

Abstract
G protein-coupled receptors (GPCRs) are the largest class of eukaryotic cell-surface receptors and, over the last decade, it has become clear that they are capable of dimerisation. Whilst many biochemical and biophysical approaches have been used to study dimerisation, fluorescence techniques, including Förster resonance energy transfer and single molecule fluorescence, have been key players. Here we review recent contributions of fluorescence techniques to investigate GPCR dimers, including dimerisation in cell membranes and native tissues, the effect of ligand binding on dimerisation and the kinetics of dimer formation and dissociation. The challenges of studying multicomponent membrane protein systems have led to the development and refinement of many fluorescence assays, allowing the functional consequences of receptor dimerisation to be investigated and individual protein molecules to be imaged in the membranes of living cells. It is likely that the fluorescence techniques described here will be of use for investigating many other multicomponent membrane protein systems.
Keywords: G protein-coupled receptor, Dimer, Oligomerisation, FRET, Fluorescence, Single molecule fluorescence
Introduction
All organisms rely on membrane proteins to interact with their environment. In particular, cell-surface receptors perform vital roles in sensing environmental changes and eliciting the correct physiological response. In eukaryotes, the largest family of cell-surface receptors are G protein-coupled receptors (GPCRs) which possess seven transmembrane helices (TMs) (Luttrell 2008). GPCRs bind a wide variety of ligands, including hormones, peptides and nucleotides, resulting in activation of heterotrimeric G proteins (Gα, Gβ and Gγ). Gα and Gβγ subunits activate various downstream signalling cascades, dependent on the G protein composition (Oldham and Hamm 2008). Involvement in a wide variety of physiological processes has resulted in GPCRs becoming attractive drug targets—approximately 30 % of pharmaceuticals target these receptors (Jacoby et al. 2006).
GPCRs were initially believed to function as monomers activating G proteins in a 1:1 stoichiometry, and a number of studies indicate that this model is possible (Leitz et al. 2006; Whorton et al. 2007). However, there is much evidence that GPCRs can form dimeric, or higher-order oligomeric, complexes and, in the case of a number of receptors, it has become clear that dimerisation can have functional consequences (Ciruela et al. 2010a). It remains to be seen whether the apparently wide-spread ability of GPCRs to dimerise is universally associated with functional significance.
Many excellent reviews focus on the history of GPCR dimerisation and the controversies surrounding this concept, as well as describing the physiological effects of dimer formation in detail (Franco et al. 2007; Gurevich and Gurevich 2008; Ciruela et al. 2010a; Fuxe et al. 2010; Palczewski 2010). In this review, we will focus on recent contributions of various fluorescence techniques to our understanding of GPCR dimerisation, also considering the limitations of these techniques and future directions. We would like to point out that much of the evidence presented here builds upon fundamental biochemical and functional studies which are beyond the scope of this review.
Demonstrating GPCR dimers by RET
Resonance energy transfer (RET) has formed a cornerstone of the experimental techniques available to study GPCR dimerisation (Fig. 1). RET is the nonradiative transfer of energy from a donor to an acceptor, and the result is a decrease in donor emission and an increase in acceptor emission. The efficiency of RET (ERET) is inversely proportional to the sixth power of the distance between donor and acceptor molecules (R):
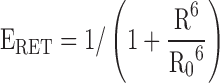 |
R0, which is the Förster distance, is the distance at which energy transfer between the donor and acceptor is 50 % efficient (Fig. 2a). Förster RET (FRET) can be used to study biological systems as many fluorophores have R0 values of approximately 5 nm and are thus sensitive over distances comparable to protein dimensions (Stryer 1978). A number of other factors affect ERET, including orientation of the donor and acceptor (Stryer and Haugland 1967), the donor quantum yield, the extinction coefficient of the acceptor (Stryer and Haugland 1967) and the spectral overlap between donor and acceptor. All of these variables influence the choice of fluorophore pairs for FRET. Analysis of FRET is complicated by photobleaching, crosstalk (direct excitation of the acceptor by the wavelength of light used to excite the donor) and bleedthrough (overlap of the donor and acceptor emission spectra). Bioluminescent RET (BRET) in which the donor is luciferase and the acceptor a variant of green fluorescent protein (GFP) eliminates some of these issues. For a comprehensive review of FRET and its variants, see Ciruela et al. (2010b).
