

Abstract
Visualization of subcellular structures and their temporal evolution is of utmost importance to understand a vast range of biological processes. Optical microscopy is the method of choice for imaging live cells and tissues; it is minimally invasive, so processes can be observed over extended periods of time without generating artifacts due to intense light irradiation. The use of fluorescence microscopy is advantageous because biomolecules or supramolecular structures of interest can be labeled specifically with fluorophores, so the images reveal information on processes involving only the labeled molecules. The key restriction of optical microscopy is its moderate resolution, which is limited to about half the wavelength of light (∼200 nm) due to fundamental physical laws governing wave optics. Consequently, molecular processes taking place at spatial scales between 1 and 100 nm cannot be studied by regular optical microscopy. In recent years, however, a variety of super-resolution fluorescence microscopy techniques have been developed that circumvent the resolution limitation. Here, we present a brief overview of these techniques and their application to cellular biophysics.
Keywords: Fluorescence microscopy, Super-resolution, Live-cell imaging, Structured illumination, Localization microscopy, Stimulated emission depletion
Introduction
Optical microscopy has been with us for more than 400 years. Early designs of compound microscopes appeared in the late 16th century and, after slow advances during the following centuries, the design of conventional light microscopes was essentially pushed to its physical limits in the second half of the 19th century, especially as a result of the work of Ernst Abbe, Otto Schott, and Carl Zeiss in Jena, Germany. Abbe, among others, realized that there is a fundamental limitation to the resolution of optical images due to the requirement for the objective lens to capture the first-order diffraction from a structure with a particular spacing (Abbe 1873). The famous Abbe resolution law,
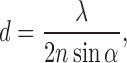 |
states that, to distinguish two objects in an image, their lateral separation must be larger than a minimal distance, d, which depends on the wavelength, λ, and the numerical aperture, n sin α, with refractive index, n, of the medium between the object and the objective lens; α is the half-angle of the objective lens aperture. This popular relation led to the widely held belief that structures smaller than 200 nm cannot be resolved by using far-field optical microscopy with visible light. Other techniques were subsequently developed for the determination of biological structures down to atomic resolution. X-ray crystallography (Friedrich et al. 1913) was applied by Kendrew (Kendrew et al. 1960) and Perutz (Perutz et al. 1968) for the determination of protein structures after solving the phase problem, and it has subsequently become a routine method to solve protein structures. Electron microscopy (Knoll and Ruska 1932) is available for high-resolution protein structure determination using two-dimensional protein crystals (Henderson and Unwin 1975) and, more recently, the structures of large biomolecular aggregates are being tackled using electron tomography (Lucic et al. 2005). Although atomic force microscopy (Binning et al. 1986) is limited to surfaces, it has also been very useful for structural studies, especially of membrane proteins (Müller et al. 2008). Even a spectroscopic method, nuclear magnetic resonance (NMR) spectroscopy, which only indirectly probes spatial scales via their effects on magnetic transitions of nuclear spins, has become a powerful tool for protein structure determination (Wüthrich 1987), which provides a wealth of information on the dynamics as well (Lange et al. 2008).
Yet, for visualization of biological structures and processes inside living organisms, light microscopy, despite its resolution limitation, is the method of choice. X-ray diffraction, NMR, and electron microscopy require special samples, mostly in-vitro preparations, and are thus not applicable to studies of living specimens at high spatial resolution. Light microscopy, by contrast, is easy to use, fast, and noninvasive, and thus ideally suited for the observation of living systems, as was already appreciated by scientists including Hooke, Malpighi, and van Leeuwenhoek in the 17th century. Later, in the 19th century, Schleiden, Schwann, Fleming, and others, laid the foundations of cell biology using optical microscopy.
Optical microscopy using fluorescence detection appears particularly powerful for cellular imaging (Pawley 2006). It relies on the reemission of light absorbed by fluorophores that are attached to structures of interest in specific ways to make them visible. Fluorescence light is delayed in time and red-shifted in wavelength and, therefore, can be very well separated from the incident and elastically scattered light, leading to excellent image contrast. Most commonly, organic dyes and fluorescent proteins are used as fluorescence markers. Organic dyes are comparatively small (1–2 nm) molecules that can be attached to cellular structures in various ways. There are a great variety of bright fluorescent dyes around that are relatively resistant to photobleaching.
In recent years, novel fluorescent biomarkers of ∼5–10 nm diameter have been developed based on nanocrystals, including metal (Zheng et al. 2007), semiconductor (Michalet et al. 2005), and nanodiamond (Fu et al. 2007) quantum dots. Although these markers have not yet found as widespread application as organic dyes, they show great promise as fluorescent markers due to their excellent brightness and photostability. Fluorescent proteins are a class of small proteins (∼3 nm) that spontaneously produce a fluorescent chromophore in their interior (Nienhaus 2008). Although the photophysical and photochemical properties of this fluorophore do not match those of synthetic dyes or nanocrystals, their key advantage is that these marker proteins are genetically encoded and, therefore, are produced by the cell itself (Shimomura 2006); no further staining steps are necessary prior to the experiment. By fusing the DNA encoding the fluorescent protein to the gene of a protein under study, the cell expresses the protein as a so-called fusion protein, which has an additional domain that renders the protein fluorescent.
Exciting new developments in light microscopy started ∼20 years ago. In addition to tremendous advances in fluorescence labeling technology, the field has enormously benefited from technical developments of key microscope components, i.e., powerful laser light sources (pulsed and cw lasers), detectors performing close to their physical limits (CCD cameras, avalanche photodiodes), optoelectronics and nanomechanical equipment for beam steering and sample positioning and, probably most importantly, computers that are capable of storing vast amounts of data and processing images in reasonable amounts of time. Since diffraction is a fundamental physical property of the wave nature of light, there is no way of literally breaking the optical resolution limit. However, researchers have introduced clever strategies that exploit the nonlinear response of fluorescent dyes to light excitation to retrieve spatial information beyond the Abbe barrier, using ingenious microscope implementations, which will be surveyed in this review.
Microscopy designs and confocal techniques
There are two basic far-field fluorescence imaging modes, wide field and confocal. In wide field epifluorescence microscopy, the excitation light impinges on the sample via the objective, which is also used to collect the reemitted fluorescence. The entire field of view is imaged with an area-sensitive detector (CCD camera). There is no discrimination of light along the optical axis; planes at all depths of the sample contribute. However, a sharp image is only formed close to the focal plane; the light emanating from the other planes produces a background blur that may obscure features in the focal plane. The researcher can resort to very thin samples (z < 2 μm), or the excitation has to occur by means of total internal reflection fluorescence (TIRF) (Thompson et al. 1981). In this microscopy technique, light is reflected off a surface to a medium of lower refractive index n. The evanescent wave in this medium decays exponentially in the axial direction and excites fluorophores only within typically 100 nm from the surface. Therefore, the technique provides excellent axial discrimination but lacks 3D capability.
Confocal microscopy is a raster scanning approach, in which a point source, by means of the microscope objective, is imaged to a point in the focal plane of the sample. The fluorescence emanating from this point is collected through a pinhole that is geometrically confocal to the source, i.e., virtually at the same position (apart from the fact that a dichroic mirror separates excitation and emission paths). Only light from the focal plane is efficiently transmitted through the pinhole, whereas light emanating from out-of-focus planes is blocked. Therefore, this method provides axial resolution (∼500–800 nm), and 3D images can be taken by scanning either the laser or the sample laterally (x-, y-directions); the z-direction is controlled by varying the distance between sample and objective. Since the point spread function (PSF, Fig. 1a), i.e., the image of a point-like object is the convolution of the PSF for excitation and emission in confocal microscopy, the method features a slightly improved lateral resolution over standard microscopy.
Fig. 1a–e.

