

Summary
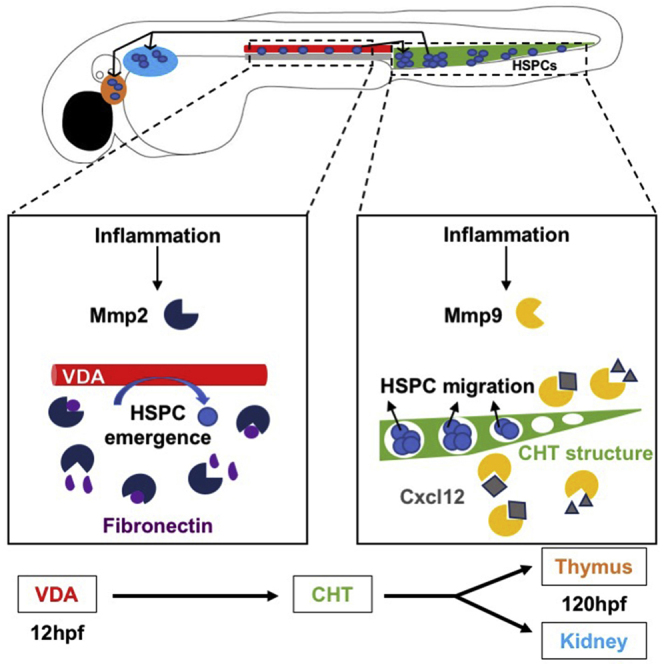
Hematopoietic stem/progenitor cells (HSPCs) are formed during ontogeny from hemogenic endothelium in the ventral wall of the dorsal aorta (VDA). Critically, the cellular mechanism(s) allowing HSPC egress and migration to secondary niches are incompletely understood. Matrix metalloproteinases (MMPs) are inflammation-responsive proteins that regulate extracellular matrix (ECM) remodeling, cellular interactions, and signaling. Here, inhibition of vascular-associated Mmp2 function caused accumulation of fibronectin-rich ECM, retention of runx1/cmyb+ HSPCs in the VDA, and delayed caudal hematopoietic tissue (CHT) colonization; these defects were absent in fibronectin mutants, indicating that Mmp2 facilitates endothelial-to-hematopoietic transition via ECM remodeling. In contrast, Mmp9 was dispensable for HSPC budding, being instead required for proper colonization of secondary niches. Significantly, these migration defects were mimicked by overexpression and blocked by knockdown of C-X-C motif chemokine-12 (cxcl12), suggesting that Mmp9 controls CHT homeostasis through chemokine regulation. Our findings indicate Mmp2 and Mmp9 play distinct but complementary roles in developmental HSPC production and migration.
Keywords: HSC/HSPC, Mmp2, Mmp9, inflammation, PGE2, zebrafish, Cxcl12, fibronectin, extracellular matrix, zebrafish
Graphical Abstract
Highlights
-
•
Mmp2 function is required for completion of EHT and HSPC egress from the VDA
-
•
Mmp2 controls EHT via remodeling fibronectin-rich extracellular matrix in the VDA
-
•
Mmp9 activity is necessary for migration between secondary hematopoietic niches
-
•
Mmp9 influences Cxcl12 signaling to control CHT vascularity and HSPC colonization
Theodore and colleagues illustrate essential yet distinct roles for inflammation-induced matrix metalloproteinase 2 (Mmp2) and Mmp9 in developmental hematopoiesis. Mmp2 facilitates hematopoietic stem and progenitor cell (HSPC) budding and egress from the ventral dorsal aorta by regulating extracellular matrix fibronectin accumulation; in contrast, Mmp9 modifies Cxcl12/Cxcr4 signaling to enable HSPC colonization of, localization within, and migration between, secondary hematopoietic niches.
Introduction
Hematopoietic stem cells (HSCs) are defined by their lifelong ability to self-renew and generate each blood cell type. In vertebrates, HSCs first emerge from the ventral wall of the dorsal aorta (VDA) in the aorta-gonad-mesonephros (AGM) region of the developing embryo (Dzierzak and Speck, 2008). The process of de novo HSC production, termed endothelial-to-hematopoietic transition (EHT), involves the specification, budding, and egress of select hemogenic endothelial cells into circulation. As this process proceeds, hemogenic endothelial cells acquire markers of HSC identity, and once EHT is complete, begin migration to secondary niches to expand in number and differentiate (Bertrand et al., 2010, Kissa and Herbomel, 2010). Endothelial cells slated to undergo EHT are specified by sequential actions of the Hedgehog and vascular endothelial growth factor (VEGF) pathways, which in turn activate Notch (Carroll and North, 2014). Notch signaling in the VDA is responsible for runx1 induction via scl and gata2; Runx1 function is required for EHT and HSC production (Chen et al., 2009, Kissa and Herbomel, 2010, North et al., 1999). While factors necessary for VDA specification and EHT are increasingly defined (Carroll and North, 2014), the mechanisms by which hemogenic endothelium is structurally remodeled to allow hematopoietic stem and progenitor cell (HSPC) budding, egress, and subsequent migration to intermediate and adult niches are incompletely understood.
In the mouse, HSCs are present in the AGM, umbilical and vitelline arteries by embryonic day 10.5 (E10.5), followed shortly by the placenta. Newly formed HSCs migrate via the circulation to the fetal liver (FL), a transient niche, to mature and proliferate. After leaving the FL, HSCs migrate to sites of adult hematopoiesis, seeding the thymus at E11.5, the spleen at E12.5, and finally the bone marrow (BM) by E15 (Dzierzak and Speck, 2008). In zebrafish embryos, de novo specified HSCs begin to bud from the VDA by 28 hours post fertilization (hpf), then migrate to the caudal hematopoietic tissue (CHT; mammalian fetal liver equivalent) from 36 to 72 hpf to proliferate and differentiate into committed progenitors. Shortly thereafter, HSPCs leave the CHT and populate the adult sites of hematopoiesis, the thymus and kidney marrow (Carroll and North, 2014). This general pattern of sequential migrations between hematopoietic niches is evolutionarily conserved (Dzierzak and Speck, 2008) and presumed to be essential for proper HSPC development and function. We and others have determined that sterile inflammatory signaling regulates HSPC production in vertebrate embryos (Espín-Palazón et al., 2014, He et al., 2015, Li et al., 2014, Sawamiphak et al., 2014). Similarly, we have previously shown that the well-known inflammatory factor prostaglandin E2 (PGE2) increases HSC numbers (North et al., 2007) and affects CXCL12/CXCR4-mediated HSPC homing to the adult niche across species (Goessling et al., 2011). However, the detailed mechanism(s) by which inflammatory signals influence HSPC development, including EHT and migratory behavior, are largely unknown.
The extracellular matrix (ECM) is a network of secreted proteins that function in concert to structurally support cells and regulate cellular processes (Rozario and DeSimone, 2010). The ECM is an important component of the BM hematopoietic microenvironment, where it can sequester cytokines and regulate their downstream signaling (Davis and Senger, 2005); developmental niches such as the AGM and fetal liver are also ECM rich (Marshall et al., 1999). ECM structure is highly dynamic and actively remodeled by degrading proteases known as matrix metalloproteinases (MMPs) (Heissig et al., 2003). MMPs are well-characterized inflammatory mediators (Parks et al., 2004) that perform a variety of distinct and overlapping biological functions depending on their spatiotemporal localization. In particular, MMP2 and MMP9, which represent the gelatinase family, have been implicated in angiogenic ECM remodeling downstream of Notch and VEGF signaling (Funahashi et al., 2011). As Notch and VEGF are essential for HSPC formation, and given the connections between inflammatory activity and HSC production, we sought to investigate the functional requirement(s) for gelatinase activity during embryonic HSPC development.
Here, we demonstrate that Mmp2 and Mmp9 are induced in response to inflammatory signals to perform necessary, but spatially and temporally distinct, functions during definitive hematopoiesis. Mmp2 facilitates the completion of EHT, including egress from the VDA and HSPC migration from the AGM to the CHT, by modulating levels of fibronectin-rich ECM. In contrast, Mmp9 influences embryonic Cxcl12 activity, affecting the physical structure of the CHT niche as well as HSPC colonization and retention therein, to affect subsequent migration to adult niches. These in vivo studies highlight the essential role of Mmp2 and Mmp9 in regulating HSPC production and migration to establish lifelong hematopoiesis during embryonic development.
