

Abstract
Neuronal nitric oxide synthases (nNOS) are Ca2+/calmodulin-activated enzymes that synthesize the gaseous messenger nitric oxide (NO). nNOSμ and the recently described nNOSβ, both spliced nNOS isoforms, are important enzymatic sources of NO in skeletal muscle, a tissue long considered to be a paradigmatic system for studying NO-dependent redox signaling. nNOS is indispensable for skeletal muscle integrity and contractile performance, and deregulation of nNOSμ signaling is a common pathogenic feature of many neuromuscular diseases. Recent evidence suggests that both nNOSμ and nNOSβ regulate skeletal muscle size, strength, and fatigue resistance, making them important players in exercise performance. nNOSμ acts as an activity sensor and appears to assist skeletal muscle adaptation to new functional demands, particularly those of endurance exercise. Prolonged inactivity leads to nNOS-mediated muscle atrophy through a FoxO-dependent pathway. nNOS also plays a role in modulating exercise performance in neuromuscular disease. In the mdx mouse model of Duchenne muscular dystrophy, defective nNOS signaling is thought to restrict contractile capacity of working muscle in two ways: loss of sarcolemmal nNOSμ causes excessive ischemic damage while residual cytosolic nNOSμ contributes to hypernitrosylation of the ryanodine receptor, causing pathogenic Ca2+ leak. This defect in Ca2+ handling promotes muscle damage, weakness, and fatigue. This review addresses these recent advances in the understanding of nNOS-dependent redox regulation of skeletal muscle function and exercise performance under physiological and neuromuscular disease conditions.
Keywords: Nitric oxide, nNOS, Ryanodine receptor, Nitrosylation, Dystrophin, Fatigue
Introduction
Humans have a remarkable capacity for long distance running in hot climates that is unique among primates and compares favorably with that of other mammals. This capacity for endurance exercise is thought to have significantly shaped human evolution and enabled australopithecines, the ape-like ancestors of modern humans, to hunt prey and scavenge meat (Bramble and Lieberman 2004; Lieberman and Bramble 2007). The ability of skeletal muscles to resist fatigue is critical for running long distances. In modern times, endurance running is not required for survival, rather it is practiced by professional and recreational athletes. Elucidating the mechanisms governing endurance exercise performance and fatigue resistance is important for understanding human uniqueness amongst animals as well as human athletic potential.
The mechanisms regulating exercise capacity are not just relevant to understanding the limits of athletic performance, they are inextricably linked with human longevity. Moderate exercise or activity has well-known beneficial effects on human health, quality of life, and lifespan. Physical activity serves as an effective treatment for chronic age-related conditions, such as heart failure, obesity, type 2 diabetes, neurodegeneration, and sarcopenia (the age-related loss of muscle mass and strength) (Handschin and Speigelman 2008; Crimi et al. 2009). Central to the capacity for exercise is the ability of skeletal muscles to resist fatigue. Skeletal muscle fatigue is defined here as the reversible decline in force output with repeated and intensive use. Debilitating fatigue is often associated with heart failure and neuromuscular disease and leads to an inability to perform everyday tasks, profoundly decreasing quality of life (Crimi et al. 2009; Lou et al. 2010). Thus, it is important to determine the pathways controlling skeletal muscle fatigue and identify targets that promote fatigue resistance.
The mechanisms that regulate skeletal muscle fatigue have been intensively investigated, with a prominent role emerging for free radical-mediated redox signaling. Reactive oxygen species (ROS) are important regulators of muscle contractility and fatigue (Allen et al. 2008; Powers and Jackson 2008). The free radical nitric oxide (NO) synthesized by neuronal nitric oxide synthase (nNOS) is also a critical regulator of skeletal muscle exercise performance. nNOSμ appears to act as an activity sensor, with the modulation of nNOS signaling amplitude forming part of the adaptive reprogramming of muscle to endurance exercise or prolonged inactivity. Current evidence suggests a role for nNOS signaling in the regulation of skeletal muscle fatigue. Paradoxically, in dystrophic diseased muscle, nNOSμ appears to gain a pathogenic activity that contributes to muscle dysfunction. This review will focus on the regulation of exercise capacity by nNOS under both physiological and neuromuscular disease contexts.
nNOS splice variants in skeletal muscle
nNOS is the major source of NO in skeletal muscle cells (Stamler and Meissner 2001; Percival et al. 2010). It is a Ca2+/calmodulin-activated enzyme that catalyzes the NADPH- and O2-dependent synthesis of NO from L-arginine (Bredt and Snyder 1990; Bredt et al. 1991). The Ca2+/calmodulin sensitivity of nNOS facilitates its coupling to muscle contraction. The increases in intracellular Ca2+ that occur upon muscle excitation increase basal nNOS activity several fold during contraction (Stamler and Meissner 2001).
Skeletal muscles express two alternatively spliced nNOS isoforms, nNOSμ and nNOSβ, both of which are expressed in fast and slow muscle types (Silvagno et al. 1996; Stamler and Meissner 2001; Percival et al. 2010, Figure 1). nNOSμ is scaffolded to the subsarcolemmal dystrophin glycoprotein complex (DGC) and neuromuscular junction (Brenman et al. 1995, 1996; Adams et al. 2000). The sarcolemmal nNOSμ association requires α-syntrophin, dystrophin, and α-dystrobrevin (Brenman et al. 1995, 1996; Grady et al. 2000; Adams et al. 2000, 2008). In mice, nNOSμ is not only localized at the sarcolemma, but is also normally expressed at relatively high levels in the cytoplasm (Chang et al. 1996; Kameya et al. 1999; Thomas et al. 2003). However, in human muscle approximately 75% of nNOSμ is associated with the sarcolemmal DGC, suggesting species-specific differences in nNOSμ distribution (Chang et al. 1996). A pool of nNOSμ is associated with the ryanodine receptor 1 Ca2+ release channel (RyR1) at the sarcoplasmic reticulum (SR) Ca2+ store where it can S-nitrosylate and regulate RyR1 channel activity (Xu et al. 1999; Stamler and Meissner 2001; Salanova et al. 2008; Li et al. 2011a, Figure 1). nNOSμ association and regulation of the ryanodine receptor is also seen in the heart (Gonzalez et al. 2007). nNOSβ associates with the subsarcolemmal Golgi complex (Percival et al. 2010), suggesting the presence of a high concentration of nNOS-derived NO from both nNOSμ and nNOSβ just under the plasma membrane.
Fig. 1.
