. Acad. Sci. U.S.A. 111, 11715–11720 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rosen L., Baron S., Bell J. A., Four newly recognized adenoviruses. Proc. Soc. Exp. Biol. Med. 107, 434–437 (1961). [DOI] [PubMed] [Google Scholar]
- 10.Kasel J. A., Evans H. E., Spickard A., Knight V., Conjunctivitis and enteric infection with adenovirus types 26 and 27: Responses to primary, secondary and reciprocal cross-challenges. Am. J. Hyg. 77, 265–282 (1963). [DOI] [PubMed] [Google Scholar]
- 11.Turner M. A., Middha S., Hofherr S. E., Barry M. A., Comparison of the life cycles of genetically distant species C and species D human adenoviruses Ad6 and Ad26 in human cells. J. Virol. 89, 12401–12417 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Baden L. R., Walsh S. R., Seaman M. S., Tucker R. P., Krause K. H., Patel A., Johnson J. A., Kleinjan J., Yanosick K. E., Perry J., Zablowsky E., Abbink P., Peter L., Iampietro M. J., Cheung A., Pau M. G., Weijtens M., Goudsmit J., Swann E., Wolff M., Loblein H., Dolin R., Barouch D. H., First-in-human evaluation of the safety and immunogenicity of a recombinant adenovirus serotype 26 HIV-1 Env vaccine (IPCAVD 001). J. Infect. Dis. 207, 240–247 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Weaver E. A., Barry M. A., Low seroprevalent species D adenovirus vectors as influenza vaccines. PLOS ONE 8, e73313 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Senac J. S., Doronin K., Russell S. J., Jelinek D. F., Greipp P. R., Barry M. A., Infection and killing of multiple myeloma by adenoviruses. Hum. Gene Ther. 21, 179–190 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen C. Y., Weaver E. A., Khare R., May S. M., Barry M. A., Mining the adenovirus virome for oncolytics against multiple solid tumor types. Cancer Gene Ther. 18, 744–750 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Waddington S. N., McVey J. H., Bhella D., Parker A. L., Barker K., Atoda H., Pink R., Buckley S. M. K., Greig J. A., Denby L., Custers J., Morita T., Francischetti I. M. B., Monteiro R. Q., Barouch D. H., van Rooijen N., Napoli C., Havenga M. J. E., Nicklin S. A., Baker A. H., Adenovirus serotype 5 hexon mediates liver gene transfer. Cell 132, 397–409 (2008). [DOI] [PubMed] [Google Scholar]
- 17.Alba R., Bradshaw A. C., Parker A. L., Bhella D., Waddington S. N., Nicklin S. A., van Rooijen N., Custers J., Goudsmit J., Barouch D. H., McVey J. H., Baker A. H., Identification of coagulation factor (F)X binding sites on the adenovirus serotype 5 hexon: Effect of mutagenesis on FX interactions and gene transfer. Blood 114, 965–971 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Di Paolo N. C., van Rooijen N., Shayakhmetov D. M., Redundant and synergistic mechanisms control the sequestration of blood-born adenovirus in the liver. Mol. Ther. 17, 675–684 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tian J., Xu Z., Smith J. S., Hofherr S. E., Barry M. A., Byrnes A. P., Adenovirus activates complement by distinctly different mechanisms in vitro and in vivo: Indirect complement activation by virions in vivo. J. Virol. 83, 5648–5658 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Doronin K., Flatt J. W., Di Paolo N. C., Khare R., Kalyuzhniy O., Acchione M., Sumida J. P., Ohto U., Shimizu T., Akashi-Takamura S., Miyake K., MacDonald J. W., Bammler T. K., Beyer R. P., Farin F. M., Stewart P. L., Shayakhmetov D. M., Coagulation factor X activates innate immunity to human species C adenovirus. Science 338, 795–798 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xu Z., Qiu Q., Tian J., Smith J. S., Conenello G. M., Morita T., Byrnes A. P., Coagulation factor X shields adenovirus type 5 from attack by natural antibodies and complement. Nat. Med. 19, 452–457 (2013). [DOI] [PubMed] [Google Scholar]
- 22.San Martin C., Latest insights on adenovirus structure and assembly. Viruses 4, 847–877 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang Y., Bergelson J. M., Adenovirus receptors. J. Virol. 79, 12125–12131 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wodrich H., Guan T., Cingolani G., Von Seggern D., Nemerow G., Gerace L., Switch from capsid protein import to adenovirus assembly by cleavage of nuclear transport signals. EMBO J. 22, 6245–6255 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mangel W. F., McGrath W. J., Toledo D. L., Anderson C. W., Viral DNA and a viral peptide can act as cofactors of adenovirus virion proteinase activity. Nature 361, 274–275 (1993). [DOI] [PubMed] [Google Scholar]
