

Abstract
While numerous maladies are associated with hypobaric hypoxia, muscle protein loss is an important under studied topic. Hence, the present study was designed to investigate the mechanism of muscle protein loss at HH. SD rats were divided into normoxic rats, while remaining rats were exposed to simulated hypoxia equivalent to 282-torr pressure (equal to an altitude of 7620 m, 8% oxygen), at 25 °C for 6, 12, and 24 h. Post-exposure rats were sacrificed and analysis was performed. Ergo, muscle loss-related changes were observed at 12 and 24 h post-HH exposure. An increased reactive oxygen species production and decreased thiol content was observed in HH-exposed rats. This disturbance caused substantial protein oxidative modification in the form of protein carbonyl content and advanced oxidation protein products. The analysis showed increase levels of bityrosine, oxidized tryptophan, lysine conjugate, lysine conjugate with MDA, protein hydroperoxide, and protein-MDA product. These changes were also in agreement with increase in lipid hydroperoxides and MDA content. HSP-70 and HSP-60 were upregulated significantly, and this finding is corroborated with increase in ER stress biomarker, GRP-78. Overloading of cells with misfolded proteins further activated degradative machinery. Consequently, pro-apoptotic signaling cascade, caspase-3, and C/EBP homologous protein were also activated in 24-h HH exposure. Release of tryptophan and tyrosine was also increased with 24-h HH exposure, indicated protein degradation. Elevation in resting intracellular calcium ion, [Ca2+]i, was also observed at 12- and 24-h HH exposure. The present study provides a detailed mechanistic representation of muscle protein loss during HH exposure.
Electronic supplementary material
The online version of this article (doi:10.1007/s12192-017-0795-8) contains supplementary material, which is available to authorized users.
Keywords: High altitude; Hypobaric hypoxia (HH); Protein modifications; Muscle loss, proteostasis; Calcium
Introduction
High altitude is associated with a number of adverse factors such as decreased ambient pressure of oxygen, decreased temperature, humidity, and increased UV radiation. In comparison with all the above, the prime contributing factor at high altitude is hypobaric hypoxia, i.e., fall in partial pressure of oxygen. In 1973, hypoxia was defined as the state in which oxygen in the lung, blood, and/or tissues is abnormally low, compared with that of a normal resting man that breaths air at sea level (Bligh and Johnson 1973). However, hypoxia is a condition not only associated with high altitude but also with a number of pathologies such as anemia, vascular abnormalities, heart failure, chronic obstructive pulmonary disease, or sleep apnea. High altitude exerts considerable effects on physiological function and physical performance of an individual. While numerous high-altitude studies have been conducted, one pathophysiological effect is still untouched, i.e., muscle protein loss and decrease in physical performance.
Skeletal muscle is metabolically active, the most voluminous, and major protein reservoir tissue of the body. Loss of muscle mass with decreased muscle fiber size is a result of typical mountaineering expeditions to Himalayas (Hoppler and Desplanches 1992; Hoppler et al. 1990). After a 10-month stay at high altitude (4100 or 3750 m), Indian soldiers had a muscle mass loss of 1.74 and 1.38 kg, respectively (Bharadwaj et al. 2000). Consequences of hypoxia include the initial loss of water and subsequent loss of muscle and fat mass (Butterfield 1990). The balance between new protein synthesis and degradation is necessary to maintain the steady state level of protein in our bodies. In vitro studies suggested that attenuated energy turnover and loss of muscle mass may be developed at hypoxia exposure due to a decrease in overall protein turnover rate (Heerlein et al. 2005; Hochachka et al. 1996; Koritzinsky et al. 2006; Kraggerud et al. 1995). Climbers exposed to extreme altitude for prolonged time exhibit evidence of oxidative stress in skeletal muscles probably due to overproduction of reactive oxygen species (ROS) and an increase in muscle lipofuscin level (Martinelli et al. 1990). Muscle biopsies show relevant evidence of muscle oxidative stress including oxidized proteins, oxidized lipids, and sensitivity in relation to vitamin E supplementation (Magalhaes et al. 2005). Several studies also report muscle mass loss and reduced performance at high altitude (Mathieu-Costello 2001; Edwards et al. 2010; Hoppler and Vogt 2001). Also, a brief reduction in oxygen supply due to hypoxia leads to acutely diminished muscle mass and performance (Calbet et al. 2009). In our previous study, elevated protein degradation leading to skeletal muscle atrophy under chronic hypobaric hypoxia was observed (Chaudhary et al. 2012a, b).
High altitude can cause increased cellular oxidative stress that consequently damage lipids, proteins, and DNA (Bailey et al. 2001; Magalhaes et al. 2004; Singh et al. 2001; Vigano et al. 2008; Rathor et al. 2015). Highlighting proteins under hypoxic conditions, elevated protein carbonylation in skeletal muscle fiber was observed during oxidative stress (Barreir and Hussain 2010). Along with this, oxidation of aromatic amino acid residues, reduction of methionine, and protein-protein cross-linkages was also noted (Barreir and Hussain 2010). Another important aspect of antioxidant status, viz. total thiol content of proteins, is also altered during muscle fatigue that disrupts redox status of tissues (Ferreira and Reid 2008). With the above facts in our mind, the present study was designed to unravel the immediate cellular and molecular cascades for protein modifications, degradation, and apoptosis under acute hypobaric hypoxia in skeletal muscle.
Material and Methods
Ethics statement
Male Sprague-Dawley rats weighing 220 ± 20 g were taken (n = 5) for the experiment. Rats were maintained three rats per cage at 25 ± 2 °C in the Animal Facility, DIPAS, India, and given food and water ad libitum under 12-h day–night cycle. The study was approved by the Animal Ethical Committee of the Institute in accordance with Committee for the Purpose of Control and Supervision of Experiments on Animals (CPCSEA) of the Government of India.
Hypobaric hypoxia exposure
A total of 20 male Sprague-Dawley rats were used in the study. The rats were divided into four groups of five rats each:
| Group 1 | Unexposed and normoxic rats served as control |
| Group 2 | Hypobaric hypoxia (HH) exposure (25,000 ft) for 6 h |
| Group 3 | Hypobaric hypoxia (HH) exposure (25,000 ft) for 12 h |
| Group 4 | Hypobaric hypoxia (HH) exposure (25,000 ft) for 24 h |
Simulated high-altitude exposure was performed in an animal decompression chamber maintained at pressure of 282 torr (equivalent to an altitude of 7620 m, 8% oxygen), coupled to mercury barometer, at 25 °C for hypoxic group (Decibel Instruments, India). The airflow in the chamber was 2 l/min with relative humidity maintained at 50 to 55%. Control group rats were maintained in the normoxic condition within the same laboratory.
After exposure, animals were anesthetized with sodium pentobarbital (50 mg/kg, i.p.) and skeletal muscles were excised from hind limb of rats for different biochemical and histopathological analyses. For biochemical analysis, muscle tissue was immediately stored at −80 °C for further use.
Biochemical assays and histopathological analysis
For biochemical estimations, the tissue was homogenized in 0.154 M KCl-EDTA buffer and skeletal muscle tissues of rats were fixed in 4% formalin for histopathological analysis.
Oxidative stress marker
ROS
Free radical generation was determined by fluorescence using 2,7-dichlorofluoroscein diacetate (DCFH-DA) (Cathcart et al. 1983). DCFH-DA passively diffuses through cellular membranes, is cleaved by an esterase, and the resultant DCFH can be oxidized to the highly fluorescent dichlorofluorescein (DCF), emitting fluorescence in the presence of ROS. Briefly, 150 μl of tissue homogenate was incubated with 10 μl of 100 μM DCFH-DA for 30 min in dark. Fluorescence was read using a fluorimeter (Perkin-Elmer, UK) with excitation at 485 nm and emission at 535 nm. Readings were obtained in arbitrary fluorometric units, and results were expressed as fold change in free radical generation.
Antioxidant activity in terms of thiol content
Thiol content was considered as primary defense system in the body, and its oxidation could be correlated with the production of oxidative stress in the body. The concentrations of total SH groups (T-SH), protein-bound SH groups (Pr-SH), and non-protein SH groups (Npr-SH) were estimated based on their reactivity with 5,5′-dithiobis (2-nitrobenzoate) (DTNB) by the method of Sedlak and Lindsay (1968). For determination of T-SH, a 0.125-ml aliquot of homogenate was added to 0.375 ml of standard incubation medium (40 mM Tris, 2 mM EDTA, 100 mM KCl, pH 8.0). After addition of 25 μl of DTNB (10 mM in methanol) and 2.5 ml methanol, the mixture was incubated for 30 min and centrifuged at 2500 rpm for 10 min. The concentration of Npr -SH was determined after addition of 0.5 ml homogenate to 1.75 ml H2O and 0.25 ml trichloroacetic acid (50%). Following centrifugation, 25 μl DTNB and 1 ml 0.4 M Tris (pH 8.9) were added to 0.5 ml supernatant and incubated for 5 min. The absorbance of the 2-nitro-5-mercaptobenzoic acid formed was measured at 412 nm. GSH served as a standard. The concentration of Pr-SH was calculated by subtracting values for Npr-SH from that of T-SH.