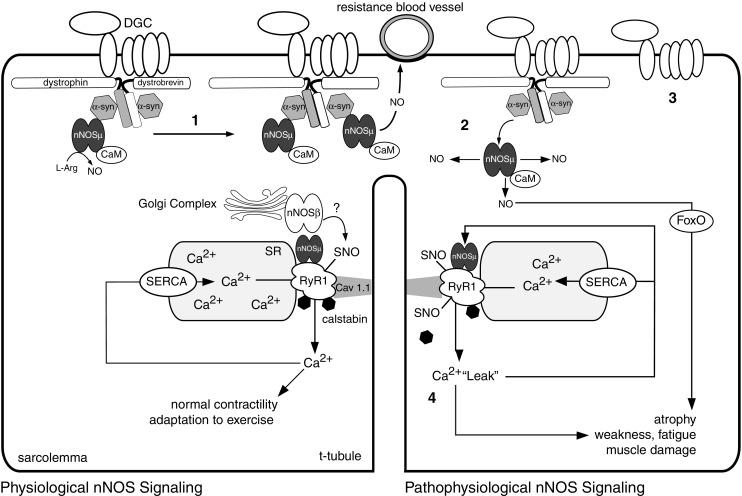
Model for the activity-dependent control of Ca2+/calmodulin (CaM) regulated neuronal nitric oxide synthase (nNOS) signaling and function. nNOSμ localizes to the dystrophin glycoprotein complex by binding a-syntrophin (a-syn), where it opposes blood vessel constriction during exercise. The spliced nNOS isoforms nNOSμ and nNOSβ (?) may regulate muscle fatigue and exercise performance by promoting normal ryanodine receptor 1 (RyR1) activity. SNO represents S-nitrosylation of regulatory cysteine thiols. RyR1 releases Ca2+ into the cytosol during contraction, while a sarcoplasmic reticulum Ca2+-ATPase (SERCA) pumps Ca2+ from the cytosol back into the sarcoplasmic reticulum (SR). 1 nNOSμ signaling is increased by endurance type exercise. 2 Prolonged inactivity leads to dissociation of nNOSμ from the sarcolemma inducing atrophy through a forkhead transcription factor (FoxO) mediated pathway. 3 In Duchenne muscular dystrophy, loss of dystrophin destabilizes the dystrophin glycoprotein complex (DGC), and impairs the vasoregulatory function of nNOSμ, leading to RyR1 destabilization, pathogenic nNOSμ-mediated RyR1 hypernitrosylation and Ca2+ leak. 4 RyR1 Ca2+ leak can lead to muscle damage, weakness, and fatigue. RyR1 Ca2+ leak can further inappropriately activate nNOSμ, leading to further RyR1 nitrosylation and progressively more damaging Ca2+ leakage in a severely detrimental feed-forward loop
In summary, skeletal muscle has at least three spatially distinct nNOS signaling compartments defined by the association of nNOSμ with the DGC and with RyR1 at the SR as well as Golgi complex-associated nNOSβ. Tight spatial control of nNOS enzyme localization appears to be critical for facilitating isoform-specific function and the diversity of NO signaling in muscle.
nNOS and skeletal muscle adaptation to exercise
In response to exercise, skeletal muscle undergoes adaptive remodeling to meet the new functional and metabolic demand. This tremendous plasticity results from the reprogramming of signaling pathways (particularly Ca2+-dependent pathways) that modulate the contractile and metabolic properties of muscle (Bassel-Duby and Olson 2006). A large body of evidence now suggests that nNOSμ upregulation is an important part of the adaptation of mammalian skeletal muscle to intense endurance exercise.
Most human studies suggest that endurance-type exercise leads to increased nNOSμ expression (Frandsen et al. 2000; Rudnick et al. 2004; McConell et al. 2007). For example, endurance-trained athletes have significantly higher (approx. 60%) skeletal muscle nNOSμ basal expression than the general population (McConell et al. 2007). Intensive short-term cycling exercise can also acutely increase nNOSμ expression by approximately 50% in sedentary individuals (McConell et al. 2007). Thus, increased basal expression of nNOSμ appears to be an adaptation to endurance training in humans.
Studies in rats also suggest that endurance-type exercise increases skeletal muscle nNOSμ expression. nNOSμ expression and activity increased in the hindlimb and respiratory muscles of rats subjected to long-term treadmill running or swimming (Balon and Nadler 1997; Tatchum-Talom et al. 2000; Vassilakopoulos et al. 2003). Skeletal muscle nNOSμ expression declines with age in rats, suggesting that sarcopenia may be linked with the downregulation of nNOSμ (Song et al. 2009), but long-term treadmill running has been found to reverse this age-related decrease in nNOSμ expression (Song et al. 2009). These data suggest the intriguing possibility that nNOSμ signaling may contribute to the beneficial effects of exercise in aged muscle.
Thus, in agreement with findings in humans, rat skeletal muscle also exhibits increased nNOSμ signaling in response to endurance training. Taken together, the results of rodent and human studies suggest that the reprogramming of nNOS expression and activity is an important event in the adaptation of skeletal muscle to endurance exercise.
Inactivity-dependent regulation of nNOS expression and catalytic function
Not only does skeletal muscle adapt to increases in activity, it also adapts to decreases in activity. In humans, inactivity leads to the loss of skeletal muscle bulk and strength, commonly known as “use it or lose it”. Prolonged inactivity or immobilization leads to skeletal muscle wasting or atrophy. Inhibition of atrophy pathways has clinical application to the alleviation of muscle wasting associated with sarcopenia, neuromuscular disease, cancer (cachexia), prolonged bed rest, and space flight. While nNOSμ expression can be increased by endurance exercise activity, its normal basal expression and localization are also activity-dependent. Human subjects confined to bed for 90 days were found to experience atrophy of the vastus lateralis (thigh) and soleus (calf) muscles. This atrophy was accompanied by reduced nNOSμ expression and sarcolemmal localization in the vastus lateralis only, suggesting that atrophic regulation of nNOSμ occurs by unidentified muscle-specific mechanisms (Rudnick et al. 2004). Thigh muscle size and nNOSμ localization are preserved if bed rest is accompanied by ergometer exercise, which also increases nNOSμ expression above pre-bed rest levels. These data are consistent with reports that nNOSμ expression is induced by long-term exercise.
As in humans, nNOSμ also acts as an inactivity sensor in rodents. In mice, nNOS may trigger atrophy in response to prolonged immobilization. Hindlimb suspension (unloading) causes muscle atrophy, delocalization, and posttranscriptional downregulation of nNOSμ protein to approximately 50% of wild-type levels (Tidball et al. 1998; Suzuki et al. 2007). Full nNOSμ expression requires its localization at the sarcolemma (Adams et al. 2000). Suzuki et al. (2007) reported that cytosolic nNOS activity is increased by unknown mechanisms and promotes hindlimb muscle atrophy through the activation of the forkhead transcription factor FoxO3 pathway (Figure 1). FoxO3 is a pivotal transcription factor involved in the regulation of muscle mass (Glass 2010). Elimination of all nNOSμ (but not sarcolemmal nNOSμ alone) attenuates muscle wasting and grip strength weakness in immobilized mice. Thus, under hindlimb unloading conditions, cytosolic nNOSμ promotes muscle atrophy through a FoxO3 pathway, making nNOSμ a therapeutic target for disuse-induced muscle atrophy (Suzuki et al. 2007). In agreement with an important role for nNOSμ regulating atrophy signaling, nNOSμ also mediates FoxO3-dependent muscle atrophy and autophagy resulting from the knockdown of sarcolemmal dihydropyridine receptor (Pietri-Rouxel et al. 2010).