Principle of 4Pi microscopy. a When using only a single objective, fluorescence photons can only be collected from less than one half the total solid angle (2π), and the PSF of a standard confocal microscope is extended in the axial direction. By using two objectives in an interferometric arrangement, coverage of the full solid angle (4π) is approximated more closely. An interference pattern arises with a sharp maximum in the axial direction. b The limited aperture angles of the objectives give rise to side lobes that can be removed by mathematical deconvolution. c Confocal image of membrane-stained (DiI) E. coli bacteria. Scale bar 500 nm. d Raw 4Pi image of the same specimen; the effects of the side lobes are clearly visible. e 4Pi image after deconvolution
To achieve depth discrimination, one may also resort to two-photon excitation. Fluorescence in the optical spectrum can be excited by using two infrared photons of half the energy (or twice the wavelength), provided that the spatial and temporal density of photons is high enough that the process can take place with a suitable yield. The temporal density is ensured by using pulsed laser sources, typically titanium-sapphire (Ti:Sa) lasers. Due to the quadratic dependence of the fluorescence emission on the excitation intensity, the intensity can be adjusted such that the spatial density is only high enough in the focus of the objective but not anymore in neighboring axial planes. Consequently, out-of-focus emission is attenuated, leading to optical sectioning in the axial direction (Denk et al. 1990). Additionally, the imaging depth is increased due to the lower Rayleigh scattering of infrared light, allowing for deep tissue studies (Helmchen and Denk 2002).
To further improve the axial resolution of the confocal microscope and, at the same time, to collect more of the incoherent photon emission emanating from the sample in all directions (into the entire solid angle, 4π), an interferometric arrangement of two opposing objectives is implemented in 4Pi confocal microscopy (Hell and Stelzer 1992). The two counterpropagating wavefronts are coherently superimposed at the excitation spot or at the detector or at both, resulting in an observation volume that is sharpened in the axial direction by a two-source interference pattern (Fig. 1b). Side lobes appear in the 4Pi image that can be suppressed fairly efficiently by two-photon excitation and confocal detection and finally be removed by postprocessing, i.e., using a mathematical deconvolution algorithm on the raw data. As an example, we compare confocal, 4Pi raw and 4Pi deconvoluted images of Escherichia coli bacteria in Figure 1c–e.
In 4Pi confocal microscopy, an axial resolution of ∼100 nm is typically achieved (Gugel et al. 2004; Glaschick et al. 2007). Although the lateral resolution is unchanged, structures appear more clearly because of the excellent sectioning in the axial direction. I5M also utilizes this interferometric approach, although in a wide field design (Gustafsson 1999). One of the major applications of 4Pi and I5M microscopy has been the study of subcellular organelles in 3D (Nagorni and Hell 1998; Gustafsson 1999; Egner et al. 2004; Medda et al. 2006; Ivanchenko et al. 2007). These two concepts have subsequently been combined with techniques that improve the lateral resolution so as to improve the resolution in all three dimensions, 4Pi-STED (Dyba et al. 2003) and I5S (Shao et al. 2008), as will be discussed below.
Reversible saturable optical fluorescence transitions-based super-resolution microscopy
Stimulated emission depletion (STED) microscopy (Hell and Wichmann 1994), or more generally, reversible saturable optical fluorescence transitions (RESOLFT) microscopy (Hofmann et al. 2005; Bossi et al. 2006) is based on a targeted, point-scanning approach in a raster-scanning confocal microscope. STED, the earliest realization of super-resolution microscopy (Hell and Wichmann 1994), is closely related to saturated structured illumination microscopy (SSIM, see below), in which regular illumination patterns are used instead of point-scanning (Hell 2009; Heintzmann and Gustafsson 2009). All these techniques have in common that they utilize nonlinear responses of the fluorophores to light irradiation, i.e., switching between dark and bright states.
In a STED microscope, the exciting focused spot of a confocal microscope is spatially overlayed with a depletion beam that has an annular shape in the focal plane, with zero intensity in the center. Consequently, fluorophores that do not reside close to the center are efficiently deexcited by stimulated emission of photons in the direction of the depletion beam, so they do not reach the detector. Only fluorophores near the center can thus emit spontaneous fluorescence photons (Fig. 2a). Therefore, the effective size of the excitation PSF is smaller than the usual diffraction-limited PSF, and the higher the intensity of the depletion beam, the more the fluorescence is confined to the central region (Hell 2007). The achievable resolution can be expressed as
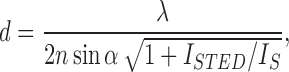 |
where λ is the wavelength, n the refractive index, α the half-angle under which the fluorescence is collected, I S the characteristic saturation intensity of the fluorophore, and I STED the intensity of the STED beam. STED is a method that relies on general physical principles of light-matter interaction; no special switching capabilities of the fluorescence markers are needed. However, the markers must show excellent photostability because the fluorophores undergo a large number of excitation-depletion cycles while both excitation and STED beams are scanned across the sample (Fig. 2b). Figure 2c depicts a STED image of vimentin filaments in PtK2 cells labeled with primary and secondary antibodies (Moneron et al. 2010). These images were recorded with continuous-wave fiber lasers, which avoids the need for temporal synchronization of the excitation and the depletion beam when using laser pulses (Willig et al. 2007). With a combination of wave plates as beam shaping devices, the excitation and depletion beams are intrinsically overlayed and do not require additional alignment (Reuss et al. 2010). STED microscopy can be further combined with 4Pi microscopy to improve the axial as well as the lateral resolution (Dyba et al. 2003). One can also shape the pattern imprinted on the wavefront of the depletion beam that extends the depletion to the axial direction, thus avoiding the complexity introduced by the use of two objectives (Wildanger et al. 2009). STED has already been successfully applied to various biological applications. For example, live cell imaging was performed by using photostable variants of fluorescent proteins (Hein et al. 2008; Nägerl et al. 2008). The movement of synaptic vesicles in living neurons could be studied at video frame rates using fast beam scanners (Westphal et al. 2008). Two-color STED has also been implemented and applied to studies of synaptic proteins in neurons at a resolution of 30 nm (Meyer et al. 2008) and the distribution of proteins in the mitochondrial membrane (Donnert et al. 2007). Additionally, since a STED microscope is confocal, fluorescence correlation spectroscopy (FCS) can be performed with a significantly reduced confocal volume (Kastrup et al. 2005). STED-FCS was applied in the direct observation of the nanoscale dynamics of membrane lipids in the plasma membrane of a living cell (Eggeling et al. 2009). The potentially unlimited resolution of STED microscopy has been confirmed by the localization of fluorescent nitrogen-vacancy centers in diamond with sub-nanometer precision (Han et al. 2009). Other principles than stimulated emission can be used to deplete the active state of the fluorescent markers around the center of the excitation beam, including pumping of a triplet state (Hell and Kroug 1995; Bretschneider et al. 2007) or exploiting a photoswitching mechanism, e.g., cis-trans isomerization of fluorophores between bright and dark states (Hell 2003; Hofmann et al. 2005; Dedecker et al. 2007). This generalization has been termed reversible saturable optical fluorescence transitions (RESOLFT) (Hofmann et al. 2005; Bossi et al. 2006).
Fig. 2a–e.

Reversible saturable optical fluorescence transition (RESOLFT) microscopy. a A fluorophore can be switched reversibly between a dark state and a bright state using light. A special illumination pattern can be applied that reduces the effective size of the point spread function (PSF), for example, by stimulated emission depletion (STED). b Sketch of the scanning procedure. The excitation beam is overlayed with the donut-shaped depletion beam with zero intensity in the center. Thus, all molecules further away from the center (red dots) are efficiently deexcited by stimulated emission before they fluoresce; only those molecules close to the center emit fluorescence (green dots). c STED image of vimentin filaments in PtK2 cells labeled with primary and secondary antibodies. d Confocal image of the region marked by the white frame in (c). e STED image of the same region, the cross-section marked by a white bar features a width of 68 nm; scale bars 1 μm. Reprinted with kind permission (Moneron et al. 2010)
Structured-illumination microscopy
Structured-illumination microscopy (SIM) utilizes the possibility of transforming local information into global information by using a Fourier transform (Gustafsson 2000). Lack of detail, i.e., resolution, in real space corresponds to the absence of high spatial frequencies in Fourier space. When illuminating fine structures in the sample (Fig. 3a) with a pattern, for example a line grid (Fig. 3b), a beat pattern (Moiré fringes) with lower spatial frequency than the original structures emerges in the resulting image (Fig. 3c), containing information about the higher spatial frequencies. Patterned illumination is used in combination with wide field imaging. To acquire an image, a set of raw images is recorded by shifting and rotating the illumination pattern so as to maximize coverage of frequency space. Based on the known illumination pattern, high resolution images can be reconstructed by using a mathematical algorithm. Because of diffraction, the periodicity and thus the spatial frequency of the illumination pattern is limited, so that the resolution gain of structured illumination in the linear mode is only a factor of two (as is the case for confocal microscopy). SIM has been extended to the third dimension by using three coherent beams (3D-SIM) (Gustafsson et al. 2008) and by combining SIM with I5M into a technique called I5S, for which an axial resolution of ∼100 nm has been reported (Shao et al. 2008). SIM works with all types of fluorophores and is easily extended to multicolor applications (Schermelleh et al. 2008). An example is shown in Fig. 3, where a conventional wide field image (Fig. 3d) is compared with a SIM image (Fig. 3e) of a primary chicken fibroblast.