Results
Gelatinase Expression Is Regulated by Sterile Inflammatory Signaling
We previously reported that exposure of BM or umbilical cord blood to PGE2 induced expression of a number of inflammatory intermediates, including matrix remodeling enzymes of the MMP family (Goessling et al., 2009). As ECM remodeling is essential in the BM microenvironment (Davis and Senger, 2005) and inflammation influences HSC production, we sought to determine whether inflammation-induced Mmp activity affected the onset or progression of embryonic HSPC development. Consistent with prior observations (Esain et al., 2015), exposure to CAY10397 (10 μM, 12–36 hpf), a selective 15-hydroxyprostaglandin dehydrogenase (15-PGDH) inhibitor that stabilizes endogenous PGE2 levels, enhanced expression of the conserved HSPC markers runx1 and cmyb by whole-mount in situ hybridization (WISH) (Figures S1A and S1B). In addition to a potent effect on runx1 (p < 0.01), CAY10397 also significantly increased the expression of mmp2 (p < 0.05) and mmp9 (p < 0.01) by qPCR at 42 hpf (Figure 1A). This effect was confirmed by WISH, whereby CAY10397 notably enhanced mmp2 and mmp9 expression over controls at 36 hpf (Figure 1B). In contrast, indomethacin (10 μM), a pan-cyclooxygenase inhibitor that inhibits PGE2 production and HSPC formation (North et al., 2007), decreased both mmp2 and mmp9 expression by qPCR and WISH (Figures 1A and 1B). Together, these data indicate that endogenous PGE2-associated basal inflammatory activity affects embryonic gelatinase expression.
Figure 1.

Inflammatory Activity Regulates Mmp2 and Mmp9 Expression in the VDA
(A) runx1, mmp2, and mmp9 expression was enhanced following exposure (36–42 hpf) to CAY10397 (10 μM) but decreased by indomethacin (10 μM), as determined by qPCR (∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001; 40 pooled embryos/condition × 3 replicates).
(B) CAY10397 (12–36 hpf) increased mmp2 and mmp9 expression, while indomethacin (Indo) diminished expression by WISH (n ≥ 20/condition).
(C) WISH time course (18–72 hpf) for mmp2 and mmp9 expression (n value as in B).
Arrowheads mark mmp9+ cells; brackets mark dorsal/ventral boundaries of the VDA and CV. Scale bars, 100 μm.
To investigate the cellular associations of mmp2 and mmp9 expression with developmental hematopoiesis, we performed time-course WISH analysis (18–72 hpf). Prominent mmp2 expression was seen in the trunk, including the vasculature and adjacent mesenchyme, at all time points (Figure 1C), consistent with prior reports (Detry et al., 2012). qPCR analysis of fluorescence-activated cell sorting (FACS)-isolated fractions from Tg(kdrl:dsred;cmyb:gfp) embryos confirmed mmp2 expression in the Flk1:dsRed+cMyb:GFP− vasculature as well as double-negative fraction (Figure S1C). In contrast, by WISH, mmp9 expression was punctate in appearance, primarily associated with cells within the vasculature, and noticeably enriched in the tail during the developmental windows examined (Figure 1C). As qPCR of the sorted fractions showed broad mmp9 expression (Figure S1C) and HSPCs are not robustly present in the circulation at 30 hpf, RNA sequencing (RNA-seq) analysis was used to further clarify the main cellular source of mmp9 expression. Primitive macrophages and neutrophils were isolated from Tg(mpeg1:mcherry;mpx:gfp) embryos by FACS at 72 hpf (1,000 pooled embryos, >50,000 cells/fraction). While low mmp9 expression was detected in Mpeg1+ macrophages, consistent with prior observations (Travnickova et al., 2015), mmp9 levels were nearly 200-fold higher in Mpx+ neutrophils (Figure S1D). To confirm these results, we employed a targeted ablation strategy using tissue-specific expression of nitroreductase (NfsB) (Curado et al., 2008): Tg(mpeg1:gal4;uas:nfsb-mcherry) (macrophage) or Tg(mpx:gal4;uas:nfsb-mcherry) (neutrophil) embryos were exposed to metronidazole (Mtz; 24–48 hpf), then assayed for mmp9 expression by WISH. Loss of Mpeg1+ macrophages had no visible impact on mmp9 levels (Figures S1E and S1F); in contrast, ablation of Mpx+ neutrophils dramatically reduced mmp9 in the majority of embryos examined. Together, these data indicate that inflammatory-responsive Mmp2 and Mmp9 are present in discrete cellular subsets in the hematopoietic niche during embryonic HSPC production.
Mmp2 Inhibition Alters HSPC Egress from the VDA
To assess the role of gelatinase activity in HSPC specification and budding, we exposed zebrafish embryos to pan-gelatinase inhibitors prinomastat (AG-3340) and SB-3CT from 12 to 36 hpf. While later exposure was tolerated, SB-3CT was toxic at published doses (Travnickova et al., 2015) during the earliest stages of HSPC development (Figures S2A–S2H), precluding evaluation. In contrast, compared with controls, prinomastat (20 μM) treatment altered expression of runx1/cmyb in the AGM at 36 hpf (Figures 2A and 2B); however, the Flk1:GFP+ trunk vasculature (Figure 2C) and ephrinb2+ VDA (Figure S2I), appeared structurally and developmentally normal. Prinomastat exposure changed the normal “beads on a string” pattern, consisting of a continuous row of flattened runx1+ hemogenic endothelial cells and occasional newly formed cmyb+ buds of 1–2 cells, to a disrupted and clumped arrangement (Figures 2A and 2B). To determine which gelatinase contributed to this phenotype, we exposed embryos to either the selective MMP2 inhibitor ARP-101 (ARP; 10 μM) or the MMP9 inhibitor MMP9-I (5 μM). ARP treatment (12–36 hpf) phenocopied the effect of prinomastat on runx1/cmyb patterning, with expression gaps and clumps of HSPCs in the VDA (Figures 2A and 2B), but normal vessel patterning (Figures 2C and S2I). In contrast, MMP9-I had no effect on the AGM at this stage of development. Importantly, neither prinomastat nor ARP altered total Flk1:dsRed+/cMyb:GFP+ HSPC number by FACS (Figure 2D), nor was there any effect on cell death in the AGM compared with controls by acridine orange analysis at 36 hpf (Figures S2J and S2K).
Figure 2.

Inhibition of Mmp2, but Not Mmp9, Function Affects EHT in the VDA
(A) Exposure to prinomastat (20 μM) or ARP-101 (10 μM; 12–36 hpf) caused abnormal runx1/cmyb patterning in the VDA, while MMP9-I (5 μM) had no effect.
(B) Qualitative phenotypic distribution of embryos from (A) scored with normal or abnormal runx1/cmyb expression (n ≥ 20/condition).
(C) In vivo imaging of Flk1:GFP at 36 hpf indicated that MMP inhibitor (12–36 hpf) treatment did not affect physical vasculature structure (n ≥ 5 embryos/condition).
(D) FACS analysis of double-positive HSPCs in Tg(kdrl:dsred/cmyb:gfp) embryos showed no difference in HSPCs after MMP inhibitor exposure (12–36 hpf; 5 embryos/sample, ≥3 replicates/condition).
(E) MO knockdown of mmp2 or mmp9 phenocopied effects of chemical inhibition on runx1/cmyb WISH at 36 hpf.
(F) Phenotypic distribution of embryos from (E) scored for runx1/cmyb expression (as in A) in the AGM (n value as in B).
(G) MO knockdown of mmp2 or mmp9 had no impact on Flk1:GFP+ endothelium (n value as in C).
(H) FACS analysis of Flk1:dsRed+/cMyb:GFP+ HSPCs showed no significant difference between mmp2 or mmp9 morphants and controls at 36 hpf (n value as in D).
Arrowheads mark HSPC clusters. Error bars denote mean ± SD. Scale bars, 100 μm.
