

Abstract
Cadmium (Cd) is a carcinogen with several well-described toxicological effects in humans, but its molecular mechanisms are still not fully understood. Overexpression of heat shock protein 27 (HSP27/HSPB1)—a multifunctional protein chaperone—has been shown to protect cells from oxidative damage and apoptosis triggered by Cd exposure. The aims of this work were to investigate the potential use of extracellular recombinant HSP27 to prevent/counteract Cd-induced cellular toxicity and to evaluate if peroxynitrite was involved in the development of Cd-induced toxicity. Here, we report that the harmful effects of Cd correlated with changes in oxidative stress markers: upregulation of reactive oxygen species, reduction in nitric oxide (NO) bioavailability, increment in lipid peroxidation, peroxynitrite (PN), and protein nitration; intracellular HSP27 was reduced. Treatments with Cd (100 μM) for 24 h or with the peroxynitrite donor, SIN-1, decreased HSP27 levels (~50%), suggesting that PN formation is responsible for the reduction of HSP27. Pre-treatments of the cells either with Nω-nitro-l-arginine methyl ester hydrochloride (L-NAME) (a pharmacological inhibitor of NO synthase) or with recombinant HSP27 (rHSP27) attenuated the disruption of the cellular metabolism induced by Cd, increasing in a 55 and 52%, respectively, the cell viability measured by CCK-8. Cd induced necrotic cell death pathways, although apoptosis was also activated; pre-treatment with L-NAME or rHSP27 mitigated cell death. Our findings show for the first time a direct relationship between Cd-induced toxicity and PN production and a role for rHSP27 as a potential therapeutic agent that may counteract Cd toxicity.
Electronic supplementary material
The online version of this article (doi:10.1007/s12192-017-0768-y) contains supplementary material, which is available to authorized users.
Keywords: Heat shock protein 27 (HSP27), Cytoprotection, Signaling cadmium toxicity, Oxidative stress
Introduction
Cadmium (Cd) is a non-essential, carcinogenic metal that can harm human health. In humans, exposure to this dangerous chemical typically occurs through the occupational environment, diet, smoking, and environmental pollution, causing various health problems, ranging from kidney and liver damage to cancer. Nowadays, Cd is still widely used in industrial processes, for example, as an anti-corrosive agent, a stabilizer in PVC products, a color pigment, a neutron absorber in nuclear power plants, and as a component of Ni–Cd batteries, television screens, lasers, and in some cosmetics; before the 1960s, it was also used with zinc to weld seals in lead water pipes (Bernhoft 2013; Salazar and McNutt 2012). Phosphate fertilizers also contain a large Cd load (Al-Shawi 1999; Lambert et al. 2007). Although some Cd-containing products can be recycled, a large portion of Cd waste makes its way back into the environment through landfill disposal and incineration of Cd-containing waste (Järup 2003; Järup and Åkesson 2009). Therefore, Cd pollution has awakened an increasing worldwide concern. Currently, the treatments available for individuals exposed to sub-lethal doses of Cd rely on the use of chelators (Bernhoft 2013), supplemented with vitamin D in the presence of bone disease symptoms (Andersen and Nielsen 1988; Cantilena 1982; Gil et al. 2011; Jones and Cherian 1990; Medicine ICoI 2010; Tandon et al. 2003). Nevertheless, some authorities do not recommend the use of chelators such as calcium–EDTA because symptoms of pulmonary and renal injuries may typically worsen (even with cessation of Cd exposure) and, subsequently, lessen survival (Nordberg et al. 2014). Additionally, this therapy may increase kidney tubules damage (Gilman et al. 1946).
Heavy metal-induced toxicity produces deleterious consequences in the ecosystem (de Vries et al. 2007) as well as in human health (Organization WH and Joint 2007). Exposure to Cd, either from the environment (e.g., smoking habits, diet) or occupational hazards produces a variety of toxicological and carcinogenic effects depending on the doses and period of exposure (Joint et al. 2011; Organization WH and Humans IWGotEoCRt 1993). The toxicological effects include the following: damage in kidney (Nordberg et al. 2002; Nordberg et al. 2012; Nordberg et al. 2009), liver (Kang et al. 2013; Koyu et al. 2006), and stomach and induction of several disorders involving the respiratory system (Cancer IAfRo et al. 1993; Kirschvink et al. 2006), bone (Satarug and Moore 2004; Takebayashi et al. 2000), and the nervous system (Sinha et al. 2008; Viaene et al. 2000). The carcinogenic effects of Cd has been described in vitro and in vivo for several cancers (Nordberg et al. 2014; Organization WH and Humans IWGotEoCRt, 1993); in humans, Cd plays a role in hormone dependent cancer (prostate, breast, and endometrial cancer) (Cho et al. 2013). An increased Cd concentration has been found in tissue and biological media of patients with breast cancer (Ionescu et al. 2006; Strumylaite et al. 2011; Strumylaite et al. 2014). Additionally, in post-menopausal women, dietary chronic exposures to low Cd doses have been linked to increased risk of endometrial and breast cancer in several studies (Åkesson and Julin 2008; Gallagher and Chen 2010; Itoh et al. 2014).
Currently, several indirect mechanisms have been implicated in Cd toxicity and carcinogenicity. Cd ion can inactivate a series of enzymes and proteins containing metals through direct binding in their active sites and/or perturbation in the enzyme topography, damaging cellular membranes. Other mechanisms implicate reactive oxygen species (ROS) production, even though Cd cannot catalyze typical Fenton-type reactions (because it does not accept or donate electrons under physiological conditions) (Casalino et al. 2002; Szuster-Ciesielska et al. 2000). Additionally, depletion of glutathione, a key intracellular antioxidant, has been described due to Cd binding to sulfhydryl groups (Valko et al. 2005). A single oral dose exposure to soluble Cd salts, in ranges of 30–40 mg, is lethal in humans (Dressler et al. 2002; Taylor et al. 1999). Nevertheless, most of the cases of intoxication are due to a long-term accumulation because only about 0.001% of Cd in the body is excreted per day, and therefore, Cd can persist for a period of >30 years in different organs and tissues (Friberg et al. 1992; Järup and Åkesson 2009).
The ability of cells to adapt to metabolic, proteotoxic, oxidative, and DNA-damaging stresses is crucial in maintaining the organism’s health. However, when the normal stress responsive clearance pathways are overloaded, toxic effects leading to disease such as cancer or chronic degenerative disorders may be inevitable. One major, evolutionarily conserved adaptive mechanism is the heat shock response, which involves the upregulation of a family of stress-inducible proteins, known as heat shock proteins (HSPs). This class of proteins helps to preserve cellular homeostasis by preventing protein aggregation and apoptosis. In particular, there is evidence showing that one member of the group of small heat shock proteins, heat shock protein-27 (HSP27 also known as HSPB1), if overexpressed (using transfection technology), can enhance resistance to the toxic effects of cadmium chloride (CdCl2) as well as to other metals in vitro (Wu and Welsh 1996). Although transfection of HSP27 is effective in vitro, it is not feasible to use this approach for treating humans exposed to Cd. To overcome this problem, in this work, we suggest using recombinant HSP27 (rHSP27), a protein that can be internalized in cells and possibly be further regulated at the cellular level by the organism. In the cell, HSP27 forms large oligomers capable of protecting cells from oxidative stress and heat shock (Arrigo 2001; Rogalla et al. 1999). It also interacts with key components involved in caspase activation and with the apoptotic signaling pathway promoting cell survival (Bruey et al. 2000; Concannon et al. 2001; Paul et al. 2002). Based on its cytoprotective effects, we hypothesize that rHSP27 may be a good alternative to the current treatments for Cd toxicity.
Although the potential application of rHSP27 as a cytoprotective molecule was very exciting, several studies have showed an overexpression of HSP27 in various cancers, including cancer of the breast, ovary, and endometrium. The precise role of HSP27 in these cancers is unclear; however, it is argued that the anti-apoptotic effect of HSP27 may enhance tumorigenic properties and the resistance to anti-cancer therapies. High levels of this protein have been observed in metastatic tissues compared to non-metastatic tissues in prostate cancer (HSP26 MAPKAPK2). In those cases, HSP27 expression was found to be dysregulated in tumor tissues. HSP27 (HSPB1) is overexpressed in breast cancers and may affect the disease outcome as well as the sensitivity of tumors to chemotherapy and radiotherapy (Ciocca et al. 2010b; Fanelli et al. 2008). In prostate cancer, HSPB1 expression was also seen in a high percentage of hormone-refractory patients and correlated with poor clinical outcome and shorter survival (Ciocca et al. 2010a). In our study, the administration, doses, and time of exposure to rHSP27 were regulated. Therefore, to explore the use of rHSP27 to prevent Cd toxicity, we explore not only the cytoprotective effect of rHSP27 but also its capability to enhance cell migration, a necessary step for cancer invasion and metastasis. According to our results, treatment with rHSP27 did not enhance the migration capabilities of the cells and protected the cells against Cd toxicity.
Our results, similar to previous findings (Oh and Lim 2006; Szuster-Ciesielska et al. 2000), indicated that Cd induced production of ROS. More specifically, we found for the first time evidence that Cd induced a strong oxidant species: peroxynitrite (ONOO−; “PN”), which is formed from the reaction of nitric oxide (NO) with superoxide (O2 −) (Pryor and Squadrito 1995). PN as well as Cd, subsequently modulated HSP27 levels. On the other hand, when NO formation (and, consequently, PN formation) was blocked with Nω-nitro-l-arginine methyl ester hydrochloride (L-NAME), the reduction in HSP27 levels was delayed and the cellular viability was preserved. Summarizing this work showed a correlation between Cd and PN production, the reduction of HSP27 due to PN production confirming the role of HSP27 in Cd resistance. We used, in this case, a recombinant extracellular protein, rHSP27, to abrogate Cd toxic effects. The use of the extracellular proteins will allow, in the future, further evaluation in vivo and in vitro, making rHSP27 a promising candidate for future therapies.