Fig. 3a–e.

Structured illumination microscopy (SIM). a Specimen with structures that are too close to be resolvable by conventional optical microscopy. b A periodic pattern with a repeat distance at the diffraction limit is used for sample illumination. c In the overlay of the two patterns, the unresolvable sample structures become visible as a Moiré pattern whose spatial frequency can be resolved. d Conventional wide field image and e SIM image of a primary chicken fibroblast with immunofluorescence staining of F-actinin (green) and microtubuli (red). Scale bar 1 μm. The images were acquired with Zeiss technology ‘Superresolution mit ELYRA (http://www.zeiss.de/elyra)’
Recently, the SIM method has been extended to provide, in principle, unlimited resolution. By optically saturating fluorescence markers in the sample, a nonlinear response of the fluorescence signal to the excitation intensity is achieved. Consequently, the periodic pattern contains higher harmonics of its fundamental frequency, which is key to the resolution enhancement (Heintzmann et al. 2002). For saturated SIM (SSIM), a lateral resolution of ∼50 nm was demonstrated (Gustafsson 2005). One of the main advantages of the SIM principle is the fast image acquisition. A frame rate of up to 11 Hz has been reported (Kner et al. 2009), making SIM an excellent tool for live cell imaging experiments (Hirvonen et al. 2009).
Localization-based super-resolution microscopy
Single-molecule, localization-based super-resolution microscopy uses photoactivatable fluorochromes to disperse spatial information in the time domain. It has long been known that the position of single fluorescence emitters can be determined with a precision that significantly exceeds the width of the PSF, which governs the resolution in standard imaging. Indeed, individual steps of motor proteins along filaments were analyzed with 1 nm accuracy using single-molecule optical imaging assays (Yildiz et al. 2003; Yildiz and Selvin 2005). In conventional fluorescence microscopy, a high density of fluorescence markers is required to faithfully image fine structures, but many fluorophores emit at the same time. Their PSFs overlap in the resulting image, so they cannot be localized individually. With the advent of photoactivatable fluorophores, the emission properties of which can be controlled externally by light, we finally have tools at hand that allow us to localize individual fluorophores with high precision. We start with all markers in their inactive (off) state, and then we activate them sparsely so that in each image frame only a few appear, which then can be localized individually. By repeated acquisition (10–100 frames per second), we collect a large number of frames (102–104) that are all analyzed individually. Finally, we reconstruct an image from the measured loci in all frames. For these experiments, photoactivatable fluorescent proteins (PAFPs) have been shown to be very convenient, as they can be used as fusion markers for a particular protein under study. EosFP is a popular photoactivatable fluorescent protein, which changes its fluorescence irreversibly from green to red upon illumination with ∼400 nm light (Wiedenmann et al. 2004; Nienhaus et al. 2006; Wiedenmann and Nienhaus 2006). By appropriate adjustment of the 400-nm activating laser intensity, only a few molecules are converted to red emitters and registered in the red color channel during acquisition of a CCD image for 30–100 ms. While recording a series of images, the activating laser is adjusted such that photoconversion and photobleaching are balanced until the supply of fluorescent markers is depleted. The positions of the fluorophores in each individual image are determined by software, and the final reconstructed image is a density map depicting the distribution of emitters at a resolution in the range of a few tens of nanometers. The entire process is illustrated in Fig. 4a. The 2D localization precision, i.e., the standard error of the mean,
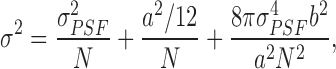 |
depends crucially on the number of photons, N, detected from each fluorophore; σ PSF is the standard deviation of the PSF, a is the camera pixel size, and b the background noise (Thompson et al. 2002). Thus, there are three contributions to the localization error: the first term represents photon statistics, the second term comes from the finite size of the detector pixels, and the last term arises from the background noise. The equation indicates that the uncertainty in the determination of the mean of the distribution decreases and approaches zero as the number of collected photons increases.
Fig. 4a–d.

Photoactivation localization microscopy (PALM). a Schematic depiction of the principle. A small, spatially well separated subset of fluorophores is photoactivated to a fluorescent state by weak illumination with light of a specific wavelength. The activated molecules are imaged by using a laser that excites their fluorescence until photobleaching occurs. Because the individual PSFs do not overlap, each fluorophore can be localized with a precision of a few ten nanometers. Up to several thousand image frames are typically acquired sequentially so that a large number of fluorophores can be localized. A final high resolution image is reconstructed from all the fluorophore locations, revealing the distribution of fluorophores within the specimen. b Total internal reflection microscopy (TIRFM) image (right) and PALM image (left) overlay of a live HeLa cell expressing an α-actinin-mIrisFP fusion construct. Scale bar 5 μm. Close-up of the region marked by the white box in (b) imaged by c PALM and d TIRFM. Scale bar 1 μm
A variety of localization-based concepts have been developed independently, such as photoactivated localization microscopy (PALM) (Betzig et al. 2006), fluorescence photoactivation localization microscopy (FPALM) (Hess et al. 2006), and stochastic optical reconstruction microscopy (STORM) (Rust et al. 2006). In general, synthetic dyes are more photostable than fluorescent proteins and, therefore, provide a higher localization precision. For example, ∼6,000 photons per molecule were reported per switching cycle using the photoswitchable fluorophore pair Cy3-Cy5 (Bates et al. 2005, 2007), whereas tdEosFP, one of the brightest PAFPs for PALM imaging, yields only ∼2,600 photons per molecule (Shroff et al. 2007), and monomeric FPs often only a few hundred photons (Fuchs et al. 2010). However, that number still suffices for a ten-fold resolution enhancement, and FPs are easier to use and ideally suited for in vivo experiments because they are expressed by the cell itself (Hess et al. 2007; Shroff et al. 2008). Synthetic dyes, by contrast, require specific buffer conditions involving high concentrations of reductants for photoswitching, and additional labeling procedures that limit their application to fixed specimens (Bates et al. 2005; Heilemann et al. 2005). PALM with independently running acquisition (PALMIRA) (Egner et al. 2007) is a simplified version of PALM that uses only a single laser line, and direct STORM (dSTORM) is a simplified version of STORM that avoids the second dye molecule for activation (Heilemann et al. 2008). Blink microscopy (BM) uses oxidizing and reducing agents for continuous switching of single oxazine dyes so as to control the off- and on-times, respectively (Vogelsang et al. 2009). By introducing a simultaneous two-color stroboscopic illumination (S-PALM), the speed of PALM imaging was increased by fast switching of variants of the fluorescent protein Dronpa (Flors et al. 2007). In ground state depletion followed by individual molecule return (GSDIM), dye molecules are shelved in the triplet state from which they return spontaneously (Fölling et al. 2008). In point accumulation for imaging in nanoscale topography (PAINT), fluorescent molecules are in their nonfluorescent off-state while diffusing freely and switch to the on-state upon binding to the structure to be imaged (Sharonov and Hochstrasser 2006). Super-resolution optical fluctuation imaging (SOFI) relies on higher-order statistical analysis of temporal fluctuations, e.g., fluorescence blinking of quantum dots, to obtain subdiffraction optical resolution in all three dimensions (Dertinger et al. 2009).
