

Abstract
Background
Colorectal cancer is the third leading cause of cancer-related mortality in most developed countries. This mortality is mainly due to the metastatic progression to the liver with the frequent recurrence. Colorectal cancer remains therapeutic challenge and this has intensified a search for a new drug target. In an effort to establish a novel targeted-therapy, we studied the molecular mechanisms of cancer stem cell inhibitor salinomycin.
Materials and Methods
Co-immunoprecipitation was performed to examine STAT3-STAT1 protein interactions. Telomerase activity was measured by PCR-Elisa assays. Apoptosis and cell stress arrays were analyzed to identify important proteins responding to salinomycin treatments.
Results
IL-6 and TNF-α induced STAT3 and STAT1 interactions, however the interactions were abolished by salinomycin challenge. Salinomycin reduced cancer stem cell phenotype and decreased telomerase activity of colorectal cancer cells.
Conclusion
Our work uncovers a new mechanism by which salinomycin inhibits cancer stemness suggesting a novel targeted-therapy for metastatic colorectal cancer.
Keywords: Cancer stem cells, STAT3, STAT1, Telomerase, Salinomycin, MST-312
Introduction
Colorectal cancer (CRC) is the third leading cause of mortality in United States and major cause of death worldwide (1). CRC mortality is primarily due to the hepatic metastasis. Over 50% of patients are diagnosed with hepatic metastases, either at the time of initial presentation or as a result of disease recurrence (2). There have been no major breakthrough in the treatment of metastatic CRC for decades. Multiple new FDA-approved therapies have been tried, nevertheless the 5-year survival remains very poor. Conventional chemotherapy efficiently targets tumor bulk, but there exists a small subpopulation of cells that contributes to resistance to the therapy and tumor regrowth. These cells are termed cancer stem cells (CSCs). Increasing evidences suggest that CSCs are responsible for the initiation and maintenance of tumors, resistance to cytotoxic drugs and disease recurrence (3). Therefore, targeting cancer stem cell population became essential to improve colorectal cancer prognosis.
Salinomycin is a polyether antibiotic isolated from Streptomyces albus that acts as a potassium ionophore. It was identified from the high throughput chemical library screening for CSC inhibitor and showed higher efficacy than the conventional chemotherapeutic agents (4). Salinomycin treatment induced cell death from various solid tumors. In mechanism study, salinomycin was shown to induce apoptosis from prostate cancer via the accumulated reactive oxygen species and mitochondrial membrane depolarization (5). For breast cancer, it has been reported salinomycin promoted anoikis and decreased the CD44(+)/CD24(−) cancer stem cell population via inhibition of STAT3 (signal transducer and activator of transcription 3) in MDA-MB-231 cells (6). However, it is still ill-defined by which mechanisms salinomycin specifically inhibits colon cancer stem cell populations.
STAT3 is a latent transcription factor involved in immune response and aberrantly activated in the various cancer stem cell populations (7). We have demonstrated that IL-6 treatment induced STAT3 activation and concurrent epithelial-mesenchymal transition (EMT) in colorectal cancer (8). Notably, STAT3 downstream target genes including cyclin D, Bax, VEGF and MMP2 contribute to cancer cell proliferation, drug resistance and metastasis (9).
MST-312 (telomerase inhibitor IX) is a synthetic compound that functions as a telomerase inhibitor (10). Telomerase is an enzyme which lengthens telomeres in DNA strands and activated in more than 90% of human malignancies. Increasing clinical cases revealed that telomerase was specifically activated in various human malignancies including colorectal cancer (11). Furthermore, clinicians reported that the prognosis of the patients with high telomerase activity was significantly worse than that of patients with moderate or low telomerase activity (P< 0.01) (12). These reports suggest telomerase may serve as a good prognosis marker for CRC as well as a good potential cellular target for CRC treatment.
In addition to its role in telomeric DNA polymerization, hTERT (human telomerase reverse transcriptase) has been implicated for multiple essential roles for oncogenesis (13~15). Ectopic expression of hTERT promoted the malignant transformation independently of telomere lengthening (16). Interestingly, hTERT was shown to stimulate EMT and induce stemness in human gastric cancer cells, thereby promote cancer metastasis and recurrence (17). These reports suggest hTERT modulates not only telomerase but also regulates CSC generation and maintenance in aggressive tumors. We hypothesized that salinomycin might inhibit telomerase via STAT3 inactivation in colon cancer stem cells. Herein, we report a new molecular mechanism by which salinomycin abolishes STAT3 and STAT1 interactions and decreases telomerase. Our results suggest that targeting STAT3 and telomerase can be an efficient novel therapy for metastatic colorectal cancer.
Material and Methods
Cell culture and reagents
ATCC CRL-1541 normal colon fibroblast, HT-29 and SW480 cancer cell lines were purchased from the American Type Culture Collection (ATCC, Manassas, VA, U.S.A.). They were maintained in a monolayer culture in DMEM/F12 (Dulbecco’s modified Eagle medium) with 10% fetal bovine serum, 2.5% L-Glutamine and 0.5% Penicillin/Streptomycin. Salinomycin (Sigma, catalog number: S4526), MST-312 (Sigma, catalog number: M3949) and IL-6 (Sigma, catalog number: I3268) were purchased from Sigma company (St. Louis, MO, USA).
