

Abstract
The control of TB is a global health priority. Over the last decade, considerable progress has been made in the field of TB vaccines with numerous vaccine candidates entering the clinic and two candidates now in Phase IIb efficacy trials. Nevertheless, the lack of predictive animal models and biomarkers of TB vaccine efficacy prevents rational vaccine down-selection and necessitates prolonged and expensive clinical efficacy trials in target populations. Advances in molecular technology and progress in the development of human as well as animal mycobacterial challenge models make the identification of one or more immune correlates of protection a genuine prospect over the next decade. Moreover, the increasing pace, extent and coordination of global research efforts in TB promises to broaden understanding and inform the next generation of vaccine candidates against TB as well as related globally important pathogens.
Keywords: clinical, development, knowledge, progress, TB, vaccine
The field of TB vaccine research has reached a pivotal moment. Ten years ago, the prospect of obtaining data to suggest that a new vaccine could protect humans against TB was poor. Not one new TB vaccine had entered clinical trials since the BCG vaccine, known to be inconsistently protective against pulmonary TB, was first administered in 1921 [1]. Progress has gathered pace since 2002 when the first novel TB vaccine entered Phase I clinical trials. To date, 15 different candidate vaccines have been tested clinically through many dozens of Phase I and Phase II clinical trials [101]. The two most advanced of these candidates are currently being evaluated in Phase IIb efficacy trials. Impressively, the prospect of efficacy data on a new TB vaccine, which appeared so elusive a decade ago, is only months away.
The scale of this progress could not have been anticipated. However, we are still far from the ultimate goal of licensing a universally effective pre-exposure TB vaccine strategy with which to eliminate TB. Whether or not the first-generation vaccines currently advancing through the development pipeline exhibit any protective efficacy, it is unclear whether a single vaccine will be capable of preventing all stages of TB in all populations. Mycobacterium tuberculosis is a complex pathogen and its interactions with the human immune system are multifaceted and incompletely understood. It might not have been necessary to fund the progression of several novel vaccine candidates as far down the development pipeline as Phase I and II clinical trials if immune correlates of protection or predictive animal models were available for early stage vaccine evaluation. Thus, numerous challenges remain before we have a new TB vaccine that can begin to exert a positive impact upon the global burden of TB [2].
The current context of efforts to develop improved TB vaccines could not be more relevant. Presently, approximately 1.5 million people continue to die each year from TB and 9 million new cases are identified. The current decline in incidence is estimated to be as little as 1% per year, while the absolute number of cases worldwide continues to rise [102]. The World Health Assembly declared TB a global health emergency in 1990, which brought an infectious disease that had been neglected for many decades into sharp focus. The United Nations’ Sixth Millennium Development Goal, announced in 2000, is to reverse TB incidence by 2015 [103]. This was followed by the launch of the WHO Global Plan to Stop TB in 2006, which additionally targets a halving of global prevalence and mortality rates by 2015 compared with 1990 levels, and by 2050, a reduction in the global incidence of TB disease to less than one per million of the population, thus eliminating TB as a public health problem [104]. However, despite these calls to action, and most notably the partial success of programs to improve upon poor access to, and monitoring of, short course anti-TB drug therapy, the rising incidence of TB has not yet reversed in all world regions. An alarmingly high number of TB cases and deaths are attributable to HIV infection, with multidrug and extensively drug-resistant TB intensifying the crisis. BCG, the only licensed TB vaccine, has a highly variable protective efficacy and is unsafe in HIV-infected people.
There is a powerful case for continued efforts to develop a universally efficacious TB vaccination strategy. Immunization is considered the most cost-effective health intervention for tackling the TB epidemic and new vaccines are a critical component in the Global Plan to Stop TB. An efficacious pre-exposure vaccine is deemed crucial for eliminating TB [3]. It is notable that to date, the only successful global eradication of an infectious disease (smallpox) was achieved by vaccination. The near-success of the polio eradication campaign has also been achieved by effective vaccination.
The goal of this review is to describe the current status of the field of TB vaccine development and address the key priorities, challenges and potential solutions on the horizon in the next decade of research.
Current status of the TB vaccine pipeline
According to the first Blueprint for Tuberculosis Vaccine Development published in 2000, “A long-term commitment to develop an effective vaccine against TB is required, with the recognition that it will be a major and difficult scientific challenge” [4]. At that time, a number of vaccine candidates were emerging and being screened in small animal models but none had moved beyond the preclinical stage. This burst of activity was in part driven by the sequencing of the M. tuberculosis genome a few years earlier, which had identified an enormous range of antigens for screening as potential vaccine immunogens, and also by the availability of recombinant technology [5]. In September 2002, a recombinant modified vaccinia virus Ankara (MVA) expressing Antigen 85A, MVA85A, was the first novel TB vaccine to enter clinical development. Since 2002, 14 other candidate TB vaccines have followed MVA85A into clinical development and ten pre-exposure vaccine candidates are currently being actively evaluated [101]. These comprise one recombinant live mycobacterial vaccine designed to replace current BCG and nine subunit vaccines for boosting BCG and are listed in Table 1 . Two further vaccines have recently received support for clinical evaluation.
Table 1. Pre-exposure candidate tuberculosis vaccines in current active clinical development.
| Type of vaccine | Product | Description | Current phase of development |
|---|---|---|---|
| Recombinant live |
VPM1002 |
Recombinant BCG Prague strain expressing listeriolysin and urease deletion mutation |
Phase IIa |
| Fusion protein |
ID93/GLA-SEHybrid 1 + CAF01HyVac 4/Aeras†-404 + IC31Hybrid 56 + IC31M72 + AS01Hybrid 1 + IC31 |
Antigens Rv2608, Rv3619, Rv3620, Rv1813 plus adjuvant GLA-SEAntigens 85B and ESAT-6 plus adjuvant CAF01Antigens 85B and TB10.4 plus adjuvant IC31Antigens 85B, ESAT-6 and Rv2660 plus adjuvant IC31Antigens Rv1196 and Rv0125 (32 and 39 kDa antigens) plus adjuvant AS01Antigens 85B and ESAT-6 plus adjuvant IC31 |
Phase IPhase IPhase IPhase IPhase IIaPhase IIa |
| Viral vectored | AdAg85AAeras-402/Crucell Ad35MVA85A | Adenovirus 5 vector expressing antigen 85AAdenovirus 35 vector expressing antigens 85A, 85B and TB10.4MVA vector expressing antigen 85A | Phase IPhase IIbPhase IIb |
†A nonprofit organization.