The popularity of localization-based high resolution microscopy has led to various new developments in the field of fluorescence markers, especially in the field of photoactivatable fluorophores. Different emission spectra allow for dual color PALM experiments (Bates et al. 2007; Bock et al. 2007; Schönle and Hell 2007; Shroff et al. 2007; Andresen et al. 2008; van de Linde et al. 2009). Cellular dynamics has been studied (Shroff et al. 2008), and high resolution pulse-chase experiments have also been implemented (Fuchs et al. 2010). A great advantage of these localization-based super-resolution methods is that they do not rely on any special microscopy hardware. In principle, these techniques can be implemented on any existing wide field microscope that is equipped with a fast and sensitive CCD camera as well as lasers for excitation and photoactivation of the fluorophores. Only software is required in addition, for localization of the individual fluorophores and for reconstruction of the final, high-resolution images. Frequently, a TIRF microscope is preferred for its limited depth of view, so that contrast is improved in comparison to standard wide field microscopy. This technique was applied when observing live HeLa cells expressing an α-actinin-mIrisFP fusion construct shown as a TIRF/PALM overlay in Fig. 4b.
The localization of fluorophores has been extended to the third dimension in various ways. In the astigmatism-based method (Huang et al. 2008), a cylindrical lens is introduced into the detection path to distort the shape of the PSF, and the distance to the focal plane can be extracted from the shape of the distorted PSF. Alternatively, a spatial light modulator (SLM) is placed in the Fourier plane of the imaging system (Pavani et al. 2009). Thus, every object point is convoluted with two double-helical lobes, with the angular orientation of the lobes depending on the axial location of the object above or below focus. In biplane PALM (BP-PALM), the z-position of a fluorophore between the two focal planes is computed by fitting a three-dimensional model of the PSF to the two images of this fluorophore (Juette et al. 2008). Axial sectioning can also be achieved by using two-photon excitation (Fölling et al. 2007) or interferometric designs (iPALM) (Shtengel et al. 2009). In iPALM, two opposing objectives collect photons emitted by the fluorophores in an interferometric fashion, as in 4Pi microscopy. Due to the photons’ self-interference for equal path lengths, the distance of the emitter from the focal plane can be determined with a very high precision of 10–20 nm. Additionally, twice the number of photons is collected, which further improves the lateral localization precision. With this technique, the vertical composition of focal adhesions was studied in great detail (Shtengel et al. 2009). In virtual volume super-resolution microscopy (VVSRM), a tilted mirror is positioned close to the sample to capture a side view in addition to the front view, thus yielding isotropic 3D resolution (Tang et al. 2010).
Recent years have also witnessed great improvements in PALM image analysis. Careful image analysis is required to generate excellent high resolution pictures by localization of single molecules. These calculations are computationally demanding, and until very recently, image reconstruction took much longer than data acquisition. Consequently, researchers were only able to assess the quality of their data long after the measurement. By using a triangulation algorithm (Andersson 2008), localization can be performed during data acquisition. An instant high-resolution preview is thus available, making the data acquisition procedure more intuitive and interactive (Hedde et al. 2009). By parallelizing molecule localization using modern graphics processing units (GPUs) instead of the conventional central processing units (CPUs), localization can be speeded up by orders of magnitude while maintaining a high localization precision (Smith et al. 2010; Quan et al. 2010). These developments are welcome to keep up with even faster cameras that will become available in the near future. Localization software is also available as an ImageJ plugin for an out-of-the-box application (Henriques et al. 2010), which even includes a 3D reconstruction algorithm.
Considerations and limitations
In general, the higher the required resolution in super-resolution microscopy, the higher are the demands on the fluorophores. 4Pi, I5M, and SIM do not rely on special dye characteristics and are, therefore, not more demanding on the dyes than conventional fluorescence microscopy techniques. Note, however, that at higher resolution with subsequently reduced pixel/voxel size, more photons are required to reach a certain signal-to-noise level for mere statistical reasons. For localization-based techniques, the number of photons emitted by a single fluorophore is essential for the localization precision and, consequently, the achievable resolution (Thompson et al. 2002). An even more pertinent issue is the lifetime of the dark and bright states of the fluorescent markers. The PSFs of the single emitters must not overlap. Therefore, the molecules must be maintained in their dark state considerably longer than in their bright state (van de Linde et al. 2010). Another criterion is the dynamic range, i.e., the intensity ratio between the bright and dark states. The probability of the fluorophore emitting a photon while in its nonfluorescent state must be sufficiently small to achieve a reasonable contrast. In RESOLFT microscopy, the brightness of the individual molecule is less important than a high resistance to switching fatigue. The fluorescence markers have to undergo many transitions between the fluorescent and nonfluorescent states before contributing appreciably to the image. In STED microscopy, the high intensity of the depletion beam (>1 GW/cm²) may cause photodamage in living specimens (Klar et al. 2000). A common aspect of all high-resolution fluorescence microscopy techniques is that the labeling density has to be at least twice as large as the desired resolution (Shannon 1949).
The ultimate resolution limitation in super-resolution optical microscopy is posed by the ability to precisely label the structure of interest. Evidently, the fluorescent marker cannot reside where the structure itself is located, it has a certain size, and linkers are frequently introduced to specifically attach the fluorophore to a target. In live-cell imaging, too dense labeling may interfere with those processes that are to be studied. When imaging dynamic processes, conditions are naturally even more demanding. Localization-based techniques require the acquisition of a large number of individual frames, from which a single high-resolution image is finally reconstructed. With current CCD camera technology, the frame collection time is limited to the millisecond timescale. Thus, acquisition of super-resolved images presently takes a few dozen seconds at the minimum, processes on the subsecond timescale are too fast to be studied. STED microscopy of live cells at video rate has been demonstrated but is as yet confined to micron-sized regions of interest (Westphal et al. 2008).
Fluorophores
Except for STED and SSIM microscopy, for which regular dyes can be used, the key to super-resolution imaging is photoactivatable fluorophores. They should be bright in their activated states, i.e., feature both large fluorescence quantum yields and extinction coefficients to produce strong signals above background. Also, spontaneous interconversion between active and inactive forms should be slow compared with light-induced activation, which is especially important for RESOLFT imaging.
Photoactivatable fluorescent proteins are very popular as markers for super-resolution imaging, especially for studies of living cells and organisms. We distinguish two different forms of photoactivation, reversible photoactivation between a bright and a dark state, also known as photoswitching, and irreversible photoactivation, also known as photoconversion, which involves a permanent photochemical modification of the fluorescent protein (Wiedenmann and Nienhaus 2006). The first photoconvertible fluorescent protein, photoactivatable green fluorescent protein (PA-GFP) was generated by mutagenesis of the original GFP (Patterson and Lippincott-Schwartz 2002), which emits green fluorescence only after intense light irradiation at 400 nm. Later on, a monomeric red fluorescent protein PA-RFP-1-1 became available (Verkhusha and Sorkin 2005). PA-GFP has been used in FPALM imaging to determine the diffusion coefficient of hemagglutinin in live fibroblasts (Hess et al. 2007). However, the low contrast ratio of both photoactivatable proteins restricts their application in super-resolution imaging. In a second class of photoconverting fluorescent proteins, the emission color irreversibly changes from green to red upon irradiation with 400 nm light as in Kaede (Ando et al. 2002), the engineered KikGR (Tsutsui et al. 2005), and EosFP (Wiedenmann et al. 2004). Reversible photoswitchers are required for RESOLFT imaging. The first reported application was with asFP595 (Lukyanov et al. 2000) followed by Dronpa (Ando et al. 2004). PS-CFP2 was employed in dual-color super-resolution imaging (Shroff et al. 2007), and rsCherry (Stiel et al. 2008) was the first monomeric red fluorescent protein that was photoswitchable. Lately, a fluorescent protein has become available that combines the two photoactivation modes, namely IrisFP (Adam et al. 2008; Fuchs et al. 2010). It is reversibly switchable in the green and, after photoconversion with 400 nm light, turns into a reversibly switchable red fluorescent protein.
Organic dyes can also exhibit a reversible photoswitching mechanism suitable for super-resolution imaging, including photochromic rhodamines (Fölling et al. 2007) and cyanine dyes, which were employed in the first STORM experiment (Rust et al. 2006). Photoswitchable dyes are much brighter than fluorescent proteins, but their application in living cells is restricted because they may not be able to traverse the plasma membrane. Moreover, photoswitching requires the presence of thiol reagents at high concentrations, which may not be compatible with the live cell environment. Irreversibly photoactivatable dyes for super-resolution imaging are also available, e.g., Q-rhodamine (Mitchison et al. 1998; Gee et al. 2001).