Material and methods
Cell lines and treatment
HeLa cells obtained from ATCC were cultured in RPMI or DMEM media with the addition of 10% fetal bovine serum (FBS) (Gibco). When they reached 85% of confluence, the cells were subjected to serum-free media containing the following treatments: 0, 5, 50, and 100 μM Cd during different times, the selected doses were previously described by our working group (Cuello-Carrión et al. 2015) and coincided with the doses used in other recent publications (Odewumi et al. 2015; Panjehpour et al. 2010; Rusanov et al. 2015). Cd was added to the culture media from a stock solution of 1 mM CdCl2. Treatment with L-NAME (Sigma) was performed since 24 h before and during Cd treatment at a final concentration of 500 μM in DMEM 10% BFS. Treatment with 3-morpholinosydnonimine hydrochloride (SIN-1; Sigma) was performed for 3 h in serum-free media at a final concentration ranging from 1 to 5 mM; after calibration, we use 1 mM SIN-1 as positive control.
Establishment of permanently transfected HeLa cells
HeLa wild type was plate in six-well plate at 60% confluence and transfected with 2 μl of X-treme Gene HP transfection reagent (Roche) and 1 μg of a plasmid pRNAi-H1-hyg (Biosettia) containing the following sequences: Scramble: AAAAGCTACACTATCGAGCAATTTTGGATCCAAAATTGCTCGATAGTGTAGC for HeLa pScr and 762: AAAAGCCGCCAAGTAAAGCCTTATTGGATCCAATAAGGCTTTACTTGGCGGC for HeLa p762. The cells were incubated ON and selected with 200 μg/ml hygromycin. After selection, HeLa p762 reaches a 50% of downregulation for HPS27 and the resistant phenotype was maintained with 50 μg/ml of the antibiotic.
Generation of recombinant proteins
N-terminal His-tagged HSP27 DNA was constructed into a pET-21a vector, and the plasmid was transformed into an Escherichia coli expression strain as previously described (Salari et al. 2013). For the negative control, we used the C-terminal fragment (rC1); this truncated inactive form of HSP-27, which spans the C-terminal amino acids 90–205, was cloned using the aforementioned strategy. Recombinant proteins were purified by with Ni–NTA resin. The purity of the final recombinant proteins were determined to be more than 95% by SDS–PAGE with a concentration lower than 5 endotoxin units/mg protein. For the treatments, the rHSP27 or rC1 was diluted to 100 μg/ml in DMEM with or without 10% FBS (when used combined with Cd the solution was prepared in serum-free media, when administrated alone the recombinant proteins were diluted in DMEM with 10% FBS). The dose of rHSP27 used in this work was chosen from previous in vitro and in vivo analysis performed by our group (Chen et al. 2009; Salari et al. 2013).
ROS determination
The ROS indicator assay was performed using a cell-permeable 2′,7′-dichlorodihydrofluorescein diacetate (H2DCFDA) agent (Life Technologies) following the manufacturer’s protocol. Briefly, 2 × 105 cells were seeded in a 96-well plate for 24 h. Then, they were incubated with the reagent for 40 min, washed with PBS, and treated with Cd or 50 μM H2O2 (positive control) for the indicated times. Upon cleavage of the acetate groups by intracellular esterases and oxidation, the non-fluorescent H2DCFDA is converted to the highly fluorescent 2′,7′-dichlorofluorescein (DCF) and the fluorescence measurements were recorded at excitation/emission λ = 495/525 nm using BioTek Synergy Mx microplate reader
Peroxynitrite determination
Peroxynitrite was assayed with the fluorescent probe 1 that was kindly provided by Dr. Youngmi Kim (Department of Chemistry, Institute of Nanosensor and Biotechnology, Dankook University, Korea). It was designed for selective detection of cellular peroxynitrite as was previously described by Kim et al. (2014). Briefly, the probe was diluted in phosphate buffer (10 mM, pH = 7.4) at 37 °C and used at a final concentration of 10 μM. HeLa cells (105) were seeded in a 96-well plate, allowed to attach for 24 h, pre-loaded with probe 1 for 20 min, washed with PBS, and then treated according to the following groups: untreated (cells with culture medium), treated (with 5–100 μM Cd or Cd + L-NAME as previously described), and positive control with 1 mM SIN-1. The reactions were monitored at 37 °C, and fluorescence signals for each well were measured at emission λ = 540 nm (excitation signals for each well were measured λ = 430 nm) at every minute for 30 min.
TBARS determination
The samples from HeLa cells, treated with different Cd concentrations, were used for the thiobarbituric acid–reactive substance (TBARS) assay, and the levels of lipid peroxidation products, mainly malondialdehyde (MDA), were determined spectrophotometrically as described by Buege et al. (Buege and Aust 1978). Briefly, HeLa cells were grown in six-well plates and then treated with different Cd concentrations. The samples were homogenized at pH 7.4, and the proteins were precipitated with 20% trichloroacetic acid (TCA, Sigma-Aldrich Co.). The supernatant containing MDA, the end product of the lipid peroxidation, was incubated with a 0.7% thiobarbituric acid solution (TBA, Sigma-Aldrich Co.) to measure the TBARS content. An acid hydrolysis product of 1,1,3,3-tetramethoxy propane (TMP) was used as standard.
Measurement of NO2
Nitric oxide (NO) formation was measured indirectly by assaying nitrite, a stable product of NO oxidation as was described by Schulz et al. (1999). Briefly, nitrites were determined alone, using the Griess reagent and then measuring the optical density (OD) at 540 nm. The level of NO was expressed in micromoles of NO2 per milliliters after being equilibrated with a solution of NaNO2 (10 mM).
Cytotoxicity assay
Cells (105) were grown in 96-well plates for 24 h; the media were replaced with serum-free media and exposed to Cd alone or Cd + L-NAME treatment as described. To analyze the effects of the recombinant proteins, the following protocol was performed: (a) For the co-treatment experiment, a combination of rHSP27 or rC1 (100 μg/ml) plus Cd (5, 50, or 100 μM) diluted in DMEM was used. (b) To analyze the preventive effect of the recombinant proteins, a 24-h pre-treatment with rHSP27 or rC1 (100 μg/ml prepared in DMEM plus 10% FBS) was performed followed by Cd solution prepared in DMEM and (c) exposure to Cd solution diluted in DMEM for 3 h followed by treatment with rHSP27 or rC1 (100 μg/ml prepared in DMEM plus 10% FBS) for 24 h. The CCK-8 assay was carried out according to the provided protocol (Promega Corporation, Madison, USA).
Immunofluorescence
Cells (3 × 105) were seeded on glass coverslips in 24-well plates and exposed to the different Cd concentrations, Cd + L-NAME or with SIN-1 as indicated in each study. After washing, the cells were fixed in 4% paraformaldehyde in 1× phosphate buffered saline (PBS buffer; VWR). Fixed cells were permeabilized with 0.5% (v/v) Triton X-100 for 5 min, washed carefully, blocked in 10% goat serum, and incubated overnight with mouse monoclonal antibody nitrotyrosine (Abcam 1:500) and Alexa fluor 488-conjugated goat anti-mouse (Invitrogen, 1:500) as secondary antibody.
Western blotting
After the treatments, cells were washed once with PBS and collected by scraping in 200 μl of lysis buffer [50 mM HEPES, 0.5 M sodium chloride, 1.5 mM magnesium chloride, 1 mM EDTA, 10% (v/v) glycerol, 1% Triton X-100, and Protease Inhibitor Cocktail (Roche)]. The lysates were incubated on ice followed by centrifugation at 1000 rpm for 10 min at 4 °C. Proteins were quantified using Bradford (Sigma) micromethod according to the manufacturer’s protocol. Equal amounts (10 μg) of protein from each treatment group were diluted with loading buffer and boiled. Proteins were separated in 10% SDS–PAGE and transferred to a nitrocellulose membrane using IBlot’s (Life Technologies) 7-min program according to the manufacturer’s protocol. Blots were then probed with primary mouse monoclonal anti-HSP27 antibody (Millipore, 1:8000) or mouse monoclonal anti-beta-actin antibody (NOVUS Biologicals, 1:10,000), followed by incubation with the secondary antibody anti-mouse conjugated with peroxidase (1:10,000). Finally, they were incubated with ECL Amersham detection reagent according to manufacturer’s recommendations and visualized with “ImageQuant LAS 4000” (GE Healthcare Life Sciences).
Flow cytometry cell death assessment
Annexin V-FITC apoptosis detection kit (BD Biosciences) was used according to the manufacturer’s protocol. Briefly, HeLa cells were grown in a six-well plate, treated with Cd, L-NAME, rHSP27, or rC1 as indicated. The media and the cells harvested with trypsin were centrifuged and then resuspended in binding buffer ×1 and counted. An aliquot containing 105 cells was incubated with 5 μl of Annexin V-FITC and 5 μl of propidium iodide (PI) for 15 min in the dark, and 400 μl binding buffer (1×) was added to each sample. The stained cells were analyzed with flow cytometry using BD LSRII (BD Biosciences, San Jose, CA).
Cell migration assay
The transwell insert for 24-well plate (8 μm-pore size, Corning, NY, USA) was used to measure the migratory ability of cells. For transwell migration assays, HeLa cells (20,000) were seeded and lead to attach for 6 h into the transwell insert (upper chamber) with culture medium with 10% FCS, and serum-free media was added into the lower chamber (the space between the well bottom and the insert). Then, the upper chamber media were replaced with rHSP27 or rC1 (100 μg/ml) in culture media for 12 h. Next, both upper and lower chamber media were removed and washed once with PBS, then Cd-containing media or serum-free media was added to the upper chamber; culture medium with 10% FCS was added in the lower chamber as chemo-attractant. After 24 h incubation, the cells were fixed with 4% paraformaldehyde for 2 h at room temperature and stained with Hoechst 10 min. The non-migratory cells were carefully removed from the upper surface of the insert membrane with cotton wool. The number of cells migrated to the lower surface of the membrane, 5–10 randomly selected fields of view, was pictured under the microscope, and the migratory cells were determined by counting with ImageJ software. Data were expressed as the percentage of invasion of treated cells as compared to the control cells.