Ad: Adenovirus; CAF: Cationic adjuvant formulation; ESAT-6: Early secretory antigenic target 6; GLA-SE: Glucopyranosyl lipid adjuvant-stable emulsion; IC: Intercell; MVA: Modified vaccinia virus Ankara; VPM: Vakzine Projekt Management.
Live & subunit TB vaccines in current development
The purpose of a recombinant live whole mycobacterial vaccine is to replace existing BCG with either an improved version of BCG, an attenuated strain of M. tuberculosis, or another recombinant mycobacterial species. The leading vaccine product in active clinical development is VPM1002, which is a strain of BCG Prague engineered to express listeriolysin from Listeria monocytogenes, together with deletion of urease expression, the purpose of which is to enhance cross-priming of CD8+ T cells by BCG through the MHC class 1 pathway [6]. This candidate vaccine has progressed to Phase IIa trials in South African infants (ClinicalTrials.gov identifier: NCT01479972) [105]. Two other recombinant BCG vaccines have entered Phase I trials, rBCG30 and Aeras 422, but these candidates are not currently active.
In contrast with live vaccines, subunit vaccines are intended to be administered after BCG and designed to boost the priming effect of BCG by delivering one or more potentially protective mycobacterial antigens, also present in BCG, in an immunogenic construct such as a fusion protein with adjuvant or a recombinant viral vector (e.g., poxvirus or adenovirus). This strategy is termed heterologous prime–boost immunization. MVA85A, the most advanced candidate TB vaccine along the pipeline, consists of a MVA attenuated poxvirus vector that is replication deficient and engineered to express Antigen 85A, an immunodominant protein secreted by M. tuberculosis and conserved in all other mycobacteria including BCG. Initial Phase I trials were designed to demonstrate safety and immunogenicity in small numbers of BCG-naive M. tuberculosis-uninfected healthy UK adults before progressing into BCG-vaccinated individuals, M. tuberculosis-infected individuals, TB endemic populations, HIV-infected people and simultaneously through younger age groups into infants [7–9]. Vaccination with MVA85A gives rise to strong and sustained antigen-specific CD4+ T-cell immunity [10]. In 2009, MVA85A entered the first Phase IIb efficacy trial of a novel TB vaccine in BCG-vaccinated infants in South Africa (ClinicalTrials.gov identifier: NCT00953927) [105]. The study is powered to detect a 60% decrease in the incidence of TB disease compared with BCG alone. In 2011, this vaccine candidate entered a second Phase IIb efficacy trial in HIV-infected adults in Senegal and South Africa (ClinicalTrials.gov identifier: NCT01151189) [105].
After MVA85A, the next most advanced vaccine candidate is Aeras-402, which has recently entered a Phase IIb efficacy trial in BCG-vaccinated infants (ClinicalTrials.gov identifier: NCT01198366) [105]. It is a recombinant adenovirus 35 vector that expresses three antigens: 85A, 85B and 10.4. Adenoviral constructs are known to induce high levels of CD8+ T cells, which are considered important for protection. However, a disadvantage with the use of recombinant adenoviral strains is that circulating wild-type human adenoviruses can lead to natural infection and pre-existing neutralizing antibodies and cellular immunity in the population, which could abrogate immune responses to vaccination. Selection of low seroprevalence adenoviral subtypes such as Ad35 may partially overcome this problem [11]. Ultimately, a combination of MVA and adenoviral vector vaccines may be preferential to induce satisfactory levels of both CD4+ and CD8+ T cells, and such prime–boost regimens have been successfully optimized in malaria vaccine development [12].
Recombinant fusion proteins also permit efficient delivery of M. tuberculosis antigens, shared with BCG, to the immune system. They consist of one or more fused antigens rationally selected for their specific immunogenic properties and disease-stage expression profile. However, these products rarely induce sufficiently potent and durable immunity alone unless formulated with an adjuvant. Fortunately, new adjuvant preparations have recently emerged that incorporate immunostimulators (e.g., pattern recognition receptor ligands, cytokines and bacterial cell wall components) in novel vehicles (e.g., oil-in-water emulsions, particulates and multimers) to achieve tailored enhancement of Th1-type cell-mediated immunity. One example is Intercell’s (Vienna, Austria) proprietary IC31 adjuvant, which is employed in three separate fusion protein candidates: HyVac 4, Hybrid 56 and Hybrid 1. Hybrid 1 is also being evaluated with another liposomal adjuvant formulation called cationic adjuvant formulation 01. Ongoing safety and immunogenicity trials will determine which of these fusion protein–adjuvant combinations will be most suitable for Phase IIb efficacy assessment. Of note, Hybrid 1 and Hybrid 56 may be less attractive because they both contain ESAT-6, which could confound the diagnosis of latent M. tuberculosis infection [13]. There are several fusion protein candidates in clinical development. The most advanced is M72, which is a fusion protein of the two M. tuberculosis antigens Rv1196 and Rv0125, being developed by GlaxoSmithKline. The adjuvant used was initially AS02 but is now being used with AS01, a liposomal compound containing 3-O-desacyl-4´-monophosphoryl lipid A [14]. The next novel protein expected to enter clinical trials is the 21-kDa heparin-binding hemagglutinin purified from BCG [101].