Fig. 1.

Fluorescence techniques for studying G protein-coupled receptors (GPCR) dimerisation. In each case, a dimeric GPCR complex is shown with the fluorophores used to monitor receptor association. See the main text for detailed explanations of the techniques. FRET Förster resonance energy transfer, BRET bioluminescent RET, TR-FRET time-resolved FRET, homoFRET FRET between identical fluorophores, CODA-RET complemented donor–acceptor RET, BiFC bimolecular fluorescence complementation, CFP cyan, YFP yellow fluorescent protein, Luc luciferase, C- C-terminal, – N-terminal, Gα alpha subunit heterotrimeric G protein, Snap SNAP-tags
Fig. 2.

FRET as a molecular ruler. a FRET efficiency (E RET; and hence the signal) is proportional to the distance between the fluorophores. R 0 Förster distance, R distance between donor and acceptor molecules. b Schematic representation of a GPCR dimer in which monomers are labelled at the intracellular surfaces of transmembrane helix 3 (red star) or 5 (green star). Intracellular loops are shown between helices. Multiple pairs of fluorophores at different locations can be used to build a model of the dimer and observe conformational changes upon ligand binding
In early GPCR FRET experiments, receptors were fused to either cyan (CFP) or yellow fluorescent protein (YFP), both derived from GFP. FRET (Fig. 1a; see, for example, Overton and Blumer 2000; Wurch et al. 2001; Latif et al. 2002; Dinger et al. 2003) and BRET (Fig. 1b; see, for example, Angers et al. 2000; James et al. 2006) experiments both demonstrated dimerisation/oligomerisation of GPCRs (reviewed by Milligan et al. 2003). One drawback of these early experiments is the inability to distinguish between cell-surface and intracellular receptors, a particular issue with heterologously expressed receptors which may be at higher concentrations than in native systems. An alternative can be to use purified receptors reconstituted into native lipids (Harding et al. 2009), which enables precise control of protein concentration and lipid composition so that the lipid dependence of dimerisation can be investigated. However this approach is not possible for many GPCRs due to limitations with expression and purification.
Monitoring cell-surface dimerisation of GPCRs
Studies of cell-surface receptor oligomerisation have made use of time-resolved FRET (TR-FRET) with Eu3+ as donor and Alexa Fluor 647, allophycocyanin or d2 as acceptor. The emission fluorescence of Eu3+ cryptate is long-lived, allowing FRET measurement after endogenous fluorophores have finished emitting, and it has a very limited overlap with acceptor emission, dramatically increasing the signal:noise ratio. Receptor-specific antibodies labelled with either Eu3+ or an appropriate acceptor produce TR-FRET upon binding to GPCR dimers (Fig. 1c), and extracellular N-terminal epitopes ensure that only cell-surface receptors are probed. δ-Opiod receptor dimers (McVey et al. 2001) and γ-aminobutyrate B1 and B2 receptor heterodimers (Maurel et al. 2004) have been detected using this approach. As with all fluorescence techniques, there is a need for caution when using TR-FRET. Firstly, bivalent antibodies may lead to the stabilisation of large complexes. Secondly, immunoglobulins are large (approx. 150 kDa, 160 Å long), which may sterically hinder oligomer assembly or, conversely, increase FRET due to random collisions. Finally, the location of the fluorophore on the antibody rather than directly on the GPCR increases the uncertainty of the distance between fluorophores, which is likely to depend upon the orientation of the antibody–receptor complex.