The results of these human and rodent studies suggest that nNOSμ signaling reflects the external physiological exercise demands placed upon skeletal muscle, particularly from long-term activity (endurance exercise) or prolonged inactivity. nNOSμ levels thus directly reflect the exercise capacity of skeletal muscle. The molecular pathways linking muscle activity to nNOS expression remain to be determined. nNOSμ signaling pathways appear to play a pivotal role as a sensor of the external exercise demands placed on the skeletal muscles of both rodents and humans.
nNOS splice variants regulate skeletal muscle fatigue resistance
The data presented so far provide compelling evidence that skeletal muscle nNOSμ acts as an activity sensor. But why would increasing nNOS enhance muscle endurance? One explanation arises from recent reports that both nNOSμ and nNOSβ play a pivotal role in promoting skeletal muscle fatigue resistance. The nNOS regulation of fatigue has been studied using a minimally invasive in situ approach that preserves muscle innervation, vascularization, temperature, and tissue O2 concentrations (Percival et al. 2008, 2010). This approach is widely considered to be the most physiologically relevant method for measuring skeletal muscle fatigue and is especially useful given the known vasomodulatory function and exquisite oxygen sensitivity of nNOS enzymatic activity (Thomas et al. 1998; Eu et al. 2003; Stuehr et al. 2004; Allen et al. 2008).
In situ studies suggest that both nNOSμ and nNOSβ play a pivotal role in skeletal muscle fatigue resistance. When subjected to a fatigue protocol, nNOSμ-deficient tibialis anterior (TA) muscles from mice of a mixed B6/129 genetic background showed a 20% decrease in force-generating capacity, independent of muscle size (Percival et al. 2008). nNOSμ-deficient muscle also exhibited reduced muscle strength soon after fatigue, indicating impaired post-exercise recovery. However, nNOSμ-deficient muscles did eventually exhibit a full force recovery that was consistent with force deficits resulting from fatigue and not some other cause. Thus, nNOSμ appears to be necessary for sustaining muscle force output during and immediately after exercise. These findings are in agreement with Eu et al. (2003) who concluded that nNOSμ promotes contractility under low O2 concentrations characteristic of working muscle. These data suggest that the adaptive increases in nNOSμ expression in response to endurance exercise may enhance skeletal muscle fatigue resistance and thus promote exercise performance.
While nNOSμ-deficient muscles in a B6/129 strain were found to be susceptible to fatigue, the nNOSμ-deficient muscles from mice on a C57/Bl6 background exhibited normal fatigue resistance (Percival et al. 2010). These data suggest an important role for strain-specific genetic modifiers that modulate fatigue in the absence of nNOSμ. In agreement with these findings, murine exercise performance varies dramatically and in an unpredictable manner between wild-type mice from different genetic backgrounds (Lerman et al. 2002). The use of matched strain controls is thus crucial when measuring skeletal muscle fatigue. The α-syntrophin null mouse (C57/Bl6) that lacks sarcolemmal nNOSμ also exhibits normal skeletal muscle fatigue, despite well-established defects in attenuation of α-adrenergic receptor-mediated sympathetic vasoconstriction (Thomas et al. 2003; Percival et al. 2010). Thus, the inability to resist exercise-induced sympathetic vasoconstriction has no impact on skeletal muscle fatigue resistance (at least in the C57/Bl6 strain), arguing that the vasoregulatory function of nNOSμ does not play a significant role in muscle fatigue.
However, while muscles lacking nNOSμ from C57/Bl6 mice showed normal fatigability, Percival et al. (2010) observed that muscles lacking both nNOSμ and nNOSβ exhibited excessive fatigue, with an average 60% depression of maximal force output during an in situ fatigue protocol. While the loss of nNOSμ had no impact on fatigue in C57/Bl6 mice, the additional loss of nNOSβ also led to significant and prolonged post-exercise weakness after completion of the fatigue protocol. These data suggest an important and previously unrecognized role for nNOSβ in the regulation of skeletal muscle fatigue resistance in vivo.
Mechanisms by which nNOS splice variants regulate skeletal muscle fatigue
It is evident that during exercise, nNOS splice variants orchestrate two simultaneous and complementary pathways in active muscle. Sarcolemmal nNOSμ opposes sympathetic vasoconstriction and promotes blood delivery, while nNOSβ simultaneously promotes sustained force output. It is likely that nNOSβ deficiency negatively impacts the tightly controlled balance between energy supply and demand in working skeletal muscle by, for example, impacting mitochondrial integrity (Percival et al. 2010). Exaggerated fatigue can be caused by many factors, such as defects in metabolic energy pathways, shifts in fiber type, and aberrant cellular Ca2+ handling (Allen et al. 2008). What possible mechanisms could contribute to decreased resistance to fatigue in nNOSμ and nNOSβ-deficient skeletal muscle?
Fiber type composition is well known to influence skeletal muscle fatigue resistance. Skeletal muscle tissue is a mixture of muscle fiber types, namely, types I, IIa, IIx, and IIb (IIb in rodents only). These fiber types are identified by their myosin heavy chain isoform expression and have different metabolic and functional properties (Schiaffino et al. 2007). Slow type I fibers are generally more fatigue resistant than fast type II fibers due to their higher oxidative metabolic capacity. Although murine nNOSμ deficiency substantially increased type IIx fiber number at the expense of more oxidative type IIa fibers, this had no impact on muscle fatigue (Percival et al. 2010). Importantly, in contrast to mice, human type IIx fibers are the most prone to fatigue, leading to the prediction that human muscle lacking normal nNOSμ expression would be highly susceptible to fatigue (Keyser 2010). There was a 50% increase in fatigable type IIb fibers in murine muscles lacking both nNOSμ and nNOSβ, again at the expense of type IIa fibers that may contribute to fatigue (Percival et al. 2010). Together these data suggest that nNOSμ and nNOSβ promote an oxidative type II fiber type composition, but that these changes in fiber composition make little if any contribution to muscle fatigue in mice. Therefore, nNOS appears to regulate muscle fatigue through other mechanisms. Interestingly, mitochondrial defects, such as aberrant swelling suggestive of mitochondrial dysfunction, accompanied the shifts in type II fiber composition in nNOS-deficient muscle (Percival et al. 2010). Defective modulation of mitochondrial ATP energy supply offers a more plausible mechanism to account for the severe muscle fatigue in mice lacking all active nNOS.