Cell viability assay
Cell viability was determined using the Cell Titer 96 Aqueous One Solution Cell Proliferation Assay kit [MTS, 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium] (Promega, Madison, WI, USA catalog number: G3580) following the manufacturer’s instructions. The quantity of formazan product was determined by measuring absorbance at 450 nm using a Promega Glo Max-Multi detection system (Promega, Madison, WI).
Western blot analyses
Monolayer cultures of respective cell lines at 80–90% confluence were lysed using 100 μl of RIPA buffer (Thomas Scientific Inc. Swedesboro, NJ). Tris-glycine (Bio-Rad, Irvine, CA) gels were loaded with 50–100 μg of cell lysates. After electrophoresis, the gel was transferred to a nitrocellulose membrane for 2 hours. The membrane was blocked for 1 hour in 5% BSA or 5% skim milk at 4°C. The membrane was then washed 3 times with 1xTTBS and incubated overnight with the primary antibody at 4°C. Primary antibodies of STAT3, pSTAT3, P38, β-catenin and β-actin were purchased from Cell Signaling Technology (Danvers, MA). After incubation with the secondary antibodies conjugated with horseradish peroxidase (HRP), the protein bands were developed with the chemiluminescent reagents.
Co-immunoprecipitation
Cancer cells were stimulated with IL6 (10 ng/ml) and TNF-α (25 ng/ml) for 48 hours. For inhibition, salinomycin (3 μM) or MST-312 (5 μM) were challenged during the cytokine stimulation. Next, cells were washed once with PBS buffer and lysed in immunoprecipitation lysis buffer (Thomas Scientific Company). Antibodies to STAT3 (Cell signaling technology) were added to the cell lysates and incubated for one hour at 4°C. Protein A agaroses (Santa cruz biochnology) were then added to the cell lysates 20 μl each, incubated for overnight at 4°C. After the immunoprecipiation, cell lysates were briefly spun and washed three times with PBS buffer. Immuno-pellets were resuspended in 100 μl of loading dye (Bio Rad) and resolved on a 10 % polyacrylamide gel electrophoresis. Immunoblotting was performed with STAT1 antibody to confirm the STAT3-STAT1 interactions.
Colonosphere formation assay
Matrigel (BD, Cambridge, MA) 200 μl was spread as a thick layer on wells of a 24 well plate and allowed to polymerize at 37°C for 15 minutes. 2 × 104 cancer cells grown in monolayer were trypsinized to single cells and plated on top of the pre-coated matrigel. Plates were incubated at 37°C to allow cells to fully settle down before media was replaced with appropriate culture media containing 5% matrigel.
Stress and Apoptosis Antibody Array
The Stress and Apoptosis Signal Antibody Array Kit was purchased from Cell Signaling Technology (Cell Signaling Technology, Beverly, MA; Catalog number:12856) and followed the manufacturer’s instructions.
Telomerase Activity Assay
Cells were processed according to the manufacturer’s protocol for the TeloTAGGG Telomerase PCR ELISA kit (Roche, Orange, CA. Catalog number: 11854666910). Briefly, cell pellets were thawed in lysis reagent, incubated on ice for 30 minutes, and centrifuged at 16,000g for 20 minutes at 4 °C. Telomerase activity was immediately measured in the resultant supernatant using the telomeric repeat amplification protocol in which telomerase, if present in the cell lysate, adds telomeric repeats to the 3′ end of a biotin-labeled synthetic P1-TS primer. Samples were amplified by polymerase chain reaction (PCR), with P1-TS and P2 primers creating an elongated telomere. The PCR product was denatured and hybridized to a digoxigenin-labeled probe that detects telomeric repeats in a subsequent enzyme-linked immunosorbent assay (ELISA). Telomerase assays were performed three times independently and P values less than 0.05 were considered statistically significant.
Statistical analysis
Student t-tests were used to evaluate the significance of changes in all combination treatment assays compared to controls. Differences were considered statistically significant if P < 0.05.
Results
Salinomycin challenge reduced cancer cell proliferation, inhibited STAT3 phosphorylation and P38 and β-catenin expressions
First of all, we tested if salinomycin specifically inhibited colon cancer cells with a low-toxicity to normal human colon cells. To this end, ATCC CRL-1541 normal colon fibroblast cell line was used. As shown in Fig. 1, salinomycin inhibited HT-29 and SW480 colorectal cancer cell growth effectively over the range of 1, 2 and 5 μM for 48 hours (Fig. 1A). However, the normal colon cell line CRL-1541 retained ~ 80% cell proliferation with the 2 μM salinomycin concentration. HT-29 and SW480 cancer cells showed 22% and 17% cell proliferation at 2 μM salinomycin, respectively. Our results confirmed the specific colon cancer cell growth inhibition by salinomycin treatments.
Figure 1. Salinomycin inhibited STAT3 activation and down-regulated P38 and β-catenin in colorectal cancer cells.