As an alternative to antibodies, SNAP-tags can be engineered into the N-terminus of the receptor (Maurel et al. 2008). SNAP-tags covalently react with benzyl-guanines (BG) which can have fluorophores bound to the benzyl group (Fig. 1c). Provided that these modified BGs are membrane-impermeable, only cell-surface receptors are labelled. Through strict control of fluorophore–BG concentrations, it is possible to label equally a population of SNAP–GPCRs with two different fluorophores, allowing TR-FRET. SNAP-tags are approximately two-thirds the size of GFP (much smaller than immunoglobulins), thereby reducing the possibility of artefactual effects. This technology enabled confirmation of the obligate heterodimerisation of gamma-aminobutyric acid (GABA) B receptors and demonstration of the dimerisation of these heterodimers, in contrast to metabotropic glutamate receptors (mGluR) which form only heterodimers (Maurel et al. 2008). In this study, various Class A receptors produced TR-FRET in an analogous system giving a two- to threefold difference in signal compared to GABA receptors. This observation could be explained by variation in the distance between the fluorophores but could also indicate that only a proportion of Class A receptors are dimeric, compared to obligate GABA heterodimers.
Constitutive dimerisation of a serotonin 5-HT1A-eYFP receptor fusion was demonstrated via homoFRET (Paila et al. 2011), i.e. FRET in which the same fluorophore is used as both donor and acceptor (Fig. 1d). The excitation and emission spectra of the fluorophore must have considerable overlap (a small Stokes’ shift), and homoFRET is revealed by a new decay process in TR-fluorescence anisotropy kinetics and an associated decrease in steady state fluorescence anisotropy. The rotational correlation time of eYFP in this construct was estimated to be 11 ns, which is in excess of the fluorescence lifetime of 3.3 ns, ensuring that changes in anisotropy are due to homoFRET (Paila et al. 2011). One caveat is that fluorescence anisotropy is affected by the rotational dynamics of the fluorophore itself which is further influenced by fluorophore size and environment viscosity.
One disadvantage of FRET-based techniques is the difficulty in distinguishing dimers from higher-order oligomers. To address this, a technique based on fluorescence recovery after photobleaching (FRAP) was developed (Dorsch et al. 2009). FRAP measures the diffusion of non-photobleached fluorescent molecules into a region which has been bleached irreversibly (Fig. 3). Association of the fluorescent component with another molecule will retard diffusion and decrease the rate and extent of FRAP. β-adrenergic receptors (β-AR) were tagged with N-terminal YFP or C-terminal CFP/Cerulean and YFP-tagged receptors immobilised via a polyclonal anti-YFP antibody (monoclonal antibodies were not sufficient to immobilise receptors, presumably due to 1:1 stoichiometry) and areas of membrane photobleached. FRAP of the co-expressed YFP-βAR and βAR-CFP/Cerulean was monitored post-bleaching. β1-AR-CFP showed FRAP whereas, as expected, only a small fraction of YFP-β1-AR was mobile after antibody immobilisation. Immobilisation of YFP-β1-AR did result in approximately a 15 % restriction in mobility of β1-AR-CFP, indicating a specific, but unstable and transient, interaction. Neither YFP-β2-AR nor β2-AR-Cerulean showed significant FRAP after antibody treatment and photobleaching, indicating a stable interaction. This interaction could be oligomeric since even a 3.5× excess of β2-Cerulaean was effectively immobilised by the antibody-YFP-β2-AR complex.
Fig. 3.