Next-generation vaccines
The next generation of vaccine products is likely to feature both variations on existing technology and new approaches. Novel vectors such as lymphocytic choriomeningitis virus, parainfluenza virus type 2 and Sendai virus are appearing on the preclinical horizon, while the portfolio of live vaccines is set to expand with numerous mycobacterial species and BCG strains as well as other recombinant bacteria undergoing preclinical testing [15,101]. Rapid, systematic and unbiased methods of antigen identification and characterization should accelerate and refine rational selection of immunogenic candidates from a wider repertoire. This will be crucial as multiple stimulatory, regulatory and inhibitory epitopes may be necessary to achieve protection. Concurrently, adjuvants and delivery systems for these antigens are expected to continue to expand in number and intricacy. Furthermore, elegant basic science discoveries by pioneering investigators at the cutting edge are also opening new avenues, as recently exemplified by Sweeney et al., who experimentally replaced the ESX3 secretion system protein in wild-type Mycobacterium smegmatis with the orthologous ESX3 from M. tuberculosis [16]. This unexpectedly conferred robust protection and even sterilizing immunity in M. tuberculosis challenged mice without restoring virulence. Known as IKEPLUS, this innovative vaccine still requires extensive preclinical evaluation before clinical investigations can begin. Another promising approach has been the development of live auxotrophic mutant strains of M. tuberculosis as potential vaccine platforms. The leading candidate includes targeted PhoP and fadD26 gene deletions resulting in a highly attenuated M. tuberculosis strain that exhibits protective efficacy in mice, guinea pigs and nonhuman primates [17–19]. This vaccine candidate is soon expected to begin Phase I clinical trials.
The journey so far
Having summarized the current status of TB vaccines, the authors now briefly consider the past decade of progress, which can be represented by the annual change since 2002 in the number of candidate TB vaccines within each phase of active clinical development. This is depicted in Figure 1 .
Figure 1. Cumulative number of novel preventative tuberculosis vaccines in Phase I, IIa and IIb clinical development each year since 2002.
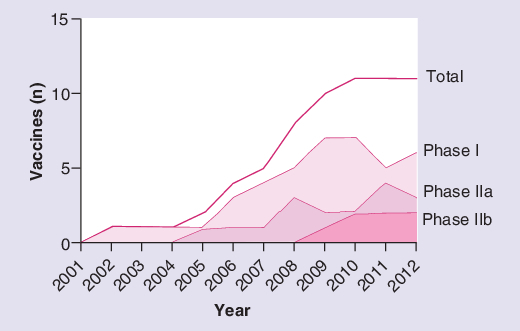
Vaccines no longer in clinical development are included in the data until their year of discontinuation. Vaccines solely being developed for therapeutic indications are excluded.
The sigmoidal shape of the graph suggests that while the total number of vaccine candidates in active clinical development is now higher than ever, this is not matched by continued growth at all phases of the pipeline. It shows an initial period of exponential growth in vaccine candidates undergoing Phase I and then Phase II trials (for reasons discussed above) followed by a slowdown in new products entering the clinical pipeline in recent years and a flattening off in the number of vaccines progressing to the next advanced phase of development. This is despite the healthy number of vaccine candidates poised at the preclinical stage. The StopTB pipeline 2011 lists five products currently in preclinical development that are now being manufactured under good manufacturing practice conditions, in preparation for clinical assessment, and a further 35 next-generation vaccine candidates in early stage evaluation, more than in the previous 4 years [101].
Multiple factors may underlie this observation. First, not all vaccines have progressed equally successfully. One vaccine has been withdrawn for safety concerns and development is no longer active for at least one more candidate. Second, supply factors are at play. The current financial downturn has been cited as a contributory factor; there was a 29% decline in funding dedicated to TB vaccine research recorded between 2009 and 2010 [104]. The 2010 funding figure for TB vaccines was US$78.4 million, a small sum considering the estimated US$3 million required to take a new vaccine through Phase I development, US$18 million required for Phase IIa and US$48 million for Phase IIb (Phase III trials are estimated to cost up to US$265m) [20–22]. Alongside the financial bottleneck is an important capacity bottleneck with a shortage of Phase IIb trial capacity in high-incidence endemic countries. These issues will impact upon the rate of progress at each level of vaccine evaluation.
Third, the slowdown in pipeline growth may represent an appropriate intellectual response given our current limitations in scientific knowledge. Without a correlate of protection or a predictive animal model with which to select new candidates for evaluation, it would be understandable if investigators and funders did not actively push forward with next-generation Phase I testing on the basis of preclinical findings alone, and the initiation of new Phase II trials may be seen as unwise at a time when the first human efficacy data are imminent. Whether the first efficacy results turn out to be positive, negative or intermediate, they will surely provide insight and guidance for the next generation of vaccines as well as those already in the pipeline. Paradoxically, only through efficacy trials will proposed correlates be validated. Thus, the shape of cumulative vaccine development in the last 10 years may be explained by fundamental gaps of scientific understanding as well as by resource limitations. These are the knowledge gaps that need to be urgently targeted in the coming years in order to transform the nascent TB vaccine pipeline into a more informed, coordinated and iterative process.
The next 10 years: addressing the knowledge gaps
Protection from most vaccine-preventable infectious diseases is mediated by neutralizing antibody induced by vaccination or natural exposure to the pathogen, which usually achieves sterilizing immunity. Vaccine efficacy for these diseases can therefore be predicted by assaying this simple surrogate biomarker, and is epitomized by a group C meningococcal vaccine which has been successfully licensed in the UK prior to availability of efficacy data on the basis of a serum bacteriocidal assay alone [23]. However, several obstacles prevent a similar approach being adopted for TB.