- 26.Wiethoff C. M., Wodrich H., Gerace L., Nemerow G. R., Adenovirus protein VI mediates membrane disruption following capsid disassembly. J. Virol. 79, 1992–2000 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Maier O., Galan D. L., Wodrich H., Wiethoff C. M., An N-terminal domain of adenovirus protein VI fragments membranes by inducing positive membrane curvature. Virology 402, 11–19 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Moyer C. L., Wiethoff C. M., Maier O., Smith J. G., Nemerow G. R., Functional genetic and biophysical analyses of membrane disruption by human adenovirus. J. Virol. 85, 2631–2641 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mangel W. F., San Martin C., Structure, function and dynamics in adenovirus maturation. Viruses 6, 4536–4570 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.San Martin C., Glasgow J. N., Borovjagin A., Beatty M. S., Kashentseva E. A., Curiel D. T., Marabini R., Dmitriev I. P., Localization of the N-terminus of minor coat protein IIIa in the adenovirus capsid. J. Mol. Biol. 383, 923–934 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Reddy V. S., Natchiar S. K., Stewart P. L., Nemerow G. R., Crystal structure of human adenovirus at 3.5 Å resolution. Science 329, 1071–1075 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Everitt E., Lutter L., Philipson L., Structural proteins of adenoviruses. XII. Location and neighbor relationship among proteins of adenovirion type 2 as revealed by enzymatic iodination, immunoprecipitation and chemical cross-linking. Virology 67, 197–208 (1975). [DOI] [PubMed] [Google Scholar]
- 33.Everitt E., Sundquist B., Pettersson U., Philipson L., Structural proteins of adenoviruses: X. Isolation and topography of low molecular weight antigens from the virion of adenovirus type 2. Virology 52, 130–147 (1973). [DOI] [PubMed] [Google Scholar]
- 34.Stewart P. L., Fuller S. D., Burnett R. M., Difference imaging of adenovirus: Bridging the resolution gap between X-ray crystallography and electron microscopy. EMBO J. 12, 2589–2599 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fabry C. M. S., Rosa-Calatrava M., Conway J. F., Zubieta C., Cusack S., Ruigrok R. W. H., Schoehn G., A quasi-atomic model of human adenovirus type 5 capsid. EMBO J. 24, 1645–1654 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Snijder J., Benevento M., Moyer C. L., Reddy V., Nemerow G. R., Heck A. J. R., The cleaved N-terminus of pVI binds peripentonal hexons in mature adenovirus. J. Mol. Biol. 426, 1971–1979 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Reddy V. S., Natchiar S. K., Gritton L., Mullen T.-M., Stewart P. L., Nemerow G. R., Crystallization and preliminary X-ray diffraction analysis of human adenovirus. Virology 402, 209–214 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Benevento M., Di Palma S., Snijder J., Moyer C. L., Reddy V. S., Nemerow G. R., Heck A. J. R., Adenovirus composition, proteolysis and disassembly studied by in-depth qualitative and quantitative proteomics. J. Biol. Chem. 289, 11421–11430 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kucukelbir A., Sigworth F. J., Tagare H. D., Quantifying the local resolution of cryo-EM density maps. Nat. Methods 11, 63–65 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cowtan K., The Buccaneer software for automated model building. 1. Tracing protein chains. Acta Crystallogr. D Biol. Crystallogr. 62, 1002–1011 (2006). [DOI] [PubMed] [Google Scholar]
- 41.Jones T. A., Zou J.-Y., Cowan S. W., Kjeldgaard M., Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr. 47 (Pt. 2), 110–119 (1991). [DOI] [PubMed] [Google Scholar]
- 42.Brunger A. T., Adams P. D., Clore G. M., DeLano W. L., Gros P., Grosse-Kunstleve R. W., Jiang J. S., Kuszewski J., Nilges M., Pannu N. S., Read R. J., Rice L. M., Simonson T., Warren G. L., Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr. D Biol. Crystallogr. 54, 905–921 (1998). [DOI] [PubMed] [Google Scholar]
- 43.Zubieta C., Schoehn G., Chroboczek J., Cusack S., The structure of the human adenovirus 2 penton. Mol. Cell 17, 121–135 (2005). [DOI] [PubMed] [Google Scholar]
- 44.Liu H., Wu L., Zhou Z. H., Model of the trimeric fiber and its interactions with the pentameric penton base of human adenovirus by cryo-electron microscopy. J. Mol. Biol. 406, 764–774 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cao C., Dong X., Wu X., Wen B., Ji G., Cheng L., Liu H., Conserved fiber-penton base interaction revealed by nearly atomic resolution cryo-electron microscopy of the structure of adenovirus provides insight into receptor interaction. J. Virol. 86, 12322–12329 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ilca S. L., Kotecha A., Sun X., Poranen M. M., Stuart D. I., Huiskonen J. T., Localized reconstruction of subunits from electron cryomicroscopy images of macromolecular complexes. Nat. Commun. 6, 8843 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.van Raaij M. J., Mitraki A., Lavigne G., Cusack S., A triple β-spiral in the adenovirus fibre shaft reveals a new structural motif for a fibrous protein. Nature 401, 935–938 (1999). [DOI] [PubMed] [Google Scholar]