Schematic representation of fluorescence recovery after photobleaching (FRAP). Anti-YFP antibodies added to cells containing YFP-GPCR and GPCR–CFP fusions cause immobilisation of the YFP-tagged receptors. A region of the cell is then photobleached (dashed circle) and, after a period of time, the bleached region is monitored for fluorescent molecules which have diffused from outside. In the case of monomeric receptors, FRAP of the GPCR–CFP molecules, but not of the YFP–GPCR molecules, will occur. For dimeric receptors, no FRAP will be observed as the GPCR–CFP molecules are immobilised via the YFP-tagged receptors
Fluorescent ligands for probing GPCR dimerisation in native tissues
Given the controversial nature of GPCR dimerisation, it is vital to demonstrate dimerisation in native cells. Many expression systems result in non-physiological receptor concentrations, and engineering cell lines to introduce tagged receptors is challenging. Without the ability to label the cytoplasm/TM regions, an alternative is to use labelled ligands. The availability and suitability of fluorescent ligands depends on two main factors—the chemistry to manufacture them and the ability to label whilst retaining affinity and efficacy. A large number of fluorescent ligands are available for GPCRs (Kuder and Kiec-Kononowicz 2008), but there will no doubt be receptors for which production of such ligands is challenging. It should be noted that in this type of experiment it is impossible to study dimers in the unliganded state, a condition which may be of particular significance when ligand binding can promote or inhibit dimer formation (Lukasiewicz et al. 2007; Pioszak et al. 2010). Finally, it may not be possible to study receptors expressed at low levels or cells which express multiple receptors for the same ligand. Nevertheless, the use of tagged receptors remains a remarkably attractive approach for studying physiologically relevant dimerisation (Ambrosio and Lohse 2010).
In a recent study, Albizu et al. (2010) validated the use of ligand–ligand TR-FRET to detect receptor dimers (Fig. 1e). Ligands carried either the lanthanide donor or Alexa acceptor. Cells heterologously expressing vasopressin or oxytocin receptors produced TR-FRET upon binding of two differentially tagged antagonists, but much less TR-FRET upon exposure to two differentially tagged agonists. This result is consistent with previous data demonstrating that a single agonist binds to these dimeric GPCRs (Albizu et al. 2006). These data show the importance of validating a number of ligands—for example, if only one agonist binds per dimer, FRET will not be observed with a labelled agonist even if dimeric receptors are present. Crucially, this study also demonstrated dimerisation in native tissues. TR-FRET was clearly observed when isolated membrane fractions from mammary glands of lactating rats, which express the oxytocin receptor at a relatively high level (1–3 pmol/mg protein), were exposed to the fluorescent antagonists. Organ patches were also analysed to ensure that the receptors were at the cell membrane. This would appear to be a convincing demonstration of GPCR dimerisation in native tissues.
Effects of ligand binding on GPCR dimerisation
GPCR dimerisation may be promoted or inhibited by different ligands. Although many GPCR dimers appear to be unaffected by agonists or antagonists (e.g. Herrick-Davis et al. 2007; Kasai et al. 2011; Paila et al. 2011), there are reports of ligand-dependent oligomerisation (e.g. Harikumar and Miller 2008). It should also be noted that different methods, such as crosslinking compared to FRET, can show different ligand-dependence, the reasons for which are not always obvious.
The parathyroid hormone receptor (PTH1R) is a Class B GPCR with a large extracellular domain (ECD). A whole-cell BRET approach with PTH1R fused to C-terminal luciferase or YFP revealed that PTH1R forms a constitutive dimer which is mediated by the ECD (Pioszak et al. 2010). Addition of the agonist PTH disrupted the dimer, suggesting that monomers are the signalling unit. Although BRET monitors the entire cell, due to the occurrence of ligand-dependence, it is likely that dimers are present in the membrane. This was in fact confirmed by FRET between CFP/YFP-tagged PTH1R at the cell surface.
Fluorescence lifetime microscopy has been used to investigate the ligand-dependence of adenosine A2a and serotonin 5-HT1A receptors (Lukasiewicz et al. 2007). The fluorescence lifetime is the time a fluorophore remains excited before it emits a photon (frequently a few nanoseconds). This lifetime is independent of the number of fluorescent molecules present and is altered by the inter-probe distance but also by the energy transfer between the probe and the environment. Lukasiewicz et al. (2007) measured fluorescence lifetime using time-correlated single photon counting and calculated FRET efficiency, revealing receptor homo-oligomerisation, which was diminished by antagonists (reduced FRET efficiency) and enhanced by agonists (increased FRET efficiency). These results are in contrast with those obtained in homoFRET studies (Paila et al. 2011).