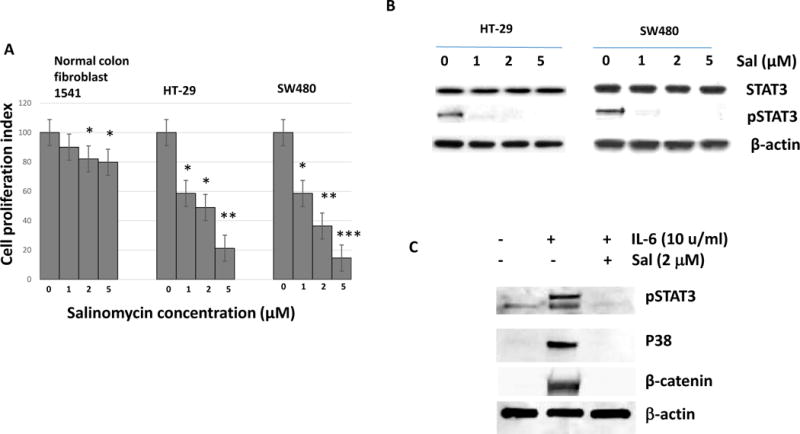
(A) Cells were treated with salinomycin 1, 2, 5 μM for 48 hours and measured for cell viability. Data are presented as average ± SD (n=3 in each group) *p < 0.05, ** p < 0.01 ***, p < 0.001 vs. untreated control. (B) HT-29 and SW480 cells were treated with salinomycin 1, 2, 5 μM for 48 hours and phospho-STAT3 levels were measured. *p < 0.05, ** p < 0.01 vs. untreated control. (C) HT-29 cells were treated with IL6 in the presence or absence of salinomycin (2 μM), then subject to western analysis of pSTAT3, P38 and β-catenin.
Salinomycin was originally identified from the high throughput screening for breast cancer stem cell inhibitor. STAT3 was previously shown to be activated selectively in various cancer stem cell populations. We hypothesized salinomycin may inhibit STAT3 activation in a dose dependent manner in colorectal cancer cells. After exposure to salinomycin (1~ 5 μM) for 48 hours, phospho-STAT3 protein levels were reduced in a dose-dependent manner (Fig. 1B). Salinomycin inhibited the STAT3 phosphorylation from 1 μM in HT-29 and SW480 (Fig. 1B). However, total STAT3 amount was not affected. Besides pSTAT3, we have examined P38 and β-catenin activation with IL6. As shown in figure, both P38 and β-catenin were activated with IL6 stimulation (Fig. 1C). However, salinomycin challenge has abolished the P38 and β-catenin upregulation with pSTAT3 inhibition. Our data suggest that salinomycin inhibits cancer cell proliferation and STAT3 signaling in colorectal cancer cells.
Salionomycin suppressed epithelial-mesenchymal transition in colorectal cancer cells
IL-6 treatment induced epithelial-mesenchymal transition (EMT) through STAT3 activation in colon cancer cells (18). We next examined whether salinomycin challenge suppressed the IL-6 induced EMT. Typical EMT markers E-cadherin was reduced upon IL-6 treatment (10 u/ml) for 72 hours and vimentin started to express in colon cancer cells, indicating EMT is induced (Fig. 2A). However, in the presence of salinomycin 2 μM, E-cadherin expression has been restored whereas vimentin expression has decreased, implicating the EMT has been suppressed. Corresponding histograms were presented with statistical differences (Fig. 2B). These results indicate that salinomycin acts as a negative regulator of STAT3 during the EMT process.
Figure 2. Salinomycin suppressed epithelial-mesenchymal transition.
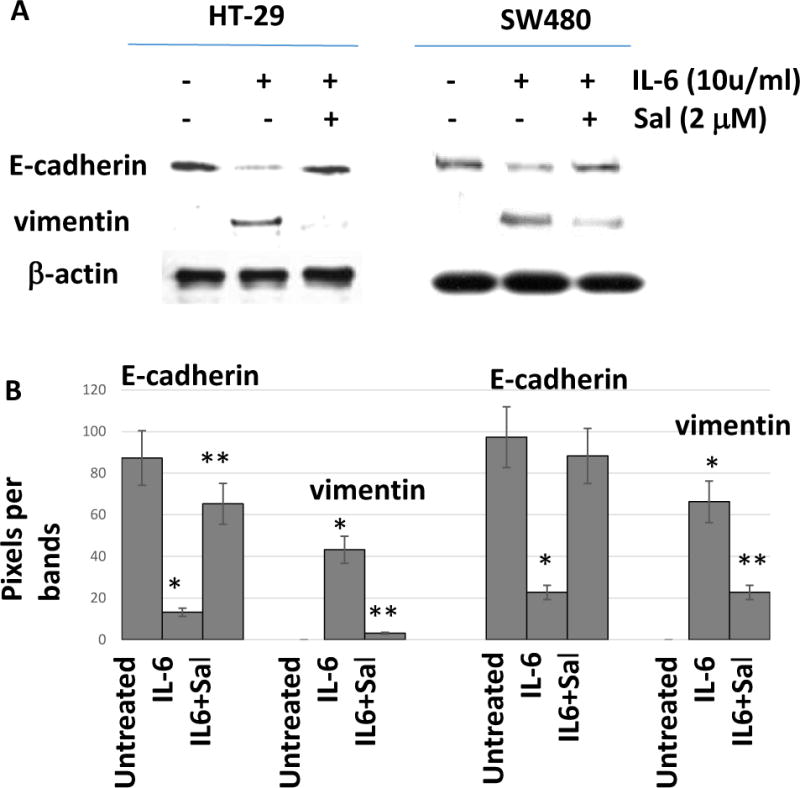
(A) HT-29 and SW480 cells were treated with IL-6 (10 u/ml) and salinomycin (2 μM) to monitor the EMT markers. (B) Histograms were presented from the EMT induced by IL-6 and suppressed by salinomycin. Data are presented as average ± SD (n=3 in each group) *p < 0.05, ** p < 0.01 vs. untreated control.