Understanding natural infection
Natural exposure to M. tuberculosis does not induce sterilizing immunity. While a significant proportion of exposed individuals may achieve rapid clearance of M. tuberculosis after exposure to infected household contacts, many individuals progress to chronic latent infection, of which 10% are ultimately susceptible to active TB disease. In the other 90%, it is not understood how latent TB infection appears to confer nonsterilizing protection against the development of active disease. It is remarkable that successfully treated TB patients remain susceptible to exogenous reinfection, possibly more so than the general population [24,25]. Detailed modeling of the complexities of natural M. tuberculosis transmission and precise definition of the different disease entities is therefore critical for vaccine development. Population studies in at-risk cohorts have been hampered by moderate sample sizes, the quirks of the tuberculin skin test and a lack of diagnostic biomarkers for distinguishing between infection, clearance of infection and active disease. Thus, accurate pinpointing of innate and adaptive human immune responses which are important for rapid clearance or control of M. tuberculosis, and which could be of great relevance for a pre-exposure vaccine, has not been possible. Innovative approaches to modeling disease transmission are now being made. A controlled guinea pig model of natural infection using exhaust air from TB inpatient wards has begun to shed light on rates of acute resolution of infection and disease progression [26]. Meanwhile, through a mouse model of TB reinfection, insights are being gained into the subsets of immune cells mediating susceptibility to exogenous reinfection [27].
Understanding protective immunity
Another obstacle preventing the application of the classical approach to TB vaccines is an incomplete understanding of which components of the host immune response are essential, and which are sufficient, to achieve protection; unlike for most licensed vaccines, an important role for antibodies has not been identified in vaccine-induced protection against TB. Over the last decades, an ‘outline map’ of the key components of protective immunity to TB has been delineated through observational studies of mycobacterial susceptibility in humans coupled with experimental studies in small animals. Th1-type cytokines including IFN-γ and TNF-α, and T-cell subtypes including CD4+ T cells and CD8+ T cells as well as multiple cytokine-producing T cells have been proposed as essential components of protection. These individual elements are, however, not sufficient and other cell subsets and cytokines, including regulatory and inhibitory pathways, have been identified as playing important roles. Recently, peripheral blood profiling of BCG-vaccinated adult and infant cohorts and TB patients has brought into question the strength of association between protection and antigen-specific T-cell IFN-γ secretion or polyfunctional T-cell frequencies [28]. The protective immune response to TB is complex and multidimensional, and ultimately may not be quantifiable by linear scales of arbitrary immune response categories in the same way as antibody-mediated diseases.
An additional layer of complexity is added by the fact that human cellular immune responses are highly variable between individuals. A standard dose of an investigational TB vaccine typically induces ‘lowest’ and ‘highest’ IFN-γ ELISpot responses which differ by up to 100-fold [29,30]. The optimal level of immunogenicity within this range and the threshold level (analogous to antibody titer) around which protection may succeed or fail is not known. On the one hand, this heterogeneity opens many potential avenues for investigating the genetic and other determinants of vaccination, and increases the prospects of a range of different vaccines working through different mechanisms being effective in different individuals. On the other hand, it confounds researchers’ efforts to integrate, quantify and compare immune responses in small clinical trials. Comparison between vaccine candidates would also be ameliorated if greater harmonization of immunogenicity assays could be achieved in line with published findings and WHO panel recommendations [31,32].
Identifying correlates of protection
The third obstacle is that no models or biomarkers exist for predicting vaccine efficacy. This knowledge gap is intimately linked to our ignorance of protective host immune responses and is possibly the greatest roadblock in the journey ahead. It is a realistic prospect that the next 10 years could herald a novel licensed TB vaccine demonstrating protective efficacy without knowing its mechanism of protection and without the availability of a validated surrogate biomarker of protection. This would be the opposite scenario to that of the meningococcal vaccine mentioned earlier, but business as usual for TB because after 90 years, the mechanism by which the BCG vaccine mediates protection against disseminated disease is still not understood.
As the map of host M. tuberculosis immune networks identified through both animal and human studies continues to increase in complexity, innovative approaches will be needed to define immune mechanisms and identify biomarkers of protection. For one, the next years are likely to feature an expansion in the number of studies using unbiased transcriptomic approaches. These studies will take advantage of high-throughput microarray technology and pathway discovery databases to characterize TB and vaccine-induced changes in gene expression ex vivo at multiple time points. The differentially expressed biosignatures (genes or gene clusters) which associate strongly with protection are taken forward for further investigation. Not only could they shed light on new protective immune pathways worthy of study, they could also be proposed and ultimately qualify as correlates or surrogates of clinical TB outcomes [33], BCG vaccination outcomes [Fletcher HA et al., Manuscript in Preparation] or novel vaccine candidate outcomes [Matsumiya M et al., Manuscript in Preparation]. Such an unbiased, systematic, hypothesis-generating strategy brings clear advantages over and above classical a priori modes of investigation, and can be applied across the breadth of the host–pathogen relationship. Moreover, the small blood volumes required are appropriate for infants, which are the focus of most BCG and advanced-phase novel TB vaccine studies. There is considerable hope that transcriptomic and metabolomic approaches can help bridge the knowledge gap between the responses needed for protection and the responses vaccines are inducing [34]. Judging by the last 10 years, the next decade of technological advances will undoubtedly enable enormous progress in molecular biology and a harmonized strategy should be used in TB and parallel fields to maximize the sharing and interpretation of this powerful science.
As well as molecular methods, in vitro mycobacterial growth inhibition assays (GIAs) will have an increasing role to play in the coming years. In GIAs, peripheral blood (or components) of either infected or vaccinated subjects is incubated with mycobacteria and the extent of growth inhibition (i.e., mycobacterial killing) is quantified. These GIAs avoid immune mechanism selection bias because they assay the sum of all the parts of cellular immunity. As such, they may be expected to correlate with in vivo growth inhibition (i.e., efficacy), and could be developed for screening early stage vaccine candidates. They may also correlate with in vivo M. tuberculosis challenge in both small and large animal models, which could bridge another knowledge gap, namely our understanding of the utility of animal models for predicting human vaccine efficacy. A number of GIA protocols are in development and a current challenge is to standardize these for optimal comparison of data in future projects [35–38] [Fletcher HA et al., Manuscript in Preparation].