Defects in Ca2+ release from the SR intracellular Ca2+ store play a critical role in skeletal muscle fatigue (Allen et al. 2008). The RyR1 plays a pivotal role in Ca2+ release from the SR during excitation–contraction coupling and is an important target of nNOS-derived NO in skeletal muscle (Lanner et al. 2010, Figure 1). RyR1 co-immunoprecipitates and colocalizes with cytoplasmic nNOSμ in human skeletal muscle (Salanova et al. 2008). NO can act directly on RyR1 through S-nitrosylation of cysteine thiols or indirectly through canonical cGMP-dependent pathways that activate cGMP-dependent kinase (PKG), which can in turn phosphorylate RyR1 (Suko et al.1993; Sun et al. 2001; Meissner 2010). Under basal conditions, RyR1 is endogenously nitrosylated (Eu et al. 2000, Figure 1). Nitrosylation is thought to activate RyR1, although this is still being debated (Stoyanovsky et al.1997; Eu et al. 2000; Cheong et al. 2005; Meissner 2010). The cytoplasmic amino terminus of RyR1 acts as a scaffold for regulatory proteins such as calstabin1, also known as FK506 binding protein 12 (FKBP12) (Kushnir et al. 2010). FKBP12 is required for normal exercise performance and is thought to stabilize RyR1 by increasing its closed state probability, thereby preventing “Ca2+ leak” (Bellinger et al. 2008). Pathogenic RyR1 Ca2+ release or a “Ca2+ leak” can lead to global perturbation of muscle Ca2+ handling, including depletion of SR Ca2+ stores and muscle damage and fatigue (Wang et al. 2005; Bellinger et al. 2008, Figure 1).
Mice subjected to a strenuous swimming regimen or humans subject to intensive cycling exercise exhibit similar RyR1 complex remodeling, including increased protein kinase A-mediated RyR1 phosphorylation and reduced of RyR1-associated phosphodiesterase 4D3 (PDE4D3) and FKBP12. Bellinger et al. (2008) observed that depletion of both PDE4D3 and FKBP12 led to impaired exercise performance. Exercise led to a progressive increase in RyR1 nitrosylation that was thought to promote FKBP12 disassociation, correlating well with expected increases in nNOSμ expression and activity. Reduction of the RyR1-associated FKBP12 association was suggested to destabilize RyR1, leading to the Ca2+ leak and resulting in protease-mediated muscle damage, weakness, and impaired exercise performance (Aracena et al. 2005; Bellinger et al. 2008, Figure 1). Although S-nitrosylation of RyR1 reduced the binding affinity of FKBP12 (calstabin1) for RyR1 in vitro, it did not prevent FKBP12 reassociation with the RyR1 complex in vivo (Aracena et al. 2005; Bellinger et al. 2008). Interestingly, both RyR2 hyponitrosylation resulting from nNOSμ deficiency and RyR1 hypernitrosylation resulting from excessive nNOS activity promote Ca2+ leakage (Gonzalez et al. 2007; Durham et al. 2008).
While the precise role of exercise-induced S-nitrosylation of RyR1 on exercise performance remains to be determined, these data in combination with the results from nNOS knockout mice suggest a possible mechanism by which nNOS-derived NO serves to regulate skeletal muscle fatigue resistance (Figure 1). During normal exercise, nNOS may be required to appropriately S-nitrosylate RyR1, thus promoting normal Ca2+ release and fatigue resistance. However, over-training exercise or pathophysiological stress (e.g., chronic activation of the fight-or-flight response or disease) results in excessive RyR1 complex destabilization, causing the Ca2+ leak (Bellinger et al. 2008). Abnormal Ca2+ release or leakage may also inappropriately lead to pathogenic nNOS activation, causing further oxidative and nitrosative stress including RyR1 hypernitrosylation, which in turn causes more Ca2+ leakage (in a feed-forward loop), leading to muscle damage, weakness, and exaggerated fatigue (Durham et al. 2008, Figure 1). In summary, the evidence to date suggests that both too little and too much nitrosylation may cause RyR leakage, and that both may cause skeletal muscle fatigue.
nNOSμ function in neuromuscular disease
Exercise intolerance is a common and debilitating feature of neuromuscular diseases, including Duchenne and Becker muscular dystrophies (DMD and BMD, respectively) and α-, β-, and γ-sarcoglycanopathies (Kobayashi et al. 2008; Lou et al. 2010). Becker patients often exhibit excessive exercise intolerance in response to mild exercise that is thought to result from sarcolemmal nNOSμ deficiency (Kobayashi et al. 2008). Duchenne patients struggle with everyday levels of activity, but especially with long periods of walking or stair climbing. Progressive muscle cell loss and weakness is a major contributing factor to reduced exercise capacity. Mild exercise intolerance is recapitulated by the mdx mouse model of DMD (Carter et al. 1995). DMD, BMD, α-, β-, and γ-sarcoglycanopathies as well as other neuromuscular diseases are characterized by reduced nNOSμ expression, particularly sarcolemmal nNOSμ, due to disruption of the dystrophin glycoprotein complex. (Brenman et al. 1995; Chang et al. 1996; Crosbie et al. 2002; Kobayashi et al. 2008; Finanger-Hedderick et al. 2011).
Recent gene therapy studies in the mdx mouse model of DMD show that nNOSμ deficiency contributes to impaired exercise performance. Minidystrophin cDNAs that restore sarcolemmal nNOSμ expression improved the ability of mdx muscle to oppose sympathetic vasoconstriction, improving active muscle perfusion and substantially increasing treadmill running exercise performance (Lai et al. 2009). Improved muscle perfusion reduced contraction-induced ischemic muscle damage and inflammation. A very similar minidystrophin cDNA lacking the nNOSμ targeting sequence did not enhance exercise performance to the same degree (Lai et al. 2009). The dystrophin minigenes that restore nNOSμ and thus enhance exercise performance are the most therapeutically effective DMD gene therapy constructs to date. In a separate study, cytosolic muscle-specific overexpression of nNOSα, an nNOS splice variant not normally expressed in skeletal muscle, substantially increased voluntary treadmill running times of mdx mice (Wehling-Hendricks et al. 2009). Taken together, these results suggest that cytosolic and sarcolemmal nNOS signaling pathways promote the exercise performance of mdx mice.
The exercise intolerance shown by DMD patients appears to arise in part from vascular dysfunction, which has long been suspected to be a critical pathogenic mechanism in DMD (Asai et al. 2007; Kobayashi et al. 2008; Lai et al. 2009; reviewed in Percival et al. 2011). Sarcolemmal nNOSμ deficiency leads to unopposed α-adrenergic receptor-mediated vasoconstriction in the active hindlimbs of mice mdx and contracting forearms of DMD patients (Thomas et al. 1998; Sander et al. 2000). As such, during exercise, active muscles lacking dystrophin and sarcolemmal nNOSμ experience repeated ischemic damage, including focal necrosis. Thus, nNOSμ acts to prevent ischemic muscle damage during muscle activity and may be one mechanism contributing to the exercise intolerance of DMD patients. From a therapeutic point of view, vascular dysfunction can be targeted with vasodilating drugs. Pharmacological augmentation of NO-cGMP signaling using phosphodiesterase 5 inhibitors to increase the blood supply to working mdx muscles can reduce post-exercise muscle damage and enhance nNOS-dependent mild exercise-induced inactivity in mdx mice (Asai et al. 2007; Kobayashi et al. 2008). Together, these data highlight the importance of the vasomodulatory function of sarcolemmal nNOSμ in promoting exercise performance in dystrophic muscle.