Semiconductor nanocrystalline quantum dots exhibit superior photostability and brightness in comparison with synthetic dyes and are commonly used in single-molecule imaging. Recently, RESOLFT imaging has been reported using direct light driven modulation of Mn-doped ZnSe quantum dot fluorescence (Irvine et al. 2008). The blinking of quantum dots, which can be a nuisance in single-particle tracking, has been taken advantage of in a first application of the SOFI method of superresolution imaging (Dertinger et al. 2009).
Outlook
In recent years, a multitude of super-resolution light microscopy techniques has emerged that will be helpful for unraveling the molecular details underlying biological processes in the cell. These variants have very specific properties; they, e.g., provide multi-color, 3D-, or video-rate imaging, but not all of these features are available simultaneously with the same technique in one and the same microscope. Currently, there is a development toward simplified designs that may allow for a wider application of these techniques, especially by nonspecialists. Of crucial importance are efficient and easy-to-handle labeling systems, and brighter, more photostable and smaller fluorescent markers. The first commercial versions of STED microscopy and a combined SIM/PALM microscope have been introduced to the market, and exciting further developments will soon appear on the horizon.
Acknowledgments
This work was supported by the Deutsche Forschungsgemeinschaft and the State of Baden-Württemberg through the Center for Functional Nanostructures (CFN), by Deutsche Forschungsgemeinschaft grant NI 291/9 and by the Fonds der Chemischen Industrie. Prof. M. Bastmeyer, Karlsruhe Institute of Technology, Germany, kindly provided the images acquired with the Zeiss technology “Superresolution mit ELYRA (http://www.zeiss.de/elyra).”
References
- Abbe E. Beiträge zur Theorie des Mikroskops und der mikroskopischen Wahrnehmung. Arch Mikrosk Anat. 1873;9:413–468. doi: 10.1007/BF02956173. [DOI] [Google Scholar]
- Adam V, Lelimousin M, Böhme S, Desfonds G, Nienhaus K, Field MJ, Wiedenmann J, McSweeney S, Nienhaus GU, Bourgeois D. Structural characterization of IrisFP, an optical highlighter undergoing multiple photo-induced transformations. Proc Natl Acad Sci USA. 2008;105:18343–18348. doi: 10.1073/pnas.0805949105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersson SB. Localization of a fluorescent source without numerical fitting. Opt Express. 2008;16:18714–18724. doi: 10.1364/OE.16.018714. [DOI] [PubMed] [Google Scholar]
- Ando R, Hama H, Yamamoto-Hino M, Mizuno H, Miyawaki A. An optical marker based on the UV-induced green-to-red photoconversion of a fluorescent protein. Proc Natl Acad Sci USA. 2002;99:12651–12656. doi: 10.1073/pnas.202320599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ando R, Mizuno H, Miyawaki A. Regulated fast nucleocytoplasmic shuttling observed by reversible protein highlighting. Science. 2004;306:1370–1373. doi: 10.1126/science.1102506. [DOI] [PubMed] [Google Scholar]
- Andresen M, Stiel AC, Fölling J, Wenzel D, Schönle A, Egner A, Eggeling C, Hell SW, Jakobs S. Photoswitchable fluorescent proteins enable monochromatic multilabel imaging and dual color fluorescence nanoscopy. Nat Biotechnol. 2008;26:1035–1040. doi: 10.1038/nbt.1493. [DOI] [PubMed] [Google Scholar]
- Bates M, Blosser TR, Zhuang X. Short-range spectroscopic ruler based on a single-molecule optical switch. Phys Rev Lett. 2005;94:108101. doi: 10.1103/PhysRevLett.94.108101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bates M, Huang B, Dempsey GT, Zhuang X. Multicolor super-resolution imaging with photo-switchable fluorescent probes. Science. 2007;317:1749–1753. doi: 10.1126/science.1146598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betzig E, Patterson GH, Sougrat R, Lindwasser OW, Olenych S, Bonifacino JS, Davidson MW, Lippincott-Schwartz J, Hess HF. Imaging intracellular fluorescent proteins at nanometer resolution. Science. 2006;313:1642–1645. doi: 10.1126/science.1127344. [DOI] [PubMed] [Google Scholar]
- Binning G, Quate C, Gerber C. Atomic force microscope. Phys Rev Lett. 1986;56:930–933. doi: 10.1103/PhysRevLett.56.930. [DOI] [PubMed] [Google Scholar]
- Bock H, Geisler C, Wurm CA, von Middendorff C, Jakobs S, Schönle A, Egner A, Hell SW, Eggeling C. Two-color far-field fluorescence nanoscopy based on photoswitchable emitters. Appl Phys B. 2007;88:161–165. doi: 10.1007/s00340-007-2729-0. [DOI] [Google Scholar]
- Bossi M, Fölling J, Dyba M, Westphal V, Hell SW. Breaking the diffraction resolution barrier in far-field microscopy by molecular optical bistability. New J Phys. 2006;8:275. doi: 10.1088/1367-2630/8/11/275. [DOI] [Google Scholar]
- Bretschneider S, Eggeling C, Hell SW. Breaking the diffraction barrier in fluorescence microscopy by optical shelving. Phys Rev Lett. 2007;98:218103. doi: 10.1103/PhysRevLett.98.218103. [DOI] [PubMed] [Google Scholar]
- Dedecker P, Hotta J, Flors C, Sliwa M, Uji-i H, Roeffaers MB, Ando R, Mizuno H, Miyawaki A, Hofkens J. Subdiffraction imaging through the selective donut-mode depletion of thermally stable photoswitchable fluorophores: numerical analysis and application to the fluorescent protein Dronpa. J Am Chem Soc. 2007;129:16132–16141. doi: 10.1021/ja076128z. [DOI] [PubMed] [Google Scholar]
- Denk W, Strickler JH, Webb WW. Two-photon laser scanning fluorescence microscopy. Science. 1990;248:73–76. doi: 10.1126/science.2321027. [DOI] [PubMed] [Google Scholar]
- Dertinger T, Colyer R, Iyer G, Weiss S, Enderlein J. Fast, background-free, 3D super-resolution optical fluctuation imaging (SOFI) Proc Natl Acad Sci USA. 2009;106:22287–22292. doi: 10.1073/pnas.0907866106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donnert G, Keller J, Wurm CA, Rizzoli SO, Westphal V, Schönle A, Jahn R, Jakobs S, Eggeling C, Hell SW. Two-color far-field fluorescence nanoscopy. Biophys J. 2007;92:L67–L69. doi: 10.1529/biophysj.107.104497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dyba M, Jakobs S, Hell SW. Immunofluorescence stimulated emission depletion microscopy. Nat Biotechnol. 2003;21:1303–1304. doi: 10.1038/nbt897. [DOI] [PubMed] [Google Scholar]
- Eggeling C, Ringemann C, Medda R, Schwarzmann G, Sandhoff K, Polyakova S, Belov VN, Hein B, von Middendorff C, Schönle A, Hell SW. Direct observation of the nanoscale dynamics of membrane lipids in a living cell. Nature. 2009;457:1159–1162. doi: 10.1038/nature07596. [DOI] [PubMed] [Google Scholar]
- Egner A, Verrier S, Goroshkov A, Söling HD, Hell SW. 4Pi-microscopy of the Golgi apparatus in live mammalian cells. J Struct Biol. 2004;147:70–76. doi: 10.1016/j.jsb.2003.10.006. [DOI] [PubMed] [Google Scholar]
- Egner A, Geisler C, von Middendorff C, Bock H, Wenzel D, Medda R, Andresen M, Stiel AC, Jakobs S, Eggeling C, Schönle A, Hell SW. Fluorescence nanoscopy in whole cells by asynchronous localization of photoswitching emitters. Biophys J. 2007;93:3285–3290. doi: 10.1529/biophysj.107.112201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flors C, Hotta J, Uji-i H, Dedecker P, Ando R, Mizuno H, Miyawaki A, Hofkens J. A stroboscopic approach for fast photoactivation-localization microscopy with Dronpa mutants. J Am Chem Soc. 2007;129:13970–13977. doi: 10.1021/ja074704l. [DOI] [PubMed] [Google Scholar]