Another approach to achieving early stage vaccine down-selection, and to discovering a biomarker which correlates with protection, is the development of a human mycobacterial challenge model. Human pathogen challenge models exist for evaluating vaccines for a number of other infectious diseases including malaria, influenza, typhoid and dengue [39–42]. Although the use of virulent M. tuberculosis as an experimental challenge in humans raises several ethical and safety concerns, M. bovis BCG, a closely related live Mycobacterium, is currently licensed for human use. Thus, a BCG challenge model is being developed which uses intradermal BCG as a surrogate for aerosol M. tuberculosis, and which assumes that a vaccine that protects against M. tuberculosis should also quantifiably inhibit intradermal BCG growth. The latter is assayed by skin biopsy at the BCG ‘challenge’ site. A preliminary study demonstrated a significant reduction in BCG CFUs by quantitative PCR in subjects with prior BCG vaccination compared with controls [43]. Further optimization and evaluation of this promising model, and translation into experimental animals (nonhuman primate, mouse) and naturally exposed animals (cattle), is ongoing. In parallel, recent efforts to engineer specifically attenuated strains of M. tuberculosis safe enough for human use could eventually lead to an aerosol human M. tuberculosis challenge model.
Optimizing vaccine delivery
Identifying a product that demonstrates satisfactory protective efficacy is a central but not exclusive goal of the TB vaccine pipeline. Numerous other parameters will need to be addressed before an optimized product can be recommended for mass deployment. These include determination of the best dose, dosing intervals, formulation (including constituents and adjuvants), route of administration as well as potential interactions with other vaccines. If a combination of two or more novel vaccines might predictably enhance efficacy, their combined administration will also need to be evaluated. The coadministration of MVA85A with vaccines in the WHO’s Expanded Programme on Immunization (EPI) has been assessed in Gambian infants [44]. This showed no effect of MVA85A on the level of humoral immunity induced by existing licensed vaccines in the EPI schedule, but did show a reduction in the cellular immunogenicity of MVA85A. The clinical significance of this reduced immunogenicity is unclear as the coadministered group still developed Antigen-85A-specific immune responses. Whether a higher MVA85A dose could overcome this and whether this effect may be generic amongst T-cell-inducing vaccines is not known.
The choice of delivery route for a new TB vaccine may significantly impact on its immunogenicity and efficacy. Established options include intradermal injection by the Mantoux technique like BCG, and intramuscular injection as employed by the majority of currently licensed vaccines. Of particular relevance to TB is the prospect of vaccine delivery by the pulmonary route. Matching route of infection to route of immunization makes strong biological sense and is backed up by both preclinical challenge experiments of TB vaccines and by clinical experience with other mucosally administered vaccines including polio, influenza and measles. Needle-free strategies such as inhaled aerosol vaccination are highly attractive. The first ever clinical trial of inhaled aerosol delivery of a viral vector vaccine is currently ongoing (ClinicalTrials.gov identifier: NCT01497769) [105].
Adjuvant technology is also experiencing a period of growth and novel recombinant protein TB vaccines are being reformulated in new generation adjuvants as they progress through the clinical pipeline [14,45]. As questions about formulation, delivery and dosage begin to be addressed alongside primary product evaluation, it will be of enormous benefit to the field as a whole in the coming years if the conclusions can in part be generalized between vaccines and vaccine types, preventing the need for repetitive noninferiority studies. This highlights the need for both positive and negative results to be widely shared in the research community, and pinpoints another crucial role for a future surrogate or correlate of protection that could be used to make multiple early stage comparisons between all the relevant variables prior to embarking on a full scale clinical development program. A further consideration is that diverse correlates may ultimately be required for assessing different constructs in different target populations.
Adapting to the future
As if the current obstacles facing the TB field were not sufficiently challenging, future research agendas will also need to be highly adaptable. The next 10 years are likely to see changes in TB (and HIV) epidemiology, further evolution and diversity of M. tuberculosis strains (including drug-resistant strains), modifications of BCG policy, the introduction of new vaccines and therapies for other relevant infectious diseases as well as transformation of socioeconomic and political climates in the highest burden countries. Novel research tools will also transform the field. It will be crucially important to avoid thinking of M. tuberculosis in isolation, but instead do so in the changing context of other mycobacteria, helminths, globally important pathogens such as HIV and malaria, any new vaccines introduced as well as the enormous diversity of its natural hosts.
The next 10 years: addressing the resource & strategy gaps
The first Phase IIb efficacy trials of novel TB vaccines are very likely to be completed in the next few years. This will be a remarkable achievement considering the multiple barriers to success and will represent an important proof-of-principle milestone for the field. As well as informing the design and feasibility of subsequent Phase IIb studies, the true cost and effort required for even larger scale Phase III trials will be brought into focus. This is crucial, for as long as the early down-selection of vaccine candidates is hampered by a lack of immunological tools, vaccines with promising Phase I safety and immunogenicity will need to be directed undifferentiated down the development pipeline. Progress along this translational research pathway would be facilitated by a long-term coordinated program of capacity building in field sites of high TB incidence. The pathway will need to integrate with upstream preclinical and Phase I facilities, as well as with downstream stakeholders including regulatory bodies, manufacturers and national public health organizations. Examples of priority areas might include improving regulatory pathways to shorten review timelines, standardizing clinical and immunological end points for trials, determining pricing, distribution and access solutions for a marketable product and embedding basic science research (particularly on correlates of protection) within all efficacy trials.
At the same time, a globally concerted TB vaccine research program needs to address deficiencies in knowledge of TB pathogenesis and immunology in conjunction with next-generation vaccine design. This will require a higher degree of effective collaboration and harmonization without compromising the enormous richness of innovative potential or diversity of scientific approaches. Nevertheless, academic institutions, industrial partners and governments ultimately share an objective in common, which is to deploy a protective TB vaccine in a resource-poor setting. Therefore, improved efforts to acquire, share and translate knowledge should be made between individual investigators and institutions. The united agenda must include investment in multisite prospective studies of TB infection and disease and/or vaccination in both infants and adults. Their purpose will be to characterize and integrate clinicopathological and immunogenetic phenotypes in sample sizes large enough and over long enough time intervals to accurately and completely define the host M. tuberculosis relationship [46]. Human tissue samples from these studies will need to be carefully planned, collected, stored and apportioned in a maximally informative way. The most efficient approach may be to incorporate these studies within anticipated Phase III trials of novel TB vaccines, for which enormous financial and political motivation will be required.