Conformation of GPCR oligomers
Despite recent GPCR crystal structures (Rasmussen et al. 2007; Jaakola et al. 2008; Lebon et al. 2011; Rasmussen et al. 2011a, b; Rosenbaum et al. 2011; Warne et al. 2011; Xu et al. 2011) little is known of the structure of GPCR dimers. One crystal structure features both parallel and (non-physiological) antiparallel dimers (Hanson et al. 2008), although it is uncertain how dimerisation may be influenced by crystal packing. Fluorescence techniques are beginning to resolve the oligomer architecture.
Single-cysteine mutants of the β2 adrenergic receptor have been purified, labelled with Cy3 or Cy5 fluorophores and reconstituted into lipid vesicles (Fung et al. 2009). Three different labelling sites were selected to allow triangulation of helices within the dimer (Fig. 2). FRET efficiency between labels on TM6 and helix 8 increased a small amount upon the addition of agonist or neutral antagonist, but the inverse agonist produced a much larger increase. Increases in FRET efficiency can be multifactorial (see above), but in this case it seems most likely due to reorientation of the protomers (or small movements within the protomers themselves) or an increase in the number of receptors per oligomer. FRET saturation experiments (altering the donor:acceptor ratio) demonstrated that, in the case of the inverse agonist, this increase is likely due to the formation of higher-order oligomers, which may exclude other signalling components from the receptors, thereby providing a possible mechanism for inverse agonism.
A similar experiment used the constitutively dimeric mGluR as a model (Yanagawa et al. 2011). Introduction of mCerulean or Venus fluorophores as fusions into the cytoplasmic loops produced FRET upon dimerisation. Agonist-binding altered FRET intensity due to reorganisation of the dimer; all loops become closer upon activation. Heterodimers, in which one receptor is unable to bind the agonist, revealed a synergistic effect on dimer rearrangement with the binding of two glutamates. The addition of relatively large fluorophores may cause abnormal conformations in the dimer due to steric hindrances, but this approach is justified due to the inherent technical limitations of labelling proteins in the cytoplasm of living cells compared to the relative flexibility of in vitro labelling.
Functional effects of dimerisation
Using confocal microscopy and a perfusion system to achieve infinite dilution, allosteric interactions at adenosine-A3 receptors have been demonstrated in live cells (May et al. 2011). The dissociation rate of the fluorescent agonist ABA-X-BY630 was increased by nine- to 19-fold upon the addition of orthosteric agonists or antagonists, indicating allosterism across the dimer—the addition of orthosteric ligands will not affect dissociation kinetics at monomers. Co-expression of a non-binding A3 receptor mutant decreased this effect, indicating true allosterism. Such allosterism is likely to be of significance in pharmacology and drug development.
As well as forming homodimers, many GPCRs are capable of heterodimerisation, and evidence of the functional relevance of this process is emerging (Birdsall 2010). A recent study of the dopamine D1R and D2R receptors used CODA-RET (complemented donor-acceptor RET; Fig. 1f) to directly monitor the effects of heterodimerisation (Urizar et al. 2011). Two domains of luciferase were split between the C-termini of two receptors so only heterodimers produce functional donor. The mVenus acceptor was engineered into the Gα sequence in a position where BRET between receptors and Gα correlates with activation. The authors found that the potency of the agonist to activate Gαi1 was increased by approximately tenfold at the D1R–D2R heterodimers compared to D2R homodimers. This direct demonstration of allosteric effects at the dimer interface indicates that heterodimers are likely to play significant physiological roles in modulating signal transduction. An extension to this technique has been proposed (Piehler 2011) using a split fluorescent protein as the donor, allowing FRET and visualisation of the signalling complexes.
GPCRs can also form heterodimers of dimers, such as μ- and δ-opiod receptors (μOR/δOR) (Golebiewska et al. 2011). Prolonged exposure to morphine causes the desensitisation of μOR, although FRET approaches show no large effects on plasma membrane localisation or G protein-coupling of the receptors. In contrast, if δORs are co-expressed, morphine exposure increases the diffusion rate of eYFP–μOR. Using single cell fluorescence imaging on live cells along with fluorescence correlation spectroscopy, the authors demonstrate that eYFP–μOR and eCFP–δOR form a tetrameric arrangement which is dissociated by morphine, thereby providing a possible role for heterodimerisation in morphine sensitivity.