Protein oxidation and modification markers
Protein carbonyl content
Oxidative modifications of amino acid residues include derivatization of amino acid residues such as proline, arginine, and lysine to reactive carbonyl derivatives. Briefly, 2,4-dinitrophenylhydrazine reacts with protein carbonyl forming a Schiff base to produce the corresponding hydrazone that can be analyzed spectrophotometrically (Levine et al. 1990). Muscle tissue was homogenized in ice-cold 50 mM phosphate buffer (pH 7.2) containing 1 mM EDTA and protease inhibitor cocktail. The homogenate was centrifuged at 10,000×g for 15 min, and the supernatant was checked for presence of nucleic acids by measuring the absorbance at 260 and 280 nm. A ratio of 280/260 nm more than 1 indicated that the sample was free of nucleic acid contamination. To 200 μl of sample, 600 μl 10 mM 2,4-dinitrophenylhydrazine (DNPH) in 2 N HCl was added; 600 μl of 2 N HCl was added as a blank control. The mixture was incubated for 1 h at room temperature. The protein was precipitated with an equal volume of 20% trichloroacetic acid and was washed three times with ethanol/ethyl acetate (1:1 v/v). The final precipitate was dissolved in 400 μl of 6 M guanidine hydrochloride (pH 2.3), and the insoluble debris was removed by centrifugation. The absorbance of the DNPH derivatives was measured at 360 nm. The concentration of carbonyl groups was calculated by using an absorbance coefficient 22 nM/cm and expressed as nmol carbonyl per mg of protein.
Advanced oxidation protein products
Advance oxidation protein product was considered to be a relevant marker for oxidant-induced protein damage. It is formed during oxidative stress by the action of chlorinated oxidants, mainly hypochlorous acid and chloramines. Determination of advanced oxidation protein product (AOPP) (i.e., some oxidation products with characteristic absorbance) was based on spectrophotometric detection according to Witko-Sarsat et al. (1996) modified for muscle tissue. Briefly, tissue homogenates, prepared in 0.154 M KCl-EDTA, were diluted 1:5 with phosphate-buffered saline (PBS), pH 7.4. For standard curve, 200 μl of chloramin T (0–100 μmol/l) and 200 μl of PBS as blank were applied on a microtiter plate. Similarly, 200 μl of diluted samples was applied. Ten microliters of 1.16 M potassium iodide and 20 μl of acetic acid were added to each well, and absorbance at 340 nm was measured immediately. Concentrations of AOPPs were obtained in chloramines units and expressed as μmol chloramine/mg protein.
Free MDA, protein-MDA, protein, and lipid hydroperoxides
A simple method for the simultaneous quantification of the main lipid and protein peroxidation product was done after their initial fractionation. Free malondialdehyde (FrMDA), protein-bound malondialdehyde (PrMDA), total lipid hyderoperoxides (LOOH), and protein hyderoperoxides (PrOOH) based on the spectrophotometric and spectroflourimetric analyses were done according to Grintzalis et al. (2013). FrMDA and PrMDA (released from proteins by alkaline hydrolysis) were measured after the reaction of MDA with thiobarbituric acid (TBA) under acidic conditions by the specific fluorimetric quantification of the resulting MDA–(TBA)2 adduct chromophore. The measurement of LOOH and PrOOH is based on the reaction of Fe+3 (resulting from the reaction of LOOH and PrOOH with Fe+2) with xylenol orange (XO) and the photometric quantification of the resulting XO–Fe complex. Concentrations are expressed as nanomoles.
Oxidative protein content by fluorescent measurement
Muscle tissue was homogenized in 0.3 M sucrose and 10 mM HEPES, pH 7.4. Homogenate was centrifuged (1200×g for 10 min) to remove the debris (total membrane fraction). Protein concentration was determined by the Bradford (1994) method. Fluorescence measurements were performed in solution containing 50 μg of homogenate protein per ml, 10 mM HEPES, and 100 mM KCl, pH 7.0 at 25 °C on a Perkin-Elmer fluorimeter. Fluorescence emission spectra (300 to 450 nm, slit width 2 nm) of tryptophan were measured with excitation at 295 nm (slit width 1 nm) (Dousset et al. 1994). Fluorescence emission spectra of bityrosine, product of tyrosine oxidation, were recorded in range of 380 to 440 nm, at excitation wavelength of 325 nm (Giulivi and Davies 1994). Emission spectra of Lys conjugates with lipid peroxidation products (425 to 480 nm) were recorded at 365 nm, and excitation spectra of conjugates of Lys (325 to 380 nm) were measured with emission at 440 nm (Dousset et al. 1994).
Protein degradation pathways
Calpain assay
Calpains were measured in the homogenate using N-succinyl-Leu-Tyr-7-amido-4-methylcoumarin (SLY-AMC) as a substrate (Mastrocola et al. 2008). A stock solution of 50 mM SLY-AMC was prepared in dimethyl sulfoxide and stored at −20 °C. Muscle extract was incubated for 60 min at 37 °C in a buffer solution (pH 7.4) containing 25 mM HEPES (pH 7.5), 0.1% CHAPS, 10% sucrose, 10 mM DTT, 0.1 mg/ml ovalbumin, and substrate. Fluorescence of the liberated AMC was monitored in a Perkin-Elmer fluorimeter (LS45) at excitation 380 nm and emission 460 nm.
The 20S proteasome activity of Ub-proteasome pathways
The ubiquitin proteasome pathway was studied by assaying the chymotrypsin-like enzyme activity of 20S proteasome, as described earlier (Hepple et al. 2008). The muscle extracts containing 60 μg protein were incubated for 30 min at 37 °C in 50 μl of a buffer containing 100 mM Tris-HCl (pH 8.0), 1 mM DTT, 5 mM MgCl2, 1 mM Suc-LLVY-AMC, 2 mg/ml ovalbumin, and 0.07% SDS. The reaction was terminated by 25 μl of 10% SDS and diluted by 2 ml of 0.1 M Tris-HCl (pH 9.0). Fluorescence of the liberated AMC was monitored in a Perkin-Elmer fluorimeter at excitation 380 nm and emission 460 nm.
Degradation of tyrosine and tryptophan release
Frozen tissues were homogenized in ice-cold phosphate buffer (0.1 M, pH 7.4) containing 140 mM KCl, 1 mM EDTA, and 1 mM PMSF. The homogenate was centrifuged at 960g for 10 min and the supernatant was used. All steps were carried out at 4 °C. Sodium dodecyl sulfate was added to sample aliquots (final concentration 0.1%). The tryptophan content within solubilized proteins was determined fluorimetrically at excitation and emission wavelengths of 280 and 345 nm, respectively (Bondy 1996). The tyrosine content within solubilized proteins was determined fluorimetrically at excitation and emission wavelengths of 277 and 320 nm, respectively (Gusow et al. 2002).
Intracellular free calcium
Intracellular calcium was measured according to the method of Meder et al. (1997). The intracellular free calcium was determined by fluorescent calcium indicator dye Fura-2/AM. Fura-2/AM crosses the membrane, and is hydrolyzed by the esterases to Fura-2 that binds free ionic calcium to give fluorescence, that is proportional to the amount of free calcium. The cytosolic fraction was incubated with 5 M Fura-2/AM for 40 min at 37 °C. This was followed by centrifugation at 20,000g for 20 min, and the pellet was washed with Ca2+-free physiological buffer (133 mM NaCl, 4.8 mM KCl, 1.2 mM Na2HPO4, 1.2 mM MgSO4, 10 mM HEPES, and 10 mM glucose, pH 7.4) and recentrifuged. The pellet was resuspended in physiological buffer, and fluorescence (F) at 340- to 380-nm excitation and 510-nm emission was measured, and [Ca2+]i was calculated as follows:
[Ca2+]i = [(F − F min)/F max − F] ⋅ K d
The K d value for Ca2+ Fura-2 complex was 225 nm. Maximal fluorescence (F max) was measured after lysis of plasma membrane of skeletal muscles with SDS, and minimal fluorescence (F min) was measured in the presence of 5 mM EGTA. The results were expressed as nanomolar of free intracellular calcium.