If the annual number of research articles published about TB vaccines can be taken as a crude indicator of overall progress in the field, the recent decline is disturbing (Figure 2). Funding for TB research has also declined recently and contrasts poorly with research for other potentially vaccine-preventable pathogens such as HIV and malaria [47]. In the current turbulent economic context, traditional donors are less likely to increase funding. Yet global health and economic growth are intimately related, and TB control can be thought of as an indicator of global developmental progress [48]. Thus, increased commitment from international organizations and governments as well as private funders is warranted. Innovative sources of financing also need to be identified to secure continued investment over the next 10 years of TB vaccine research and sustain the momentum gathered in the last ten. Partnerships across vaccine-preventable communicable diseases should be strengthened to maximize efficiency and increase knowledge sharing. Crucially, without a substantial scale-up of coordinated research activities, the ambitious Millennium Development Goal and Global Plan to Stop TB targets will not be met.
Figure 2. Cumulative and annual number of publications about tuberculosis vaccines each year since 1995.
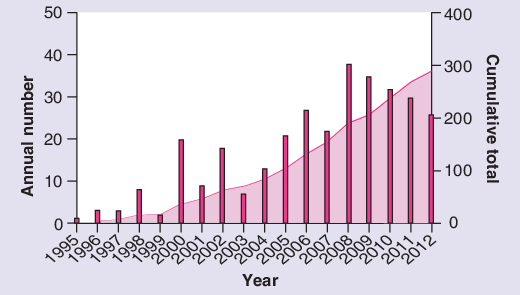
Publications were counted if they appeared in a PubMed search for terms ‘tuberculosis vaccine’ in either title or abstract for each given year of publication, using the search query (tuberculosis vaccine [Title/Abstract]) AND (‘YYYY’[Date - Publication]). Note that the 2012 figure includes up to end of October 2012.
Expert commentary & five-year view
By virtue of the achievements of the last decade of TB vaccine research, the field is now poised to bridge the void between immunogenicity of novel vaccines and potential efficacy for the first time. The vaccine development pipeline of the next 5–10 years will be shaped partly by the success or failure of the leading candidates, and partly by the extent of progress in addressing the fundamental knowledge gaps highlighted in this review. Our objectives will be honed as a result of a deeper understanding of the science of the host–pathogen relationship, pinpointing exactly what is immunologically required of an effective novel vaccine and how correlates of protection could be developed. Although it is too early to anticipate how many more vaccine candidates and clinical trials will be required, the need for continued and increased financial and infrastructural investment to ultimately control TB by vaccination is justified. Subtle changes to the ways we work and interact as scientists, centralizing our efforts while championing individual creative endeavor, also need to be explored. The first Blueprint for Tuberculosis Vaccine Development published in 2000 predicted “at least 20 years until the candidate vaccines that are most likely to be safe and protective are implemented globally” [4]. If this speculation is to be believed, then the next decade, the ‘Decade of Vaccines’, will herald even greater achievements than the last [106].
Key issues
• Major progress in TB vaccines has been made in the last 10 years.
• Approximately a dozen TB vaccine candidates are currently in clinical evaluation.
• TB vaccine candidates include live mycobacterial vaccines to replace BCG and subunit vaccines to boost BCG.
• The next generation of TB vaccine candidates includes innovative vectors, antigens and adjuvants.
-
• Major challenges over the next 10 years include:
– Identifying immunological correlates of vaccine-induced protection;
– Determining the predictive value of preclinical animal models;
– Optimizing aspects of vaccine delivery;
– Increasing capacity to carry out and fund efficacy trials.
• Continued progress requires sustained financial investment, technological advancement and synergistic collaboration.
Financial & competing interests disclosure
H McShane is a named inventor in a patent filing related to MVA85A and is a shareholder in a joint venture, Oxford Emergent Tuberculosis Consortium, formed for the future development of this vaccine. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
References
Papers of special note have been highlighted as:
• of interest
•• of considerable interest
- 1. Calmette A. Preventive vaccination against tuberculosis with BCG. Proc. R. Soc. Med. 24(11), 1481–1490 (1931). [PMC free article] [PubMed] [Google Scholar]
- 2. Brennan MJ, Thole J. Tuberculosis vaccines: a strategic blueprint for the next decade. Tuberculosis (Edinb.) 92(Suppl. 1), S6–13 (2012). [DOI] [PubMed] [Google Scholar]; •• This detailed plan outlines a comprehensive strategy for developing and introducing TB vaccines over the next 10 years.
- 3. Dye C, Williams BG. Eliminating human tuberculosis in the twenty-first century. J. R. Soc. Interface 5(23), 653–662 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ginsberg AM. A proposed national strategy for tuberculosis vaccine development. Clin. Infect. Dis. 30(Suppl. 3), S233–S242 (2000). [DOI] [PubMed] [Google Scholar]
- 5. Cole ST, Brosch R, Parkhill J et al Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature 393(6685), 537–544 (1998). [DOI] [PubMed] [Google Scholar]
- 6. Desel C, Dorhoi A, Bandermann S, Grode L, Eisele B, Kaufmann SH. Recombinant BCG ΔureC hly+ induces superior protection over parental BCG by stimulating a balanced combination of type 1 and type 17 cytokine responses. J. Infect. Dis. 204(10), 1573–1584 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. McShane H, Pathan AA, Sander CR et al Recombinant modified vaccinia virus Ankara expressing antigen 85A boosts BCG-primed and naturally acquired antimycobacterial immunity in humans. Nat. Med. 10(11), 1240–1244 (2004). [DOI] [PubMed] [Google Scholar]; • The first clinical trial of BCG-prime modified vaccinia virus Ankara 85A-boost, demonstrating safety and significant antigen-specific T-cellular immunogenicity in healthy adult subjects in the UK.