- 48.Schoehn G., El Bakkouri M., Fabry C. M. S., Billet O., Estrozi L. F., Le L., Curiel D. T., Kajava A. V., Ruigrok R. W. H., Kremer E. J., Three-dimensional structure of canine adenovirus serotype 2 capsid. J. Virol. 82, 3192–3203 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cheng L., Huang X., Li X., Xiong W., Sun W., Yang C., Zhang K., Wang Y., Liu H., Huang X., Ji G., Sun F., Zheng C., Zhu P., Cryo-EM structures of two bovine adenovirus type 3 intermediates. Virology 450–451, 174–181 (2014). [DOI] [PubMed] [Google Scholar]
- 50.Rosa-Calatrava M., Grave L., Puvion-Dutilleul F., Chatton B., Kedinger C., Functional analysis of adenovirus protein IX identifies domains involved in capsid stability, transcriptional activity, and nuclear reorganization. J. Virol. 75, 7131–7141 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Campos S. K., Parrott M. B., Barry M. A., Avidin-based targeting and purification of a protein IX-modified, metabolically biotinylated adenoviral vector. Mol. Ther. 9, 942–954 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Condezo G. N., Marabini R., Ayora S., Carazo J. M., Alba R., Chillón M., San Martín C., Structures of adenovirus incomplete particles clarify capsid architecture and show maturation changes of packaging protein L1 52/55k. J. Virol. 89, 9653–9664 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pérez-Berná A. J., Marabini R., Scheres S. H. W., Menéndez-Conejero R., Dmitriev I. P., Curiel D. T., Mangel W. F., Flint S. J., San Martín C., Structure and uncoating of immature adenovirus. J. Mol. Biol. 392, 547–557 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pérez-Berná A. J., Ortega-Esteban A., Menéndez-Conejero R., Winkler D. C., Menéndez M., Steven A. C., Flint S. J., de Pablo P. J., San Martín C., The role of capsid maturation on adenovirus priming for sequential uncoating. J. Biol. Chem. 287, 31582–31595 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Martinez R., Schellenberger P., Vasishtan D., Aknin C., Austin S., Dacheux D., Rayne F., Siebert A., Ruzsics Z., Gruenewald K., Wodrich H., The amphipathic helix of adenovirus capsid protein VI contributes to penton release and postentry sorting. J. Virol. 89, 2121–2135 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Silvestry M., Lindert S., Smith J. G., Maier O., Wiethoff C. M., Nemerow G. R., Stewart P. L., Cryo-electron microscopy structure of adenovirus type 2 temperature-sensitive mutant 1 reveals insight into the cell entry defect. J. Virol. 83, 7375–7383 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Baniecki M. L., McGrath W. J., McWhirter S. M., Li C., Toledo D. L., Pellicena P., Barnard D. L., Thorn K. S., Mangel W. F., Interaction of the human adenovirus proteinase with its 11-amino acid cofactor pVIc. Biochemistry 40, 12349–12356 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Snijder J., Borst A. J., Dosey A., Walls A. C., Burrell A., Reddy V. S., Kollman J. M., Veesler D., Vitrification after multiple rounds of sample application and blotting improves particle density on cryo-electron microscopy grids. J. Struct. Biol. 198, 38–42 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Glaeser R. M., Typke D., Tiemeijer P. C., Pulokas J., Cheng A., Precise beam-tilt alignment and collimation are required to minimize the phase error associated with coma in high-resolution cryo-EM. J. Struct. Biol. 174, 1–10 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Suloway C., Pulokas J., Fellmann D., Cheng A., Guerra F., Quispe J., Stagg S., Potter C. S., Carragher B., Automated molecular microscopy: The new Leginon system. J. Struct. Biol. 151, 41–60 (2005). [DOI] [PubMed] [Google Scholar]
- 61.Li X., Mooney P., Zheng S., Booth C. R., Braunfeld M. B., Gubbens S., Agard D. A., Cheng Y., Electron counting and beam-induced motion correction enable near-atomic-resolution single-particle cryo-EM. Nat. Methods 10, 584–590 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Campbell M. G., Veesler D., Cheng A., Potter C. S., Carragher B., 2.8 Å resolution reconstruction of the Thermoplasma acidophilum 20S proteasome using cryo-electron microscopy. eLife 4, e06380 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mindell J. A., Grigorieff N., Accurate determination of local defocus and specimen tilt in electron microscopy. J. Struct. Biol. 142, 334–347 (2003). [DOI] [PubMed] [Google Scholar]