Morpholino (MO) knockdown confirmed the chemical inhibition experiments and highlighted compound specificity: mmp2 morphants displayed altered runx1/cmyb expression (Figures 2E and 2F), similar to ARP treatment, while mmp9 morphants had no VDA phenotype at 36 hpf. Again, neither mmp2 nor mmp9 knockdown significantly affected overall vascular structure (Figure 2G) or total Flk1:dsRed+/cMyb:GFP+ HSPC numbers (Figure 2H). Together, these data indicate that gelatinase function is not essential for gross vascular development or specification, but suggest that Mmp2 modifies AGM HSPC emergence.
Mmp2 Inhibition Leads to Abnormal HSPC Accumulation in the AGM
To further characterize the cellular defect caused by Mmp2 inhibition, we employed several transgenic HSPC reporter lines. Analysis of Tg(runx1P2:GFP) (Lam et al., 2010) embryos following ARP exposure (12–36 hpf) mirrored the WISH results (Figure 3A). Evaluation of a novel HSPC line, Tg(runx1+23(144−378):egfp) (referred to hereafter as Runx1+144) (Figures S3A and S3B), which exhibits comparatively higher early AGM fluorescence intensity (Figure S3C) allowing longitudinal evaluation from embryonic specification to adulthood (Figure S3D), likewise showed altered patterning with ARP exposure (Figures 3B and 3C) and no change in total Runx1+144:GFP+/Flk1:dsRed+ HSPCs (Figure 3D). Confocal imaging revealed that Runx1+144+/Flk1+ HSPCs in the VDA at 36 hpf primarily exist as singlets or doublets (Figures 3E and 3F). Occasional aggregates consisting of more than three HSPCs were observed in controls; in contrast, following ARP exposure, a significant increase was observed in both the number of Runx1+144+/Flk1+ aggregates per embryo (p < 0.05; Figure 3F) and the total cells in each cluster (p < 0.05; Figure 3G).
Figure 3.

Mmp2 Inhibition Causes Abnormal HSPC Accumulation in the VDA
(A) In vivo imaging for Runx1:GFP confirmed altered HSPC development in the VDA with Prinomastat (20 μM) and ARP (10 μM) (12–36 hpf; n ≥ 20 embryos/condition).
(B) In vivo imaging for Runx1+144:GFP phenocopied effects seen in 3A.
(C) Qualitative phenotypic distribution of embryos from (B) scored with normal or abnormal Runx1+144:GFP expression in the VDA at 36 hpf (n value as in A).
(D) FACS analysis of Runx1+144:GFP+/Flk:dsRed+ HSPCs showed no significant difference with MMP inhibitor exposure (12–36 hpf; 5 embryos/sample, ≥3 replicates/condition).
(E) Representative confocal images of Tg(runx1+23(144−378):egfp/kdrl:dsred) embryos, showing: (top) double-positive HSPC clusters in the VDA with ARP treatment (12–54 hpf) compared with control; (bottom) high magnification of a select cluster (n > 10 embryos/condition).
(F) Absolute counts of Runx1+144+/Flk+ HSPC clusters (of >3 cells) in the VDA in embryos from (E) showed a significant increase in total clusters after ARP treatment (n > 10 embryos/condition; ∗p < 0.05).
(G) Absolute counts of Runx1+144+/Flk1+ HSPC clusters (of >3 cells) in the VDA in embryos from (E) showed a significant increase in cells per cluster with ARP treatment (n > 10 embryos/condition; ∗p < 0.05).
(H) Still images from the time-lapse analysis (2:45–3:10 min time stamp; see Movies S3 and S4) showed altered budding and egress of Runx1+144+Flk1+ HSPCs in the presence of ARP exposure (12–54 hpf; 2 replicates/condition) compared with controls.
Arrows mark HSPC clusters; brackets mark dorsal/ventral boundaries of the VDA and CV. Error bars denote mean ± SD. Scale bars, 100 μm (A and B) and 50 μm (E and H).
Time-lapse imaging (42–54 hpf) confirmed that Runx1+144+ cells were specified, but failed to complete EHT and/or egress from the VDA without Mmp2 activity. Unlike loss of runx1 (Kissa and Herbomel, 2010), emerging HSPCs did not “explode” following initiation of EHT; rather, instead of budding, detaching, and immediately entering circulation as seen in controls (Movie S1), in the absence of Mmp2 activity, HSPCs accumulated at discrete intervals throughout the VDA as clusters of increasing size (Movie S2). High magnification of individual clusters from replicate comparisons (Figure 3H) showed newly specified HSPCs actively formed protrusions, but were restricted in their mobility within the subaortic space with loss of Mmp2 function (Movies S3 and S4). To confirm whether expansion of individual clusters was primarily driven by accumulation of HSPCs, we examined proliferation using the cell-cycle reporter line, Tg(EF1:mAG-zGEM(1/100)) (Sugiyama et al., 2009), crossed to Tg(kdrl:dsred): while Flk1+ cells in the VDA actively proliferate at 36 hpf, no differences were seen between ARP-treated embryos and controls (Figures S3E and S3F). Together, these findings suggest that with reduced Mmp2 function, HSPCs cannot properly exit the AGM, leading to the formation of expanded hematopoietic clusters.
HSPC Egress from the VDA Is Inhibited by ECM Accumulation in the Absence of Mmp2
In the adult vasculature, MMP2 proteolytically digests ECM proteins, such as gelatin, collagen, and fibronectin, to allow cellular movements such as extravasation (Heissig et al., 2003, Parks et al., 2004). Confocal imaging confirmed that large trunk vessels are lined with fibronectin-rich ECM during the window of EHT (Figure 4A). Furthermore, fibronectin+ ECM was found to envelop rounded cells embedded within the VDA, which could be readily identified as Cd41:GFP+ HSPCs by 48 hpf (Figure 4B). Given these observations, we sought to determine whether the Mmp2-associated HSPC budding defect was correlated with ECM accumulation in the hemogenic niche. Indeed, ARP exposure (12–36 hpf) resulted in enhanced fibronectin staining in the VDA compared with controls (Figure 4C). Similar results were obtained with the pan-gelatinase inhibitor SB-3CT (32–48 hpf) (Figure S4A). Of note, fibronectin accumulation was particularly evident in the extravascular space between artery and vein, where HSPCs normally enter circulation, as confirmed by cross-section analysis (Figure S4B), consistent with their inability to properly exit the AGM region.
Figure 4.

Mmp2 Facilitates HSPC Budding and Migration via Extracellular Matrix Digestion
(A) Confocal images show fibronectin-rich ECM in the VDA (31 hpf): α-fibronectin antibody, green; DAPI (nuclei) staining, blue. Arrows point to cells between the dorsal aorta (DA) and posterior cardinal vein (PCV).
(B) High-magnification imaging of Tg(itga2b:gfp) embryos at 48 hpf shows Cd41:GFP+ HSPCs embedded in fibronectin-rich ECM between the DA and PCV.
(C) ARP (10 μM) treatment (12–36 hpf) increased fibronectin staining in the AGM (right) compared with controls at 36 hpf (left) (n ≥ 10 embryos/condition). Arrows mark areas of high fibronectin staining.
(D) ARP exposure (12–72 hpf) caused sustained cmyb AGM expression in WT siblings, not in fibronectin (fn1−/−) mutants at 72 hpf. Brackets mark anterior/posterior extent of AGM region.
(E) Qualitative phenotypic distribution of embryos from (D) scored for presence or absence of cmyb in the AGM at 72 hpf (n ≥ 20/condition).
WT, wild-type. Scale bars, 20 μm (A), 10 μm (B), 30 μm (C), and 100 μm (D).
To further investigate whether ECM remodeling by Mmp2 was necessary to complete EHT and then egress from the VDA and intravascular space into circulation, we examined fibronectin (fn1) mutant zebrafish (Trinh and Stainier, 2004), which lack proper ECM organization. In both wild-type siblings and fn1−/− mutants, cmyb expression was localized primarily within the CHT by 72 hpf; significantly, whereas Mmp2 inhibition by ARP resulted in maintenance of cmyb+ HSPCs in the VDA at 72 hpf, this effect was not observed in fn1−/− embryos (Figures 4D and 4E). Together, these findings imply that Mmp2 promotes remodeling of fibronectin-rich ECM in the VDA to allow HSPC egress from the AGM and subsequent migration to secondary sites of hematopoiesis.