Statistical and data analysis
Analysis of WB and photographs were performed using ImageJ software (Ounis et al. 2005). Statistical analyses were completed using the Prism computer program (GraphPad Software, San Diego, CA). The comparison between groups was carried out by one-way and two-way ANOVA with Dunnett and Bonferroni post-tests, respectively.
Results
Cd treatment upregulates ROS and lipid peroxidation, reduces NO bioavailability, and induces peroxynitrite production and protein nitration
One of the mechanisms of heavy metal cytotoxicity is partly related to the generation of oxidative stress, through a heterogeneous mechanism that includes both the reduction of anti-oxidative defense and the production of ROS by mitochondrial damage (Sandbichler and Höckner 2016). Numerous findings have shown that Cd exposure produces O2 − and NO. Under stress, the cells can produce large amounts of both species which quickly react with each other to generate a series of nitrosating agents, the most potently toxic one being PN (Beckman 1996). Thus, to establish times and doses in which Cd induced an oxidative status on the cells, first we used the fluorescent probe H2-DC-FDA which detects several ROS species, including peroxynitrite (Glebska and Koppenol 2003; Possel et al. 1997) and superoxide (LeBel et al. 1992). Cd treatment resulted in significant increase in ROS production, after 80 min with 100 μM Cd treatment (*p < 0.05, **p < 0.01, ***p < 0.001) and after 160 min with 50 μM Cd (*p < 0.05), while 5 μM Cd did not show significant differences at least for the first 3 h of treatment (Fig. 1a). A biological indicator of cellular damage from free radical generation and propagation is lipid peroxidation; we measured the formation of TBARS, a lipid peroxidation product. Significant increases in TBARS were found 1 h after 5 or 100 μM Cd exposure (***p < 0.001; Fig. 1b). Additionally, NO formation was measured using a nitrite assay (Schulz et al. 1999); after 1 h, 50 and 100 μM Cd treatment showed significant reductions (***p < 0.001; Fig. 1c); however, 5 μM Cd did not modify NO production. Taken together, all these findings suggest that Cd could be inducing PN production, a highly reactive oxidant species capable to induce cellular toxicity.
Fig. 1.

Cd treatment induces oxidative stress. a ROS assay measured in HeLa cells pre-loaded with H2DCFDA reagent for 40 min, washed, and then treated with Cd or H2O2 for the indicated time and concentrations and recorded at Ex/Em 495/525 nM. The results were expressed as relative fluorescence units (RFU). Values represent the means of three independent experiments ±SD (*p < 0.05; **p < 0.01; ***p < 0.001). b Lipid peroxidation as measured by TBARS formation after 1 h Cd treatment. c NO production in HeLa cells exposed to the indicated concentrations of Cd for 1 h (***p < 0.001). The results represent mean ± SD of three independent experiments (***p < 0.001). d Peroxynitrite production assayed in HeLa cells pre-loaded with probe 1 exposed to Cd. The spectra were obtained each minute during a period of 30 min, and fluorescence intensity at 540 nm was measured with excitation at 430 nm. Positive control was performed with SIN-1 and negative control with H2O2 (10 μM) as shown in Supp. Fig. 1a. Values represent the means of three independent experiments ± SD. e Confocal images of nitrotyrosine labeling in HeLa cells after treatment with 0, 5, and 100 μM Cd after 6 h. Green: nitrotyrosine; blue: DAPI; bar: 8 nm (color figure online)
To test whether PN was involved in the toxic effects of Cd, we used a fluorescent boronate-based probe which specifically detects PN in cells (known as “probe 1”) (Kim et al. 2014); we confirmed that PN production was detected only when cells were treated with the PN donor, SIN-1, but not with H2O2 (Supplemental Fig. 1a). Treatments with 50 or 100 μM Cd clearly tended to increase PN levels after 20–30 min of exposures, although due to the limitation of the probe (max. 30 min), this tendency could not be further examined in longer times (Fig. 1d). Therefore, we tested the cells for indirect evidence of PN by evaluating nitration as a footprint of its production in vivo. Once in cells, PN interacts with Cu–Zn and Mn–superoxide dismutase which catalyze the nitration of the proteins in tyrosine residues forming 3-nitrotyrosine (NT) (Beckman et al. 1992) (Beckman 1996). This modification can induce inhibition of the enzymes (Alvarez et al. 2004; Beckman 1996; Beckman et al. 1992; Ischiropoulos et al. 1992; MacMillan-Crow et al. 1998) and changing its normal protein function/activities (Greenacre and Ischiropoulos 2001; Ischiropoulos 2003). Despite that all cancer cells have a basal level of protein nitration, after 6 h treatment with Cd, HeLa cells showed a notable accumulation of NT evaluated by immunofluorescence labeling with anti-NT antibodies (Fig. 1e).
Cd-induced peroxynitrite/nitrotyrosine formation and reduction in HSP27 was prevented by L-NAME, a NOS inhibitor
In order to confirm if Cd was producing PN, cells were treated with L-NAME (a pharmacological inhibitor of nitric oxide synthase (NOS), which is required for the generation of NO) and PN/NT formation was assessed after Cd exposure. Initially, we performed a time course experiment, measuring PN generation in cells pre-treated with L-NAME followed by 10, 20, or 30 min of Cd exposure (Supplemental Fig. 1b, c). Next, we evaluated NT formation after Cd + L-NAME treatment, using the PN donor, SIN-1, as a positive control (Fig. 2a). Pre-treatment of cells with L-NAME lowered NT formation after 3 h of Cd treatment (5 and 100 μM); however, this effect was less pronounced after 6 h, suggesting that L-NAME pre-treatment delays PN and NT formation. As expected, while L-NAME treatment alone yielded similar NT immunofluorescent labeling as with the control, incubation of cells with increasing concentrations of SIN-1 correlated with more NT fluorescence labeling (Fig. 2a).
Fig. 2.

SIN-1 and Cd lowered the levels of HSP27 in HeLa cells. a Nitrotyrosine images were obtained by confocal microscopy. Top panel: Representative images of untreated HeLa cells (basal control), cells pre-treated with L-NAME (500 μM; similar to untreated), and cells treated with SIN-1 (1–3 mM) for 3 h plus 24 h recovery (positive control) are shown. Bottom panel: Nitrotyrosine was evaluated in HeLa cells exposed to Cd (5 or 100 μM) ± L-NAME pre-treatment. Green: nitrotyrosine; blue: DAPI; bar: 8 nm. b Western blots showing HSP27 expression in HeLa cells exposed to 100 μM Cd (3–24 h) + pre-treatment with L-NAME. c Western blots of HeLa cells exposed to 1 mM SIN-1 for 3 h and then assayed for HSP27 expression after 1–24 h of recovery. c, d Densitometries of the HSP27/beta-actin bands were measured and graph values represent the means of three independent experiments ± SD (*p < 0.05; **p < 0.01; ***p < 0.001) (color figure online)
Since different stresses modulate HSP27, and its overexpression correlates with resistance to Cd’s toxic effects (Wu and Welsh 1996), which include oxidative stress (Arata et al. 1995), as well as protection against Cd-induced morphological changes (Lee et al. 2005), we next analyzed if acute Cd exposure alters HSP27 expression. Although treatment with 5 μM Cd did not alter HSP27 expression (data not shown), 100 μM Cd reduced HSP27 levels after 6–9 h of exposure (Fig. 2c). Similar to its effects in attenuating PN and NT formation, pre-treatment with L-NAME partially prevented the reduction in HSP27 (Fig. 2b). Based on these results, we hypothesized that the generation of PN after Cd treatment was directly involved in reducing HSP27 levels. To support this, HeLa cells were exposed to the PN donor, SIN-1 (1 mM), for 3 h and then assayed for HSP27 following increasing recovery times (1–24 h). The treatment progressively reduced the expression of HSP27 as shown by western blot (Fig. 2c; *p < 0.05, **p < 0.01, ***p < 0.001); we also compared the levels of HSP27 after 24 h with the different treatments (Supplemental Fig. 2). All these findings together suggest that one mechanism by which Cd lowers HSP27 levels is through generation of PN (Supplemental Fig. 3).
Viability was protected by L-NAME and pre-treatment with rHSP27
To support the role of PN in Cd toxicity, first we established the effect of Cd in the cell viability of wild type HeLa cells, HeLa pScr (the transfection control), and HeLa p762 (downregulated for HSP27 in a 50%) using the CCK-8 assay, which is an indicator of metabolic activity (Supplemental Fig. 4). Cd caused a significant time and doses dependent reduction in the viability with 50–100 μM Cd, but not with 5 μM Cd treatment; therefore, we select 100 μM Cd as representative of cytotoxic dose for further studies. Next, we blocked NO formation by treating cells with L-NAME and compare the viability after Cd exposure. We found that L-NAME restored cell viability for all the concentrations tested (Fig. 3a), which further suggested that PN is one of the species responsible for Cd toxicity.
Fig. 3.

Viability was restored by L-NAME and by pre-treatment with rHSP27. Viability of HeLa cells measured as metabolic activity with CCK-8. A HeLa cells were grown in 96-well plates and then treated with or without L-NAME for 24 h, and then the cells were exposed to the indicated doses of Cd or Cd + L-NAME during different times (3–24 h). B-1 HeLa cells were co-treated with Cd + rHSP27 or Cd + rC1 for the indicated times, which improved viability. B-2 HeLa cells were pre-treated with rHSP27 or rC1 for 24 h and then exposed to Cd for the indicated time. The pre-treatment restored viability. B-3 HeLa cells were exposed to different doses of Cd (5, 50, or 100 μM) for 3 h and then treated with rHSP27 or rC1 for 24 h with post-treatment improving viability. All the values are representative of the means of three independent experiments ± SD (*p < 0.05; **p < 0.01; ***p < 0.001; n = 9 for each data point)
Considering that toxic doses of Cd diminished HSP27 levels (Fig. 2c, d) and keeping in mind the association of HSP27 with resistance to Cd toxicity (Wu and Welsh 1996), we hypothesized that recombinant HSP27 (rHSP27) could abrogate the toxicity of Cd. We evaluated the doses of rHSP and rC1 to use on the cells first by CCK-8 (data not shown) and performed the following: (a) co-treatment of HeLa cells with rHSP27 or rC1 (a C-terminal HSP27 fragment with chaperoning activity) and Cd combined (Fig. 3b (1)), (b) pre-treatment with rHSP27 or rC1 followed by Cd (Fig. 3b (2)), and (c) exposure to Cd (5, 50, or 100 μM) for 3 h followed by rHSP27 or rC1 treatment for 24 h (Fig. 3b (3)). All treatments with rHSP27 improved cell viability, but only the pre-treatment with rHSP27/rC1 completely restored the metabolic activity of the cells to control levels.