- 8. Minassian AM, Rowland R, Beveridge NE et al A Phase I study evaluating the safety and immunogenicity of MVA85A, a candidate TB vaccine, in HIV-infected adults. BMJ Open 1(2), e000223 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Scriba TJ, Tameris M, Smit E et al A Phase IIa trial of the new tuberculosis vaccine, MVA85A, in HIV- and/or Mycobacterium tuberculosis-infected adults. Am. J. Respir. Crit. Care Med. 185(7), 769–778 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Beveridge NE, Price DA, Casazza JP et al Immunisation with BCG and recombinant MVA85A induces long-lasting, polyfunctional Mycobacterium tuberculosis-specific CD4+ memory T lymphocyte populations. Eur. J. Immunol. 37(11), 3089–3100 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Abbink P, Lemckert AA, Ewald BA et al Comparative seroprevalence and immunogenicity of six rare serotype recombinant adenovirus vaccine vectors from subgroups B and D. J. Virol. 81(9), 4654–4663 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sheehy SH, Duncan CJ, Elias SC et al ChAd63-MVA-vectored blood-stage malaria vaccines targeting MSP1 and AMA1: assessment of efficacy against mosquito bite challenge in humans. Mol. Ther. 20(12), 2355–2368 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Aagaard C, Hoang T, Dietrich J et al A multistage tuberculosis vaccine that confers efficient protection before and after exposure. Nat. Med. 17(2), 189–194 (2011). [DOI] [PubMed] [Google Scholar]
- 14. Leroux-Roels I, Forgus S, De Boever F et al; The M72 Study Group Improved CD4(+) T cell responses to Mycobacterium tuberculosis in PPD-negative adults by M72/AS01 as compared to the M72/AS02 and Mtb72F/AS02 tuberculosis candidate vaccine formulations: a randomized trial. Vaccine doi:10.1016/j.vaccine.2012.05.035 (2012). (Epub ahead of print). [DOI] [PubMed] [Google Scholar]
- 15. Flatz L, Hegazy AN, Bergthaler A et al Development of replication-defective lymphocytic choriomeningitis virus vectors for the induction of potent CD8+ T cell immunity. Nat. Med. 16(3), 339–345 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sweeney KA, Dao DN, Goldberg MF et al A recombinant Mycobacterium smegmatis induces potent bactericidal immunity against Mycobacterium tuberculosis Nat. Med. 17(10), 1261–1268 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]; • Preclinical study demonstrating the ability of a recombinant Mycobacterium smegmatis strain expressing the ESX3 gene from M. tuberculosis known as IKEPLUS to protect mice against M. tuberculosis challenge.
- 17. Martin C, Williams A, Hernandez-Pando R et al The live Mycobacterium tuberculosis phoP mutant strain is more attenuated than BCG and confers protective immunity against tuberculosis in mice and guinea pigs. Vaccine 24(17), 3408–3419 (2006). [DOI] [PubMed] [Google Scholar]
- 18. Verreck FA, Vervenne RA, Kondova I et al MVA.85A boosting of BCG and an attenuated, phoP deficient M. tuberculosis vaccine both show protective efficacy against tuberculosis in rhesus macaques. PLoS ONE 4(4), e5264 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Williams A, Hatch GJ, Clark SO et al Evaluation of vaccines in the EU TB Vaccine Cluster using a guinea pig aerosol infection model of tuberculosis. Tuberculosis (Edinb.) 85(1-2), 29–38 (2005). [DOI] [PubMed] [Google Scholar]
- 20. Walker R. Aeras Global Tuberculosis Vaccine Foundation. A Product Development Partnership for New TB Vaccines. Presented at: HIV Vaccines Trial Network Conference. Washington, DC, USA, 6 May 2010. [Google Scholar]
- 21. Kupferschmidt K. Infectious disease. Taking a new shot at a TB vaccine. Science 334(6062), 1488–1490 (2011). [DOI] [PubMed] [Google Scholar]
- 22. Beresford B, Sadoff JC. Update on research and development pipeline: tuberculosis vaccines. Clin. Infect. Dis. 50(Suppl. 3), S178–S183 (2010). [DOI] [PubMed] [Google Scholar]
- 23. Miller E, Salisbury D, Ramsay M. Planning, registration, and implementation of an immunisation campaign against meningococcal serogroup C disease in the UK: a success story. Vaccine 20(Suppl. 1), S58–S67 (2001). [DOI] [PubMed] [Google Scholar]
- 24. Chiang CY, Riley LW. Exogenous reinfection in tuberculosis. Lancet Infect. Dis. 5(10), 629–636 (2005). [DOI] [PubMed] [Google Scholar]
- 25. Verver S, Warren RM, Beyers N et al Rate of reinfection tuberculosis after successful treatment is higher than rate of new tuberculosis. Am. J. Respir. Crit. Care Med. 171(12), 1430–1435 (2005). [DOI] [PubMed] [Google Scholar]
- 26. Dharmadhikari AS, Basaraba RJ, Van Der Walt ML et al Natural infection of guinea pigs exposed to patients with highly drug-resistant tuberculosis. Tuberculosis (Edinb.) 91(4), 329–338 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]; • Innovative preclinical approach aiming to model human TB disease patterns by studying a cohort of guinea pigs exposed to exhaust air from a hospital TB inpatient ward.
- 27. Henao-Tamayo M, Obregón-Henao A, Ordway DJ, Shang S, Duncan CG, Orme IM. A mouse model of tuberculosis reinfection. Tuberculosis (Edinb.) 92(3), 211–217 (2012). [DOI] [PubMed] [Google Scholar]
- 28. Kagina BM, Abel B, Scriba TJ et al; other members of the South African Tuberculosis Vaccine Initiative Specific T cell frequency and cytokine expression profile do not correlate with protection against tuberculosis after bacillus Calmette-Guérin vaccination of newborns. Am. J. Respir. Crit. Care Med. 182(8), 1073–1079 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]; •• Clinical study in South African infants demonstrating no correlation between BCG-induced protection and antigen-specific T-cell cytokine expression including IFN-γ.