Mechanisms other than aberrant nNOSμ-mediated vasomodulation contribute to the poor exercise performance of dystrophin-deficient muscle. In humans, profound muscle wasting and generalized weakness means that muscles are unable to generate sufficient force for routine activity. In addition, muscles must work at a higher fraction of their maximal force-generating capacity, which will also contribute to exercise intolerance. The loss of normal nNOS expression and strict spatial control of activity may contribute to the loss of muscle bulk, particularly through the induction of the FoxO-mediated atrophy pathway (Suzuki et al. 2007; Percival et al. 2008; Percival et al. 2010). The loss of nNOSμ substantially increases type IIx fibers, which are more easily fatigued in humans. Thus, reductions in nNOSμ may promote more fatigue-susceptible muscles in dystrophy. Also, type IIx fibers are more susceptible to degeneration and are preferentially lost in DMD patients (Webster et al. 1988; Smerdu et al. 1994). Thus, another consequence of nNOSμ reduction may be to accelerate muscle wasting, thus indirectly decreasing exercise tolerance.
While nNOSμ deficiency does not impact muscle strength in nNOS knockouts, the loss of cytosolic nNOSμ expression increases muscle strength in dystrophin-deficient mdx skeletal muscle (Li et al. 2011a). This result suggests a negative gain-of-function for cytoplasmic nNOSμ in a dystrophic environment. Negative gain-of-function or toxic activity of nNOS is well described in other pathophysiological conditions, such as stroke and neurodegenerative disease, which are also characterized by abnormal Ca2+ handling and oxidative stress (Huang et al. 1994; Ayata et al. 1997).
One consequence of excessive nNOS activity is nitrosative stress resulting from hypernitrosylation of normal protein targets (Foster et al. 2009). nNOSμ-mediated RyR1 hypernitrosylation was observed in mdx mice and correlated with muscle weakness (Bellinger et al. 2009; Li et al. 2011a; Li et al. 2011b). nNOSμ depletion restored RyR1 nitrosylation to basal levels and enhanced muscle strength (Li et al. 2011a), suggesting that aberrant activation of cytosolic nNOSμ was leading to excessive nitrosylation of RyR1 and, consequently, suppressing muscle strength. RyR1 hypernitrosylation was also observed in mdx mice (Bellinger et al. 2009). Increased RyR1 nitrosylation led to reduced FKBP12 association, promoting the RyR1 channel Ca2+ leak (Bellinger et al. 2009). The remodeling of the RyR1 macromolecular complex in mdx muscle was similar to that observed in over-trained muscle (Bellinger et al. 2008, 2009). Pharmacological stabilization of the RyR1 channel with S107 increased FKBP12 association and reduced Ca2+ spark frequency to wild-type levels, which decreased calpain-mediated muscle damage and enhanced fatigue resistance (Bellinger et al. 2009). Interestingly, the RyR1 leak may lead to excessive nNOS activation and RyR1 hypernitrosylation, leading to even more Ca2+ leak and progressive muscle damage and weakness (Durham et al. 2008, Figure 1). Thus, nNOSμ mediated hypernitrosylation of RyR1 leading to calstabin 1 depletion and Ca2+ leak may provide a potent pathogenic mechanism in muscular dystrophy that contributes to a reduced exercise capacity of skeletal muscle.
Conclusion
In summary, nNOSμ and nNOSβ regulate fatigue, oxidative capacity, and blood delivery that are important for the optimal exercise capacity of skeletal muscle. Although the precise mechanisms for these roles played by nNOS remain to be established, it is likely that nNOS-mediated nitrosylation of RyR1 is important in many of these processes. In addition to its vasomodulatory and fatigue resistance roles, nNOSμ also acts as an activity sensor, mediating different signaling programs in response to sustained exercise or prolonged inactivity. Thus, nNOSμ appears to play an important role in the adaptation of muscle to new metabolic and functional demands. Under pathophysiological conditions, such as those found in the mdx mouse, nNOSμ has both protective and toxic activities. Sarcolemmal nNOSμ-deficiency in mdx mice causes ischemic damage and restricts exercise capacity. Cytosolic nNOSμ contributes to the RyR1 hypernitrosylation, enhancing the pathogenic Ca2+ leak. This defect in Ca2+ handling promotes progressive muscle damage, weakness, and fatigue. These data support a prominent role for nNOS in dystrophic pathogenesis and exercise intolerance, with implications for the treatment of muscular dystrophy. In conclusion, strict control of nNOS signal transduction is necessary for the peak exercise performance of skeletal muscle.
Acknowledgments
I wish to thank Drs Kimberley Craven, Stanley Froehner, Marvin Adams, and Nicholas Whitehead for insightful discussions and critical comment. Funding sources include the Muscular Dystrophy Association (69075), Parent Project Muscular Dystrophy, and NIH grants R01 AR056221, R01 NS33145, and PO1 NS046788.