- Fölling J, Belov V, Kunetsky R, Medda R, Schönle A, Egner A, Eggeling C, Bossi M, Hell SW. Photochromic rhodamines provide nanoscopy with optical sectioning. Angew Chem Int Ed Engl. 2007;46:6266–6270. doi: 10.1002/anie.200702167. [DOI] [PubMed] [Google Scholar]
- Fölling J, Bossi M, Bock H, Medda R, Wurm CA, Hein B, Jakobs S, Eggeling C, Hell SW. Fluorescence nanoscopy by ground-state depletion and single-molecule return. Nat Methods. 2008;5:943–945. doi: 10.1038/nmeth.1257. [DOI] [PubMed] [Google Scholar]
- Friedrich W, Knipping P, Laue M. Eine quantitative Prüfung der Theorie für die Interferenzerscheinungen bei Röntgenstrahlen. Ann Phys. 1913;41:989–1002. [Google Scholar]
- Fu CC, Lee HY, Chen K, Lim TS, Wu HY, Lin PK, Wei PK, Tsao PH, Chang HC, Fann W. Characterization and application of single fluorescent nanodiamonds as cellular biomarkers. Proc Natl Acad Sci USA. 2007;104:727–732. doi: 10.1073/pnas.0605409104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuchs J, Böhme S, Oswald F, Hedde PN, Krause M, Wiedenmann J, Nienhaus GU. Imaging protein movement in live cells with super-resolution using mIrisFP. Nat Methods. 2010;7:627–630. doi: 10.1038/nmeth.1477. [DOI] [PubMed] [Google Scholar]
- Gee KR, Weinberg ES, Kozlowski DJ. Caged Q-rhodamine dextran: a new photoactivated fluorescent tracer. Bioorg Med Chem Lett. 2001;11:2181–2183. doi: 10.1016/S0960-894X(01)00421-8. [DOI] [PubMed] [Google Scholar]
- Glaschick S, Röcker C, Deuschle K, Wiedenmann J, Oswald F, Mailänder V, Nienhaus GU. Axial resolution enhancement by 4Pi confocal fluorescence microscopy with two-photon excitation. J Biol Phys. 2007;33:433–443. doi: 10.1007/s10867-008-9084-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gugel H, Bewersdorf J, Jakobs S, Engelhardt J, Storz R, Hell SW. Cooperative 4Pi excitation and detection yields sevenfold sharper optical sections in live-cell microscopy. Biophys J. 2004;87:4146–4152. doi: 10.1529/biophysj.104.045815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gustafsson MG. Extended resolution fluorescence microscopy. Curr Opin Struct Biol. 1999;9:627–634. doi: 10.1016/S0959-440X(99)00016-0. [DOI] [PubMed] [Google Scholar]
- Gustafsson MG. Surpassing the lateral resolution limit by a factor of two using structured illumination microscopy. J Microsc. 2000;198:82–87. doi: 10.1046/j.1365-2818.2000.00710.x. [DOI] [PubMed] [Google Scholar]
- Gustafsson MG. Nonlinear structured-illumination microscopy: wide-field fluorescence imaging with theoretically unlimited resolution. Proc Natl Acad Sci USA. 2005;102:13081–13086. doi: 10.1073/pnas.0406877102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gustafsson MG, Shao L, Carlton PM, Wang CJ, Golubovskaya IN, Cande WZ, Agard DA, Sedat JW. Three-dimensional resolution doubling in wide-field fluorescence microscopy by structured illumination. Biophys J. 2008;94:4957–4970. doi: 10.1529/biophysj.107.120345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han KY, Willig KI, Rittweger E, Jelezko F, Eggeling C, Hell SW. Three-dimensional stimulated emission depletion microscopy of nitrogen-vacancy centers in diamond using continuous-wave light. Nano Lett. 2009;9:3323–3329. doi: 10.1021/nl901597v. [DOI] [PubMed] [Google Scholar]
- Hedde PN, Fuchs J, Oswald F, Wiedenmann J, Nienhaus GU. Online image analysis software for photoactivation localization microscopy. Nat Methods. 2009;6:689–690. doi: 10.1038/nmeth1009-689. [DOI] [PubMed] [Google Scholar]
- Heilemann M, Margeat E, Kasper R, Sauer M, Tinnefeld P. Carbocyanine dyes as efficient reversible single-molecule optical switch. J Am Chem Soc. 2005;127:3801–3806. doi: 10.1021/ja044686x. [DOI] [PubMed] [Google Scholar]
- Heilemann M, van de Linde S, Schuttpelz M, Kasper R, Seefeldt B, Mukherjee A, Tinnefeld P, Sauer M. Subdiffraction-resolution fluorescence imaging with conventional fluorescent probes. Angew Chem Int Ed Engl. 2008;47:6172–6176. doi: 10.1002/anie.200802376. [DOI] [PubMed] [Google Scholar]
- Hein B, Willig KI, Hell SW. Stimulated emission depletion (STED) nanoscopy of a fluorescent protein-labeled organelle inside a living cell. Proc Natl Acad Sci USA. 2008;105:14271–14276. doi: 10.1073/pnas.0807705105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heintzmann R, Gustafsson MG. Subdiffraction resolution in continuous samples. Nat Photonics. 2009;3:362–364. doi: 10.1038/nphoton.2009.102. [DOI] [Google Scholar]
- Heintzmann R, Jovin TM, Cremer C. Saturated patterned excitation microscopy–a concept for optical resolution improvement. J Opt Soc Am A Opt Image Sci Vis. 2002;19:1599–1609. doi: 10.1364/JOSAA.19.001599. [DOI] [PubMed] [Google Scholar]
- Hell SW. Toward fluorescence nanoscopy. Nat Biotechnol. 2003;21:1347–1355. doi: 10.1038/nbt895. [DOI] [PubMed] [Google Scholar]
- Hell SW. Far-field optical nanoscopy. Science. 2007;316:1153–1158. doi: 10.1126/science.1137395. [DOI] [PubMed] [Google Scholar]
- Hell SW. Microscopy and its focal switch. Nat Methods. 2009;6:24–32. doi: 10.1038/nmeth.1291. [DOI] [PubMed] [Google Scholar]
- Hell SW, Kroug M. Ground-state depletion fluorescence microscopy, a concept for breaking the diffraction resolution limit. Appl Phys B. 1995;60:495–497. doi: 10.1007/BF01081333. [DOI] [Google Scholar]
- Hell SW, Stelzer EHK. Fundamental improvement of resolution with a 4Pi-confocal microscope using two-photon excitation. Opt Commun. 1992;93:277–282. doi: 10.1016/0030-4018(92)90185-T. [DOI] [Google Scholar]
- Hell SW, Wichmann J. Breaking the diffraction resolution limit by stimulated emission: stimulated-emission-depletion fluorescence microscopy. Opt Lett. 1994;19:780–782. doi: 10.1364/OL.19.000780. [DOI] [PubMed] [Google Scholar]
- Helmchen F, Denk W. New developments in multiphoton microscopy. Curr Opin Neurobiol. 2002;12:593–601. doi: 10.1016/S0959-4388(02)00362-8. [DOI] [PubMed] [Google Scholar]
- Henderson R, Unwin PN. Three-dimensional model of purple membrane obtained by electron microscopy. Nature. 1975;257:28–32. doi: 10.1038/257028a0. [DOI] [PubMed] [Google Scholar]
- Henriques R, Lelek M, Fornasiero EF, Valtorta F, Zimmer C, Mhlanga MM. QuickPALM: 3D real-time photoactivation nanoscopy image processing in ImageJ. Nat Methods. 2010;7:339–340. doi: 10.1038/nmeth0510-339. [DOI] [PubMed] [Google Scholar]
- Hess ST, Girirajan TP, Mason MD. Ultra-high resolution imaging by fluorescence photoactivation localization microscopy. Biophys J. 2006;91:4258–4272. doi: 10.1529/biophysj.106.091116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hess ST, Gould TJ, Gudheti MV, Maas SA, Mills KD, Zimmerberg J. Dynamic clustered distribution of hemagglutinin resolved at 40 nm in living cell membranes discriminates between raft theories. Proc Natl Acad Sci USA. 2007;104:17370–17375. doi: 10.1073/pnas.0708066104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirvonen LM, Wicker K, Mandula O, Heintzmann R. Structured illumination microscopy of a living cell. Eur Biophys J. 2009;38:807–812. doi: 10.1007/s00249-009-0501-6. [DOI] [PubMed] [Google Scholar]