IL6 and TNF-α stimulation enhanced STAT3 and STAT1 interactions, however, salinomycin abolished the protein-protein interactions
Pro-inflammatory cytokines IL6 and TNF-α activated STAT3 and STAT1, respectively. We hypothesized that STAT3 and STAT1 may interact each other, and when IL6 and TNF-α were treated, the protein-protein interactions might be enhanced. To test this, we performed co-immunoprecipation assay where we pulled down STAT3 proteins and resolved the immune-pellets on western blot to identify the presence of STAT1 protein. As shown in the figure, STAT1 was bound to STAT3 protein and the interactions were increased upon IL6 and TNF-α combined treatments (Fig. 3A). Our results suggest that STAT3 binds STAT1 and the interactions are enhanced with cytokine stimulation.
Figure 3. STAT3 binds STAT1 and the interactions were enhanced with IL6 and TNF-α co-treatments and abolished with salinomycin challenge.
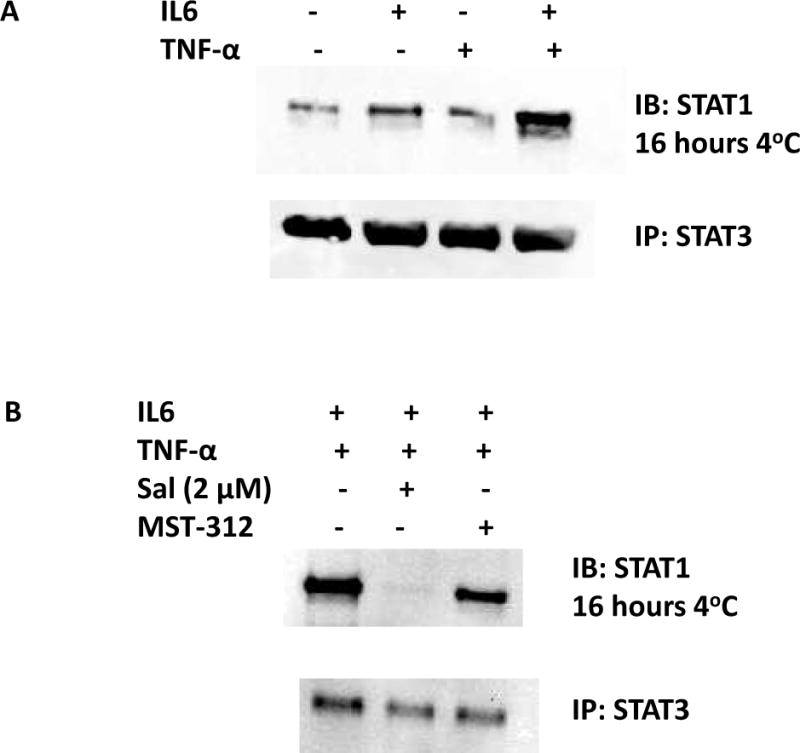
(A) Co-immunoprecipitation revealed that STAT3 binds STAT1. When co-treated with IL6 and TNF-α, the interactions were enhanced. (B) Salinomycin challenge has abolished the STAT3-STAT1 interactions.
Next, we tested if salinomycin or MST-312 challenge can abolish the STAT3-STAT1 interactions. Our previous data showed salinomycin inhibited STAT3 phosphorylation (Fig. 1B). MST-312 is a telomerase inhibitor and does not inhibit STAT3 phosphorylation. Salinomycin challenge abolished the STAT3-STAT1 protein-protein interactions but MST-312 did now inhibit the interactions (Fig. 3B). STAT3-STAT1 interactions were likely enhanced via activated STAT3 phosphorylation. Our data suggest that salinomycin abolished the STAT3 binding with STAT1 by inhibiting its phosphorylation step.
Salinomycin treatment inhibited colonosphere formation and wound healing ability of colorectal cancer cells
As salinomycin challenge suppressed epithelial-mesenchymal transition, we next focused on the cancer stem cell phenotype. To examine the CSC traits, we studied the colonosphere formation ability and wound healing capacity. In colonosphere culture condition, HT-29 and SW480 cells formed colonospheres in 7 days. However, when we pre-treated cells for 24 hours with salinomycin (1~5 μM), the colonosphere forming capacity has been significantly reduced (Fig. 4A). In HT-29, pre-treatment with salinomycin 1 μM for 24 hours reduced the colonosphere forming fold ratio to 0.66 compared with untreated control. Salinomycin 2 μM and 5 μM also reduced the fold changes to 0.42 and 0.18, respectively. SW480 cell line showed a similar pattern with the salinomycin challenge.
Figure 4.