Caspase-3 substrate cleavage assay
Caspase-3 is an enzyme activated during the induction of apoptosis. The activity of caspase-3 in skeletal muscle was estimated using colorimetric substrate, Ac-Asp-Glu-Val-Asp p-nitroaniline, and Ac-DEVD-pNA (Calbiochem) by previously described method of Li et al. (2004). Muscle tissue was homogenized in 10 volume of ice-cold lysis buffer (50 mM HEPES, pH 7.4, 0.1% CHAPS, 5 mM DTT, 2 mM EDTA, 2 mM EGTA, 0.1% Triton X-100) containing 1 mM PMSF and protease inhibitors. Homogenates were centrifuged at 14,000g for 15 min, and the supernatants were used for assay of caspase-3 activity. Sample protein (100 mg) was diluted with assay buffer (50 mM HEPES, pH 7.4, 100 mM NaCl, 0.1% CHAPS, 10 mM DTT, 2 mM EDTA, 2 mM EGTA, 0.1% Triton X-100) and incubated at 25 °C with the substrate Ac-DEVD-pNA, 200 μM final concentration, in a 96-well microtiter plate. Cleavage of the p-nitroaniline (pNA) dye from the peptide substrate was determined by the measure of absorbance of pNA at 405 nm in a microplate reader (Biotek, VT, USA). Results were calibrated with known concentrations of p-NA and expressed as nmol p-NA per minute per mg protein.
HSP-60 and HSP-70 ELISA
HSP60 and HSP70 were quantified in muscle by commercially available rat ELISA (Cusabio ELISA Kit, China) according to the manufacturer’s instructions. The intensity of the color reaction was read spectrophotometrically in a plate reader, and the concentration was expressed in ng/mg protein.
Histopathology
Muscle was removed from rats from all the four groups, and the morphological features of the muscle were analyzed using digital images of the stained section. Images are acquired using a bright-light microscope at ×20 magnification, digital camera, and image capture software (Lieca DM RBE microscope). The ×20 high-power photomicrograph of muscle biopsy from control showed transverse cut muscle fibers that have a polygonal shape with peripheral nuclei, intact sarcolemma, and non-fragmented sarcoplasm. Intact muscle tissue also shows homogenous fiber size distribution with thin, delicate endomysial connective tissue with little space among them.
Immunoblotting
Preparation of cytosolic extracts
For cytosolic fractionation, muscle tissue was homogenized in an ice-cold buffer (0.5 M sucrose, 10 mM HEPES, 10 mM KCl, 1.5 mM MgCl2, 10% glycerol, 1 mM EDTA, 1 mM DTT, 1 mM PMSF fortified with protease inhibitors). Homogenates were kept on ice for 15 min, 0.6% Nonidet P-40 was added, and then centrifuged for 20 min at 5000g at 4 °C. The supernatant with cytosolic fraction was collected and stored at −80 °C for further analysis. Total protein concentrations were determined using the Bradford (1994) method.
Western blotting
Protein (50 μg) was separated by 10% SDS-polyacrylamide gel electrophoresis, based on the molecular weight of the protein of interest and transferred onto a nitrocellulose membrane (Millipore, Billerica, USA). The membranes were blocked with 3% bovine serum albumin in PBS containing 0.1% Tween 20 (Sigma), washed, and probed with respective mouse/rabbit monoclonal antibodies. Primary antibodies GRP78, caspase-3, and C/EBP homologous protein (CHOP) were obtained from Sigma (St. Louis, MO, USA). The membranes were then incubated with anti-mouse/rabbit-IgG HRP conjugate (Sigma). The membrane was washed and incubated with chemiluminescent substrate (Sigma), and the bands were developed using X-ray films (Kodak, Rochester, NY, USA). Quantification was performed by densitometry using ImageJ software.
Statistical analysis
All the experiments were performed on a minimum of three different occasions, and data are presented as mean ± SEM. One-way analysis of variance with post hoc Bonferroni analysis was used to determine statistical significance between groups. All analysis was conducted using GraphPad Prism version 6.00 software (GraphPad, CA, USA). The p value of <0.05, with a 95% confidence interval, was considered significant.
Results
Acute hypobaric hypoxia induces oxidative stress via induction of ROS and depletion of thiol contents
Hypobaric hypoxia leads to generation of free radicals and depletion of thiol contents. ROS levels in muscle due to hypobaric hypoxia exposure were increased significantly by 3.68-, 3.88-, and 4.27-fold at 6-, 12-, and 24-h HH exposure, respectively, as compared to control rats (Fig. 1a).
Fig. 1.

Hypobaric hypoxia induced oxidative damage via ROS generation and affects intracellular sulfhydryl content in rat muscle. a Reactive oxygen species. b Total thiol content (T-SH). c Protein thiol content (Pr-SH). d Non-protein thiol content (NPr-SH). Data represents the mean ± SE; N = 5. Different symbols, double dagger, asterisk, and dagger indicate the significant differences between experimental groups (p < 0.05), while groups with matching symbols denote no difference (p ≥ 0.05)
Oxidative homeostasis is maintained by thiol content of the body and may be consider a first-line defense system. Cysteine and methionine are the main thiol containing amino acids involved in maintenance of homeostasis and also have the ability to undergo oxidation during stress conditions. As in the method described by Sedlak and Lindsay (1968), the levels of T-SH groups and NPr-SH groups were determined directly; the concentration of Pr-SH groups was calculated as a difference between above two indices for each animal. The total sulfhydryl content was decreased significantly at HH exposure, and maximum decrement was observed at 24-h HH exposure (Fig. 1b). A similar decline was seen for protein and non-protein thiol contents (Fig. 1c, d) after HH exposure.
Disturbed oxidative homeostasis induces oxidative protein modifications and the amino acid oxidation
HH exposure induced increased ROS and decreased thiol content and resulted in further protein oxidation. The levels of protein carbonyl derivatives in HH-exposed group were found higher in 6-, 12-, and 24-h HH-exposed rats as compared to control rats, and the increment was directly proportional to exposure time (Fig. 2a).
Fig. 2.

Free radical generation under hypobaric hypoxic stress resultant into protein modification and the amino acid oxidation in skeletal muscle tissue homogenate by fluorescence excitation-emission spectra. a Protein carbonylation. b Advanced oxidation protein products (AOPPs). c Formation of protein-protein cross-linkage (bityrosine formation) due to presence of free radical species shows characteristic emission spectra at 380–440 nm. d Emission spectra due to the oxidation of aromatic amino acid tryptophan under hypoxic condition, showing a shift in intensity at the peak emission wavelength. e Excitation spectra of lysine conjugates measured at 325–380 nm. f Emission spectra at 425–480 nm for the lysine conjugates with lipid peroxidation products like malondialdehyde, 4-hydroxynenal through covalent attachment designated as reactive carbonyl species (RCS)-derived protein carbonylation. Data represents the mean ± SE; N = 5. Different symbols, double dagger, asterisk, and dagger indicate the significant differences between experimental groups (p < 0.05), while groups with matching symbols denote no difference (p ≥ 0.05)
Another known and proven marker for oxidant induced protein damage is AOPP. Figure 2b shows AOPP formation in 6-, 12-, and 24-h HH-exposed animals; a significant increment was observed in all HH-exposed animals, but maximum AOPP formation (2.57-fold) was found at 24-h HH exposure (2.57 ± 0.106 μmol chloramine T/mg protein; p < 0.05) with comparison to control rats ((0.727 ± 0.062 μmol chloramine T/mg protein) (Fig. 2b).
The free radicals have a short half-life, high reactivity, modifying and damaging activity of cellular proteins. To evaluate the oxidative protein modification due to acute hypobaric hypoxia, we measured the oxidation of aromatic amino acids, primarily tyrosine and tryptophan, through the measurement of fluorescence emission spectra. Free radicals tend to attack aromatic amino acids like tyrosine and tryptophan that are a component of polypeptides. A shift in fluorescence emissions of cellular proteins is indicative of bityrosine product and oxidized tryptophan formation. Our results showed a shift of fluorescence emission spectra that was significantly higher in 24-h HH exposure as compared to control rats.
Beside this, oxidized tryptophan and bityrosine products were also measured by fluorescence emission spectra (Table 1). As shown in Fig. 2c, there was marked increase in bityrosine products in HH-exposed rats. Figure 2d shows the oxidized tryptophan which was found to be significantly higher in 24-h HH-exposed rats as compared to control rats. Along with these effects of free radicals, lysine was also oxidized, which further forms into lysine-MDA-lysine diimine cross-links with lipid peroxidation products such as malondialdehyde (MDA) and 4-hydroxynonenal. Quantifying the fluorescence emission for such products, the increment was observed highest at 24-h HH-exposed rats (1.118 ± 0.01 for lysine oxidation and 0.864 ± 0.03 for lysine conjugate with LPO) (Fig. 2e, f).