Pre-treatment with HSP27 or L-NAME protects against necrotic cell death due to Cd toxicity
To evaluate if rHSP27 pre-treatment protects cells from death, we performed an Annexin V assay, using flow cytometry (Fig. 4e). First, we established that the toxic effects of 100 μM Cd induced cell death mainly by necrosis p < 0.001 after 12 h (Fig. 4a). Next, we treated cells with rHSP27, C1 or L-NAME. Pre-treatment with rHSP27 but not its truncated form, C1, attenuated necrosis induced by Cd at 12 h, but this became similar to Cd at 24 h; in contrast, L-NAME was effective for a period up to 24 h (Fig. 4b). L-NAME also significantly diminished apoptosis induced by Cd at 3 h (Fig. 4c). Pre-treatment with rHSP27, C1, or L-NAME was protected against total cell death induced by Cd at 12 h, although only L-NAME was protective at 24 h (Fig. 4d, e).
Fig. 4.

Pre-treatment with HSP27 or L-NAME protects against cell death due to Cd toxicity. Cell death was evaluated by flow cytometry. a Annexin V/PI was measured in untreated control cells and in HeLa cells exposed to 100 μM Cd during different times. Graphs represent the percentages of necrotic, apoptotic or early apoptotic cells with regard to the total cells. b Percentage of necrotic cells in HeLa cells pre-treated with rHSP27, rC1, L-NAME, or vehicle and then exposed to 100 μM Cd. c Percentage of apoptotic cells in HeLa cells pre-treated with rHSP27, rC1, L-NAME, or vehicle and then exposed to 100 μM Cd. d Percentage of total number of dead cells after pre-treatment with rHSP27, rC1, L-NAME, or vehicle and then exposed to Cd 100 μM. All the values are representative of the means of three independent experiments ± SD (*p < 0.05; **p < 0.01; ***p < 0.001). e Schematic graph of flow cytometry: Q1 necrotic cells, Q2 apoptotic cells, Q3 live cells, Q4 early apoptotic cells
Pre-treatment with rHSP27 does not enhance the migration capabilities of tumoral HeLa cells
Even though rHSP27/rC1 proteins showed a beneficial effect increasing cellular metabolism (an indicator of viability; Fig. 3), only rHSP27 was capable of reducing necrosis (Fig. 4). This could be related to the known association between full length-HSP27 and proteins implicated in apoptosis and necrosis regulation (Arriazu et al. 2006; Bruey et al. 2000). In order to test if these recombinant proteins could be used safely in the cases of individuals with tumors, we analyzed the migratory activity of HeLa cells (Fig. 5). First, we established the basal migration capability of the cells without a chemo-attractant agent (basal control, line 1), and then we use 10% FBS in the lower chamber to evaluate the maximum migratory capability under normal conditions (positive control: line 2). After the treatments, we found that (1) Cd exposure reduced cell migration (line 2 vs 3); (2) rHSP27 does not affect cell migration (line 2 vs 4), which means that it does not promote invasive behavior; (3) rHSP27 did not enhance the migration capabilities after Cd treatment (line 5 vs line 3); and (4) even though rC1 alone did not induce changes in migration (compare line 6 with 2 and 4), it protected the migratory capabilities in HeLa cells after Cd treatment (compare line 7 with 5). Due to the lack of regulatory amino acids in rC1 suitable to be phosphorylated, rC1 may not be considered a potential therapeutic agent.
Fig. 5.

Pre-treatment with rHSP27 does not enhance the migration capabilities of tumor HeLa cells. The migration was assayed using transwell analysis. Five fields were obtained from each well, and then the migrating cells were counted using ImageJ software and then analyzed. HeLa cells were seeded, attached for 12 h, and then treated as indicated. Line 1: basal control represents cells without any treatment (upper and lower chamber), Line 2: cells without any treatment (Cd 0) in the upper chamber and media +10% SFB in the lower chamber. Line 3: cells treated with Cd (50 μM Cd in DMEM upper chamber) for 24 h and media +10% SFB in the lower chamber. Lines 4 and 5: cells were pre-treated with rHSP27 (100 μg/ml) for 12 h (upper chamber) and then exposed to the indicated Cd concentration for 24 h (upper chamber and media +10% SFB in the lower chamber). Lines 6 and 7: cells were pre-treated with rC1 for 12 h (100 μg/ml) in the upper chamber and then exposed to the indicated Cd concentration for 24 h in the upper chamber and media +10% SFB in the lower chamber. The data were expressed as means ± S.D. *p < 0.05; **p < 0.01; ***p < 0.001 vs control
Discussion
Although the pathological toxic effects of Cd in humans have been exhaustively studied (Taylor et al. 1999), there are not many treatment options to counteract the unhealthy effects induced by this metal nor an effective drug available for removing the accumulated Cd from the body. According to the Third National Report on Human Exposure to Environmental Chemicals (NHANES), Cd exposure is widespread in the general population and the need for specific treatment against this metal is imperative. Exposure to dietary amounts of Cd is enough to negatively influence human health and with a narrow margin of safety (Thomas et al. 2009; Järup and Åkesson 2009). Recently, an experimental drug indicated reductions in Cd accumulated after several weeks in mice and rats kidneys (Tang et al. 2016). However, we still need a drug capable of counteracting or ameliorating Cd’s toxic effects. The focus of this work was to understand one of the molecular mechanisms involved in Cd toxicity in order to provide the basis for possible future therapeutic approaches. Our results corroborated that the harmful effects of Cd are related to the induction of oxidative stress, in particular, we described for the first time a link between the peroxynitrite production, the Cd exposure, and the reduction of cellular HSP27 content.
PN is formed spontaneously from the following reaction: O2 − + NO ➔ ONOO− (Pryor and Squadrito 1995). Under stress conditions, the concentration of these substrates may be modulated and PN formation may be increased (Pacher et al. 2007). The PN produced may be detrimental and cause damage to biomolecules through nitration and oxidations (Radi et al. 1991a; Radi et al. 1991b; Speckmann et al. 2016; Yamakura et al. 1998). Protein nitration, hence, is a good marker used to infer the presence of PN (see Supplemental Fig. 2) (Radi 2004; Speckmann et al. 2016). The nitrated proteins, in turn, may lose their activities or become prone to proteolytic degradation, resulting in modulation of the protein turnover and abnormal signaling cascades (MacMillan-Crow et al. 1998), (Ischiropoulos 2003). For example, nitration has been found in several stressed tissues such as human rejected kidney allografts (MacMillan-Crow et al. 1996), atherosclerotic lesions (Leeuwenburgh et al. 1997), aging (VINER et al. 1999), diabetes (Turko et al. 2001), and Parkinson’s disease (Giasson et al. 2000), among others.
As PN is formed quickly without any enzyme involved, there are no currently direct inhibitors to stop the reaction and there are no robust methods to measure the formation of PN; therefore, in order to restrain PN production, it is necessary to modulate the bioavailability of either nitric oxide or superoxide. In the present work, we used L-NAME as a non-selective inhibitor of NOS to reduce the availability of NO and SIN-1 (a peroxynitrite donor) to perform the studies. Our work showed that after Cd treatment, protein nitration peaked at approximately 6 h and then the levels returned to control values at 24 h, while pre-treatment with L-NAME delayed NT formation and enhanced viability and survival of the cells exposed to high doses of Cd. Even though the performance of this inhibitor in vitro is good, it is not suitable for using in vivo because NO systems are required for normal cellular function (Cals-Grierson 2004; Massion et al. 2003). Hence, we looked at possible downstream molecular targets which could interfere with the propagation of PN-mediated toxicity. The addition of SIN-1 to the culture medium reduced HSP27 levels, similar to the treatment with 100 μM Cd during 24 h, suggesting that PN could be involved in the reduction of HSP27. For cells exposed to harmful molecules, the modulation of HSP27 could have noxious consequences, because, in such conditions, this chaperone is a key factor to maintain cellular balance and the cell viability. Whether the protein nitration contributes to the cadmium-mediated HSP27 downregulation remains unknown and it will be a focus of further studies. A relevant example described in a recent work showed that after oxidative stress, HSP60 is nitrated and then released from cells in exosomes (Campanella et al. 2015). At this point, it remains unclear if the reduction in HSP27 is due to exosome-mediated release or degradation.
Regarding HSP27 expression after Cd treatment, there is a range of results that depend on the doses used. Previous investigations showed increased expression of HSP27 after sub-toxic Cd exposures (<50 μM) (Bonham et al. 2003; Croute et al. 2005; Roccheri et al. 2004), while in our study, using ≥50 μM Cd, HSP27 levels were reduced, suggesting that the effects on the expression of this protein could be modulated by the level of stress induced. When high levels of ROS are formed, the high oxidation of macromolecules such as proteins, lipids, and DNA induces necrosis (Arrigo 2001). It is known that Cd can modulate ROS state altering antioxidant molecules such as reduced glutathione (Gaubin et al. 2000) and that this molecule is the first defense against Cd toxicity (Singhal et al. 1987). Moreover, HSP27 can activate (glucose-6-phosphate dehydrogenase (G6PD), upregulating the levels of reduced glutathione in the cells and, thus, the capability of the cells to fight oxidative stress (Préville et al. 1999). Normally, when the cells are exposed to moderate levels of ROS, reduced glutathione depletion occurs and apoptosis is triggered, but if the levels of oxidative stress are too high, necrosis occurs (Arrigo 2001; Zucker et al. 1997). In our study, Cd-induced cell death occurred mainly through necrosis, although apoptosis was minimally observed.