Mmp2 and Mmp9 Affect HSPC Colonization of Secondary Niches
To explore whether HSPCs exhibit delayed egress from AGM in the absence of Mmp2 activity, we further examined colonization of the CHT. In addition to elevated expression of cmyb in the VDA by WISH, exposure to ARP (12–72 hpf) caused a concomitant decrease in expression the CHT at 72 hpf (Figures 5A and 5C). This effect was phenocopied by WISH for cmyb in both mmp2 morphants (Figures 5B and 5C) and mutants (Figures S5A and S5B) at 72 hpf (penetrance of retention phenotype in mmp2+/− incross = 28.0%). To further quantify the impact on CHT migration and colonization, we physically separated ARP-treated embryos into trunk versus tail regions, containing AGM and CHT, respectively, using the yolk sac extension (YSE) as a reference point, then used FACS to analyze Flk1:dsRed+cMyb:GFP+ HSPC content. Consistent with the WISH data, Mmp2 inhibition increased the ratio of HSPCs in anterior (trunk/AGM) versus posterior (tail/CHT) fractions (Figure 5D; p < 0.05) compared with controls, implying that Mmp2 is necessary for HSPCs to transit from the VDA and colonize the CHT.
Figure 5.

Gelatinases Play Distinct Roles in HSPC Colonization of the CHT and Thymus
(A) ARP exposure (12–72 hpf) caused abnormal cmyb expression in the AGM. Brackets mark anterior/posterior extent of AGM region.
(B) mmp2 morphants phenocopied the effect shown in (A) at 72 hpf.
(C) Qualitative phenotypic distribution of embryos from (A) and (B) scored for abnormal retention of cmyb+ HSPCs in the AGM at 72 hpf (n ≥ 20 embryos/condition).
(D) FACS analysis of Flk1:dsRed+/cMyb:GFP+ HSPCs showed a significant enrichment in the ratio of trunk to tail HSPCs in ARP-exposed embryos at 72 hpf (10 embryo sections/sample, 5 replicates/condition; ∗p < 0.05).
(E) MMP9-I exposure (5 μM, 12–72 hpf) did not cause maintenance of cmyb AGM expression at 72 hpf.
(F) mmp9 morphants had normal levels of cmyb in the AGM at 72 hpf.
(G) Qualitative phenotypic distribution of embryos from (E) and (F) scored for cmyb+ HSPCs in the AGM at 72 hpf (n value and scoring as in C).
(H) FACS analysis of Flk1:dsRed+/cMyb:GFP+ HSPCs showed no change in the trunk-to-tail ratio of HSPCs in MMP9-I-treated embryos at 72 hpf (10 embryo sections/sample, 5 replicates/condition).
(I) ARP exposure from 12 to 120 hpf decreased thymic rag1+ expression.
(J) Qualitative phenotypic distribution of embryos from (I) scored with low, medium, or high thymic rag1+ expression at 120 hpf (n ≥ 20/condition).
(K) ImageJ measurement of rag1+ thymus area revealed a significant decrease in thymus size with Mmp2 inhibition (12–120 hpf) (n = 8/condition; ∗p < 0.05).
(L) FACS for Rag2:dsRed+ lymphoid progenitors showed a significant decrease with Mmp2 inhibition (12–120 hpf) (5 embryos/sample, >3 replicates/condition; ∗∗∗∗p < 0.0001).
(M) MMP9-I exposure from 12 to 120 hpf decreased thymic rag1+ expression.
(N) Qualitative phenotypic distribution of embryos from (M) scored for rag1 expression in the thymus at 120 hpf (n value and scoring as in J).
(O) ImageJ measurement of rag1+ thymus area revealed a significant decrease in thymus size with Mmp9 inhibition (12–120 hpf) (n = 10/condition; ∗p < 0.05).
(P) FACS for Rag2:dsRed+ lymphoid progenitors showed a significant decrease with Mmp9 inhibition (12–120 hpf) (5 embryos/sample, >3 replicates/condition; ∗p < 0.05).
Arrowheads in (I and M) mark paired thymii. Error bars denote mean ± SD. Scale bars, 100 μm.
To clarify whether increased cmyb expression in the AGM at 72 hpf reflected delayed HSPC migration or prolonged competence, we examined the effects of Mmp2 inhibition at an intermediate time point: at 48 hpf, the AGM is still competent to produce HSPCs (Carroll and North, 2014), but seeding of the CHT is actively under way. Compared with wild-type siblings, cmyb+ expression in the VDA was noticeably elevated at 48 hpf in ARP-exposed embryos (Figures S5C and S5D); notably, however, this effect was concurrent with a visible decrease in HSPCs in the CHT compared with controls, indicating that movement of HSPCs from the AGM to the CHT is slowed in the absence Mmp2 activity. In contrast, neither MMP9-I exposure (12–72 hpf) nor MO-mediated mmp9 knockdown resulted in a “lagging” HSPC phenotype in the AGM (Figures 5E–5H). Furthermore, as no difference in blood flow, as evaluated by Gata1:dsRed+ erythrocyte transit in Flk1:GFP+ vasculature, was noted (Figure S5E), these findings imply that HSPC are actively delayed in migration to the CHT in the absence of normal Mmp2 function.
To determine whether retention and subsequent colonization defects induced by Mmp2 inhibition were of lasting impact, we evaluated HSPC-derived lymphoid progenitors in the thymus. WISH analysis following ARP exposure (12–120 hpf) showed decreased expression of rag1 compared with controls (Figures 5I–5L); this effect was confirmed as significant by measurement of total rag1+ thymic area (p < 0.05; Figure 5K) and quantified by FACS in Tg(rag2:dsRed) embryos (p < 0.05; Figure 5L). Surprisingly, MMP9-I exposed embryos (12–120 hpf) showed a similar reduction in thymic colonization by WISH, ImageJ, and FACS analysis (Figures 5M–5P). This phenotypic convergence indicates that Mmp9 may have a role in HSPC development after AGM egress and prior to colonization of adult hematopoietic niches.
Mmp9 Regulates Vascular Niche Structure and Subsequent HSPC Seeding of the CHT
As Mmp9 had no role in specification or progression of AGM hematopoiesis, but modified lymphoid progenitor number at later developmental stages, we examined its impact on HSPCs in the CHT. Fluorescent imaging of Tg(-6.0itga2b:egfp) embryos after MMP9-I exposure (12–72 hpf) indicated abnormal enrichment of Cd41+ HSPCs in the CHT compared with controls (Figure S6A). This effect was confirmed by manual cell counts, revealing that Mmp9 inhibition caused a significant increase in Cd41+ HSPCs in the CHT at 72 hpf (p < 0.05; Figure S6B). In contrast, whole embryo FACS analysis determined that total Cd41:GFP+/Gata1:DsRed− HSPC number remained unchanged after MMP9-I exposure (p = 0.328; Figure S6C), implying that Mmp9 influences HSPC localization or retention in the CHT.
Comparative WISH analysis from timed exposures to MMP9-I indicated that whether treatment occurred during the full window of HSPC production and colonization (12–72 hpf), or in early (12–36 hpf) versus late (36 hpf+) stages of EHT and CHT migration, each resulted in enhanced cmyb CHT expression at 72 hpf (Figures 6A and 6B). Analysis of Runx1+144:GFP expression confirmed enrichment of HSPCs in the CHT at 72 hpf after Mmp9 inhibition compared with controls, regardless of time of incubation (Figures 6C and 6D). Intriguingly, however, a differential effect of early versus late treatment was noted in regard to HSPC localization within the CHT itself: with early treatment onset (12–72 hpf or 12–36 hpf), HSPCs were enriched in the anterior half of the CHT (blue bar in Figure 6C), closest to the YSE, compared with the posterior end (orange bar in Figure 6C). In contrast, when exposure was initiated at 36 hpf after the onset of EHT and CHT colonization, HSPCs were increased throughout the CHT more uniformly. While no circulation defects were noted, imaging of the tail vasculature revealed that early MMP9-I exposure (12–72 hpf or 12–36 hpf) produced a hypovascular niche (Figure 6E), characterized by fewer fenestrated pockets in the CHT. Abnormal colonization and distribution within the niche with early Mmp9 inhibition was quantified by counts of discrete Cd41:GFP+ HSPCs and confirmed a significant shift to the anterior (blue) versus posterior (orange) end of the CHT at 72 hpf (Figures S6A and S6D).