Table 1.
Measurement of protein oxidation by fluorescent spectra in response to acute hypobaric hypoxia
| Control | 6-h HH exposure | 12-h HH exposure | 24-h HH exposure | |
|---|---|---|---|---|
| Bityrosine products | 0.599 ± 0.011‡ | 0.707 ± 0.023* | 0.806 ± 0.01* | 0.967 ± 0.03† |
| Tryptophan oxidation | 27.61 ± 0.108‡ | 26.35 ± 0.131‡ | 31.97 ± 0.17* | 43.33 ± 0.21† |
| Lysine conjugates | 0.221 ± 0.0191‡ | 0.485 ± 0.021* | 0.885 ± 0.010† | 1.118 ± 0.01† |
| Lysine conjugate with LPO | 0.244 ± 0.0124‡ | 0.439 ± 0.021* | 0.574 ± 0.02* | 0.864 ± 0.03† |
Data represents the mean ± SE; N = 5. Double dagger, asterisk, and dagger indicate that differences between values with matching symbol notations within each column are not statistically significant at 5% level of probability
Acute hypobaric hypoxia enhances total, free, and protein malonaldehyde and the hydroperoxides and lipid hydroperoxides
Figure 3a shows the level of total MDA, free MDA (Fr-MDA), and protein MDA (Pr-MDA) in muscle. The results indicate increased total MDA in 6-, 12-, and 24-h HH exposure, while Pr-MDA was increased on 12- and 24-h HH exposure and free MDA increased on 24-h HH exposure.
Fig. 3.
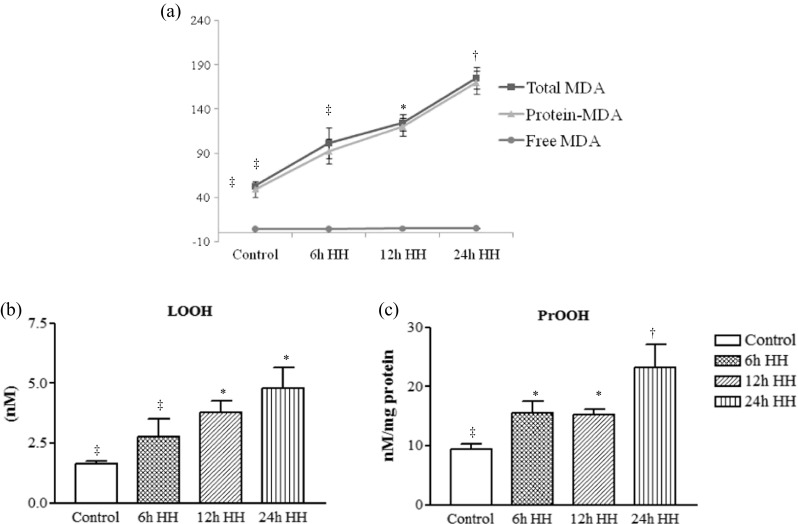
Determination of lipid peroxidation product, PrOOH, and LOOH by the spectophotometric method, which is based on the reaction of TBA with MDA and Fe3+ with xylenol orange (XO) under acidic conditions and the formation of XO-Fe complex absorbing at 532 nm, respectively. a Total MDA (strikethrough square), protein MDA (Pr-MDA) (strikethrough triangle), and free MDA (Fr-MDA) (strikethrough circle). b LOOH represents lipid and other hydrophobic peroxides. c PrOOH representing protein hyderoperoxides. Data represents the mean ± SE; N = 5. Different symbols, double dagger, asterisk, and dagger indicate the significant differences between experimental groups (p < 0.05), while groups with matching symbols denote no difference (p ≥ 0.05)
LOOH and PrOOH are biomarkers of oxidative stress-induced lipid peroxidation and protein oxidation. The results indicate the increase in PrOOH in 24-h HH-exposed rats, while LOOH increased significantly at 12- and 24-h HH-exposed animals with comparison to control rats (Fig. 3b, c).
Acute hypobaric hypoxia affects heat shock proteins and ER chaperone expression
Acute hypobaric hypoxia exposure leads to protein modification and misfolding. To rectify these modifications, heat shock proteins and ER chaperones get activated. In our study, we studied cytosolic heat shock protein HSP70, heat shock protein HSP60, and ER chaperone GRP78/BiP in relation to hypobaric hypoxic condition and found that HSP70, HSP60, and GRP78 expressions increased significantly with the exposure time (Fig. 4a–d) in rats as compared to control rats.
Fig. 4.
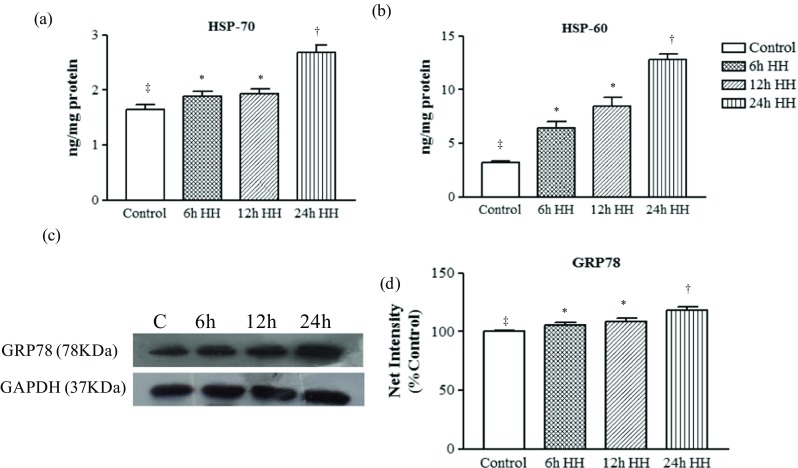
Misfolded/oxidized proteins activate the heat shock proteins and ER chaperones in response to acute hypoxic stress. Graph shows the significant increase of heat shock proteins in acute hypoxic insult in muscle tissue homogenate. a HSP70. b HSP60. c Representative western blot identifying the expression of ER chaperone (GRP-78) in cytosolic tissue extract. Loading control was normalized with reference to GAPDH. d Graph represents the semiquantitative densitometric analysis of the protein expression. The net intensity was analyzed by using the ImageJ software. Data represents the mean ± SE; N = 5. Different symbols, double dagger, asterisk, and dagger indicate the significant differences between experimental groups (p < 0.05), while groups with matching symbols denote no difference (p ≥ 0.05)
Increased oxidized proteins lead to cellular degradation due to elevated resting intracellular [Ca2+]
Increased expressions of chaperones clearly indicate the overloading of cells with misfolded/oxidized proteins. Accumulation of misfolded proteins thus induces the increased activity of 20S proteasome, a catalytic core of 26S proteasome. The activity was assayed by fluorogenic substrate, Suc-LLVY-AMC. A significant increase in chymotrypsin-like protease activity of 20S proteasome was observed in 12- and 24-h HH-exposed rats, while no significant change was noted in 6-h HH-exposed rats as compared to control rats (Fig. 5a).
Fig. 5.

Presence of oxidized proteins in the cell activates several degradative machinery, release of aromatic amino acids after degradation, and elevation of intracellular [Ca2+]i in muscle. a The 20S proteasome activity was analyzed by using fluorogenic substrate Succ-LLVY-AMC. b Calcium ion-dependent calpain activity was also analyzed fluorometrically by using substrate SLY-AMC. c, d Hypobaric hypoxia causes elevation of release of aromatic amino acids tyrosine and tryptophan residues after the degradation through proteasome and calpain activity. e Resting intracellular [Ca2+]i in muscle. Data represents the mean ± SE; N = 5. Different symbols, double dagger, asterisk, and dagger indicate the significant differences between experimental groups (p < 0.05), while groups with matching symbols denote no difference (p ≥ 0.05)
Further, in the series, calpain activity was also measured using fluorogenic substrate for calpain, Suc-LVY-AMC. The results show that calpain activity increased significantly in 12- and 24-h HH exposure, while calpain activity remain unchanged in 6-h HH-exposed rats in comparison to unexposed rats (Fig. 5b).
Based on the above results, we conclude that both calpain- and proteasome-mediated proteolysis initiated by hypoxic stress leads to muscle protein degradation. Once the proteasome pathway is activated, it initiated degradation of proteins. The animals exposed with HH for 24 h showed significant increase in the fluoroscence emissions due to released tyrosine and tryptophan residues (2.42- and 2.28-fold, respectively; p < 0.05) as compared to control animals (Fig. 5c).