As we proposed that HSP27 was downregulated by PN (trigger by Cd), we hypothesized that loading the cells with a recombinant protein able to restore the cellular HSP27 could have a positive effect in resistance and viability. Consistent with this, a recent study showed that treatment with pro-inflammatory cytokines reduced HSP27 levels through a PN-dependent mechanism and this event was associated with apoptosis in human capillary endothelial cells (Nahomi et al. 2014). Our results showed that even though both treatments—either with rHSP27 or with rC1 before Cd exposure—completely restored cell metabolism to control levels, rC1 was less efficient in protecting against cell death. Also, rHSP27 did not enhance the migration capabilities of the cells, suggesting that it will not enhance malignant capabilities in cancer cells, making rHSP27 an interesting molecule to counteract Cd toxicity. These findings suggest that in order to protect cells from Cd toxicity, HSP27 needs both its molecular chaperone’s capabilities (found in rC1) and its capacity to interact with different client proteins and to become phosphorylated and regulated by cells, which is not possible in the case of rC1. Although more studies are necessary to confirm the possibility of using rHSP27 as a form of therapy in living organisms, due to the current lack of effective treatments for people at high risk for Cd exposure, the potential impact of a therapeutic approach using rHSP27 to counteract Cd toxicity is very high. A future therapeutic strategy could involve screening high-risk individuals (smokers, industrial workers) and those with elevated Cd levels could be candidates for administering pre-treatment with rHSP27 to avoid further damage.
In conclusion, Cd induced an oxidative and nitrosative stress that reduced HSP27 levels, affecting cell function and survival. The use of recombinant HSP27 instead of plasmids or DNA carrying vector could be a suitable, easily regulable alternative to overexpress a mis-regulated protein during Cd toxicity. This work constitutes one step forward to understand the mechanism of Cd toxicity and a promising alternative.
Electronic supplementary material
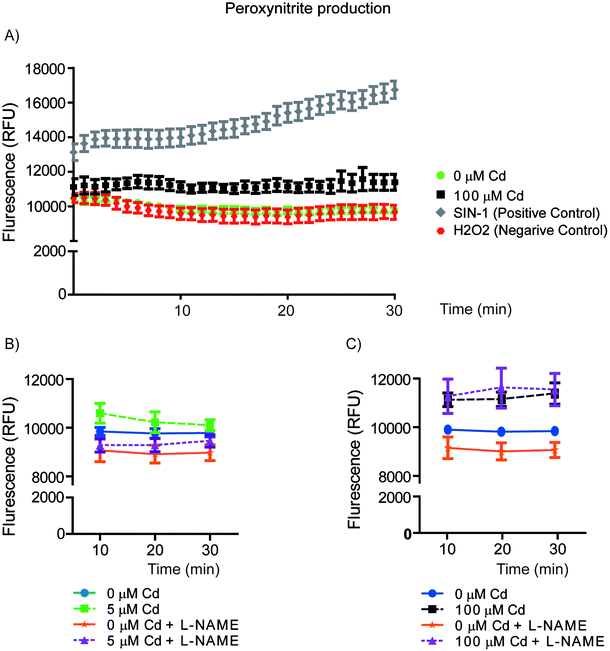{kind=link}
Peroxynitrite production A) HeLa cells pre-loaded with probe-1 were exposed either to Cd, SIN-1 (positive control, donor of NO and superoxide) or 10 μM H2O2 (which induces superoxide species but not peroxynitrite). The spectra were obtained each minute during a period of 30 min and fluorescence intensity at 540 nm was measured with excitation at 430 nm. B-C) Untreated control HeLa cells or pre-treated with 500 μM L-NAME were pre-loaded with probe 1 and then exposed to 5 μM Cd (B) or 100 μM Cd (C) to assay peroxynitrite production. In all cases the graph values represent the means of three independent experiments ±SD (n=6). (GIF 51 kb)
.
.
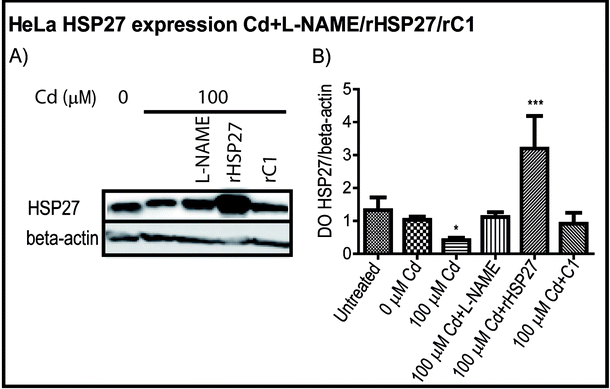{kind=link}
HSP27 content in HeLa cells. A) Western blott of HeLa cells Control (line 1), treated with 100 μM Cd during 24h (line 2), L-NAME and then Cd (line 3), pre-treated with rHSP2 or rC1 and then Cd (line 4-5 respectively). B) Optical density of HSP27/beta-actin expression. The graph values represent the means of three independent experiments ±SD (* p< 0,05; ** p< 0,01; *** p< 0,001). (GIF 39.5 kb)
.
.
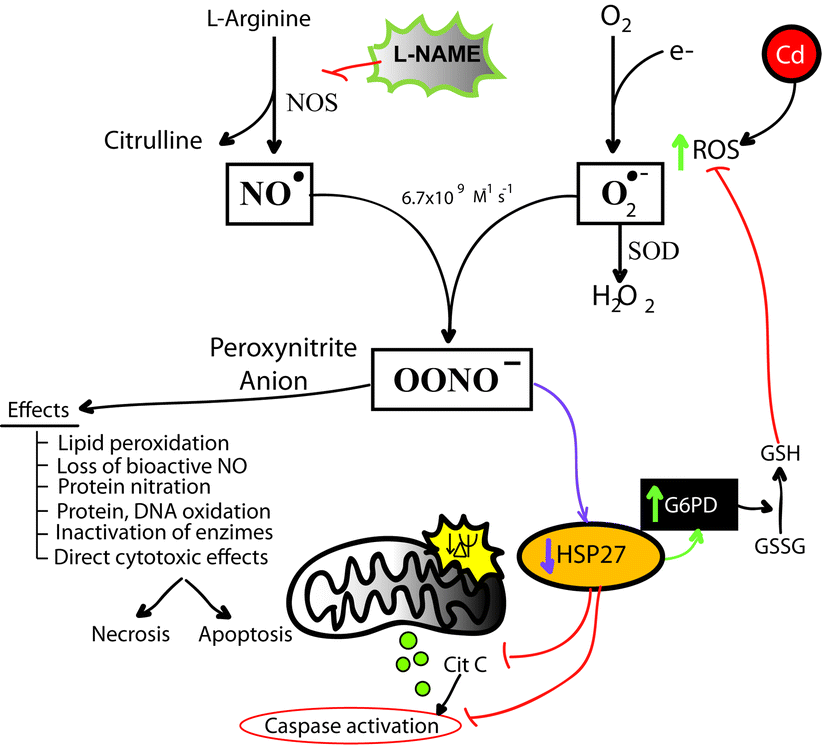{kind=link}
Peroxynitrite production and cellular effects. (GIF 66.2 kb)
.
.
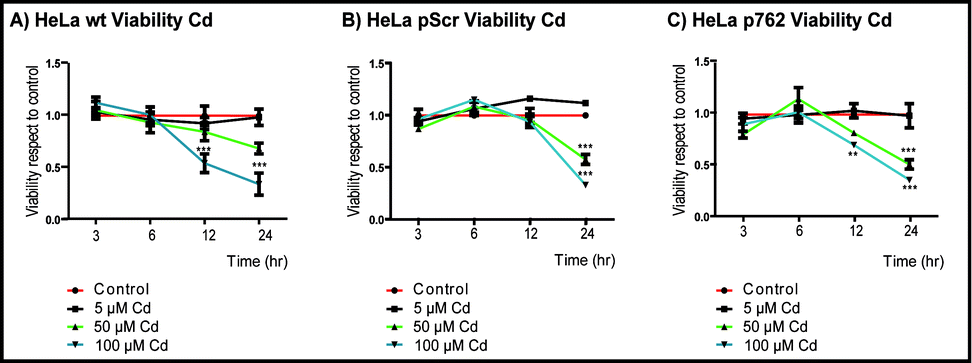{kind=link}
Viability of HeLa cells measured as metabolic activity with CCK-8. A) HeLa cells were grown in 96 well plates and then the cells were exposed to the indicated doses of Cd during different times (3-24 h). B) HeLa pScr cells were stable transfected with a scramble plasmid, selected as indicate in methodology grown in 96 well plate with hygronomycin for 24 hr and then exposed to the indicated doses of Cd in serum-hygronomycin free media. C) HeLa p762 cells (downregulation for HSP27 in a 50%), were grown in 96 well plate with hygronomycin for 24 hr and then exposed to the indicated doses of Cd in serum-hygronomycin free media. All the values are representative of the means of three independent experiments ±SD (* p< 0,05; ** p< 0,01; *** p< 0,001; n=6 for each data point). (GIF 41.4 kb)
.
.
Acknowledgements
We are grateful to Dr. Youngmi Kim (Department of Chemistry, Institute of Nanosensor and Biotechnology, Dankook University, Korea) for the gift of the fluorescent probe-1 for the peroxynitrite determination.
This work was supported by operating grants jointly funded by the Canadian Institute for Health Research (CIHR) and Medtronic Canada (ISO 110836) as well as the Heart and Stroke Foundation of Canada (G-13-0001599). CIHR and Medtronic also collectively funded a peer-reviewed research chair [grant IRC 57093] to EO. The National Research Council of Argentina (CONICET) is also thanked for financial support [PIP 11220110100836 DAS 30844] to DRC.
Footnotes
Fanelli and O’Brien contributed equally to this work.
Electronic supplementary material
The online version of this article (doi:10.1007/s12192-017-0768-y) contains supplementary material, which is available to authorized users.