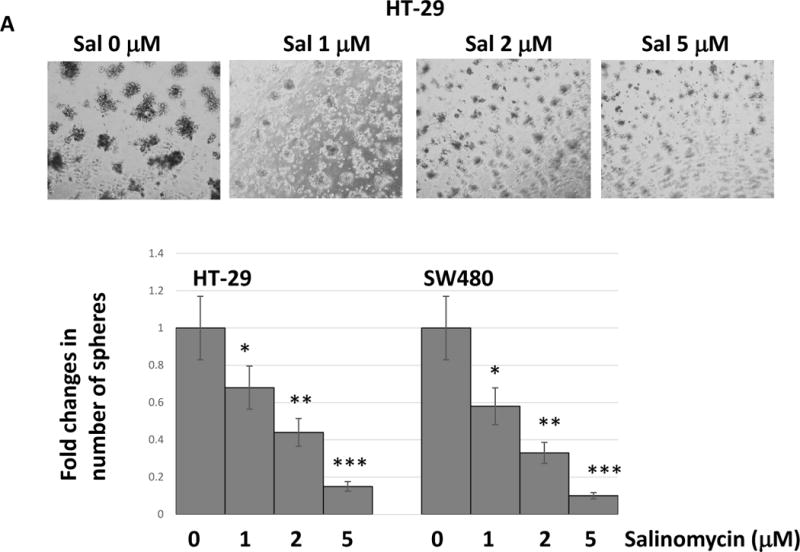
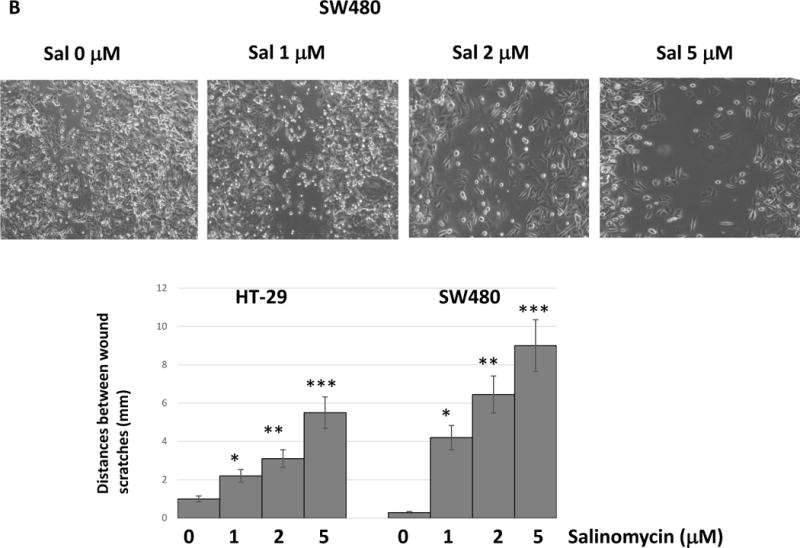
Salinomycin reduced cancer stem cell-like traits in colorectal cancer cells. (A) Cells were pre-treated with salinomycin for 24 hours and placed in the colonosphere forming wells. Colonospheres were formed for 7days. (B) Cells were pre-treated with salinomycin and applied to wound healing assays. All of data are presented as average ± SD (n=3 in each group) *p < 0.05, ** p < 0.01 ***, p < 0.001 vs. untreated control.
Next, we performed wound healing assay to measure the cell migration capability. Cancer cells were pre-treated with salinomycin 1, 2 and 5 μM for 24 hours and applied to wound scratching. In SW480, 1 μM salinomycin showed the distance of 4.1 mm between the wounds whereas untreated control showed a complete healing (Fig. 4B). Salinomycin 2 μM and 5 μM pre-treatments increased the wound distance to 6.2 and 9 mm, respectively. HT-29 showed the similar wound healing inhibition. Our data indicate that salinomycin treatments clearly inhibit the cancer stem cell phenotype.
Distinct cellular stress and apoptosis-related proteins were activated in response to salinomycin and MST-312 treatments
The observation that salinomycin challenge inhibited colorectal cancer stem cell phenotype suggests it may directly trigger certain signaling pathways. To find the underlying mechanisms of salinomycin, we studied molecular changes in the cellular stress and apoptosis proteins. We have combined salinomycin and MST-312 to accomplish the enhanced anti-cancer effects from the anti-CSC and anti-telomeric activities. To this end, the stress and apoptosis signaling antibody array kit was used. By using this array, we were able to simultaneously interrogate 19 signaling molecules that are involved in the regulation of stress response and apoptosis.
Salinomycin activated Akt (Ser 473) and downregulated Hsp27 (Ser 82) phosphorylation in HT-29 and SW480 (Fig. 5A and B). Akt is activated by stimulation of growth factor receptors and promotes anabolic growth and survival signals by targeting its broad array of substrates. In response to salinomycin challenge, cancer cells likely initiated survival signals to endure the cell stress triggered by salinomycin. Hsp27 activation was decreased in the presence of salinomycin. The main function of Hsp27 is thermo-tolerance in vivo and cytoprotection under cell stress. Salinomycin specifically inhibited the Hsp27 activation during the stress process.
Figure 5. Salinomycin and MST-312 treatments elicited stress protein activation.