Calcium is an important ion, involved with numerous activities including protein degradation via calcium-dependent calpain protease. Hence, the present study established an interesting finding that intracellular calcium ion [Ca2+]i increased significantly in 12- and 24-h HH exposure as compared with normoxic control rats (Fig. 5d).
Acute hypoxia insult affects apoptotic signaling cascade
Our study also investigated an important pro-apoptotic signaling cascade during acute hypobaric hypoxia exposure. To know underlying mechanisms for apoptosis in hypoxic muscle, caspase-3 activity, the prime enzyme involved with apoptosis was also measured in the present study. Caspase-3, a pro-apoptotic marker of 32 kDa, was upregulated in 12- and 24-h HH-exposed animals by 1.29- and 1.52-fold, respectively, as compared to control animals (Fig. 6a, b).
Fig. 6.
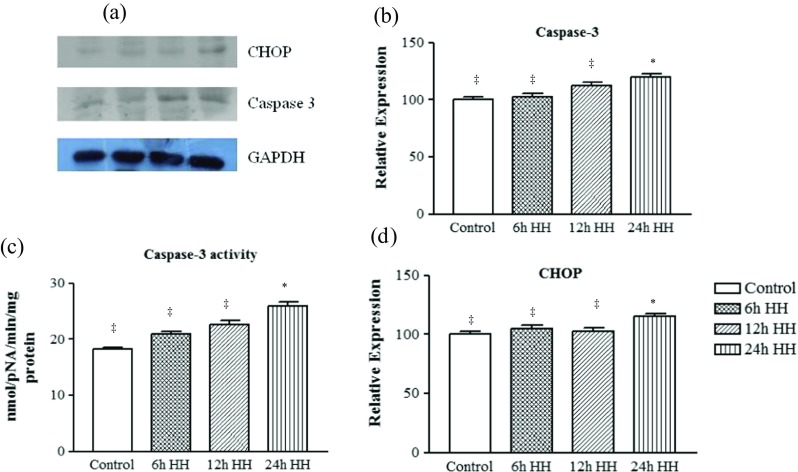
Acute hypoxic stress led to activation of pro-apoptotic markers. a Representative western blot identifying the expression of apoptotic marker of ER stress, caspase-3, and CHOP. Loading control was normalized with reference to GAPDH. b Graph represents densitometric analysis of the protein expression of caspase 3 using ImageJ software. c Caspase 3 activity. d Graph represents the densitometric analysis of CHOP using ImageJ software. Data represents the mean ± SE; N = 5. Different symbols, double dagger, asterisk, and dagger indicate the significant differences between experimental groups (p < 0.05), while groups with matching symbols denote no difference (p ≥ 0.05)
To get an indication of whether there was initiation of the apoptosis process, caspase protease activity was also estimated using the colorimetric substrate, Ac-DEVD-pNA (Fig. 6c). The protease activity of caspase was in agreement with the relative abundance of caspase (Fig. 6a, b).
When the severity of the ER stress persists, apoptosis may also involve the upregulation of CHOP. Hence, CHOP was also quantified and its upregulation was also noted after 24-h HH exposure with comparison to control rats (Fig. 6a, d).
Histopathology
The histopathology of skeletal muscle was also examined. Control muscle cut in cross section has normal morphology with peripheral nuclei and abundant myofibrils within individual muscle fibers evenly separated by the extracellular space (Fig. 7a). With expose to HH for 6, 12, or 24 h (Fig. 7b–d, respectively), there was a progressive increase in variability of muscle fiber size and shape, the extracellular space, and an apparent degradation of muscle fibers.
Fig. 7.

Myocyte architecture undergoes changes under hypobaric hypoxia exposure. Light micrographs showing hematoxylin-eosin staining of muscle; arrows indicate the area of maximal damage. a Control muscle cut in cross section has normal morphology with peripheral nuclei and abundant myofibrils within individual muscle fibers evenly separated by the extracellular space. b The 6-h HH-exposed rat muscle showing smaller in size with more space between myocytes (indicated by black arrow). c The 12-h HH-exposed rat muscle showing more space between myocytes; along with this, irregularity was also observed in myocytes (indicated by left-pointing white arrow). d Rat muscle exposed with 24-h HH exposure illustrated degeneration of muscle fibers (indicated by white triangle). The scale bar at the bottom represents 10 μm (a–d)
Discussion
Hypoxia-induced skeletal muscle atrophy as a consequence of high altitude has been well documented in past studies, but the exact molecular mechanism of hypobaric hypoxia-induced muscle loss is still unanswered. Hypoxia mediates excessive ROS generation that could be responsible for disturbed protein structure due to oxidation of amino acids. Hypoxia increases mitochondrial ROS generation at complex III that is responsible for production of oxidative stress and causing oxidative damage to biomolecules like proteins, lipids, and DNA (Chandel et al. 2000; Guzy et al. 2005). Similarly, acute and severe hypobaric hypoxia increased oxidative stress and impaired mitochondrial function in mouse skeletal muscle (Magalhaes et al. 2005). In our knowledge, our study is the first to provide a detail insight of the involvement of oxidative protein modification on muscle protein degradation due to hypoxia exposure.
The present study suggested that hypoxia insult results in an increase in ROS that cause protein oxidation. With protein oxidation, there is an incremental increase in protein carbonyl content and AOPPs as seen in the present as well as our earlier studies (Chaudhary et al. 2012b; Barreir and Hussain 2010). Increased protein carbonyl content can result from direct oxidation of protein back such as amino acids like lysine, arginine, histidine, proline, glutamic acid, and threonine (Butterfield and Stadtman 1997; Castegna et al. 2003). Other protein modifications following HH exposure include an increase in bityrosine products, tryptophan oxidation, lysine conjugates, and PrOOH (Jain et al. 2013). Dityrosine and tyrosine oxidation products are also considered as endogenous markers for proteolysis of oxidatively modified proteins (Giulivi and Davies 1993; Jain et al. 2013). ROS is also associated with an increase in free malondialdehyde, protein malondialdehyde, and LOOH (Grintzalis et al. 2013) and our results agree. Hence, our results suggest that increased free radical generation and protein modifications lead to an overload of misfolded proteins in muscle cells especially after 24-h HH exposure.
In cells under hypoxia stress, thiol content plays an important role in maintaining homeostasis. Total thiol content, protein thiol content, and non-protein thiol content were decreased significantly in a time-dependent manner in HH-exposed rats in relation to control rats. This decrease in thiol content suggests considerable elevation of thiol oxidation of proteins with simultaneous reduction in disulfide bonds (Jain et al. 2013) and appears to be associated with the oxidative stress and misfolded proteins in cells.
Our findings of increased ROS production after HH are supported by previous studies with acute and severe hypoxic conditions (Magalhaes et al. 2005; Chaudhary et al. 2012a, b) that result into changes in redox status of protein. Evidence of thiol oxidation due to oxidative stress are strongly implicated in muscle fatigue. The thiol moiety (−SH) of cysteine can undergo reversible, covalent reactions with muscle-derived oxidants to form disulfide bonds. The oxidation of cysteine leads to disturbances in protein structure and function and ultimately perturbs proteostasis (Eaton 2006). Similarly, lipid peroxidation products MDA and 4-hydroxynonenal, both of which are capable of reacting with specific amino acids including lysine under stress condition, are found to be increased and detected as a lysine conjugates with LPO fluorimetry. Thus, the present study confirms that the maximum level of protein modification through amino acid oxidations, tryptophan and tyrosine, or the cross-linking of tyrosine was seen in the HH-exposed animals in a time-dependent manner.
Furthermore, our study implied the presence of misfolded/modified protein that led to cell stress that progressed to the increased levels of cell chaperones. GRP78, HSP70, and HSP60 increased significantly in hypoxic rats as compared with control rats. GRP78 is present in endoplasmic reticulum (ER), while HSP70 and HSP60 are abundant in cytoplasm and mitochondria, respectively. Hence, the increase in these chaperones suggests the misfolded protein burden in cell, and the increase in GRP78 further suggests ER stress in myocytes. ER is a membrane-bound cell organelle responsible for proper protein folding and a critical site for quality control of proteins, calcium homeostasis, and lipid biosynthesis. Continued accumulation of unfolded proteins triggers the unfolded protein response (UPR) and activates the ER chaperones, foldases, and lectins. Few studies have focused on the essential role played by the UPR in tissue hypoxia (Kikuchi et al. 2016). ROS may cause ER stress via generation and accumulation of oxidized proteins (Van der Vlies et al. 2003). We have also analyzed the misfolded/oxidized protein accumulation in the hypoxia-exposed muscle tissues. GRP-78 is the most abundant protein in the ER lumen, and its expression is regulated by the UPR (Yoshida et al. 1998). In this regard, we identified the major ER chaperone in the skeletal muscle tissue of hypoxic animal. GRP-78 is highly upregulated in 24-h HH animals that shows the fundamental sign of the presence of a protein misfolding load in the cell. Other molecular chaperones like HSP-60 and HSP-70 are found to be upregulated in the 24-h HH animals.