Figure 6.

Mmp9 Inhibition Alters the Structure and Colonization of the CHT Niche
(A) MMP9-I treatment during discrete intervals (12–72, 12–36, or 36–72 hpf) each increased CHT cmyb expression at 72 hpf by WISH compared with controls.
(B) Qualitative phenotypic distribution of embryos from (A) scored with low, medium, or high cmyb expression in the CHT (n ≥ 20/condition).
(C) MMP9-I exposure from 12–72, 12–36, or 36–72 hpf increased appearance of Runx1+144:GFP+ cells in the CHT; early treatment (12 hpf+) favored seeding of the anterior half of the CHT (blue bar) compared with the posterior (orange bar).
(D) Qualitative phenotypic distribution of embryos from (C) scored with low, medium, or high overall Runx1+144:GFP expression in the CHT at 72 hpf (n ≥ 20/condition).
(E) In vivo imaging of Flk1:dsRed+ showed hypovascularization of the CHT after early-onset (12 hpf+) MMP9-I exposure (n ≥ 10/condition), but not with late initiation.
(F) MMP9-I treatment (12–96 hpf) retains cmyb+ HSPCs in the CHT (n ≥ 20/condition).
(G) Qualitative phenotypic distribution of embryos from (F) scored for cmyb expression in the CHT at 96 hpf (n value and scoring as in A).
(H) MMP9-I exposure (12–120 hpf) diminishes cmyb+ HSPC seeding the thymus (n ≥ 20/condition). Circles (red) highlight paired thymii.
(I) Qualitative phenotypic distribution of embryos from (H) scored for cmyb expression in the thymus at 120 hpf (n value and scoring as in A).
(J) ImageJ analysis showed significant decrease in cmyb+ thymic area after MMP9-I exposure (12–120 hpf; ∗∗∗p < 0.001).
Error bars denote mean ± SD. Scale bars, 100 μm.
We next sought to determine whether increased HSPC numbers within the CHT after Mmp9 inhibition were due to altered proliferative capacity, retention of existing HSPCs, or both. To evaluate proliferation, we processed Tg(runx1+23(144−378):egfp) embryos for 5-ethyl-2′-deoxyuridine (EdU) incorporation: MMP9-I exposure (24–72 hpf) increased the appearance of HSPCs in the CHT, as expected; however, no differences were found in relative levels of EdU labeling compared with controls (Figures S6E and S6F). To confirm and further quantify this result, we performed phosphohistone H3 (pH3) analysis: despite increased numbers of Runx1+144+ HSPCs in the CHT, there was no difference in total pH3+ cells between control and MMP9-I-treated embryos (p = 0.126; Figures S6G and S6H). To test whether the increase of HSPCs in the CHT was instead due to niche retention, we examined embryos at later developmental time points. WISH analysis of the CHT at 96 hpf revealed a sustained increase in cmyb+ cells following MMP9-I treatment (12–96 hpf) (Figures 6F and 6G); this phenotype was correlated with decreased cmyb+ HSPCs seeding the thymus, as measured by ImageJ analysis (Figures 6H–6J). Together, these data indicate that Mmp9 exerts a significant time-dependent impact on both CHT vascularity and colonization as well as HSPC migration and retention, which may result from distinct or interdependent regulation.
Mmp9 Affects Colonization and Retention in the CHT via Cxcl12/Cxcr4 Signaling
C-X-C motif chemokine-12 (CXCL12), also known as stromal cell-derived factor 1 (SDF1), is an inflammatory chemokine that influences cellular behavior via interactions with its receptor CXCR4 (Raz and Mahabaleshwar, 2009). In adult BM, MMP9 degrades CXCL12 to modulate CXCL12/CXCR4 signaling and influence HSC migration (Jin et al., 2008). In the zebrafish embryo, Cxcl12 overexpression causes hypovascularization of the CHT (Torregroza et al., 2012). As Mmp9 inhibition resulted in a similar hypovascularization phenotype coinciding with abnormal HSPC localization in the CHT, an interaction with Cxcl12/Cxcr4 signaling was examined. Modified epistasis experiments indicated that MO-mediated knockdown of cxcl12a caused hypervascularization, consistent with prior reports (Torregroza et al., 2012), and also blocked the negative effect of MMP9-I on CHT structure, as determined by measurements of total vascularized CHT area (Figures 7A and 7D). Significantly, whereas cxcl12a knockdown had minimal impact on Runx1+144:GFP+ HSPC localization (Figures 7A and 7B), MMP9-I exposure-associated accumulation of Runx1+144+ HSPCs in the CHT at 72 hpf was significantly ameliorated in cxcl12a morphants as demonstrated by relative fluorescence intensity analysis (Figure 7C). To further validate this result, cxcl12a-deficient embryos (medusa) were examined (Valentin et al., 2007). Homozygous mutants were generally unhealthy by 72 hpf, precluding evaluation; however, while no colonization phenotype was seen at baseline in cxcl12a+/− embryos, reduced Cxcl12 levels partially abrogated the effect of MMP9-I (12–72 hpf) on cmyb expression in the CHT (Figures S7A and S7B).
Figure 7.

Mmp9 Affects HSPC CHT Homeostasis via Regulation of Cxcl12 Signaling
(A) Knockdown of cxcl12a in Runx1+144:GFP+/Flk1:dsRed+ embryos blocked the effect of MMP9-I exposure (12–72 hpf) on Runx1+144:GFP localization (left); MMP9-I-associated hypovascularity was also antagonized (n ≥ 10/condition; center). Merged images are on the right.
(B) Qualitative phenotypic distribution of embryos from (A) scored with low, medium, or high Runx1+144:GFP expression in the CHT at 72 hpf (n ≥ 10/condition).
(C) ImageJ analysis revealed a significant increase in Runx1+144:GFP fluorescence intensity in the CHT with MMP9-I exposure alone (mean ± SEM; two-tailed t test; ∗p < 0.05; n ≥ 10/condition), but not in cxcl12a morphants.
(D) ImageJ analysis of Flk1:dsRed at 72 hpf indicated that a significant decrease in vascularized CHT plexus area from MMP9-I exposure (mean of quadruplicate experiments ±SEM; two-tailed t test; ∗∗p < 0.01; n ≥ 10/condition) was alleviated in cxcl12a morphants.
(E) Heat-shock (37°C, 30 min at 24 hpf) overexpression of Cxcl12b enhanced cmyb expression in the CHT (left) but decreased thymic staining (right) at 72 hpf. Ovals (red) highlight paired thymii.
(F) Qualitative phenotypic distribution of embryos from (E) scored for cmyb expression in the CHT at 72 hpf (n value and scoring as in B).
(G) ImageJ analysis showed decreased cmyb+ thymic area at 72 hpf after Cxcl12b overexpression (37°C, 30 min at 24 hpf; ∗p < 0.05).
WT, wild-type. Error bars denote mean ± SD. Scale bars, 100 μm.
Increased CXCL12 levels are known to retain cells within tissues, including the BM niche (Raz and Mahabaleshwar, 2009). To correlate developmental accumulation of Cxcl12 with elevated CHT HSPC colonization, we employed Tg(hsp70l:cxcl12b-egfp) zebrafish (Li et al., 2005); high sequence homology and similar activities allow Cxcl12a and Cxcl12b to function redundantly when overexpressed (Li et al., 2004). Heat-shock induction of Cxcl12b (24–72 hpf) increased CHT cmyb expression at 72 hpf (Figures 7E and 7F). This effect was correlated with significant reduction in cmyb+ HSPC localization in the thymus (p < 0.05; Figures 7E and 7G), phenocopying the effect of Mmp9 inhibition and supporting a model in which Mmp9 regulates colonization and retention of HSPCs in secondary niches via Cxcl12/Cxcr4 modulation. In sum, our data demonstrate that Mmp2 and Mmp9 play discrete, but essential roles in the developmental production and migration of HSPCs, affecting colonization of adult sites of hematopoiesis.