Conflict of Interest
None
References
- Adams ME, Kramarcy N, Krall SP, Rossi SG, Rotundo RL, Sealock R, Froehner SC. Absence of alpha-syntrophin leads to structurally aberrant neuromuscular synapses deficient in utrophin. J Cell Biol. 2000;150:1385–1398. doi: 10.1083/jcb.150.6.1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams ME, Tesch Y, Percival JM, Albrecht DE, Conhaim JI, Anderson K, Froehner SC. Differential targeting of nNOS and AQP4 to dystrophin-deficient sarcolemma by membrane-directed alpha-dystrobrevin. J Cell Sci. 2008;121:48–54. doi: 10.1242/jcs.020701. [DOI] [PubMed] [Google Scholar]
- Allen DG, Lamb GD, Westerblad H. Skeletal muscle fatigue: cellular mechanisms. Physiol Rev. 2008;88:287–332. doi: 10.1152/physrev.00015.2007. [DOI] [PubMed] [Google Scholar]
- Aracena P, Tang W, Hamilton SL, Hidalgo C. Effects of S-glutathionylation and S-nitrosylation on calmodulin binding to triads and FKBP12 binding to type 1 calcium release channels. Antioxid Redox Signal. 2005;7:870–881. doi: 10.1089/ars.2005.7.870. [DOI] [PubMed] [Google Scholar]
- Asai A, Sahani N, Kaneki M, Ouchi Y, Martyn JA, Yasuhara SE. Primary role of functional ischemia, quantitative evidence for the two-hit mechanism, and phosphodiesterase-5 inhibitor therapy in mouse muscular dystrophy. PLoS One. 2007;2:e806. doi: 10.1371/journal.pone.0000806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayata C, Ayata G, Hara H, Matthews RT, Beal MF, Ferrante RJ, Endres M, Kim A, Christie RH, Waeber C, Huang PL, Hyman BT, Moskowitz MA. Mechanisms of reduced striatal NMDA excitotoxicity in type I nitric oxide synthase knock-out mice. J Neurosci. 1997;17:6908–6917. doi: 10.1523/JNEUROSCI.17-18-06908.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balon TW, Nadler JL. Evidence that nitric oxide increases glucose transport in skeletal muscle. J Appl Physiol. 1997;82:359–363. doi: 10.1152/jappl.1997.82.1.359. [DOI] [PubMed] [Google Scholar]
- Bassel-Duby R, Olson EN. Signaling pathways in skeletal muscle remodeling. Annu Rev Biochem. 2006;75:19–37. doi: 10.1146/annurev.biochem.75.103004.142622. [DOI] [PubMed] [Google Scholar]
- Bellinger AM, Reiken S, Dura M, Murphy PW, Deng SX, Landry DW, Nieman D, Lehnart SE, Samaru M, LaCampagne A, Marks AR. Remodeling of ryanodine receptor complex causes “leaky” channels: a molecular mechanism for decreased exercise capacity. Proc Natl Acad Sci USA. 2008;105:2198–2202. doi: 10.1073/pnas.0711074105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellinger AM, Reiken S, Carlson C, Mongillo M, Liu X, Rothman L, Matecki S, Lacampagne A, Marks AR. Hypernitrosylated ryanodine receptor calcium release channels are leaky in dystrophic muscle. Nat Med. 2009;15:325–330. doi: 10.1038/nm.1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bramble DM, Lieberman DE. Endurance running and the evolution of Homo. Nature. 2004;432:345–352. doi: 10.1038/nature03052. [DOI] [PubMed] [Google Scholar]
- Bredt DS, Snyder SH. Isolation of nitric oxide synthetase, a calmodulin-requiring enzyme. Proc Natl Acad Sci USA. 1990;87:682–685. doi: 10.1073/pnas.87.2.682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bredt DS, Hwang PM, Glatt CE, Lowenstein C, Reed RR, Snyder SH. Cloned and expressed nitric oxide synthase structurally resembles cytochrome P-450 reductase. Nature. 1991;351:714–718. doi: 10.1038/351714a0. [DOI] [PubMed] [Google Scholar]
- Brenman JE, Chao DS, Xia H, Aldape K, Bredt DS. Nitric oxide synthase complexed with dystrophin and absent from skeletal muscle sarcolemma in Duchenne muscular dystrophy. Cell. 1995;82:743–752. doi: 10.1016/0092-8674(95)90471-9. [DOI] [PubMed] [Google Scholar]
- Brenman JE, Chao DS, Gee SH, McGee AW, Craven SE, Santillano DR, Wu Z, Huang F, Xia H, Peters MF, Froehner SC, Bredt DS. Interaction of nitric oxide synthase with the postsynaptic density protein PSD-95 and alpha1-syntrophin mediated by PDZ domains. Cell. 1996;84:757–767. doi: 10.1016/S0092-8674(00)81053-3. [DOI] [PubMed] [Google Scholar]
- Carter GT, Wineinger MA, Walsh SA, Horasek SJ, Abresch RT, Fowler WM., Jr Effect of voluntary wheel-running exercise on muscles of the mdx mouse. Neuromuscul Disord. 1995;5:323–332. doi: 10.1016/0960-8966(94)00063-F. [DOI] [PubMed] [Google Scholar]
- Chang WJ, Iannaccone ST, Lau KS, Masters BS, McCabe TJ, McMillan K, Padre RC, Spencer MJ, Tidball JG, Stull JT. Neuronal nitric oxide synthase and dystrophin-deficient muscular dystrophy. Proc Natl Acad Sci USA. 1996;93:9142–9147. doi: 10.1073/pnas.93.17.9142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheong E, Tumbev V, Stoyanovsky D, Salama G. Effects of pO2 on the activation of skeletal muscle ryanodine receptors by NO: a cautionary note. Cell Calcium. 2005;38:481–488. doi: 10.1016/j.ceca.2005.07.001. [DOI] [PubMed] [Google Scholar]
- Crimi E, Ignarro LJ, Cacciatore F, Napoli C. Mechanisms by which exercise training benefits patients with heart failure. Nat Rev Cardiol. 2009;6:292–300. doi: 10.1038/nrcardio.2009.8. [DOI] [PubMed] [Google Scholar]
- Crosbie RH, Barresi R, Campbell KP. Loss of sarcolemma nNOS in sarcoglycan-deficient muscle. FASEB J. 2002;16:1786–1791. doi: 10.1096/fj.02-0519com. [DOI] [PubMed] [Google Scholar]
- Durham WJ, Aracena-Parks P, Long C, Rossi AE, Goonasekera SA, Boncompagni S, Galvan DL, Gilman CP, Baker MR, Shirokova N, Protasi F, Dirksen R, Hamilton SL. RyR1 S-nitrosylation underlies environmental heat stroke and sudden death in Y522S RyR1 knockin mice. Cell. 2008;133:53–65. doi: 10.1016/j.cell.2008.02.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eu JP, Sun J, Xu L, Stamler JS, Meissner G. The skeletal muscle calcium release channel: coupled O2 sensor and NO signaling functions. Cell. 2000;102:499–509. doi: 10.1016/S0092-8674(00)00054-4. [DOI] [PubMed] [Google Scholar]