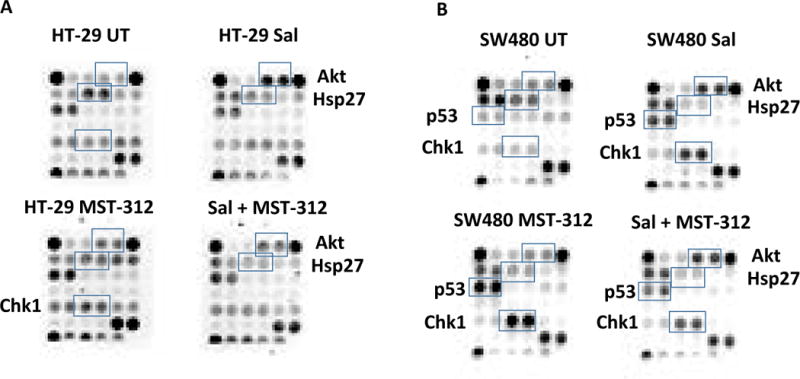
(A) HT-29 cells were treated with salinomycin alone (sal), MST-312 alone and salinomycin and MST-312 combined, then examined for the cell stress and apoptosis antibody arrays. (B) SW480 cells were treated with salinomycin alone (sal), MST-312 alone and salinomycin and MST-312 combined, then examined for the cell stress and apoptosis antibody arrays.
In SW480, p53 and Chk1 were both activated upon salinomycin and MST-312 treatment. In HT-29, Chk1 was activated upon MST-312 treatment as well. Chk1 plays a role in DNA damage check point control. Salinomycin and MST-312 induced the DNA damages, thereby promoted apoptosis in cancer cells. Chk1 likely responds to those DNA damages to control the check point more tightly. Salinomycin and MST-312 treatments induced DNA damages as p53 started to respond to the damages. P53 activation levels in HT-29 are the same in the absence and presence of salinomycin. This may reflect the differences in cancer cell line specificity.
Salinomycin down-regulated hTERT and reduced telomerase activity when combined with telomerase inhibitor
Since salinomycin inhibited STAT3 activation and reduced cancer stem cell traits, we next wished to determine whether salinomycin decreased telomerase activity. STAT3 is a known transcription factor for human telomerase reverse transcriptase (19). Thus, we first monitored hTERT and pSTAT3 expression levels with salinomycin alone and in combination with MST-312. Salinomycin 5 μM was treated for 48 hours for colorectal cancer cells. As shown in the figure, salinomycin inhibited both pSTAT3 and hTERT, indicating STAT3 inhibition may transcriptionally down-regulated hTERT (Fig. 6A). In accordance, salinomycin has decreased telomerase activities in both HT-29 and SW480, whereas chemotherapeutic drug 5-FU 5 μM for 48 hours did not affect the telomerase (Fig. 6B and C). Telomerase activity has decreased from 3.3 to 2.65 in HT-29 and from 3.35 to 2.7 in SW480. Telomerase inhibitor MST-312 treatment at 5 μM clearly reduced the telomerase activity to 1.5 and 1.3 in HT-29 and SW480, respectively. When salinomycin 5 μM and MST-312 5 μM were combined, the anti-telomerase activities were even more decreased to 1.1 in HT-29 and 0.75 in SW480. Our data revealed that salinomycin has certain anti-telomeric activities, and when combined with MST-312, it can enhance the anti-telomerase activity.
Figure 6. Salinomycin down-regulated hTERT and decreased telomerase activity.
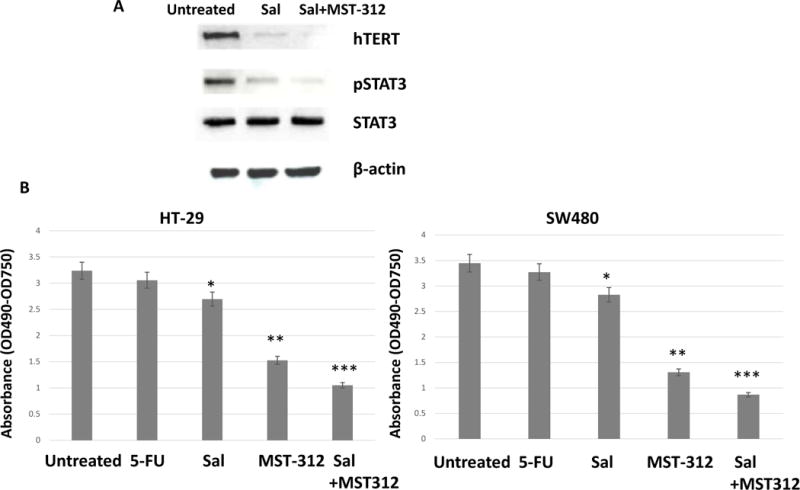
Salinomycin and MST-312 treatments decreased telomerase activity. (A) HT-29 cells were treated with salinomycin alone or combined with MST-312, then was subject to western analyses of hTERT and pSTAT3 (B) HT-29 and SW480 cells were treated with 5-FU, Salinomycin, MST-312 and salinomycin and MST-312 combined, then applied to the telomerase activity assays. Data are presented as average ± SD (n=3 in each group) *p < 0.05, ** p < 0.01 ***, p < 0.001 vs. untreated control.
Discussion
Cancer stem cells are responsible for the tumor initiation, proliferation, chemoresistance and tumor re-initiation after therapy. Therefore, it is essential to understand the signal pathways selectively activated in cancer stem cell populations so that we can devise strategies to target them. Here, we report a new molecular mechanism by which salinomycin targets cancer stem cells. Salinomycin inhibited the STAT3 phosphorylation indicating it targeted STAT3 during its action of mechanism. In accordance, cancer stem cell phenotype was inhibited in a dose-dependent manner with salinomycin treatments. STAT3 is constitutively activated in numerous human malignancies (20). Furthermore, phosphorylated STAT3 has been reported to participate in tumor invasion and metastasis (21). Our study revealed that CSC inhibitor salinomycin inactivated STAT3, abolished STAT3-STAT1 binding thereby suppressed STAT3 driven EMT process. This work has potential application to limit the tumor metastasis and recurrence in colorectal cancer.