- Hofmann M, Eggeling C, Jakobs S, Hell SW. Breaking the diffraction barrier in fluorescence microscopy at low light intensities by using reversibly photoswitchable proteins. Proc Natl Acad Sci USA. 2005;102:17565–17569. doi: 10.1073/pnas.0506010102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang B, Wang W, Bates M, Zhuang X. Three-dimensional super-resolution imaging by stochastic optical reconstruction microscopy. Science. 2008;319:810–813. doi: 10.1126/science.1153529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irvine SE, Staudt T, Rittweger E, Engelhardt J, Hell SW. Direct light-driven modulation of luminescence from Mn-doped ZnSe quantum dots. Angew Chem Int Ed Engl. 2008;47:2685–2688. doi: 10.1002/anie.200705111. [DOI] [PubMed] [Google Scholar]
- Ivanchenko S, Glaschick S, Röcker C, Oswald F, Wiedenmann J, Nienhaus GU. Two-photon excitation and photoconversion of EosFP in dual-color 4Pi confocal microscopy. Biophys J. 2007;92:4451–4457. doi: 10.1529/biophysj.106.103408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juette MF, Gould TJ, Lessard MD, Mlodzianoski MJ, Nagpure BS, Bennett BT, Hess ST, Bewersdorf J. Three-dimensional sub-100 nm resolution fluorescence microscopy of thick samples. Nat Methods. 2008;5:527–529. doi: 10.1038/nmeth.1211. [DOI] [PubMed] [Google Scholar]
- Kastrup L, Blom H, Eggeling C, Hell SW. Fluorescence fluctuation spectroscopy in subdiffraction focal volumes. Phys Rev Lett. 2005;94:178104. doi: 10.1103/PhysRevLett.94.178104. [DOI] [PubMed] [Google Scholar]
- Kendrew JC, Dickerson RE, Strandberg BE, Hart RG, Davies DR, Phillips DC, Shore VC. Structure of myoglobin: a three-dimensional Fourier synthesis at 2 A. resolution. Nature. 1960;185:422–427. doi: 10.1038/185422a0. [DOI] [PubMed] [Google Scholar]
- Klar TA, Jakobs S, Dyba M, Egner A, Hell SW. Fluorescence microscopy with diffraction resolution barrier broken by stimulated emission. Proc Natl Acad Sci USA. 2000;97:8206–8210. doi: 10.1073/pnas.97.15.8206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kner P, Chhun BB, Griffis ER, Winoto L, Gustafsson MG. Super-resolution video microscopy of live cells by structured illumination. Nat Methods. 2009;6:339–342. doi: 10.1038/nmeth.1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knoll M, Ruska E. Das Elektronenmikroskop. Z Phys. 1932;78:318–339. doi: 10.1007/BF01342199. [DOI] [Google Scholar]
- Lange OF, Lakomek NA, Fares C, Schröder GF, Walter KF, Becker S, Meiler J, Grubmüller H, Griesinger C, de Groot BL. Recognition dynamics up to microseconds revealed from an RDC-derived ubiquitin ensemble in solution. Science. 2008;320:1471–1475. doi: 10.1126/science.1157092. [DOI] [PubMed] [Google Scholar]
- Lucic V, Förster F, Baumeister W. Structural studies by electron tomography: from cells to molecules. Annu Rev Biochem. 2005;74:833–865. doi: 10.1146/annurev.biochem.73.011303.074112. [DOI] [PubMed] [Google Scholar]
- Lukyanov KA, Fradkov AF, Gurskaya NG, Matz MV, Labas YA, Savitsky AP, Markelov ML, Zaraisky AG, Zhao X, Fang Y, Tan W, Lukyanov SA. Natural animal coloration can be determined by a nonfluorescent green fluorescent protein homolog. J Biol Chem. 2000;275:25879–25882. doi: 10.1074/jbc.C000338200. [DOI] [PubMed] [Google Scholar]
- Medda R, Jakobs S, Hell SW, Bewersdorf J. 4Pi microscopy of quantum dot-labeled cellular structures. J Struct Biol. 2006;156:517–523. doi: 10.1016/j.jsb.2006.08.013. [DOI] [PubMed] [Google Scholar]
- Meyer L, Wildanger D, Medda R, Punge A, Rizzoli SO, Donnert G, Hell SW. Dual-color STED microscopy at 30-nm focal-plane resolution. Small. 2008;4:1095–1100. doi: 10.1002/smll.200800055. [DOI] [PubMed] [Google Scholar]
- Michalet X, Pinaud FF, Bentolila LA, Tsay JM, Doose S, Li JJ, Sundaresan G, Wu AM, Gambhir SS, Weiss S. Quantum dots for live cells, in vivo imaging, and diagnostics. Science. 2005;307:538–544. doi: 10.1126/science.1104274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchison TJ, Sawin KE, Theriot JA, Gee K, Mallavarapu A. Caged fluorescent probes. Methods Enzymol. 1998;291:63–78. doi: 10.1016/S0076-6879(98)91007-2. [DOI] [PubMed] [Google Scholar]
- Moneron G, Medda R, Hein B, Giske A, Westphal V, Hell SW. Fast STED microscopy with continuous wave fiber lasers. Opt Express. 2010;18:1302–1309. doi: 10.1364/OE.18.001302. [DOI] [PubMed] [Google Scholar]
- Müller DJ, Wu N, Palczewski K. Vertebrate membrane proteins: structure, function, and insights from biophysical approaches. Pharmacol Rev. 2008;60:43–78. doi: 10.1124/pr.107.07111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nägerl UV, Willig KI, Hein B, Hell SW, Bonhoeffer T. Live-cell imaging of dendritic spines by STED microscopy. Proc Natl Acad Sci USA. 2008;105:18982–18987. doi: 10.1073/pnas.0810028105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagorni M, Hell SW. 4Pi-confocal microscopy provides three-dimensional images of the microtubule network with 100- to 150-nm resolution. J Struct Biol. 1998;123:236–247. doi: 10.1006/jsbi.1998.4037. [DOI] [PubMed] [Google Scholar]
- Nienhaus GU. The green fluorescent protein: a key tool to study chemical processes in living cells. Angew Chem Int Ed Engl. 2008;47:8992–8994. doi: 10.1002/anie.200804998. [DOI] [PubMed] [Google Scholar]
- Nienhaus GU, Nienhaus K, Holzle A, Ivanchenko S, Renzi F, Oswald F, Wolff M, Schmitt F, Röcker C, Vallone B, Weidemann W, Heilker R, Nar H, Wiedenmann J. Photoconvertible fluorescent protein EosFP: biophysical properties and cell biology applications. Photochem Photobiol. 2006;82:351–358. doi: 10.1562/2005-05-19-RA-533. [DOI] [PubMed] [Google Scholar]
- Patterson GH, Lippincott-Schwartz J. A photoactivatable GFP for selective photolabeling of proteins and cells. Science. 2002;297:1873–1877. doi: 10.1126/science.1074952. [DOI] [PubMed] [Google Scholar]
- Pavani SR, Thompson MA, Biteen JS, Lord SJ, Liu N, Twieg RJ, Piestun R, Moerner WE. Three-dimensional, single-molecule fluorescence imaging beyond the diffraction limit by using a double-helix point spread function. Proc Natl Acad Sci USA. 2009;106:2995–2999. doi: 10.1073/pnas.0900245106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawley JB (2006) Handbook of biological confocal microscopy, 3rd edn. Springer, Berlin
- Perutz MF, Miurhead H, Cox JM, Goaman LC, Mathews FS, McGandy EL, Webb LE. Three-dimensional Fourier synthesis of horse oxyhaemoglobin at 2.8 A resolution: (1) x-ray analysis. Nature. 1968;219:29–32. doi: 10.1038/219029a0. [DOI] [PubMed] [Google Scholar]
- Quan T, Li P, Long F, Zeng S, Luo Q, Hedde PN, Nienhaus GU, Huang Z-L. Ultra-fast, high-precision image analysis for localization-based super resolution microscopy. Opt Express. 2010;18:11867–11876. doi: 10.1364/OE.18.011867. [DOI] [PubMed] [Google Scholar]