Contributor Information
Mariel A. Fanelli, Phone: +54-(0261)-5244153, Email: mfanelli@mendoza-conicet.gob.ar
Edward R. O’Brien, Phone: 403-944-5918, Email: ermobrie@ucalgary.ca
References
- Åkesson A, Julin B, Wolk A. Long-term dietary cadmium intake and postmenopausal endometrial cancer incidence: a population-based prospective cohort study. Cancer Res. 2008;68:6435–6441. doi: 10.1158/0008-5472.CAN-08-0329. [DOI] [PubMed] [Google Scholar]
- Al-Shawi AW, Dahl R. The determination of cadmium and six other heavy metals in nitrate/phosphate fertilizer solution by ion chromatography. Anal Chim Acta. 1999;391:35–42. doi: 10.1016/S0003-2670(99)00200-7. [DOI] [Google Scholar]
- Alvarez B, Demicheli V, Durán R, Trujillo M, Cerveñansky C, Freeman BA, Radi R. Inactivation of human Cu, Zn superoxide dismutase by peroxynitrite and formation of histidinyl radical. Free Radic Biol Med. 2004;37:813–822. doi: 10.1016/j.freeradbiomed.2004.06.006. [DOI] [PubMed] [Google Scholar]
- Andersen O, Nielsen JB, Svendsen P. Oral cadmium chloride intoxication in mice: effects of chelation. Toxicology. 1988;52:65–79. doi: 10.1016/0300-483X(88)90197-7. [DOI] [PubMed] [Google Scholar]
- Arata S, Hamaguchi S, Nose K. Effects of the overexpression of the small heat shock protein, HSP27, on the sensitivity of human fibroblast cells exposed to oxidative stress. J Cell Physiol. 1995;163:458–465. doi: 10.1002/jcp.1041630305. [DOI] [PubMed] [Google Scholar]
- Arriazu R, Pozuelo JM, Henriques-Gil N, Perucho T, Martín R, Rodríguez R, Santamaría L. Immunohistochemical study of cell proliferation, Bcl-2, p53, and caspase-3 expression on preneoplastic changes induced by cadmium and zinc chloride in the ventral rat prostate. J Histochem Cytochem. 2006;54:981–990. doi: 10.1369/jhc.5A6733.2006. [DOI] [PubMed] [Google Scholar]
- Arrigo AP. Hsp27: novel regulator of intracellular redox state. IUBMB life. 2001;52:303–307. doi: 10.1080/152165401317291156. [DOI] [PubMed] [Google Scholar]
- Beckman JS. Oxidative damage and tyrosine nitration from peroxynitrite. Chem Res Toxicol. 1996;9:836–844. doi: 10.1021/tx9501445. [DOI] [PubMed] [Google Scholar]
- Beckman JS, et al. Kinetics of superoxide dismutase-and iron-catalyzed nitration of phenolics by peroxynitrite. Arch Biochem Biophys. 1992;298:438–445. doi: 10.1016/0003-9861(92)90432-V. [DOI] [PubMed] [Google Scholar]
- Bernhoft RA (2013) Cadmium toxicity and treatment. Sci World J. doi:10.1155/2013/394652 [DOI] [PMC free article] [PubMed]
- Bonham RT, Fine MR, Pollock FM, Shelden EA. Hsp27, Hsp70, and metallothionein in MDCK and LLC-PK1 renal epithelial cells: effects of prolonged exposure to cadmium. Toxicol Appl Pharmacol. 2003;191:63–73. doi: 10.1016/S0041-008X(03)00226-6. [DOI] [PubMed] [Google Scholar]
- Bruey J-M, et al. Hsp27 negatively regulates cell death by interacting with cytochrome c. Nat Cell Biol. 2000;2:645–652. doi: 10.1038/35023595. [DOI] [PubMed] [Google Scholar]
- Buege JA, Aust SD. Microsomal lipid peroxidation. Methods Enzymol. 1978;52:302–310. doi: 10.1016/S0076-6879(78)52032-6. [DOI] [PubMed] [Google Scholar]
- Cals-Grierson M-M, Ormerod A. Nitric oxide function in the skin. Nitric Oxide. 2004;10:179–193. doi: 10.1016/j.niox.2004.04.005. [DOI] [PubMed] [Google Scholar]
- Campanella C, D’Anneo, Gammazza AM, Bavisotto CC, Barone R, Emanuele S, Cascio LF, Mocciaro E, Fais S, Macario CDE, Macario AJ, Cappello F, Lauricella M (2016) The histone deacetylase inhibitor SAHA induces HSP60 nitration and its extracellular release by exosomal vesicles in human lung-derived carcinoma cells. Oncotarget 7(20):28849–28867 [DOI] [PMC free article] [PubMed]
- Cancer IAfRo, Organization WH, Humans IWGotEoCRt (1993) Beryllium, cadmium, mercury, and exposures in the glass manufacturing industry vol 58. IARC [PMC free article] [PubMed]
- Cantilena LR, q Decreased effectiveness of chelation therapy with time after acute cadmium poisoning. Toxicol Appl Pharmacol. 1982;63:173–180. doi: 10.1016/0041-008X(82)90038-2. [DOI] [PubMed] [Google Scholar]
- Casalino E, Calzaretti G, Sblano C, Landriscina C. Molecular inhibitory mechanisms of antioxidant enzymes in rat liver and kidney by cadmium. Toxicology. 2002;179:37–50. doi: 10.1016/S0300-483X(02)00245-7. [DOI] [PubMed] [Google Scholar]
- Ciocca DR, Fanelli MA, Cuello-Carrion FD, Castro GN. Heat shock proteins in prostate cancer: from tumorigenesis to the clinic. Int J Hyperth. 2010;26:737–747. doi: 10.3109/02656731003776968. [DOI] [PubMed] [Google Scholar]
- Ciocca DR, Fanelli MA, Cuello-Carrión FD, Castro GN (2010b) Small stress proteins, biomarkers of cancer. In: Simon S, Arrigo A-P (eds) Small stress proteins and human diseases. Nova Sciences Publisher Inc.,New York, Chapter 3.1, pp 327–351
- Concannon CG, Orrenius S, Samali A. Hsp27 inhibits cytochrome c-mediated caspase activation by sequestering both pro-caspase-3 and cytochrome c. Gene Expr. 2001;9:195–201. doi: 10.3727/000000001783992605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croute F, Beau B, Murat J-C, Vincent C, Komatsu H, Obata F, Soleilhavoup J-P. Expression of stress-related genes in a cadmium-resistant A549 human cell line. Journal of Toxicology and Environmental Health, Part A. 2005;68:703–718. doi: 10.1080/15287390590925447. [DOI] [PubMed] [Google Scholar]
- Cuello-Carrión FD, et al. HER2 and β-catenin protein location: importance in the prognosis of breast cancer patients and their correlation when breast cancer cells suffer stressful situations. Clin Exp Metastasis. 2015;32:151–168. doi: 10.1007/s10585-015-9694-5. [DOI] [PubMed] [Google Scholar]
- Chen Y-X, Zhao X, McNulty M, O'Brien ER. Recombinant HSP27 therapy reduces serum cholesterol levels and experimental atherogenesis. Circulation. 2009;120:S1153–S1153. doi: 10.1161/CIRCULATIONAHA.107.751412. [DOI] [Google Scholar]
- Cho YA, Kim J, Woo HD, Kang M. Dietary cadmium intake and the risk of cancer: a meta-analysis. PLoS One. 2013;8:e75087. doi: 10.1371/journal.pone.0075087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Vries W, Lofts S, Tipping E, Meili M, Groenenberg JE, Schütze G (2007) Impact of soil properties on critical concentrations of cadmium, lead, copper, zinc, and mercury in soil and soil solution in view of ecotoxicological effects. In: Reviews of environmental contamination and toxicology. Springer, pp 47–89 [DOI] [PubMed]
- Dressler J, Schulz K, Klemm M, Schüttig R, Beuthin A, Felscher D. Lethal manganese-cadmium intoxication. A case report. Arch Toxicol. 2002;76:449–451. doi: 10.1007/s00204-002-0357-3. [DOI] [PubMed] [Google Scholar]
- Fanelli MA, et al. P-cadherin and β-catenin are useful prognostic markers in breast cancer patients; β-catenin interacts with heat shock protein Hsp27. Cell Stress Chaperones. 2008;13:207–220. doi: 10.1007/s12192-007-0007-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friberg L, Elinder C, Kjellstrom T (1992) Environmental health criteria 134: Cadmium Geneva: World Health Organization
- Gallagher CM, Chen JJ, Kovach JS (2010) Environmental cadmium and breast cancer risk Aging (Albany NY) 2:804–814 [DOI] [PMC free article] [PubMed]
- Gaubin Y, Vaissade F, Croute F, Beau B, Soleilhavoup J-P, Murat J-C. Implication of free radicals and glutathione in the mechanism of cadmium-induced expression of stress proteins in the A549 human lung cell-line. Biochim Biophys Acta. 2000;1495:4–13. doi: 10.1016/S0167-4889(99)00149-4. [DOI] [PubMed] [Google Scholar]
- Giasson BI, et al. Oxidative damage linked to neurodegeneration by selective α-synuclein nitration in synucleinopathy lesions. Science. 2000;290:985–989. doi: 10.1126/science.290.5493.985. [DOI] [PubMed] [Google Scholar]
- Gil HW, Kang EJ, Lee KH, Yang JO, Lee EY, Hong SY (2011) Effect of glutathione on the cadmium chelation of EDTA in a patient with cadmium intoxication. Hum Exp Toxicol 30(1):79–83 [DOI] [PubMed]
- Gilman A, Philips F, Allen R, Koelle E. The treatment of acute cadmium intoxication in rabbits with 2,3-dimercaptopropanol (dimercaprol) and other mercaptans. J Pharmacol Exp Ther. 1946;87:85–101. [PubMed] [Google Scholar]
- Glebska J, Koppenol WH. Peroxynitrite-mediated oxidation of dichlorodihydrofluorescein and dihydrorhodamine. Free Radic Biol Med. 2003;35:676–682. doi: 10.1016/S0891-5849(03)00389-7. [DOI] [PubMed] [Google Scholar]
- Greenacre SA, Ischiropoulos H. Tyrosine nitration: localisation, quantification, consequences for protein function and signal transduction. Free Radic Res. 2001;34:541–581. doi: 10.1080/10715760100300471. [DOI] [PubMed] [Google Scholar]