In this study, the protein degradative machinery, i.e., calpain and ubiquitine-proteasome pathways, was elevated in 24 h of HH-exposed animals, presumably to degrade the misfolded proteins from the myocytes. Calcium-dependent calpain and proteasome work in a coordinated manner to degrade muscle proteins. Our previous report (Chaudhary et al. 2012a, b) and other studies (Lemus and Goder 2014) also stated that once the protective effect of UPR fails to cope with the stress, endoplasmic reticulum employs endoplasmic reticulum-associated degradation (ERAD) to clear the aggregated misfolded or unassembled proteins.
The present study and previous studies (Chaudhary et al. 2012a, b; Jain et al. 2013) suggests that the mechanism of skeletal muscle protein degradation after HH is through the UPR, calpain, and lysosomal pathways. Calpain proteases and UPR work in sequence and a coordinated manner to degrade muscle proteins (Menconi et al. 2007; Kramerova et al. 2005; Smith and Dodd 2007). Further, release of tryptophan and tyrosine were also found significantly higher in HH-exposed rats as compare with normoxic rats that suggest that degradation of oxidized proteins is by proteasome pathway.
Calpains are intracellular non-lysosomal, Ca2+-regulated cysteine proteases (Bartoli and Richard 2005; Goll et al. 2003) that increased significantly during HH exposure. With this in mind, resting intracellular cytosolic [Ca2+]i was investigated and interestingly was found elevated in 12- and 24-h HH-exposed rats in comparison to normoxic control rats. Substantial evidences has accumulated demonstrating that ROS can influence and modulate [Ca2+]i in pulmonary arterial smooth muscle cells in response to hypoxia (Shimoda and Undem 2010). Previous studies provided evidence for a role of calcium-dependent calpain activation in various muscular pathologies (Murphy et al. 2013; Alderton and Steinhardt 2000) and increased influx of [Ca2+]i. To our knowledge, our study represents the first analysis of the elevated resting cytosolic calcium ion that could play a binary role. First, [Ca2+]i could play a synergestic role to generate ER stress, and second, it might also play an important role for calpain activation that results into protein and muscle degradation. Hence, this study suggests an interesting role of calcium in muscle protein loss in the specific case of hypobaric hypoxia exposure. Supporting this notion is the finding that altered calcium homeostasis and activation of ER stress is seen in myotonic dystrophy type 1 cells (Botta et al. 2013).
In addition to increase in proteasome and calpain protease activities, apoptosis-related markers like caspase-3 and CHOP were unregulated in 24-h HH-exposed rats. The above observation is consistent with the idea that if ER stress is prolonged or too severe, signaling switches from pro-survival to pro-death, thus triggering ER stress-induced apoptosis or cell death (Rutkowski et al. 2006; Urra et al. 2013; Sano and Reed 2013). The present study demonstrates the progressive induction of the pro-apoptotic protease, caspase-3, in 24-h HH-exposed animals. Recently, compelling evidence indicates that hypoxia-induced very low density lipoprotein receptor (VLDLr) expression triggered endothelial ER stress and apoptosis (Xie et al. 2015). With this in mind, one of the components of the ER stress-mediated apoptosis pathways, CHOP, was also analyzed and found elevated in 24-h HH-exposed rats. Similarly, CHOP protein level tended to be higher in human skeletal muscle after acute environmental hypoxia compared to normoxia (Baehr et al. 2016; D’Hulst et al. 2013).
A recent study explored the role of the transcriptional coactivator peroxisome proliferator-activated receptor-γ coactivator-1α (PGC-1α) that could also be an important factor in loss of muscle mass due to deactivation of mitochondrial biogenesis (Levett et al. 2012). Further studies are required in this direction to explore the role of PGC-1α in hypobaric hypoxia-induced muscle atrophy.
In conclusion, the present study provides insight into the mechanistic foundation of muscle loss in vivo following hypobaric hypoxia exposure. A detailed overview of protein modification is presented in this study and summarized in Fig. 8 that could be responsible for the initiation of the proteolysis process after HH. Also interesting is the intracellular calcium [Ca2+]i level enhancement that is associated with the muscle proteolysis process. Overall, our study provides a clear representation of mechanism of muscle protein loss during high-altitude exposure in which ROS activates numerous signals, an area of emerging interest that provide an understanding of muscle loss due to disturbance in protein and calcium homeostasis in cell. Our study provides a clear pathway from production of ROS to the activation of apoptosis via protein modifications and disturbance in calcium homeostasis (Fig. 8).
Fig. 8.

Diagrammatic representation of mechanism approach to elucidate the effect of hypobaric hypoxia-induced muscle loss
Electronic supplementary material
(DOC 85 kb)
Acknowledgements
The authors are thankful to Dr. (Mrs.) Shashi Bala Singh, Director, DIPAS, for her constant support and encouragement. One of the authors, Ms. Akanksha Agrawal, is thankful for obtaining Senior Research Fellowship from DRDO.
Abbreviations
- HH
Hypobaric hypoxia
- GRP
Glucose regulating protein
- HSP
Heat shock protein
- AOPP
Advanced oxidation protein product
- MDA
Malondialdehyde
Footnotes
Electronic supplementary material
The online version of this article (doi:10.1007/s12192-017-0795-8) contains supplementary material, which is available to authorized users.
References
- Alderton JM, Steinhardt RA. Calcium influx through calcium leak channels is responsible for the elevated levels of calcium-dependent proteolysis in dystrophic myotubes. J Biol Chem. 2000;275:9452–9460. doi: 10.1074/jbc.275.13.9452. [DOI] [PubMed] [Google Scholar]
- Baehr LM, West DWD, Marcotte G, Marshall AG, De Sousa LG, Baar K, Bodine SC. Age related deficits in skeletal muscle recovery following disuse are associated with neuromuscular junction instability and ER stress, not impaired protein synthesis. Ageing. 2016;8:27–146. doi: 10.18632/aging.100879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey DM, Davies B, Young IS. Intermittent hypoxic training: implications for lipid peroxidation induced by acute normoxic exercise inactive men. Clin Sci (Lond) 2001;101:465–475. doi: 10.1042/cs1010465. [DOI] [PubMed] [Google Scholar]
- Barreir E, Hussain SNA. Protein carbonylation in skeletal muscles: impact on function. Antioxid Redox Signal. 2010;12:417–429. doi: 10.1089/ars.2009.2808. [DOI] [PubMed] [Google Scholar]
- Bartoli M, Richard I. Calpains in muscle wasting. Int J Biochem Cell Biol. 2005;37:2115–2133. doi: 10.1016/j.biocel.2004.12.012. [DOI] [PubMed] [Google Scholar]
- Bharadwaj H, Prasad J, Pramanik SN, Kishnani S, Zachariah T, Chaudhary KL, Sridharan K, Srivastava KK. Effect of prolonged exposure to high altitude on skeletal muscles of Indian soldiers. Def Sci J. 2000;50:167–176. doi: 10.14429/dsj.50.3421. [DOI] [Google Scholar]
- Bligh J, Johnson KG. Glossary of terms for thermal physiology. J Appl Physiol. 1973;35:941–961. doi: 10.1152/jappl.1973.35.6.941. [DOI] [PubMed] [Google Scholar]