- Reuss M, Engelhardt J, Hell SW. Birefringent device converts a standard scanning microscope into a STED microscope that also maps molecular orientation. Opt Express. 2010;18:1049–1058. doi: 10.1364/OE.18.001049. [DOI] [PubMed] [Google Scholar]
- Rust MJ, Bates M, Zhuang X. Sub-diffraction-limit imaging by stochastic optical reconstruction microscopy (STORM) Nat Methods. 2006;3:793–795. doi: 10.1038/nmeth929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schermelleh L, Carlton PM, Haase S, Shao L, Winoto L, Kner P, Burke B, Cardoso MC, Agard DA, Gustafsson MG, Leonhardt H, Sedat JW. Subdiffraction multicolor imaging of the nuclear periphery with 3D structured illumination microscopy. Science. 2008;320:1332–1336. doi: 10.1126/science.1156947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schönle A, Hell SW. Fluorescence nanoscopy goes multicolor. Nat Biotechnol. 2007;25:1234–1235. doi: 10.1038/nbt1107-1234. [DOI] [PubMed] [Google Scholar]
- Shannon CE. Communication in the presence of noise. Proc IRE. 1949;37:10–21. doi: 10.1109/JRPROC.1949.232969. [DOI] [Google Scholar]
- Shao L, Isaac B, Uzawa S, Agard DA, Sedat JW, Gustafsson MG. I5S: wide-field light microscopy with 100-nm-scale resolution in three dimensions. Biophys J. 2008;94:4971–4983. doi: 10.1529/biophysj.107.120352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharonov A, Hochstrasser RM. Wide-field subdiffraction imaging by accumulated binding of diffusing probes. Proc Natl Acad Sci USA. 2006;103:18911–18916. doi: 10.1073/pnas.0609643104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimomura O. Discovery of green fluorescent protein. Methods Biochem Anal. 2006;47:1–13. doi: 10.1002/0471739499.ch1. [DOI] [PubMed] [Google Scholar]
- Shroff H, Galbraith CG, Galbraith JA, White H, Gillette J, Olenych S, Davidson MW, Betzig E. Dual-color superresolution imaging of genetically expressed probes within individual adhesion complexes. Proc Natl Acad Sci USA. 2007;104:20308–20313. doi: 10.1073/pnas.0710517105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shroff H, Galbraith CG, Galbraith JA, Betzig E. Live-cell photoactivated localization microscopy of nanoscale adhesion dynamics. Nat Methods. 2008;5:417–423. doi: 10.1038/nmeth.1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shtengel G, Galbraith JA, Galbraith CG, Lippincott-Schwartz J, Gillette JM, Manley S, Sougrat R, Waterman CM, Kanchanawong P, Davidson MW, Fetter RD, Hess HF. Interferometric fluorescent super-resolution microscopy resolves 3D cellular ultrastructure. Proc Natl Acad Sci USA. 2009;106:3125–3130. doi: 10.1073/pnas.0813131106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith CS, Joseph N, Rieger B, Lidke KA. Fast, single-molecule localization that achieves theoretically minimum uncertainty. Nat Methods. 2010;7:373–375. doi: 10.1038/nmeth.1449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stiel AC, Andresen M, Bock H, Hilbert M, Schilde J, Schönle A, Eggeling C, Egner A, Hell SW, Jakobs S. Generation of monomeric reversibly switchable red fluorescent proteins for far-field fluorescence nanoscopy. Biophys J. 2008;95:2989–2997. doi: 10.1529/biophysj.108.130146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang J, Akerboom J, Vaziri A, Looger LL, Shank CV. Near-isotropic 3D optical nanoscopy with photon-limited chromophores. Proc Natl Acad Sci USA. 2010;107(22):10068–10073. doi: 10.1073/pnas.1004899107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson NL, Burghardt TP, Axelrod D. Measuring surface dynamics of biomolecules by total internal reflection fluorescence with photobleaching recovery or correlation spectroscopy. Biophys J. 1981;33:435–454. doi: 10.1016/S0006-3495(81)84905-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson RE, Larson DR, Webb WW. Precise nanometer localization analysis for individual fluorescent probes. Biophys J. 2002;82:2775–2783. doi: 10.1016/S0006-3495(02)75618-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsutsui H, Karasawa S, Shimizu H, Nukina N, Miyawaki A. Semi-rational engineering of a coral fluorescent protein into an efficient highlighter. EMBO Rep. 2005;6:233–238. doi: 10.1038/sj.embor.7400361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van de Linde S, Endesfelder U, Mukherjee A, Schuttpelz M, Wiebusch G, Wolter S, Heilemann M, Sauer M. Multicolor photoswitching microscopy for subdiffraction-resolution fluorescence imaging. Photochem Photobiol Sci. 2009;8:465–469. doi: 10.1039/b822533h. [DOI] [PubMed] [Google Scholar]
- van de Linde S, Wolter S, Heilemann M, Sauer M (2010) The effect of photoswitching kinetics and labeling densities on super-resolution fluorescence imaging. J Biotechnol (in press) [DOI] [PubMed]
- Verkhusha VV, Sorkin A. Conversion of the monomeric red fluorescent protein into a photoactivatable probe. Chem Biol. 2005;12:279–285. doi: 10.1016/j.chembiol.2005.01.005. [DOI] [PubMed] [Google Scholar]
- Vogelsang J, Cordes T, Forthmann C, Steinhauer C, Tinnefeld P. Controlling the fluorescence of ordinary oxazine dyes for single-molecule switching and superresolution microscopy. Proc Natl Acad Sci USA. 2009;106:8107–8112. doi: 10.1073/pnas.0811875106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westphal V, Rizzoli SO, Lauterbach MA, Kamin D, Jahn R, Hell SW. Video-rate far-field optical nanoscopy dissects synaptic vesicle movement. Science. 2008;320:246–249. doi: 10.1126/science.1154228. [DOI] [PubMed] [Google Scholar]
- Wiedenmann J, Nienhaus GU. Live-cell imaging with EosFP and other photoactivatable marker proteins of the GFP family. Expert Rev Proteomics. 2006;3:361–374. doi: 10.1586/14789450.3.3.361. [DOI] [PubMed] [Google Scholar]
- Wiedenmann J, Ivanchenko S, Oswald F, Schmitt F, Röcker C, Salih A, Spindler KD, Nienhaus GU. EosFP, a fluorescent marker protein with UV-inducible green-to-red fluorescence conversion. Proc Natl Acad Sci USA. 2004;101:15905–15910. doi: 10.1073/pnas.0403668101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wildanger D, Medda R, Kastrup L, Hell SW. A compact STED microscope providing 3D nanoscale resolution. J Microsc. 2009;236:35–43. doi: 10.1111/j.1365-2818.2009.03188.x. [DOI] [PubMed] [Google Scholar]
- Willig KI, Harke B, Medda R, Hell SW. STED microscopy with continuous wave beams. Nat Methods. 2007;4:915–918. doi: 10.1038/nmeth1108. [DOI] [PubMed] [Google Scholar]
- Wüthrich K. Nuclear magnetic resonance–from molecules to man. Q Rev Biophys. 1987;19:3–5. doi: 10.1017/S0033583500004005. [DOI] [PubMed] [Google Scholar]
- Yildiz A, Selvin PR. Fluorescence imaging with one nanometer accuracy: application to molecular motors. Acc Chem Res. 2005;38:574–582. doi: 10.1021/ar040136s. [DOI] [PubMed] [Google Scholar]
- Yildiz A, Forkey JN, McKinney SA, Ha T, Goldman YE, Selvin PR. Myosin V walks hand-over-hand: single fluorophore imaging with 1.5-nm localization. Science. 2003;300:2061–2065. doi: 10.1126/science.1084398. [DOI] [PubMed] [Google Scholar]
- Zheng J, Nicovich PR, Dickson RM. Highly fluorescent noble-metal quantum dots. Annu Rev Phys Chem. 2007;58:409–431. doi: 10.1146/annurev.physchem.58.032806.104546. [DOI] [PMC free article] [PubMed] [Google Scholar]