Discussion
In this study, we identified distinct roles for gelatinases Mmp2 and Mmp9 in the regulation of developmental HSPC production, migration, and niche localization. We demonstrate that during zebrafish development, Mmp2 is required to remodel fibronectin-rich ECM in the VDA to facilitate HSPC egress from the AGM and subsequent migration to the CHT and thymus. In addition, we elucidate interrelated effects of Mmp9 on hematopoiesis in the CHT: via modulation of Cxcl12 activity, Mmp9 affects the structure of the vascular niche, altering HSPC localization, and promotes HSPC migration to subsequent hematopoietic niches. ECM restructuring by MMPs affects development of lungs, mammary glands, and salivary glands (Bonnans et al., 2014). Likewise, a role for MMPs in ECM remodeling and subsequent maturation occurs in the developing intestine of tadpoles (Ishizuya-Oka et al., 2000), while MMP2 in particular is required for local ECM digestion during epithelial branching (Bonnans et al., 2014). Our study reveals essential but discrete roles for Mmp2 and Mmp9 downstream of sterile inflammatory signaling during embryonic hematopoiesis, whereby each can affect HSPC migration and localization in subsequent developmental niches.
As a group, MMPs are multifunctional enzymes closely regulated by a variety of upstream signaling pathways (Parks et al., 2004). Our work specifically demonstrates a relationship between inflammatory signaling in the embryo and expression of mmp2 and mmp9. Induction of these enzymes by both endogenous and exogenous PGE2 provides further insight into the mechanisms underlying the recently described effects of inflammatory signaling on hematopoiesis (Espín-Palazón et al., 2014, He et al., 2015, Li et al., 2014, Sawamiphak et al., 2014) and suggests targets for therapeutic intervention to modulate HSPC production. Our findings, taken together with these studies, suggest that Mmp2 may be constitutively expressed downstream of endogenous signaling pathways active in the AGM, while Mmp9 is the inducible enzyme transcribed in response to external stimuli, such as inflammation, to aid HSPC expansion and/or movement in the embryo.
While our evidence points to Cxcl12 as the main modulator of embryonic HSPC localization in the CHT, the induction of Mmp2/9 by PGE2 and its potential downstream proteolytic activity imply that Mmp9 function during developmental hematopoiesis may affect inflammatory signaling on a larger scale. MMP2/9 are known to activate transforming growth factor β, a critical cytokine involved with HSPC regulation (Vaidya and Kale, 2015). Furthermore, gelatinases can work together with other MMPs to modulate cytokine gradients: for example, combinatorial MMP2/9, MMP1, and MMP3 inactivate interleukin-1β (Ito et al., 1996). Given this cooperativity, it will be interesting to investigate the relationships between MMP2/9 and the inflammatory mediators recently shown to regulate embryonic hematopoiesis.
Our findings directly interface with the role of CXCL12 (SDF1)/CXCR4 signaling in adult HSPC migration: CXCL12 levels increase in response to stress and inflammation, and influence HSPC mobilization and extravasation within the adult BM (Dar et al., 2006). As hematopoietic niches are hypoxic during HSPC specification, budding, and migration and recently shown to be under inflammatory regulation, our discovery that Mmp9-mediated modulation of Cxcl12 signaling is necessary to ensure proper HSPC maturation in the embryo illustrates functional parallels between embryonic and adult niches, despite differing cellular components. The CXCL12/CXCR4 axis has previously been therapeutically exploited to improve HSC transplantation: the CXCR4 inhibitor plerixafor (AMD3100), which prevents CXCL12 binding, is administered before BM donation to increase HSPC mobilization (Dar et al., 2006). The HSPC mobilizing agent Me6Tren also promotes HSPC migration by inducing MMP9, leading to disruption of CXCL12/CXCR4 signaling (Zhang et al., 2013). This shows the therapeutic relevance of our characterization of the relationship between chemokine signaling, MMP9, and HSPC migration, and raises the possibility that administration of CXCL12/CXCR4 modulators with known inducers of HSPC specification and expansion, such as PGE2, may ensure that HSPCs can correctly home to and seed the recipient BM niche.
Taken together, our data and the previously described roles of MMP2 and MMP9 in tissue remodeling (Giannandrea and Parks, 2014), cytokine regulation (Parks et al., 2004), and cell migration/localization (Zhang et al., 2013) suggest a model whereby these enzymes provide HSPCs with physical “permission” to leave the niches and move to other hematopoietic sites, likely affecting their maturation, expansion, or function. The wide variety of substrates cleaved by MMPs and their expression throughout embryonic development and in the adult suggests that MMP2 and MMP9 are part of a larger mechanism by which the niche provides instructive cues affecting organ development and homeostasis. Further understanding of the intersections between inflammatory signaling, ECM remodeling, and hematopoiesis could be exploited to improve in vitro and in vivo HSPC expansion efforts, as well as HSC mobilization and transplantation. Here, we find that via a combination of direct and indirect mechanisms, including ECM degradation and regulation of chemokine activity, Mmp2 and Mmp9 work to maintain HSPC equilibrium within embryonic hematopoietic niches. This work furthers our understanding of the biophysical details underlying embryonic HSPC production and function in vivo.
Experimental Procedures
Zebrafish Husbandry
Zebrafish were maintained in accordance with the Beth Israel Deaconess Medical Center and Boston Children's Hospital Institutional Animal Care and Use Committee protocols. Transgenic and mutant lines utilized are described in Supplemental Experimental Procedures. Tg(hsp70:cxcl12b-egfp) expression was induced by incubation at 37°C for 30 min.
Targeted Cell Ablation
Transgenic embryos expressing NTR exclusively in macrophages (mpeg1:gal4;uas:nfsb-mcherry) or neutrophils (mpx:gal4;uas:nfsb-mcherry) were treated with 10–12.5 mM Mtz (24–48 hpf) and fixed for WISH. As controls, transgene negative sibling embryos from the same clutch were treated with Mtz.
Chemical Exposures and WISH
Zebrafish embryos were exposed to chemical modulators in E3 fish medium in multi-well plates (see Supplemental Experimental Procedures for detailed information). Embryos were analyzed by WISH using published probes to runx1, cmyb, ephrinb2, mmp2, mmp9, and rag1, and established methods (https://zfin.org/ZFIN/Methods/ThisseProtocol.html). Images were acquired on a Zeiss Axio Imager A1 or Zeiss Discovery V8 with Axiovision LE software. WISH data were qualitatively analyzed (≥10 embryos/condition, ≥3 independent experiments) and graphically depicted as the percentage exhibiting normal versus abnormal, or high/medium/low expression compared with median profile of sibling controls.
MO Injections
MOs (Gene Tools; sequences in Supplemental Experimental Procedures) were injected at the 1-cell stage, as previously described (Cortes et al., 2016); effects were scored in comparison with matched sibling controls.
Embryo Dissociation, FACS Analysis, and Sorting
For FACS analysis, fluorescent embryos (five embryos per sample, four replicates per condition) were pooled, incubated for 1–1.5 hr at 37°C in 0.5 mg/mL Liberase (Roche), and manually dissociated. Cells were filtered through 40-μm mesh, washed in PBS, resuspended in 1× PBS, and treated with 5 nM SYTOX Red (Life Technologies) before analysis on a BD Biosciences FACSCanto II. Non-transgenic and single-color sibling controls were used for FACS gating. Data were processed using FlowJo X software (Tree Star), and statistics (two-tailed Student's t test) were run in Prism 6 (GraphPad). For FACS sorting, 1,000 embryos/condition (three replicates) were dissociated and fractionated into double-negative, Flk1+/cMyb−, and Flk1−/cMyb+ populations on a BD SORP FACSAria.
qPCR Analysis
RNA (20 pooled embryos/condition or FACS-sorted populations) was purified with an RNAqueous Total RNA isolation kit, treated with DNase-I, and used to generate cDNA using Superscript III First Strand Synthesis Supermix (Life Technologies). qRT-PCR was performed with SYBR Green PCR Master Mix (Life Technologies) on a Bio-Rad CFX384. Samples were run in triplicate with at least two biological replicate pools per condition (for primers, see Supplemental Experimental Procedures). RT-PCR Miner (http://ewindup.info/miner) was used for statistical analysis.