- Bondy SC. Evaluation of free radical-initiated oxidant events within the nervous system. In: Perez-Polo JR, editor. Methods in neuroscience 30. San Diego: Academic Press; 1996. pp. 243–259. [Google Scholar]
- Botta A, Malena A, Loro E, Del Moro G, Suman M, Pantic B, Szabadkai G, Vergani L. Altered Ca2+ homeostasis and endoplasmic reticulum stress in myotonic dystrophy type 1 muscle cells. Genes (Basel) 2013;4:275–292. doi: 10.3390/genes4020275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein–dye binding. Anal Biochem. 1994;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- Butterfield GE. The adaptations. In: Sutton JR, Coates G, Remmers JE, editors. Hypoxia. Elements of energy balance at altitude. Philadelphia: Decker BC, Inc; 1990. pp. 88–93. [Google Scholar]
- Butterfield DA, Stadtman ER. Protein oxidation processes in aging brain. Adv Cell Aging Gerontol. 1997;2:161–191. doi: 10.1016/S1566-3124(08)60057-7. [DOI] [Google Scholar]
- Calbet JA, Robach P, Lundby C. The exercising heart at altitude. Cell Mol Life Sci. 2009;66:3601–3613. doi: 10.1007/s00018-009-0148-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castegna A, Drake J, Pocernich C, Butterfield DA. Protein carbonyl levels—an assessment of protein oxidation. In: Hensley K, Floyd RA, editors. Methods in pharmacology and toxicology: Methods in biological oxidative stress. Totowa: © Humana Press Inc.; 2003. [Google Scholar]
- Cathcart R, Schwiers E, Ames BN. Detection of pico mole levels of hyderoperoxides using fluorescent dichlorofluoroscein assay. Anal Biochem. 1983;134:111–116. doi: 10.1016/0003-2697(83)90270-1. [DOI] [PubMed] [Google Scholar]
- Chandel NS, McClintock DS, Feliciano CE, Wood TM, Melendez JA, Rodriguez AM, Schumacker PT. Reactive oxygen species generated at mitochondrial complex III stabilize hypoxia-inducible factor-1α during hypoxia. A mechanic of O2 sensing. J Biol Chem. 2000;275:25130–25138. doi: 10.1074/jbc.M001914200. [DOI] [PubMed] [Google Scholar]
- Chaudhary P, Suryakumar G, Prasad R, Singh SN, Ali S, Ilavazhagan G. Effect of acute hypobaric hypoxia on skeletal muscles protein turnover. Al Ameen J Med Sci. 2012;5:355–361. [Google Scholar]
- Chaudhary P, Suryakumar G, Prasad R, Singh SN, Ali S, Ilavazhagan G. Chronic hypobaric hypoxia mediated skeletal muscle atrophy: role of ubiquitin–proteasome pathway and calpains. Mol Cell Biochem. 2012;364:101–113. doi: 10.1007/s11010-011-1210-x. [DOI] [PubMed] [Google Scholar]
- D’Hulst G, Jamart C, Van Thienen R, Hespel P, Francaux M, Deldicque L. Effect of acute environmental hypoxia on protein metabolism in human skeletal muscle. Acta Physiol. 2013;208:251–264. doi: 10.1111/apha.12086. [DOI] [PubMed] [Google Scholar]
- Dousset N, Ferretti G, Taus M, Valdiguie P, Curatola G. Fluorescence analysis of lipoprotein peroxidation. Methods Enzymol. 1994;233:459–469. doi: 10.1016/S0076-6879(94)33052-2. [DOI] [PubMed] [Google Scholar]
- Eaton P. Protein thiol oxidation in health and disease: techniques for measuring disulfides and related modifications in complex protein mixtures. Free Radic Biol Med. 2006;40:1889–1899. doi: 10.1016/j.freeradbiomed.2005.12.037. [DOI] [PubMed] [Google Scholar]
- Edwards LM, Murray AJ, Tyler DJ, Kemp GJ, Holloway, Robbins PA, Neubauer S, Levett D, Montgomery HE, Grocott MP, Clarke K. The effect of high-altitude on human skeletal muscle energetics: P-MRS results from the Caudwell Xtreme Everest expedition. PLoS One. 2010;5:5. doi: 10.1371/journal.pone.0010681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira LF, Reid MB. Muscle-derived ROS and thiol regulation in muscle fatigue. J Appl Physiol. 2008;104:853–860. doi: 10.1152/japplphysiol.00953.2007. [DOI] [PubMed] [Google Scholar]
- Giulivi C, Davies KJ. Dityrosine and tyrosine oxidation products are endogenous markers for the selective proteolysis of oxidatively modified red blood cell hemoglobin by (the 19S) proteasome. J Biol Chem. 1993;268:8752–8759. [PubMed] [Google Scholar]
- Giulivi C, Davies KJA. Dityrosine: a marker for oxidatively modified proteins and selective proteolysis. Methods Enzymol. 1994;233:363–371. doi: 10.1016/S0076-6879(94)33042-5. [DOI] [PubMed] [Google Scholar]
- Goll DE, Thompson VF, Li H, Wei W, Cong J. The Calpain system. Physiol Rev. 2003;83:731–801. doi: 10.1152/physrev.00029.2002. [DOI] [PubMed] [Google Scholar]
- Grintzalis K, Zisimopoulos D, Grune T, Weber D, Georgiou CD. Method for the simultaneous determination of free/protein malondialdehyde and lipid/protein hydroperoxides. Free Radic Biol Med. 2013;59:27–35. doi: 10.1016/j.freeradbiomed.2012.09.038. [DOI] [PubMed] [Google Scholar]
- Gusow K, Szabelski M, Rzeska A, Karolczak J, Sulowska H, Wiczk W. Photophysical properties of tyrosine at low pH range. Chem Phys Lett. 2002;362:519–526. doi: 10.1016/S0009-2614(02)01135-1. [DOI] [Google Scholar]
- Guzy RD, Hoyos B, Robin E, Chen H, Liu L, Mansfield KD, Simon MC, Hammerling U, Schumacker PT. Mitochondrial complex III is required for hypoxia-induced ROS production and cellular oxygen sensing. Cell. 2005;1(6):401–408. doi: 10.1016/j.cmet.2005.05.001. [DOI] [PubMed] [Google Scholar]
- Heerlein K, Schulze A, Hotz L, Bartsch P, Mairbaurl H. Hypoxia decreases cellular ATP demand and inhibits mitochondrial respiration of a549 cells. Am J Respir Cell Mol Biol. 2005;32:44–51. doi: 10.1165/rcmb.2004-0202OC. [DOI] [PubMed] [Google Scholar]
- Hepple RT, Qin M, Nakamoto H, Goto S. Caloric restriction optimizes the proteasome pathway with aging in rat plantaris muscle: implications for sarcopenia. Am J Physiol Regul Integr Comp Physiol. 2008;295:R1231–R1237. doi: 10.1152/ajpregu.90478.2008. [DOI] [PubMed] [Google Scholar]
- Hochachka PW, Buck LT, Doll CJ, Land SC. Unifying theory of hypoxia tolerance: molecular/metabolic defense and rescue mechanisms for surviving oxygen lack. Proc Natl Acad Sci U S A. 1996;93:9493–9498. doi: 10.1073/pnas.93.18.9493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoppler H, Desplanches D. Muscle structural modifications in hypoxia. Int J Sport Med. 1992;1:S166–S168. doi: 10.1055/s-2007-1024628. [DOI] [PubMed] [Google Scholar]
- Hoppler H, Vogt M. Muscle tissue adaptations to hypoxia. J Exp Biol. 2001;204:3133–3139. doi: 10.1242/jeb.204.18.3133. [DOI] [PubMed] [Google Scholar]
- Hoppler H, Kleinert E, Claassen C, Howald H, Kayer SR, Cerretelli P. Morphological adaptations of human skeletal muscle to chronic hypoxia. Int J Sport Med. 1990;11:S3–S9. doi: 10.1055/s-2007-1024846. [DOI] [PubMed] [Google Scholar]
- Jain K, Suryakumar G, Prasad R, Ganju L. Differential activation of myocardial ER stress response: a possible role in hypoxic tolerance. Int J Cardiol. 2013;168:4667–4677. doi: 10.1016/j.ijcard.2013.07.180. [DOI] [PubMed] [Google Scholar]
- Kikuchi D, Tanimoto K, Nakayama K. CREB is activated by ER stress and modulates the unfolded protein response by regulating the expression of IRE1a and PER. Biochem Biophys Res Commun. 2016;469:243–250. doi: 10.1016/j.bbrc.2015.11.113. [DOI] [PubMed] [Google Scholar]
- Koritzinsky M, Magagnin MG, Van den Beucken T, Seigneuric R, Savelkouls K, Dostie J, Pyronnet S, Kaufman RJ, Weppler SA, Voncken JW, et al. Gene expression during acute and prolonged hypoxia is regulated by distinct mechanisms of translational control. EMBO J. 2006;25:1114–1125. doi: 10.1038/sj.emboj.7600998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraggerud SM, Sandvik JA, Pettersen EO. Regulation of protein synthesis in human cells exposed to extreme hypoxia. Anticancer Res. 1995;15:683–686. [PubMed] [Google Scholar]