To identify signatures responding to salinomycin and MST-312 challenges, we analyzed the cellular stress and apoptosis protein arrays. Specific subset of genes that are upregulated and downregulated upon the treatments. Akt is activated to increase the survival signaling in response to the salinomycin. Cancer cells likely strengthen the survival signaling to endure the stress induced by salinomycin. Salinomycin treatment specifically downregulated Hsp27 phosphorylation which plays a role in thermos-tolerance and cell stress. Thermo-tolerance ability has been weakened by salinomycin challenge. Both Chk1 and p53 are activated to control the DNA damages elicited by salinomycin and MST-312. Chk1 and p53 activations are cellular response to control the DNA damages elicited by the treatments. This work revealed a distinct stress protein signatures that responded to the salinomycin and MST-312 challenges.
MST-312 is a telomerase inhibitor as it reduced telomerase activity and induced telomere dysfunction. MST-312 exposure to breast cancer cells elevated level of double strand breaks (DSBs) based on the presence of the ɤ-H2AX proteins (22). This acute induction of DSBs resulted in growth arrest and more evident in the metastatic breast cancer cells. We chose MST-312 because it induced growth arrest more efficiently in metastatic tumor cells. When combined with MST-312, salinomycin inhibited telomerase activity in an additive fashion. Telomerase is specifically activated in colorectal tumors. Our results suggest that salinomycin and MST-312 dual targeting cancer stem cell and telomerase can be a novel approach for curing colorectal cancer. Salinomycin inhibits cancer stem cells and MST-312 targets the bulk colorectal tumors at the same time.
We reasoned that if salinomycin inhibits STAT3 phosphorylation, it may block the STAT3 binding to the hTERT promoter because phosphorylated STAT3 mainly translocated into the nucleus. As expected, IL-6 treatment enhanced STAT3 binding to the hTERT promoter and salinomycin abolished the binding. We have shown the new anti-CSC mechanism of salinomycin where it is negatively regulating STAT3 and hTERT resulting in telomerase inhibition. STAT3-hTERT axis may contributes to cancer stem cell generation. Taken together, present work suggests that novel targeted-therapy can be implemented based on the dual targeting STAT3 and telomerase for metastatic colorectal cancer. The in vivo animal work is warranted to validate the reduction of colon tumor formation and limiting liver metastasis with the salinomycin/MST-312 co-treatment.
Acknowledgments
This work was supported by the National Institutes of Health (NIH, NCI, NIMHD, NCATS) Grants to J.V. Vadgama: U54 CA143931, U54MD007598, UL1TR000124. S. Steven Chung is a scholar supported by the Clinical Research Education and Career Development by the NIMHD R25 MD 007610, pilot project award from U54 MD 007598 and Emerging scientist award from the Urban Health Institute-CDU S21 MD 000103. Research reported in this publication was supported by Accelerating Excellence in Translational Science Pilot Grants G0812D05, pilot project award from the National Institutes of Health (NCI, NIMHD) Grants: U54 CA143931 and U54MD0075984, and NIH/NCI SC1CA200517 to Y. Wu.
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. We deeply appreciate the division of cancer research and training members for their helpful comments and suggestions.
References
- 1.Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics 2014. CA Cancer J Clin. 2014;64:9–29. doi: 10.3322/caac.21208. [DOI] [PubMed] [Google Scholar]
- 2.Luu C, Arrington K, Schoellhammer H, Singh G, Kim J. Targeted therapies in colorectal cancer: surgical considerations. J Gastrointest Oncol. 2013;4:328–336. doi: 10.3978/j.issn.2078-6891.2013.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Clarke M, Dick J, Dirks P, Eaves C, Jamieson C, Jones D, Visvader J, Weissman I, Wahl G. Cancer stem cells-perspectives on current status and future directions. Cancer Res. 2006;66:9339–9344. doi: 10.1158/0008-5472.CAN-06-3126. [DOI] [PubMed] [Google Scholar]
- 4.Gupta P, Onder T, Jiang G, Tao K, Kuperwasser C, Weinberg R, Lander E. Identification of selective inhibitors of cancer stem cells by high-throughput screening. Cell. 2009;138:645–659. doi: 10.1016/j.cell.2009.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kim K, Yu S, Lee S, Chun S, Choi Y, Park Y, Song C, Chatterjee B, Ahn S. Salinomycin-induced apoptosis of human prostate cancer cells due to accumulated reactive oxygen species and mitochondrial membrane depolarization. Biochem Biophysical Res Comm. 2011;413:80–86. doi: 10.1016/j.bbrc.2011.08.054. [DOI] [PubMed] [Google Scholar]