Immunohistochemistry
Immunohistochemistry utilized standard procedures and available antibodies (see Supplemental Experimental Procedures). Embryos, embedded in 4% agarose, were cut into 100-μm sections with a Microm HM650V vibratome (Thermo Scientific) before staining.
Confocal Microscopy and Image Analysis
Imaging was conducted using an inverted PerkinElmer Ultraview Vox or Nikon Eclipse Ti spinning disk confocal. For time-lapse microscopy (42–54 hpf), Runx1+144:GFP+/Flk1:dsRed+ embryos were embedded in 0.8%–2% low-melting agarose with tricaine (Sigma) and mounted on 35-mm center-glass dishes. Image analysis was performed using ImageJ (NIH), Volocity (PerkinElmer), or Imaris (Bitplane) software.
Cell Proliferation and Apoptosis Analysis
Runx1+144:GFP+ embryos were incubated at 72 hpf on ice for 10 min, followed by a 2-hr incubation in 400 μM EdU (4% DMSO/E3 buffer) at 28°C. Embryos were washed and fixed overnight in 4% paraformaldehyde, followed by GFP immunodetection (Millipore; 1:200). EdU labeling was performed (room temperature, 45 min) with the Click-IT Plus EdU Alexa Fluor 647 kit (Thermo Fisher) and pH3 immunostaining, as described above (1:200; Millipore). Cell death was detected by incubation (30 min) in acridine orange (5 μg/mL; Molecular Probes) in E3 embryo medium before mounting.
RNA-Seq
Double-positive Tg(mpeg1:mcherry;mpx:egfp) embryos (∼1,000) were isolated at 72 hpf, dissociated to single cells, and FACS sorted to collect >50,000 cells/population: Mpeg1:mCherry+, Mpx:EGFP+, and double-negative (remainder of the embryo). Total RNA was isolated using TRIzol LS and GenElute LPA carrier as per manufacturer’s instructions. Libraries were prepared from 50 ng of total RNA/sample as input using Ribogone (Clontech) and SMARTer Universal Low Input RNA Kit (Clontech). Sequencing was done on an Illumina Hiseq 2500.
Author Contributions
L.N.T., E.J.H., M.C., K.N., and S.Y.L. performed embryo exposures, MO injections, WISH, and fluorescence microscopy. L.N.T. and M.C. and E.J.H. conducted FACS. L.N.T. performed qPCR. E.J.H. and S.Y. analyzed RNA-seq data. E.J.H., M.L.D., M.C., S.Y.L., and J.R.P. performed immunohistochemistry and confocal microscopy. L.N.T., E.J.H., M.C., L.I.Z., and T.E.N. designed experiments, evaluated results, prepared figures and edited the manuscript. L.N.T. and T.E.N. drafted and revised the text. All authors reviewed the manuscript.
Acknowledgments
We thank: J. Gross for fn1 mutants, S. Schulte-Merker for mmp2 mutants, and G. Lieschke and S. Renshaw for the mpeg1 and mpx:gal4 lines, respectively. FACS sorting was conducted at the BIDMC/HSCI and BCH FACS cores. Confocal microscopy was done at the BCH Cellular Imaging core. This work was supported by NIH R01DK098241-01A1 (T.E.N.), R01DK098241-03S1 (T.E.N., M.C.), R01 HL04880, R01 DK53298, PO1 HL32262, P30 DK49216, U01 HL10001 and R24 DK092760 (L.I.Z.), and 1F31HL132410-01A1 (L.N.T.). E.J.H. is an HHMI Fellow of the Helen Hay Whitney Foundation. L.I.Z. is supported by the HHMI and a founder and stockholder of Fate Therapeutics, Marauder Therapeutics, and Scholar Rock.
Published: April 13, 2017
Footnotes
Supplemental Information includes Supplemental Experimental Procedures, seven figures, and four movies and can be found with this article online at http://dx.doi.org/10.1016/j.stemcr.2017.03.016.
Accession Numbers
The dataset is available at GEO: GSE93818.
Supplemental Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
References
- Bertrand J.Y., Chi N.C., Santoso B., Teng S., Stainier D.Y.R., Traver D. Haematopoietic stem cells derive directly from aortic endothelium during development. Nature. 2010;464:108–111. doi: 10.1038/nature08738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonnans C., Chou J., Werb Z. Remodelling the extracellular matrix in development and disease. Nat. Rev. Mol. Cell Biol. 2014;15:786–801. doi: 10.1038/nrm3904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll K.J., North T.E. Oceans of opportunity: exploring vertebrate hematopoiesis in zebrafish. Exp. Hematol. 2014;42:684–696. doi: 10.1016/j.exphem.2014.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen M.J., Yokomizo T., Zeigler B.M., Dzierzak E., Speck N.A. Runx1 is required for the endothelial to haematopoietic cell transition but not thereafter. Nature. 2009;457:887–891. doi: 10.1038/nature07619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortes M., Chen M.J., Stachura D.L., Liu S.Y., Kwan W., Wright F., Vo L.T., Theodore L.N., Esain V., Frost I.M. Developmental vitamin D availability impacts hematopoietic stem cell production. Cell Rep. 2016;17:458–468. doi: 10.1016/j.celrep.2016.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curado S., Stainier D.Y.R., Anderson R.M. Nitroreductase-mediated cell/tissue ablation in zebrafish: a spatially and temporally controlled ablation method with applications in developmental and regeneration studies. Nat. Protoc. 2008;3:948–954. doi: 10.1038/nprot.2008.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dar A., Kollet O., Lapidot T. Mutual, reciprocal SDF-1/CXCR4 interactions between hematopoietic and bone marrow stromal cells regulate human stem cell migration and development in NOD/SCID chimeric mice. Exp. Hematol. 2006;34:967–975. doi: 10.1016/j.exphem.2006.04.002. [DOI] [PubMed] [Google Scholar]
- Davis G.E., Senger D.R. Endothelial extracellular matrix: biosynthesis, remodeling, and functions during vascular morphogenesis and neovessel stabilization. Circ. Res. 2005;97:1093–1107. doi: 10.1161/01.RES.0000191547.64391.e3. [DOI] [PubMed] [Google Scholar]
- Detry B., Erpicum C., Paupert J., Blacher S., Maillard C., Bruyère F., Pendeville H., Remacle T., Lambert V., Balsat C. Matrix metalloproteinase-2 governs lymphatic vessel formation as an interstitial collagenase. Blood. 2012;119:5048–5056. doi: 10.1182/blood-2011-12-400267. [DOI] [PubMed] [Google Scholar]
- Dzierzak E., Speck N.A. Of lineage and legacy: the development of mammalian hematopoietic stem cells. Nat. Immunol. 2008;9:129–136. doi: 10.1038/ni1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esain V., Kwan W., Carroll K.J., Cortes M., Liu S.Y., Frechette G.M., Vedder-Sheward L.M., Nissim S., Goessling W., North T.E. Cannabinoid receptor-2 regulates embryonic hematopoietic stem cell development via PGE2 and P-selectin activity. Stem Cells. 2015;33:2596–2612. doi: 10.1002/stem.2044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espín-Palazón R., Stachura D.L., Campbell C.A., García-Moreno D., Del Cid N., Kim A.D., Candel S., Meseguer J., Mulero V., Traver D. Proinflammatory signaling regulates hematopoietic stem cell emergence. Cell. 2014;159:1070–1085. doi: 10.1016/j.cell.2014.10.