FRET approaches have also been used to demonstrate a possible role for heterodimerisation in altering muscarinic signalling in the brain (Boyer et al. 2009). This study combined TIRF microscopy with acceptor photobleaching of CFP/YFP-tagged receptors to demonstrate that M2 muscarinic receptors can heterodimerise with GABA B. In FRET-producing dimers, selective photobleaching of the YFP acceptor results in dequenching of the CFP donor. For a full review of the technical requirements for such an assay, see Boyer and Slesinger (2010). Total internal reflection fluorescence (TIRF) microscopy (Fig. 4) is an attractive technique as the evanescent field only illuminates molecules close to the coverslip, reducing intracellular fluorescence (Mattheyses et al. 2010).
Fig. 4.

Total internal reflection fluorescence (TIRF) microscopy for monitoring GPCR dimerisation. Unlike epifluorescence in which the laser is directed perpendicular to the coverslip, causing illumination of the entire sample volume, TIRF directs the laser at angle θ, resulting in illumination of only approximately 100 nm of the sample. When combined with FRET, this ensures that only molecules close to the coverslip are excited (red asterisk), as opposed to in epifluorescence when both intracellular and cell-surface populations are excited
Dynamics of GPCR dimers
TIRF microscopy and fluorophore-labelled ligands allow spatiotemporal resolution of single GPCRs in vivo. This is of particular interest in studies on the monomer–dimer equilibrium. In single molecule tracking studies, it is vital that the fluorophore chosen has a high photobleaching resistance, that the dimer be able to bind two molecules of the fluorescent ligand and that the ligand not induce receptor internalisation, causing receptors to leave the evanescent field. It should be noted that it is impossible to distinguish two molecules if they are separated by a distance below the Abbe diffraction limit (normally approx. 200 nm); additional controls are required to ensure that true dimers are formed and that the observations do not simply reflect the close proximity of two monomers.
Using such an approach, dimerisation of the M1 muscarinic receptors was studied using the antagonist telenzepine labelled with either Cy3B or Alexa 488 (Hern et al. 2010). The high affinity and low off-rate of this ligand ensured that >97 % of receptors could be labelled and the unbound ligand washed away. Receptors were expressed at near physiological levels to avoid the effects from receptor crowding. Single particles labelled with one fluorescent ligand were identified and tracked and the brightness of the spots recorded over time. Spots generally either had a normalised intensity of 1 or 2, which is consistent with the presence of monomers or dimers; no spots with an intensity of greater than 2 (oligomers) were observed. Some spots underwent 1 → 2, 2 → 1, 1 → 2 → 1 or 2 → 1 → 2 transitions, indicative of reversible dimer formation. In an extension of this experiment, both labelled antagonists were added at concentrations to result in 1:1 receptor labelling. Using dual-colour TIRF microscopy, the authors tracked spots to look for coincidence. Given random walk diffusion, the likelihood of two spots converging for 10 frames is <<1 %. However, 20 % of tracks showed such behaviour, and dual-labelled spots diffused more slowly, both behaviours consistent with dimer formation. At any one time, approximately 30 % of molecules were present as dimers, with a dissociation rate of 1.3 s-1 (dimer lifetime approx. 0.5 s).
A similar approach was used to calculate kinetic constants for the formation of N-formyl peptide receptor (FPR) dimers (Kasai et al. 2011). Using Alexa 594-labelled N-formyl hexa-amino-acid peptide (FP), the authors monitored single FPR molecules. However, as FP caused internalisation of FPR, the authors used a D71A mutant which does not activate G proteins, become phosphorylated or internalised. This study controlled for incomplete fluorescence of the labelled ligand, monomer-like populations (dimers with a single fluorescent ligand) and dimer-like populations (overlapped monomers). Analysis of spot intensity revealed monomers and dimers, and further analysis resulted in the calculation of a 2D-KD of 3.6 copies/μm2; at physiological concentrations of 2.1 copies/μm2, 41 % of receptors were in dimers. Using particle tracking, the authors calculated that monomers converted into dimers every 150 ms and dimers dissociated in 91 ms. To support the formation of receptor dimers, the authors used a bimolecular fluorescence complementation (BiFC) approach in which the N- and C-termini of YFP are fused individually to the C-terminus of FPR (Fig. 1g). FPR dimerisation was found to result in functional reconstitution of YFP—single fluorescent spots were detected in cell membranes expressing both constructs. Despite some contribution of the YFP domains to the affinity of the dimeric complex, FPR dimerisation was the major driving force in association.