- Kramerova I, Kudryashova E, Venkatraman G, Spencer MJ. Calpain 3 participates in sarcomere remodeling by acting upstream of the ubiquitin-proteasome pathway. Hum Mol Genet. 2005;14:2125–2134. doi: 10.1093/hmg/ddi217. [DOI] [PubMed] [Google Scholar]
- Lemus L, Goder V. Regulation of endoplasmic reticulum-associated protein degradation (ERAD) by ubiquitin. Cell. 2014;3(3):824–847. doi: 10.3390/cells3030824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levett DZ, Radford EJ, Menassa DA, Graber EF, Morash AJ, Hoppeler H, Clarke K, Martin DS, Ferguson-Smith AC, Montgomery HE, Grocott MP, Murray AJ, Caudwell Xtreme Everest Research Group Acclimatization of skeletal muscle mitochondria to high-altitude hypoxia during an ascent of Everest. FASEB J. 2012;26(4):1431–1441. doi: 10.1096/fj.11-197772. [DOI] [PubMed] [Google Scholar]
- Levine RL, Garland D, Oliver CN, Amici A, Climent I, Lenz AG, Ahn BW, Shaltiel S, Stadtman ER. Determination of carbonyl content in oxidatively modified protein. Methods Enzymol. 1990;186:464–478. doi: 10.1016/0076-6879(90)86141-H. [DOI] [PubMed] [Google Scholar]
- Li SY, Gomelsky M, Duan J, et al. Overexpression of aldehyde dehydrogenase-2 (ALDH2) transgene prevents acetaldehyde-induced cell injury in human umbilical vein endothelial cells: role of ERK and p38 mitogen-activated protein kinase. J Biol Chem. 2004;279:11244–11252. doi: 10.1074/jbc.M308011200. [DOI] [PubMed] [Google Scholar]
- Magalhaes J, Ascensao A, Soares JM, Neuparth MJ, Ferreira R, Oliveira J, Amado F, Duarte JA. Acute and severe hypobaric hypoxia-induced muscle oxidative stress in mice: the role of glutathione against oxidative damage. Eur J Appl Physiol. 2004;91:185–191. doi: 10.1007/s00421-003-0972-6. [DOI] [PubMed] [Google Scholar]
- Magalhaes J, Ascensao A, Soares JMC, Ferreira R, Neuparth MJ, Marques F, Duarte JA. Acute and severe hypobaric hypoxia increases oxidative stress and impairs mitochondrial function in mouse skeletal muscle. J Appl Physiol. 2005;99:1247–1253. doi: 10.1152/japplphysiol.01324.2004. [DOI] [PubMed] [Google Scholar]
- Martinelli M, Winterhalder R, Cerretelli P, Howald H, Hoppeler H. Muscle lipofuscin content and satellite cell volume is increased after high altitude exposure in humans. Experientia. 1990;46:672–676. doi: 10.1007/BF01939930. [DOI] [PubMed] [Google Scholar]
- Mastrocola R, Reffo P, Penna F, Tomasinelli CE, Boccuzzi G, Baccino FM, Aragno M, Costelli P. Muscle wasting in diabetic and in tumor-bearing rats: role of oxidative stress. Free Radic Biol Med. 2008;44:584–593. doi: 10.1016/j.freeradbiomed.2007.10.047. [DOI] [PubMed] [Google Scholar]
- Mathieu-Costello O. Muscle adaptation to altitude: tissue capillarity and capacity for aerobic metabolism. High Alt Med Biol. 2001;2:413–425. doi: 10.1089/15270290152608598. [DOI] [PubMed] [Google Scholar]
- Meder W, Fink K, Gothert M. Involvement of different calcium channels in K+- and veratridine-induced increases of cytosolic calcium concentration in rat cerebral cortical synaptosomes. Naunyn Schmiedeberg’s Arch Pharmacol. 1997;356:797–805. doi: 10.1007/PL00005120. [DOI] [PubMed] [Google Scholar]
- Menconi M, Fareed M, O’Neal P, Poylin V, Wei W, Hasselgren PO. Role of glucocorticoids in the molecular regulation of muscle wasting. Crit Care Med. 2007;35:S602–S608. doi: 10.1097/01.CCM.0000279194.11328.77. [DOI] [PubMed] [Google Scholar]
- Murphy RM, Dutka TL, Horvath D, Bell JR, Delbridge LM, Lamb GD. Ca2+-dependent proteolysis of junctophilin-1 and junctophilin-2 in skeletal and cardiac muscle. J Physiol. 2013;591:719–729. doi: 10.1113/jphysiol.2012.243279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rathor R, Sharma P, Suryakumar G, Ganju L. A pharmacological investigation of Hippophae salicifolia (H S) and Hippophae rhamnoides Turkestanica (H R T) against multiple stress (C-H-R): an experimental study using rat model. Cell Stress and Chaperones. 2015;20:821–831. doi: 10.1007/s12192-015-0603-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutkowski DT, Arnold SM, Miller CN, Wu J, Li J, Gunnison KM, Mori K, Sadighi AAA, Raden D, Kaufman RJ. Adaptation to ER stress is mediated by differential stabilities of pro-survival and pro-apoptotic mRNAs and proteins. PLoS Biol. 2006;4 doi: 10.1371/journal.pbio.0040374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sano R, Reed JC. ER stress-induced cell death mechanisms. Biochim Biophys Acta. 2013;1833:3460–3470. doi: 10.1016/j.bbamcr.2013.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sedlak J, Lindsay RH. Estimation of total, protein-bound and non-protein sulfhydrylgroups in tissue with Ellman’s reagent. Anal Biochem. 1968;25:192–205. doi: 10.1016/0003-2697(68)90092-4. [DOI] [PubMed] [Google Scholar]
- Shimoda LA, Undem C. Interactions between calcium and reactive oxygen species in pulmonary arterial smooth muscle responses to hypoxia. Respir Physiol Neurobiol. 2010;174:221–229. doi: 10.1016/j.resp.2010.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh SN, Vats P, Kumria MM, Ranganathan S, Shyam R, Arora MP, Jain CL, Sridharan K. Effect of high altitude (7,620 m) exposure on glutathione and related metabolism in rats. Eur J Appl Physiol. 2001;84:233–237. doi: 10.1007/s004210170010. [DOI] [PubMed] [Google Scholar]
- Smith IJ, Dodd SL. Calpain activation causes a proteasome-dependent increase in protein degradation and inhibits the Akt signalling pathway in rat diaphragm muscle. Exp Physiol. 2007;92:561–573. doi: 10.1113/expphysiol.2006.035790. [DOI] [PubMed] [Google Scholar]
- Urra H, Dufey E, Lisbona F, Rojas-Rivera D, Hetz C. When ER stress reaches a dead end. Biochim Biophys Acta. 2013;1833:3507–3517. doi: 10.1016/j.bbamcr.2013.07.024. [DOI] [PubMed] [Google Scholar]
- Van der Vlies D, Makkinje M, Jansens A, Braakman I, Verkleij AJ, Wirtz KW, Post JA. Oxidation of ER resident proteins upon oxidative stress: effects of altering cellular redox/antioxidant status and implications for protein maturation. Antioxid Redox Signal. 2003;15:381–387. doi: 10.1089/152308603768295113. [DOI] [PubMed] [Google Scholar]
- Vigano A, Ripamonti M, De Palma S, Capitanio D, Vasso M, Wait R, Lundby C, Cerretelli P, Gelfie C. Protein modulation in human skeletal muscles in the early phase of adaptation to hypobaric hypoxia. Proteomics. 2008;8:4668–4679. doi: 10.1002/pmic.200800232. [DOI] [PubMed] [Google Scholar]
- Witko-Sarsat V, Friedlander M, Capeillere-Blandin C, Nguyen-Khoa T, Nguyen AT, Zingraff J, Jungers P, Descamps-Latscha B. Advanced oxidation protein products as a novel marker of oxidative stress in uremia. Kidney Int. 1996;49:1304–1313. doi: 10.1038/ki.1996.186. [DOI] [PubMed] [Google Scholar]
- Xie P, Duan Y, Guo X, Hu L, Yu M. Sal a attenuates hypoxia-induced endothelial endoplasmic reticulum stress and apoptosis via down-regulation of VLDL receptor expression. Cell Physiol Biochem. 2015;35:17–28. doi: 10.1159/000369671. [DOI] [PubMed] [Google Scholar]
- Yoshida H, Haze K, Yanagi H, Yura T, Mori K. Identification of the cis-acting endoplasmic reticulum stress response element responsible for transcriptional induction of mammalian glucose regulated proteins: involvement of basic leucine zipper transcription factors. J Biol Chem. 1998;273:3741–3749. doi: 10.1074/jbc.273.50.33741. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(DOC 85 kb)