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funahashi Y., Shawber C.J., Sharma A., Kanamaru E., Choi Y.K., Kitajewski J. Notch modulates VEGF action in endothelial cells by inducing Matrix Metalloprotease activity. Vasc. Cell. 2011;3:2. doi: 10.1186/2045-824X-3-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giannandrea M., Parks W.C. Diverse functions of matrix metalloproteinases during fibrosis. Dis. Model. Mech. 2014;7:193–203. doi: 10.1242/dmm.012062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goessling W., North T.E., Loewer S., Lord A.M., Lee S., Stoick-Cooper C.L., Weidinger G., Puder M., Daley G.Q., Moon R.T. Genetic interaction of PGE2 and Wnt signaling regulates developmental specification of stem cells and regeneration. Cell. 2009;136:1136–1147. doi: 10.1016/j.cell.2009.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goessling W., Allen R.S., Guan X., Jin P., Uchida N., Dovey M., Harris J.M., Metzger M.E., Bonifacino A.C., Stroncek D. Prostaglandin E2 enhances human cord blood stem cell xenotransplants and shows long-term safety in preclinical nonhuman primate transplant models. Cell Stem Cell. 2011;8:445–458. doi: 10.1016/j.stem.2011.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Q., Zhang C., Wang L., Zhang P., Ma D., Ly J., Liu F. Inflammatory signaling regulates hematopoietic stem and progenitor cell emergence in vertebrates. Blood. 2015;125:1098–1106. doi: 10.1182/blood-2014-09-601542. [DOI] [PubMed] [Google Scholar]
- Heissig B., Hattori K., Friedrich M., Rafii S., Werb Z. Angiogenesis: vascular remodeling of the extracellular matrix involves metalloproteinases. Curr. Opin. Hematol. 2003;10:136–141. doi: 10.1097/00062752-200303000-00007. [DOI] [PubMed] [Google Scholar]
- Ishizuya-Oka A., Li Q., Amano T., Damjanovski S., Ueda S., Shi Y.B. Requirement for matrix metalloproteinase stromelysin-3 in cell migration and apoptosis during tissue remodeling in Xenopus laevis. J. Cell Biol. 2000;150:1177–1188. doi: 10.1083/jcb.150.5.1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito A., Mukaiyama A., Itoh Y., Nagase H., Thogersen I.B., Enghild J.J., Sasaguri Y., Mori Y. Degradation of interleukin 1-beta by matrix metalloproteinases. J. Biol. Chem. 1996;271:14657–14660. doi: 10.1074/jbc.271.25.14657. [DOI] [PubMed] [Google Scholar]
- Jin F., Zhai Q., Qiu L., Meng H., Zou D., Wang Y., Li Q., Yu Z., Han J., Li Q. Degradation of BM SDF-1 by MMP-9: the role in G-CSF-induced hematopoietic stem/progenitor cell mobilization. Bone Marrow Transpl. 2008;42:581–588. doi: 10.1038/bmt.2008.222. [DOI] [PubMed] [Google Scholar]
- Kissa K., Herbomel P. Blood stem cells emerge from aortic endothelium by a novel type of cell transition. Nature. 2010;464:112–115. doi: 10.1038/nature08761. [DOI] [PubMed] [Google Scholar]
- Lam E.Y.N., Hall C.J., Crosier P.S., Crosier K.E., Flores M.V. Live imaging of Runx1 expression in the dorsal aorta tracks the emergence of blood progenitors from endothelial cells. Blood. 2010;116:909–914. doi: 10.1182/blood-2010-01-264382. [DOI] [PubMed] [Google Scholar]
- Li Q., Shirabe K., Kuwada J.Y. Chemokine signaling regulates sensory cell migration in zebrafish. Dev. Biol. 2004;269:123–136. doi: 10.1016/j.ydbio.2004.01.020. [DOI] [PubMed] [Google Scholar]
- Li Q., Shirabe K., Thisse C., Thisse B., Okamoto H., Masai I., Kuwada J.Y. Chemokine signaling guides axons within the retina in zebrafish. J. Neurosci. 2005;25:1711–1717. doi: 10.1523/JNEUROSCI.4393-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y., Esain V., Teng L., Xu J., Kwan W., Frost I.M., Yzaguirre A.D., Cai X., Cortes M., Maijenburg M.W. Inflammatory signaling regulates embryonic hematopoietic stem and progenitor cell production. Genes Dev. 2014;28:2597–2612. doi: 10.1101/gad.253302.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall C.J., Moore R.L., Thorogood P., Brickell P.M., Kinnon C., Thrasher A.J. Detailed characterization of the human aorta-gonad-mesonephros region reveals morphological polarity resembling a hematopoietic stromal layer. Dev. Dyn. 1999;215:139–147. doi: 10.1002/(SICI)1097-0177(199906)215:2<139::AID-DVDY6>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- North T.E., Gu T.-L., Stacy T., Wang Q., Howard L., Binder M., Marin-Padilla M., Speck N.A. Cbfa2 is required for the formation of intra-aortic hematopoietic clusters. Development. 1999;126:2563–2575. doi: 10.1242/dev.126.11.2563. [DOI] [PubMed] [Google Scholar]
- North T.E., Goessling W., Walkley C.R., Lengerke C., Kopani K.R., Lord A.M., Weber G.J., Bowman T.V., Jang I.-H., Grosser T. Prostaglandin E2 regulates vertebrate haematopoietic stem cell homeostasis. Nature. 2007;447:1007–1011. doi: 10.1038/nature05883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parks W.C., Wilson C.L., López-Boado Y.S. Matrix metalloproteinases as modulators of inflammation and innate immunity. Nat. Rev. Immunol. 2004;4:617–629. doi: 10.1038/nri1418. [DOI] [PubMed] [Google Scholar]
- Raz E., Mahabaleshwar H. Chemokine signaling in embryonic cell migration: a fisheye view. Development. 2009;136:1223–1229. doi: 10.1242/dev.022418. [DOI] [PubMed] [Google Scholar]
- Rozario T., DeSimone D.W. The extracellular matrix in development and morphogenesis: a dynamic view. Dev. Biol. 2010;341:126–140. doi: 10.1016/j.ydbio.2009.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawamiphak S., Kontarakis Z., Stainier D.Y.R. Interferon gamma signaling positively regulates hematopoietic stem cell emergence. Dev. Cell. 2014;31:640–653. doi: 10.1016/j.devcel.2014.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugiyama M., Sakaue-Sawano A., Iimura T., Fukami K., Kitaguchi T., Kawakami K., Okamoto H., Higashijima S.-I., Miyawaki A. Illuminating cell-cycle progression in the developing zebrafish embryo. Proc. Natl. Acad. Sci. USA. 2009;106:20812–20817. doi: 10.1073/pnas.0906464106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torregroza I., Holtzinger A., Mendelson K., Liu T.-C., Hla T., Evans T. Regulation of a vascular plexus by gata4 Is mediated in zebrafish through the chemokine sdf1a. PLoS One. 2012;7:e46844–13. doi: 10.1371/journal.pone.0046844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Travnickova J., Tran Chau V., Julien E., Mateos-Langerak J., Gonzalez C., Lelièvre E., Lutfalla G., Tavian M., Kissa K. Primitive macrophages control HSPC mobilization and definitive haematopoiesis. Nat. Commun. 2015;6:6227. doi: 10.1038/ncomms7227. [DOI] [PubMed] [Google Scholar]
- Trinh L.A., Stainier D.Y.R. Fibronectin regulates epithelial organization during myocardial migration in zebrafish. Dev. Cell. 2004;6:371–382. doi: 10.1016/s1534-5807(04)00063-2. [DOI] [PubMed] [Google Scholar]
- Vaidya A., Kale V.P. TGF-β signaling and its role in the regulation of hematopoietic stem cells. Syst. Synth. Biol. 2015;9:1–10. doi: 10.1007/s11693-015-9161-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valentin G., Haas P., Gilmour D. The chemokine SDF1a coordinates tissue migration through the spatially restricted activation of Cxcr7 and Cxcr4b. Curr. Biol. 2007;17:1026–1031. doi: 10.1016/j.cub.2007.05.020. [DOI] [PubMed] [Google Scholar]
- Zhang J., Ren X., Shi W., Wang S., Chen H., Zhang B., Wang Z., Zhou Y., Chen L., Zhang R. Small molecule Me6TREN mobilizes hematopoietic stem/progenitor cells by activating MMP-9 expression and disrupting SDF-1/CXCR4 axis. Blood. 2013;123:428–441. doi: 10.1182/blood-2013-04-498535. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.