- 29. Pathan AA, Minassian AM, Sander CR et al Effect of vaccine dose on the safety and immunogenicity of a candidate TB vaccine, MVA85A, in BCG vaccinated UK adults. Vaccine 30(38), 5616–5624 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. van Dissel JT, Arend SM, Prins C et al Ag85B-ESAT-6 adjuvanted with IC31 promotes strong and long-lived Mycobacterium tuberculosis specific T cell responses in naïve human volunteers. Vaccine 28(20), 3571–3581 (2010). [DOI] [PubMed] [Google Scholar]
- 31. Beveridge NE, Fletcher HA, Hughes J et al A comparison of IFNγ detection methods used in tuberculosis vaccine trials. Tuberculosis (Edinb.) 88(6), 631–640 (2008). [DOI] [PubMed] [Google Scholar]
- 32. Hanekom WA, Dockrell HM, Ottenhoff TH et al Immunological outcomes of new tuberculosis vaccine trials: WHO panel recommendations. PLoS Med. 5(7), e145 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]; •• Describes the recommendations of a WHO panel with regard to harmonization of T-cell immunogenicity assays in Phase I and Phase IIa clinical trials of candidate TB vaccines.
- 33. Berry MP, Graham CM, McNab FW et al An interferon-inducible neutrophil-driven blood transcriptional signature in human tuberculosis. Nature 466(7309), 973–977 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Querec TD, Akondy RS, Lee EK et al Systems biology approach predicts immunogenicity of the yellow fever vaccine in humans. Nat. Immunol. 10(1), 116–125 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wallis RS, Palaci M, Vinhas S et al A whole blood bactericidal assay for tuberculosis. J. Infect. Dis. 183(8), 1300–1303 (2001). [DOI] [PubMed] [Google Scholar]
- 36. Worku S, Hoft DF. In vitro measurement of protective mycobacterial immunity: antigen-specific expansion of T cells capable of inhibiting intracellular growth of bacille Calmette-Guérin. Clin. Infect. Dis. 30(Suppl. 3), S257–S261 (2000). [DOI] [PubMed] [Google Scholar]
- 37. Kampmann B, Tena GN, Mzazi S, Eley B, Young DB, Levin M. Novel human in vitro system for evaluating antimycobacterial vaccines. Infect. Immun. 72(11), 6401–6407 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Silver RF, Li Q, Boom WH, Ellner JJ. Lymphocyte-dependent inhibition of growth of virulent Mycobacterium tuberculosis H37Rv within human monocytes: requirement for CD4+ T cells in purified protein derivative-positive, but not in purified protein derivative-negative subjects. J. Immunol. 160(5), 2408–2417 (1998). [PubMed] [Google Scholar]
- 39. Duncan CJ, Draper SJ. Controlled human blood stage malaria infection: current status and potential applications. Am. J. Trop. Med. Hyg. 86(4), 561–565 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lillie PJ, Berthoud TK, Powell TJ et al Preliminary assessment of the efficacy of a T-cell-based influenza vaccine, MVA-NP+M1, in humans. Clin. Infect. Dis. 55(1), 19–25 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Marwick C. Volunteers in typhoid infection study will aid future vaccine development. JAMA 279(18), 1423–1424 (1998). [DOI] [PubMed] [Google Scholar]
- 42. Gunther VJ, Putnak R, Eckels KH et al A human challenge model for dengue infection reveals a possible protective role for sustained interferon γ levels during the acute phase of illness. Vaccine 29(22), 3895–3904 (2011). [DOI] [PubMed] [Google Scholar]
- 43. Minassian AM, Satti I, Poulton ID, Meyer J, Hill AV, McShane H. A human challenge model for Mycobacterium tuberculosis using Mycobacterium bovis bacille Calmette–Guerin. J. Infect. Dis. 205(7), 1035–1042 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ota MO, Odutola AA, Owiafe PK et al Immunogenicity of the tuberculosis vaccine MVA85A is reduced by coadministration with EPI vaccines in a randomized controlled trial in Gambian infants. Sci. Transl. Med. 3(88), 88ra56 (2011). [DOI] [PubMed] [Google Scholar]
- 45. Billeskov R, Elvang TT, Andersen PL, Dietrich J. The HyVac4 subunit vaccine efficiently boosts BCG-primed anti-mycobacterial protective immunity. PLoS ONE 7(6), e39909 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ernst JD, Hanekom W, Hawn T, Kampmann B, Rengarajan J. Meeting report: The International Conference on Human Immunity to Tuberculosis. Tuberculosis (Edinb.) 92(5), 440–444 (2012). [DOI] [PubMed] [Google Scholar]
- 47. Ravishankar N, Gubbins P, Cooley RJ et al Financing of global health: tracking development assistance for health from 1990 to 2007. Lancet 373(9681), 2113–2124 (2009). [DOI] [PubMed] [Google Scholar]
- 48. Raviglione M, Zumla A, Marais B, Horton R, Motsoaledi A. A sustainable agenda for tuberculosis control and research. Lancet 379(9821), 1077–1078 (2012). [DOI] [PubMed] [Google Scholar]
Websites
- 101. WHO Stop TB Partnership Tuberculosis Vaccine Candidates 2011. www.stoptb.org/wg/new_vaccines/assets/documents/TB%20Vaccine%20Pipeline_rAug%202012.pdf (Accessed October 2012)
- 102. WHO Global Tuberculosis Control 2011. www.who.int/tb/publications/global_report/2011/gtbr11_full.pdf (Accessed October 2012)
- 103. WHO Millennium Development Goals. www.who.int/topics/millennium_development_goals/diseases/en/index.html (Accessed October 2012)
- 104. WHO The Global Plan to Stop TB. www.stoptb.org/assets/documents/global/plan/TB_GlobalPlanToStopTB2011-2015.pdf (Accessed October 2012)
- 105. ClinicalTrials.gov www.clinicaltrials.gov (Accessed October 2012)
- 106. The Decade of Vaccines Collaboration www.dovcollaboration.org/about-us (Accessed October 2012)