The results of both studies demonstrate the transient nature of dimeric complexes and similar proportions of receptors in the dimeric state. These kinetic parameters are likely to be extremely valuable for future physiological and computational studies. As the 2D-KD calculated (Kasai et al. 2011) is close to physiological expression levels, small changes in receptor concentration will have significant effects on dimer proportions. It is likely that different GPCRs will exist as predominantly monomers or dimers/oligomers depending on their affinity and expression levels.
Conclusions and future perspectives
A battery of fluorescence-based approaches has been applied to the study of GPCR dimers, resulting in a great deal of progress. Both RET and single molecule fluorescence studies report that molecules are within a certain distance but not necessarily in direct contact. Recent advances in microscopy and analysis have enabled resolution below the diffraction limit (Eggeling et al. 2009; Biteen 2011), which will undoubtedly be of use to the GPCR community, furthering the single molecule studies described here and extending analysis to a wider range of receptors. In general, it appears that dimers are transient, and it is critical to explore them on the single molecule level to support evidence obtained in ensemble studies.
Now that it is largely accepted that dimerisation occurs, it is vital that the functional consequences are explored, and fluorescence is at the heart of such studies. Techniques such as CODA-RET (Urizar et al. 2011) provide an exquisite method to study only dimers of interest and report upon G protein activation without the need to study downstream signalling, which can be subject to crosstalk between signalling pathways. Models of GPCR activation have moved on from “one ligand, one receptor, one G protein”, and dimerisation provides a mechanism to influence signalling in vivo. Since GPCRs are such important pharmaceutical targets, it is likely that drug development will be influenced by dimerisation and that fluorescence can be applied at all levels–ligand, receptor and G protein. The future for fluorescence studies of GPCR dimerisation is bright.
Acknowledgments
This work was supported by the Biotechnology and Biological Sciences Research Council (BBSRC; grant number BB/G019738/1).
Conflict of interest
None
References
- Albizu L, Balestre MN, Breton C, Pin JP, Manning M, Mouillac B, Barberis C, Durroux T. Probing the existence of G protein-coupled receptor dimers by positive and negative ligand-dependent cooperative binding. Mol Pharmacol. 2006;70:1783–1791. doi: 10.1124/mol.106.025684. [DOI] [PubMed] [Google Scholar]
- Albizu L, Cottet M, Kralikova M, Stoev S, Seyer R, Brabet I, Roux T, Bazin H, Bourrier E, Lamarque L, Breton C, Rives ML, Newman A, Javitch J, Trinquet E, Manning M, Pin JP, Mouillac B, Durroux T. Time-resolved FRET between GPCR ligands reveals oligomers in native tissues. Nat Chem Biol. 2010;6:587–594. doi: 10.1038/nchembio.396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ambrosio M, Lohse MJ. Microscopy: GPCR dimers moving closer. Nat Chem Biol. 2010;6:570–571. doi: 10.1038/nchembio.409. [DOI] [PubMed] [Google Scholar]
- Angers S, Salahpour A, Joly E, Hilairet S, Chelsky D, Dennis M, Bouvier M. Detection of beta 2-adrenergic receptor dimerization in living cells using bioluminescence resonance energy transfer (BRET) Proc Natl Acad Sci USA. 2000;97:3684–3689. doi: 10.1073/pnas.060590697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birdsall NJ. Class A GPCR heterodimers: evidence from binding studies. Trends Pharmacol Sci. 2010;31:499–508. doi: 10.1016/j.tips.2010.08.003. [DOI] [PubMed] [Google Scholar]