- Eu JP, Hare JM, Hess DT, Skaf M, Sun J, Cardenas-Navina I, Sun QA, Dewhirst M, Meissner G, Stamler JS. Concerted regulation of skeletal muscle contractility by oxygen tension and endogenous nitric oxide. Proc Natl Acad Sci USA. 2003;100:15229–15234. doi: 10.1073/pnas.2433468100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finanger-Hedderick EL, Simmers JL, Soleimani A, Andres-Mateos E, Marx R, Files DC, King L, Crawford TO, Corse AM, Cohn RD. Loss of sarcolemmal nNOS is common in acquired and inherited neuromuscular disorders. Neurology. 2011;76:960–967. doi: 10.1212/WNL.0b013e31821043c8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster MW, Hess DT, Stamler JS. Protein S-nitrosylation in health and disease: a current perspective. Trends Mol Med. 2009;15:391–404. doi: 10.1016/j.molmed.2009.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frandsen U, Höffner L, Betak A, Saltin B, Bangsbo J, Hellsten Y. Endurance training does not alter the level of neuronal nitric oxide synthase in human skeletal muscle. J Appl Physiol. 2000;89:1033–1038. doi: 10.1152/jappl.2000.89.3.1033. [DOI] [PubMed] [Google Scholar]
- Glass DJ. Signaling pathways perturbing muscle mass. Curr Opin Clin Nutr Metab Care. 2010;13:225–229. doi: 10.1097/MCO.0b013e32833862df. [DOI] [PubMed] [Google Scholar]
- Gonzalez DR, Beigi F, Treuer AV, Hare JM. Deficient ryanodine receptor S-nitrosylation increases sarcoplasmic reticulum calcium leak and arrhythmogenesis in cardiomyocytes. Proc Natl Acad Sci USA. 2007;104:20612–20617. doi: 10.1073/pnas.0706796104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grady RM, Zhou H, Cunningham JM, Henry MD, Campbell KP, Sanes JR. Maturation and maintenance of the neuromuscular synapse: genetic evidence for roles of the dystrophin-glycoprotein complex. Neuron. 2000;25:279–293. doi: 10.1016/S0896-6273(00)80894-6. [DOI] [PubMed] [Google Scholar]
- Handschin C, Spiegelman BM. The role of exercise and PGC1alpha in inflammation and chronic disease. Nature. 2008;454:463–469. doi: 10.1038/nature07206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Z, Huang PL, Panahian N, Dalkara T, Fishman MC, Moskowitz MA. Effects of cerebral ischemia in mice deficient in neuronal nitric oxide synthase. Science. 1994;265:1883–1885. doi: 10.1126/science.7522345. [DOI] [PubMed] [Google Scholar]
- Kameya S, Miyagoe Y, Nonaka I, Ikemoto T, Endo M, Hanaoka K, Nabeshima Y, Takeda S. alpha1-syntrophin gene disruption results in the absence of neuronal-type nitric-oxide synthase at the sarcolemma but does not induce muscle degeneration. J Biol Chem. 1999;274:2193–2200. doi: 10.1074/jbc.274.4.2193. [DOI] [PubMed] [Google Scholar]
- Keyser RE. Peripheral fatigue: high-energy phosphates and hydrogen ions. PM R. 2010;2:347–358. doi: 10.1016/j.pmrj.2010.04.009. [DOI] [PubMed] [Google Scholar]
- Kobayashi YM, Rader EP, Crawford RW, Iyengar NK, Thedens DR, Faulkner JA, Parikh SV, Weiss RM, Chamberlain JS, Moore SA, Campbell KP. Sarcolemma-localized nNOS is required to maintain activity after mild exercise. Nature. 2008;456:511–515. doi: 10.1038/nature07414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kushnir A, Betzenhauser MJ, Marks AR. Ryanodine receptor studies using genetically engineered mice. FEBS Lett. 2010;584:1956–1965. doi: 10.1016/j.febslet.2010.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai Y, Thomas GD, Yue Y, Yang HT, Li D, Long C, Judge L, Bostick B, Chamberlain JS, Terjung RL, Duan D. Dystrophins carrying spectrin-like repeats 16 and 17 anchor nNOS to the sarcolemma and enhance exercise performance in a mouse model of muscular dystrophy. J Clin Invest. 2009;119:624–635. doi: 10.1172/JCI36612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanner JT, Georgiou DK, Joshi AD, Hamilton SL. Ryanodine receptors: structure, expression, molecular details, and function in calcium release. Cold Spring Harb Perspect Biol. 2010;2:a003996. doi: 10.1101/cshperspect.a003996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lerman I, Harrison BC, Freeman K, Hewett TE, Allen DL, Robbins J, Leinwand LA. Genetic variability in forced and voluntary endurance exercise performance in seven inbred mouse strains. J Appl Physiol. 2002;92:2245–2255. doi: 10.1152/japplphysiol.01045.2001. [DOI] [PubMed] [Google Scholar]
- Li D, Yue Y, Lai Y, Hakim CH, Duan D. Nitrosative stress elicited by nNOSμ delocalization inhibits muscle force in dystrophin-null mice. J Pathol. 2011;223:88–98. doi: 10.1002/path.2799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li D, Shin J-H, Duan D. INOS ablation does not improve specific force of the extensor digitorum longus muscle in dystrophin-deficient mdx4cv mice. PLoS One. 2011;6:e21618. doi: 10.1371/journal.pone.0021618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieberman DE, Bramble DM. The evolution of marathon running: capabilities in humans. Sports Med. 2007;37:288–290. doi: 10.2165/00007256-200737040-00004. [DOI] [PubMed] [Google Scholar]
- Lou JS, Weiss MD, Carter GT. Assessment and management of fatigue in neuromuscular disease. Am J Hosp Palliat Care. 2010;27:145–157. doi: 10.1177/1049909109358420. [DOI] [PubMed] [Google Scholar]
- McConell GK, Bradley SJ, Stephens TJ, Canny BJ, Kingwell BA, Lee-Young RS. Skeletal muscle nNOS mu protein content is increased by exercise training in humans. Am J Physiol Regul Integr Comp Physiol. 2007;293:R821–R828. doi: 10.1152/ajpregu.00796.2006. [DOI] [PubMed] [Google Scholar]
- Meissner G. Regulation of ryanodine receptor ion channels through posttranslational modifications. Curr Top Membr. 2010;66:91–113. doi: 10.1016/S1063-5823(10)66005-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Percival JM, Anderson KN, Gregorevic P, Chamberlain JS, Froehner SC. Functional deficits in nNOSmu-deficient skeletal muscle: myopathy in nNOS knockout mice. PLoS One. 2008;3:e3387. doi: 10.1371/journal.pone.0003387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Percival JM, Anderson KN, Huang P, Adams ME, Froehner SC. Golgi and sarcolemmal neuronal NOS differentially regulate contraction-induced fatigue and vasoconstriction in exercising mouse skeletal muscle. J Clin Invest. 2010;120:816–826. doi: 10.1172/JCI40736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Percival JM, Adamo CM, Beavo JA, Froehner SC. Evaluation of the therapeutic utility of phosphodiesterase 5A inhibition in the mdx mouse model of duchenne muscular dystrophy. Handb Exp Pharmacol. 2011;204:323–344. doi: 10.1007/978-3-642-17969-3_14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piétri-Rouxel F, Gentil C, Vassilopoulos S, Baas D, Mouisel E, Ferry A, Vignaud A, Hourdé C, Marty I, Schaeffer L, Voit T, Garcia L. DHPR alpha1S subunit controls skeletal muscle mass and morphogenesis. EMBO J. 2010;29:643–654. doi: 10.1038/emboj.2009.366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powers SK, Jackson MJ. Exercise-induced oxidative stress: cellular mechanisms and impact on muscle force production. Physiol Rev. 2008;88:1243–1276. doi: 10.1152/physrev.00031.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudnick J, Püttmann B, Tesch PA, Alkner B, Schoser BG, Salanova M, Kirsch K, Gunga HC, Schiffl G, Lück G, Blottner D. Differential expression of nitric oxide synthases (NOS 1–3) in human skeletal muscle following exercise countermeasure during 12 weeks of bed rest. FASEB J. 2004;18:1228–1230. doi: 10.1096/fj.03-0792fje. [DOI] [PubMed] [Google Scholar]