- Ionescu JG, Novotny J, Stejskal V, Lätsch A, Blaurock-Busch E, Eisenmann-Klein M. Increased levels of transition metals in breast cancer tissue. Neuro Endocrinol Lett. 2006;27:36–39. [PubMed] [Google Scholar]
- Ischiropoulos H. Biological selectivity and functional aspects of protein tyrosine nitration. Biochem Biophys Res Commun. 2003;305:776–783. doi: 10.1016/S0006-291X(03)00814-3. [DOI] [PubMed] [Google Scholar]
- Ischiropoulos H, Zhu L, Chen J, Tsai M, Martin JC, Smith CD, Beckman JS. Peroxynitrite-mediated tyrosine nitration catalyzed by superoxide dismutase. Arch Biochem Biophys. 1992;298:431–437. doi: 10.1016/0003-9861(92)90431-U. [DOI] [PubMed] [Google Scholar]
- Itoh H, et al. Dietary cadmium intake and breast cancer risk in Japanese women: a case–control study. Int J Hyg Environ Health. 2014;217:70–77. doi: 10.1016/j.ijheh.2013.03.010. [DOI] [PubMed] [Google Scholar]
- Järup L. Hazards of heavy metal contamination. Br Med Bull. 2003;68:167–182. doi: 10.1093/bmb/ldg032. [DOI] [PubMed] [Google Scholar]
- Järup L, Åkesson A. Current status of cadmium as an environmental health problem. Toxicol Appl Pharmacol. 2009;238:201–208. doi: 10.1016/j.taap.2009.04.020. [DOI] [PubMed] [Google Scholar]
- Joint F, Organization WH, Additives WECoF (2011) Evaluation of certain food additives and contaminants: seventy-third [73rd] report of the Joint FA
- Jones MM, Cherian MG. The search for chelate antagonists for chronic cadmium intoxication. Toxicology. 1990;62:1–25. doi: 10.1016/0300-483X(90)90027-E. [DOI] [PubMed] [Google Scholar]
- Kang M-Y, Cho S-H, Lim Y-H, Seo J-C, Hong Y-C. Effects of environmental cadmium exposure on liver function in adults. Occup Environ Med. 2013;70:268–273. doi: 10.1136/oemed-2012-101063. [DOI] [PubMed] [Google Scholar]
- Kim J, Park J, Lee H, Choi Y, Kim Y. A boronate-based fluorescent probe for the selective detection of cellular peroxynitrite. Chem Commun (Camb) 2014;50:9353–9356. doi: 10.1039/C4CC02943G. [DOI] [PubMed] [Google Scholar]
- Kirschvink N, Martin N, Fievez L, Smith N, Marlin D, Gustin P. Airway inflammation in cadmium-exposed rats is associated with pulmonary oxidative stress and emphysema. Free Radic Res. 2006;40:241–250. doi: 10.1080/10715760500494657. [DOI] [PubMed] [Google Scholar]
- Koyu A, Gokcimen A, Ozguner F, Bayram DS, Kocak A. Evaluation of the effects of cadmium on rat liver. Mol Cell Biochem. 2006;284:81–85. doi: 10.1007/s11010-005-9017-2. [DOI] [PubMed] [Google Scholar]
- Lambert R, Grant C, Sauvé S. Cadmium and zinc in soil solution extracts following the application of phosphate fertilizers. Sci Total Environ. 2007;378:293–305. doi: 10.1016/j.scitotenv.2007.02.008. [DOI] [PubMed] [Google Scholar]
- LeBel CP, Ischiropoulos H, Bondy SC. Evaluation of the probe 2', 7'-dichlorofluorescin as an indicator of reactive oxygen species formation and oxidative stress. Chem Res Toxicol. 1992;5:227–231. doi: 10.1021/tx00026a012. [DOI] [PubMed] [Google Scholar]
- Lee J-S, et al. Heat shock protein 27 interacts with vimentin and prevents insolubilization of vimentin subunits induced by cadmium. Exp Mol Med. 2005;37:427–435. doi: 10.1038/emm.2005.53. [DOI] [PubMed] [Google Scholar]
- Leeuwenburgh C, Hardy MM, Hazen SL, Wagner P, Oh-ishi S, Steinbrecher UP, Heinecke JW. Reactive nitrogen intermediates promote low density lipoprotein oxidation in human atherosclerotic intima. J Biol Chem. 1997;272:1433–1436. doi: 10.1074/jbc.272.3.1433. [DOI] [PubMed] [Google Scholar]
- MacMillan-Crow L, Crow JP, Kerby JD, Beckman JS, Thompson JA. Nitration and inactivation of manganese superoxide dismutase in chronic rejection of human renal allografts. Proc Natl Acad Sci U S A. 1996;93:11853–11858. doi: 10.1073/pnas.93.21.11853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacMillan-Crow LA, Crow JP, Thompson JA. Peroxynitrite-mediated inactivation of manganese superoxide dismutase involves nitration and oxidation of critical tyrosine residues. Biochemistry. 1998;37:1613–1622. doi: 10.1021/bi971894b. [DOI] [PubMed] [Google Scholar]
- Massion P, Feron O, Dessy C, Balligand J-L. Nitric oxide and cardiac function ten years after, and continuing. Circ Res. 2003;93:388–398. doi: 10.1161/01.RES.0000088351.58510.21. [DOI] [PubMed] [Google Scholar]
- Medicine ICoI (2010) American College for Advancement in Medicine, Chelation Module. American College for Advancement in Medicine, Irvine, Calif
- Nahomi RB, Palmer A, Green KM, Fort PE, Nagaraj RH. Pro-inflammatory cytokines downregulate Hsp27 and cause apoptosis of human retinal capillary endothelial cells. Biochim Biophys Acta. 2014;1842:164–174. doi: 10.1016/j.bbadis.2013.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nordberg G, et al. Low bone density and renal dysfunction following environmental cadmium exposure in China. Ambio. 2002;31:478–481. doi: 10.1579/0044-7447-31.6.478. [DOI] [PubMed] [Google Scholar]
- Nordberg G, et al. Kidney dysfunction and cadmium exposure–factors influencing dose–response relationships. J Trace Elem Med Biol. 2012;26:197–200. doi: 10.1016/j.jtemb.2012.03.007. [DOI] [PubMed] [Google Scholar]
- Nordberg GF, Fowler BA, Nordberg M (2014) Cadmium. In: Nordberg GF, Fowler BA, Nordberg M (eds) Handbook on the toxicology of metals-Chapter 6: Toxic Metals in Food, section 2. Academic Press
- Nordberg GF, et al. Prevalence of kidney dysfunction in humans–relationship to cadmium dose, metallothionein, immunological and metabolic factors. Biochimie. 2009;91:1282–1285. doi: 10.1016/j.biochi.2009.06.014. [DOI] [PubMed] [Google Scholar]
- Odewumi C, Latinwo LM, Sinclair A, Badisa VL, Abdullah A, Badisa RB. Effect of cadmium on the expression levels of interleukin-1α and interleukin-10 cytokines in human lung cells. Mol Med Rep. 2015;12:6422–6426. doi: 10.3892/mmr.2015.4316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh S-H, Lim S-C. A rapid and transient ROS generation by cadmium triggers apoptosis via caspase-dependent pathway in HepG2 cells and this is inhibited through N-acetylcysteine-mediated catalase upregulation. Toxicol Appl Pharmacol. 2006;212:212–223. doi: 10.1016/j.taap.2005.07.018. [DOI] [PubMed] [Google Scholar]
- Organization WH, Humans IWGotEoCRt (1993) IARC monographs on the evaluation of carcinogenic risks to humans: occupational exposure of hairdressers and barbers and personal use of hair colourants; some hair dyes, cosmetic colourants, industrial dyestuffs and aromatic amines. International Agency for Research on Cancer [PMC free article] [PubMed]
- Organization WH, Joint W (2007) Health risks of heavy metals from long-range transboundary air pollution. World Health Organization Regional Office Europe
- Ounis A, Cerovic Z, Briantais J, Moya I (2005) Rasband WS, ImageJ. US National Institutes of Health, Bethesda, Maryland, USA
- Pacher P, Beckman JS, Liaudet L. Nitric oxide and peroxynitrite in health and disease. Physiol Rev. 2007;87:315–424. doi: 10.1152/physrev.00029.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panjehpour M, Taher M-A, Bayesteh M. The growth inhibitory effects of cadmium and copper on the MDA-MB468 human breast cancer cells. J Res Med Sci. 2010;15:279–286. [PMC free article] [PubMed] [Google Scholar]
- Paul C, Manero F, Gonin S, Kretz-Remy C, Virot S, Arrigo A-P. Hsp27 as a negative regulator of cytochrome C release. Mol Cell Biol. 2002;22:816–834. doi: 10.1128/MCB.22.3.816-834.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Possel H, Noack H, Augustin W, Keilhoff G, Wolf G. 2,7-Dihydrodichlorofluorescein diacetate as a fluorescent marker for peroxynitrite formation. FEBS Lett. 1997;416:175–178. doi: 10.1016/S0014-5793(97)01197-6. [DOI] [PubMed] [Google Scholar]
- Préville X, et al. Mammalian small stress proteins protect against oxidative stress through their ability to increase glucose-6-phosphate dehydrogenase activity and by maintaining optimal cellular detoxifying machinery. Exp Cell Res. 1999;247:61–78. doi: 10.1006/excr.1998.4347. [DOI] [PubMed] [Google Scholar]
- Pryor WA, Squadrito GL. The chemistry of peroxynitrite: a product from the reaction of nitric oxide with superoxide. Am J Phys. 1995;268:L699–L722. doi: 10.1152/ajplung.1995.268.5.L699. [DOI] [PubMed] [Google Scholar]
- Radi R. Nitric oxide, oxidants, and protein tyrosine nitration. Proc Natl Acad Sci U S A. 2004;101:4003–4008. doi: 10.1073/pnas.0307446101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radi R, Beckman JS, Bush KM, Freeman BA. Peroxynitrite-induced membrane lipid peroxidation: the cytotoxic potential of superoxide and nitric oxide. Arch Biochem Biophys. 1991;288:481–487. doi: 10.1016/0003-9861(91)90224-7. [DOI] [PubMed] [Google Scholar]