- 6.An H, Kim J, Oh E, Lee N, Cho Y, Seo J. Salinomycin promotes anoikis and decrease the CD44+/CD24- stem-like population via inhibition of STAT3 activation in MDA-MB-231 cells. PLOS One. 2015;10:e014919. doi: 10.1371/journal.pone.0141919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Marotta L, Almendro V, Marusyk A, Shipitsin M, Schemme J, Walker S, Bloushitain-Quimron N, Choudhury S, Maruyama R, Wu Z, Gonen M, Mulvey L, Bessarabova M, Huh S, Silver S, Kim S, Park S, Lee H, Anderson K, Richardson A, Nikolskaya T, Nikolsky Y, Liu X, Root D, Hahn W, Frank D, Polyak K. The JAK2/STAT3 signaling pathway is required for growth of CD44(+) CD24(−) stem cell-like breast cancer cells in human tumors. J Clin Invest. 2011;121:2723–2735. doi: 10.1172/JCI44745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chung S, Dwabe S, Elshimali Y, Sukhija H, Aroh C, Vadgama J. Identification of novel biomarkers for metastatic colorectal cancer using angiogenesis-antibody array and intracellular signaling array. PLOS One. 2015;10:e0134948. doi: 10.1371/journal.pone.0134948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dauer D, Ferraro B, Song L, Yu B, Mora L, Buettner R, Enkemann S, Jove R, Haura E. STAT3 regulates genes common both wound healing and cancer. Oncogene. 2005;24:3397–3408. doi: 10.1038/sj.onc.1208469. [DOI] [PubMed] [Google Scholar]
- 10.Seimiya H, Oh-hara T, Suzuki T, Naasani I, Shimazaki T, Tsukiya K, Tsuruo T. Telomere shortening and growth inhibition of human cancer cells by novel synthetic telomerase inhibitors MST-312, MST-295, and MST-1991. Mol Cancer Ther. 2002;1:657–665. [PubMed] [Google Scholar]
- 11.Bertorelle R, Rampazzo E, Pucciarelli S, Nitti D, De Rossi A. Telomeres, Telomerase and Colorectal Cancer. World J Gastroenterol. 2014;20:1940–1950. doi: 10.3748/wjg.v20.i8.1940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tatsumoto N, Hiyama E, Murakami Y, Imamura Y, Shay J, Matsuura Y, Yokoyama T. High telomerase activity is an independent prognostic indicator of poor outcome in colorectal cancer. Clin Cancer Res. 2000;6:2696–2701. [PubMed] [Google Scholar]
- 13.Schwartz H, Juliao S, Sciadini M, Miller L, Burtler M. Telomerase activity in oncogenesis in giant cell tumor of bone. Cancer. 1995;75:1094–1099. doi: 10.1002/1097-0142(19950301)75:5<1094::aid-cncr2820750507>3.0.co;2-b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Urquidi V, Tarin D, Goodison S. Role of telomerase in cell senescence and oncogenesis. Annual Rev Med. 2000;51:65–79. doi: 10.1146/annurev.med.51.1.65. [DOI] [PubMed] [Google Scholar]
- 15.Blasco M, Hahn W. Evolving views of telomerase and cancer. Trend in Cell Biol. 2003;13:289–294. doi: 10.1016/s0962-8924(03)00085-0. [DOI] [PubMed] [Google Scholar]
- 16.Stewart S, Hahn W, O’Connor B, Banner E, Lundberg A, Modha P, Mizuno H, Brooks M, Fleming M, Zimonjic D, Popescu N, Weinberg R. Telomerase contribute to tumorigenesis by a telomere length independent mechanism. Proc Nat Acad Sci. 2002;99:12606–12611. doi: 10.1073/pnas.182407599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu Z, Li Q, Li K, Chen L, Li W, Hou M, Liu T, Yang J, Lindvall C, Bjorkholm M, Jia J, Xu D. Telomerase reverse transcriptase promotes epithelial-mesenchymal transition and stem cell like traits in cancer cells. Oncogene. 2013;32:4203–4213. doi: 10.1038/onc.2012.441. [DOI] [PubMed] [Google Scholar]
- 18.Liu H, Ren G, Wang T, Chen Y, Gong C, Bai Y, Wang B, Qi H, Shen Z, Zhu L, Qian C, Lai M, Shao J. Aberrantly expressed Fra-1 by IL-6/STAT3 transactivation promotes colorectal cancer aggressiveness through epithelial-mesenchymal transition. Carcinogenesis. 2015;36:459–468. doi: 10.1093/carcin/bgv017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Konnikova L, Simeone M, Kruger M, Kotecki M, Cochran B. Signal transducer and activator of transcription 3 (STAT3) regulates human telomerase reverse transcriptase (hTERT) expression in human cancer and primary cells. Cancer Res. 2005;65:6516–6520. doi: 10.1158/0008-5472.CAN-05-0924. [DOI] [PubMed] [Google Scholar]
- 20.Turkson J, Jove R. STAT proteins: novel molecular targets for cancer drug discovery. Oncogene. 2000;19:6613–6626. doi: 10.1038/sj.onc.1204086. [DOI] [PubMed] [Google Scholar]
- 21.Kamran M, Patil P, Gude R. Role of STAT3 in cancer metastasis and translational advances. Biomed Res Int. 2013;2013:421821. doi: 10.1155/2013/421821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gurung R, Lim S, Low G, Hande M. MST-312 alters telomere dynamics, gene expression profiles and growth in human breast cancer cells. J Nutrigenetics and Nutrigenomics. 2014;7:283–298. doi: 10.1159/000381346. [DOI] [PubMed] [Google Scholar]