- Salanova M, Schiffl G, Rittweger J, Felsenberg D, Blottner D. Ryanodine receptor type-1 (RyR1) expression and protein S-nitrosylation pattern in human soleus myofibres following bed rest and exercise countermeasure. Histochem Cell Biol. 2008;130:105–118. doi: 10.1007/s00418-008-0399-6. [DOI] [PubMed] [Google Scholar]
- Sander M, Chavoshan B, Harris SA, Iannaccone ST, Stull JT, Thomas GD, Victor RG. Functional muscle ischemia in neuronal nitric oxide synthase-deficient skeletal muscle of children with Duchenne muscular dystrophy. Proc Natl Acad Sci USA. 2000;97:13818–13823. doi: 10.1073/pnas.250379497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiaffino S, Sandri M, Murgia M. Activity-dependent signaling pathways controlling muscle diversity and plasticity. Physiology (Bethesda) 2007;22:269–278. doi: 10.1152/physiol.00009.2007. [DOI] [PubMed] [Google Scholar]
- Silvagno F, Xia H, Bredt DS. Neuronal nitric-oxide synthase-mu, an alternatively spliced isoform expressed in differentiated skeletal muscle. J Biol Chem. 1996;271:11204–11208. doi: 10.1074/jbc.271.19.11204. [DOI] [PubMed] [Google Scholar]
- Smerdu V, Karsch-Mizrachi I, Campione M, Leinwand L, Schiaffino S. Type IIx myosin heavy chain transcripts are expressed in type IIb fibers of human skeletal muscle. Am J Physiol. 1994;267:C1723–C1728. doi: 10.1152/ajpcell.1994.267.6.C1723. [DOI] [PubMed] [Google Scholar]
- Song W, Kwak HB, Kim JH, Lawler JM. Exercise training modulates the nitric oxide synthase profile in skeletal muscle from old rats. J Gerontol A Biol Sci Med Sci. 2009;64:540–549. doi: 10.1093/gerona/glp021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamler JS, Meissner G. Physiology of nitric oxide in skeletal muscle. Physiol Rev. 2001;81:209–237. doi: 10.1152/physrev.2001.81.1.209. [DOI] [PubMed] [Google Scholar]
- Stoyanovsky D, Murphy T, Anno PR, Kim YM, Salama G. Nitric oxide activates skeletal and cardiac ryanodine receptors. Cell Calcium. 1997;21:19–29. doi: 10.1016/S0143-4160(97)90093-2. [DOI] [PubMed] [Google Scholar]
- Stuehr DJ, Santolini J, Wang ZQ, Wei CC, Adak S. Update on mechanism and catalytic regulation in the NO synthases. J Biol Chem. 2004;279:36167–36170. doi: 10.1074/jbc.R400017200. [DOI] [PubMed] [Google Scholar]
- Suko J, Maurer-Fogy I, Plank B, Bertel O, Wyskovsky W, Hohenegger M, Hellmann G. Phosphorylation of serine 2843 in ryanodine receptor-calcium release channel of skeletal muscle by cAMP-, cGMP- and CaM-dependent protein kinase. Biochim Biophys Acta. 1993;1175:193–206. doi: 10.1016/0167-4889(93)90023-I. [DOI] [PubMed] [Google Scholar]
- Sun J, Xin C, Eu JP, Stamler JS, Meissner G. Cysteine-3635 is responsible for skeletal muscle ryanodine receptor modulation by NO. Proc Natl Acad Sci USA. 2001;98:11158–11162. doi: 10.1073/pnas.201289098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki N, Motohashi N, Uezumi A, Fukada S, Yoshimura T, Itoyama Y, Aoki M, Miyagoe-Suzuki Y, Takeda S. NO production results in suspension-induced muscle atrophy through dislocation of neuronal NOS. J Clin Invest. 2007;117:2468–2476. doi: 10.1172/JCI30654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatchum-Talom R, Schulz R, McNeill JR, Khadour FH. Upregulation of neuronal nitric oxide synthase in skeletal muscle by swim training. Am J Physiol Heart Circ Physiol. 2000;279:H1757–H1766. doi: 10.1152/ajpheart.2000.279.4.H1757. [DOI] [PubMed] [Google Scholar]
- Thomas GD, Sander M, Lau KS, Huang PL, Stull JT, Victor RG. Impaired metabolic modulation of alpha-adrenergic vasoconstriction in dystrophin-deficient skeletal muscle. Proc Natl Acad Sci USA. 1998;95:15090–15095. doi: 10.1073/pnas.95.25.15090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas GD, Shaul PW, Yuhanna IS, Froehner SC, Adams ME. Vasomodulation by skeletal muscle-derived nitric oxide requires alpha-syntrophin-mediated sarcolemmal localization of neuronal nitric oxide synthase. Circ Res. 2003;92:554–560. doi: 10.1161/01.RES.0000061570.83105.52. [DOI] [PubMed] [Google Scholar]
- Tidball JG, Lavergne E, Lau KS, Spencer MJ, Stull JT, Wehling M. Mechanical loading regulates NOS expression and activity in developing and adult skeletal muscle. Am J Physiol. 1998;275:C260–C266. doi: 10.1152/ajpcell.1998.275.1.C260. [DOI] [PubMed] [Google Scholar]
- Vassilakopoulos T, Deckman G, Kebbewar M, Rallis G, Harfouche R, Hussain SN. Regulation of nitric oxide production in limb and ventilatory muscles during chronic exercise training. Am J Physiol Lung Cell Mol Physiol. 2003;284:L452–L457. doi: 10.1152/ajplung.00270.2002. [DOI] [PubMed] [Google Scholar]
- Wang X, Weisleder N, Collet C, Zhou J, Chu Y, Hirata Y, Zhao X, Pan Z, Brotto M, Cheng H, Ma J. Uncontrolled calcium sparks act as a dystrophic signal for mammalian skeletal muscle. Nat Cell Biol. 2005;7:525–530. doi: 10.1038/ncb1254. [DOI] [PubMed] [Google Scholar]
- Webster C, Silberstein L, Hays AP, Blau HM. Fast muscle fibers are preferentially affected in Duchenne muscular dystrophy. Cell. 1988;52:503–513. doi: 10.1016/0092-8674(88)90463-1. [DOI] [PubMed] [Google Scholar]
- Wehling-Henricks M, Oltmann M, Rinaldi C, Myung KH, Tidball JG. Loss of positive allosteric interactions between neuronal nitric oxide synthase and phosphofructokinase contributes to defects in glycolysis and increased fatigability in muscular dystrophy. Hum Mol Genet. 2009;18:3439–3451. doi: 10.1093/hmg/ddp288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu KY, Huso DL, Dawson TM, Bredt DS, Becker LC. Ntric oxide synthase in cardiac sarcoplasmic reticulum. Proc Natl Acad Sci USA. 1999;96:657–662. doi: 10.1073/pnas.96.2.657. [DOI] [PMC free article] [PubMed] [Google Scholar]