- Radi R, Beckman JS, Bush KM, Freeman BA. Peroxynitrite oxidation of sulfhydryls. The cytotoxic potential of superoxide and nitric oxide. J Biol Chem. 1991;266:4244–4250. [PubMed] [Google Scholar]
- Roccheri MC, Agnello M, Bonaventura R, Matranga V. Cadmium induces the expression of specific stress proteins in sea urchin embryos. Biochem Biophys Res Commun. 2004;321:80–87. doi: 10.1016/j.bbrc.2004.06.108. [DOI] [PubMed] [Google Scholar]
- Rogalla T, et al. Regulation of Hsp27 oligomerization, chaperone function, and protective activity against oxidative stress/tumor necrosis factor α by phosphorylation. J Biol Chem. 1999;274:18947–18956. doi: 10.1074/jbc.274.27.18947. [DOI] [PubMed] [Google Scholar]
- Rusanov A, Smirnova A, Poromov A, Fomicheva K, Luzgina N, Majouga A. Effects of cadmium chloride on the functional state of human intestinal cells. Toxicol in Vitro. 2015;29:1006–1011. doi: 10.1016/j.tiv.2015.03.018. [DOI] [PubMed] [Google Scholar]
- Salari S, et al. Extracellular HSP27 acts as a signaling molecule to activate NF-κB in macrophages. Cell Stress Chaperones. 2013;18:53–63. doi: 10.1007/s12192-012-0356-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salazar K, McNutt MK (2012) Mineral commodity summaries US Geological Survey, Reston, VA
- Sandbichler AM, Höckner M. Cadmium protection strategies—a hidden trade-off? Int J Mol Sci. 2016;17:139. doi: 10.3390/ijms17010139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satarug S, Moore MR. Adverse health effects of chronic exposure to low-level cadmium in foodstuffs and cigarette smoke. Environ Health Perspect. 2004;112:1099. doi: 10.1289/ehp.6751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz K, Kerber S, Kelm M. Reevaluation of the Griess method for determining NO/NO− 2 in aqueous and protein-containing samples. Nitric Oxide. 1999;3:225–234. doi: 10.1006/niox.1999.0226. [DOI] [PubMed] [Google Scholar]
- Singhal RK, Anderson ME, Meister A. Glutathione, a first line of defense against cadmium toxicity. FASEB J. 1987;1:220–223. doi: 10.1096/fasebj.1.3.2887478. [DOI] [PubMed] [Google Scholar]
- Sinha M, Manna P, Sil PC. Cadmium-induced neurological disorders: prophylactic role of taurine. J Appl Toxicol. 2008;28:974–986. doi: 10.1002/jat.1363. [DOI] [PubMed] [Google Scholar]
- Speckmann B, Steinbrenner H, Grune T, Klotz L-O. Peroxynitrite: from interception to signaling. Arch Biochem Biophys. 2016;595:153–160. doi: 10.1016/j.abb.2015.06.022. [DOI] [PubMed] [Google Scholar]
- Strumylaite L, Bogusevicius A, Abdrachmanovas O, Baranauskiene D, Kregzdyte R, Pranys D, Poskiene L. Cadmium concentration in biological media of breast cancer patients. Breast Cancer Res Treat. 2011;125:511–517. doi: 10.1007/s10549-010-1007-8. [DOI] [PubMed] [Google Scholar]
- Strumylaite L, Kregzdyte R, Bogusevicius A, Poskiene L, Baranauskiene D, Pranys D. Association between cadmium and breast cancer risk according to estrogen receptor and human epidermal growth factor receptor 2: epidemiological evidence. Breast Cancer Res Treat. 2014;145:225–232. doi: 10.1007/s10549-014-2918-6. [DOI] [PubMed] [Google Scholar]
- Szuster-Ciesielska A, et al. The inhibitory effect of zinc on cadmium-induced cell apoptosis and reactive oxygen species (ROS) production in cell cultures. Toxicology. 2000;145:159–171. doi: 10.1016/S0300-483X(00)00144-X. [DOI] [PubMed] [Google Scholar]
- Takebayashi S, Jimi S, Segawa M, Kiyoshi Y. Cadmium induces osteomalacia mediated by proximal tubular atrophy and disturbances of phosphate reabsorption. A study of 11 autopsies. Pathol Res Pract. 2000;196:653–663. doi: 10.1016/S0344-0338(00)80010-2. [DOI] [PubMed] [Google Scholar]
- Tandon S, Singh S, Prasad S, Khandekar K, Dwivedi V, Chatterjee M, Mathur N. Reversal of cadmium induced oxidative stress by chelating agent, antioxidant or their combination in rat. Toxicol Lett. 2003;145:211–217. doi: 10.1016/S0378-4274(03)00265-0. [DOI] [PubMed] [Google Scholar]
- Tang X et al. (2016) Mobilization and removing of cadmium from kidney by GMDTC utilizing renal glucose reabsorption pathway Toxicol Appl Pharmacol [DOI] [PMC free article] [PubMed]
- Taylor J, DeWoskin R, Ennever FK (1999) Toxicological profile for cadmium US Agency for Toxic Substances and Disease Registry, Atlanta, GA (http://www.atsdr.cdc.gov/toxpro2.html Cited 24 June 2007)
- Thomas L, Hodgson S, Nieuwenhuijsen M, Jarup L. Early kidney damage in a population exposed to cadmium and other heavy metals. Environ Health Perspect. 2009;117:181–184. doi: 10.1289/ehp.11641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turko IV, Marcondes S, Murad F. Diabetes-associated nitration of tyrosine and inactivation of succinyl-CoA: 3-oxoacid CoA-transferase. Am J Physiol Heart Circ Physiol. 2001;281:H2289–H2294. doi: 10.1152/ajpheart.2001.281.6.H2289. [DOI] [PubMed] [Google Scholar]
- Valko M, Morris H, Cronin M. Metals, toxicity and oxidative stress. Curr Med Chem. 2005;12:1161–1208. doi: 10.2174/0929867053764635. [DOI] [PubMed] [Google Scholar]
- Viaene M, Masschelein R, Leenders J, De Groof M, Swerts L, Roels H. Neurobehavioural effects of occupational exposure to cadmium: a cross sectional epidemiological study. Occup Environ Med. 2000;57:19–27. doi: 10.1136/oem.57.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viner RI, Ferrington DA, Williams TD, Bigelow DJ, Schöneich C. Protein modification during biological aging: selective tyrosine nitration of the SERCA2a isoform of the sarcoplasmic reticulum Ca2+-ATPase in skeletal muscle. Biochem J. 1999;340:657–669. doi: 10.1042/bj3400657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu W, Welsh MJ. Expression of the 25-kDa heat-shock protein (HSP27) correlates with resistance to the toxicity of cadmium chloride, mercuric chloride, cis-platinum (II)-diammine dichloride, or sodium arsenite in mouse embryonic stem cells transfected with sense or antisense HSP27 cDNA. Toxicol Appl Pharmacol. 1996;141:330–339. doi: 10.1016/S0041-008X(96)80039-1. [DOI] [PubMed] [Google Scholar]
- Yamakura F, Taka H, Fujimura T, Murayama K. Inactivation of human manganese-superoxide dismutase by peroxynitrite is caused by exclusive nitration of tyrosine 34 to 3-nitrotyrosine. J Biol Chem. 1998;273:14085–14089. doi: 10.1074/jbc.273.23.14085. [DOI] [PubMed] [Google Scholar]
- Zucker B, Hanusch J, Bauer G. Glutathione depletion in fibroblasts is the basis for apoptosis-induction by endogenous reactive oxygen species. Cell Death Differ. 1997;4:388–395. doi: 10.1038/sj.cdd.4400258. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Peroxynitrite production A) HeLa cells pre-loaded with probe-1 were exposed either to Cd, SIN-1 (positive control, donor of NO and superoxide) or 10 μM H2O2 (which induces superoxide species but not peroxynitrite). The spectra were obtained each minute during a period of 30 min and fluorescence intensity at 540 nm was measured with excitation at 430 nm. B-C) Untreated control HeLa cells or pre-treated with 500 μM L-NAME were pre-loaded with probe 1 and then exposed to 5 μM Cd (B) or 100 μM Cd (C) to assay peroxynitrite production. In all cases the graph values represent the means of three independent experiments ±SD (n=6). (GIF 51 kb)
HSP27 content in HeLa cells. A) Western blott of HeLa cells Control (line 1), treated with 100 μM Cd during 24h (line 2), L-NAME and then Cd (line 3), pre-treated with rHSP2 or rC1 and then Cd (line 4-5 respectively). B) Optical density of HSP27/beta-actin expression. The graph values represent the means of three independent experiments ±SD (* p< 0,05; ** p< 0,01; *** p< 0,001). (GIF 39.5 kb)
Peroxynitrite production and cellular effects. (GIF 66.2 kb)
Viability of HeLa cells measured as metabolic activity with CCK-8. A) HeLa cells were grown in 96 well plates and then the cells were exposed to the indicated doses of Cd during different times (3-24 h). B) HeLa pScr cells were stable transfected with a scramble plasmid, selected as indicate in methodology grown in 96 well plate with hygronomycin for 24 hr and then exposed to the indicated doses of Cd in serum-hygronomycin free media. C) HeLa p762 cells (downregulation for HSP27 in a 50%), were grown in 96 well plate with hygronomycin for 24 hr and then exposed to the indicated doses of Cd in serum-hygronomycin free media. All the values are representative of the means of three independent experiments ±SD (* p< 0,05; ** p< 0,01; *** p< 0,001; n=6 for each data point). (GIF 41.4 kb)