- 64.Roseman A. M., FindEM—A fast, efficient program for automatic selection of particles from electron micrographs. J. Struct. Biol. 145, 91–99 (2004). [DOI] [PubMed] [Google Scholar]
- 65.Lander G. C., Stagg S. M., Voss N. R., Cheng A., Fellmann D., Pulokas J., Yoshioka C., Irving C., Mulder A., Lau P. W., Lyumkis D., Potter C. S., Carragher B., Appion: An integrated, database-driven pipeline to facilitate EM image processing. J. Struct. Biol. 166, 95–102 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ludtke S. J., Baldwin P. R., Chiu W., EMAN: Semiautomated software for high-resolution single-particle reconstructions. J. Struct. Biol. 128, 82–97 (1999). [DOI] [PubMed] [Google Scholar]
- 67.Lyumkis D., Brilot A. F., Theobald D. L., Grigorieff N., Likelihood-based classification of cryo-EM images using FREALIGN. J. Struct. Biol. 183, 377–388 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Scheres S. H. W., RELION: Implementation of a Bayesian approach to cryo-EM structure determination. J. Struct. Biol. 180, 519–530 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Collaborative Computational Project, Number 4., The CCP4 suite: Programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 50, 760–763 (1994). [DOI] [PubMed] [Google Scholar]
- 70.T. A. Jones, in Molecular Replacement, E. J. Dodson, S. Gover, W. Wolf, Eds. (SERC Daresbury Laboratory, 1992), pp. 91–105. [Google Scholar]
- 71.Cowtan K., Fitting molecular fragments into electron density. Acta Crystallogr. D Biol. Crystallogr. 64, 83–89 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Laskowski R. A., MacArthur M. W., Moss D. S., Thornton J. M., PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 26, 283–291 (1993). [Google Scholar]
- 73.Pettersen E. F., Goddard T. D., Huang C. C., Couch G. S., Greenblatt D. M., Meng E. C., Ferrin T. E., UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 (2004). [DOI] [PubMed] [Google Scholar]
- 74.Weaver E. A., Hillestad M. L., Khare R., Palmer D., Ng P., Barry M. A., Characterization of species C human adenovirus serotype 6 (Ad6). Virology 412, 19–27 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Thompson J. D., Higgins D. G., Gibson T. J., CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22, 4673–4680 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Buchan D. W. A., Minneci F., Nugent T. C. O., Bryson K., Jones D. T., Scalable web services for the PSIPRED Protein Analysis Workbench. Nucleic Acids Res. 41, W349–W357 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/3/5/e1602670/DC1
table S1. Number of amino acids and sequence identity between the respective capsid proteins in HAdV-D26 and HAdV-C5.
table S2. Residues forming the HVRs in HAdV-D26 versus HAdV-C5.
table S3. Structural characteristics and interactions of the N and C termini of hexon subunits in HAdV-D26.
table S4. Fiber-PB interactions in HAdV-D26.
table S5. Protein IIIa interactions with the major and minor proteins in HAdV-D26.
table S6. Data collection and refinement statistics of HAdV-D26.
fig. S1. Resolution estimations of the cryo-EM reconstruction of HAdV-D26.
fig. S2. Estimation of local resolution of the HAdV-D26 cryo-EM reconstruction.
fig. S3. Structural similarities and differences between the hexon subunits of HAdV-D26 and HAdV-C5.
fig. S4. Structural similarities and differences between the different hexon structures in HAdV-D26.
fig. S5. Structural superposition and sequence alignment of PBs from HAdV-D26 and HAdV-C5.
fig. S6. Interactions between the conserved N terminus of the fiber and the PB in HAdV-D26.
fig. S7. The structure and analysis of the fiber molecule obtained by LAR.
fig. S8. Interactions between the protein IX triskelions and the hexons.
fig. S9. The structural overlay of the 4-HLXBs of IX from HAdV-D26 and HAdV-C5.
fig. S10. Structural superposition of the protein IX molecules.
fig. S11. Structural conservation in protein VIII molecules from HAdV-D26 and HAdV-C5.
fig. S12. Molecular clamping interactions by the protein VIII.
fig. S13. β strand formation by the unidentified densities.
fig. S14. Gluing interactions of the PPHs by protein VIII.
fig. S15. Structures, locations, and organization of minor proteins relative to the MCPs in HAdV-D26.
fig. S16. Structural similarities of the IIIa proteins from HAdV-D26 and HAdV-C5.
fig. S17. Cryo-EM density and fit of the APD of IIIa in HAdV-D26.
fig. S18. Gluing interactions involving IIIa, PPHs, and PB.