- Biteen J. Moving toward the future of single-molecule-based super-resolution imaging. Biopolymers. 2011;95:287–289. doi: 10.1002/bip.21592. [DOI] [PubMed] [Google Scholar]
- Boyer SB, Slesinger PA. Probing novel GPCR interactions using a combination of FRET and TIRF. Commun Integr Biol. 2010;3:343–346. doi: 10.4161/cib.3.4.11764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyer SB, Clancy SM, Terunuma M, Revilla-Sanchez R, Thomas SM, Moss SJ, Slesinger PA. Direct interaction of GABAB receptors with M2 muscarinic receptors enhances muscarinic signaling. J Neurosci. 2009;29:15796–15809. doi: 10.1523/JNEUROSCI.4103-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciruela F, Vallano A, Arnau JM, Sanchez S, Borroto-Escuela DO, Agnati LF, Fuxe K, Fernandez-Duenas V. G protein-coupled receptor oligomerization for what? J Recept Signal Transduct Res. 2010;30:322–330. doi: 10.3109/10799893.2010.508166. [DOI] [PubMed] [Google Scholar]
- Ciruela F, Vilardaga JP, Fernandez-Duenas V. Lighting up multiprotein complexes: lessons from GPCR oligomerization. Trends Biotechnol. 2010;28:407–415. doi: 10.1016/j.tibtech.2010.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinger MC, Bader JE, Kobor AD, Kretzschmar AK, Beck-Sickinger AG. Homodimerization of neuropeptide y receptors investigated by fluorescence resonance energy transfer in living cells. J Biol Chem. 2003;278:10562–10571. doi: 10.1074/jbc.M205747200. [DOI] [PubMed] [Google Scholar]
- Dorsch S, Klotz KN, Engelhardt S, Lohse MJ, Bunemann M. Analysis of receptor oligomerization by FRAP microscopy. Nat Methods. 2009;6:225–230. doi: 10.1038/nmeth.1304. [DOI] [PubMed] [Google Scholar]
- Eggeling C, Ringemann C, Medda R, Schwarzmann G, Sandhoff K, Polyakova S, Belov VN, Hein B, von Middendorff C, Schonle A, Hell SW. Direct observation of the nanoscale dynamics of membrane lipids in a living cell. Nature. 2009;457:1159–1162. doi: 10.1038/nature07596. [DOI] [PubMed] [Google Scholar]
- Franco R, Casado V, Cortes A, Ferrada C, Mallol J, Woods A, Lluis C, Canela EI, Ferre S. Basic concepts in G-protein-coupled receptor homo- and heterodimerization. ScientificWorldJournal. 2007;7:48–57. doi: 10.1100/tsw.2007.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fung JJ, Deupi X, Pardo L, Yao XJ, Velez-Ruiz GA, Devree BT, Sunahara RK, Kobilka BK. Ligand-regulated oligomerization of beta(2)-adrenoceptors in a model lipid bilayer. EMBO J. 2009;28:3315–3328. doi: 10.1038/emboj.2009.267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuxe K, Marcellino D, Borroto-Escuela DO, Frankowska M, Ferraro L, Guidolin D, Ciruela F, Agnati LF. The changing world of G protein-coupled receptors: from monomers to dimers and receptor mosaics with allosteric receptor-receptor interactions. J Recept Signal Transduct Res. 2010;30:272–283. doi: 10.3109/10799893.2010.506191. [DOI] [PubMed] [Google Scholar]
- Golebiewska U, Johnston JM, Devi L, Filizola M, Scarlata S. Differential response to morphine of the oligomeric state of mu-opioid in the presence of delta-opioid receptors. Biochemistry. 2011;50:2829–2837. doi: 10.1021/bi101701x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurevich VV, Gurevich EV. How and why do GPCRs dimerize? Trends Pharmacol Sci. 2008;29:234–240. doi: 10.1016/j.tips.2008.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson MA, Cherezov V, Griffith MT, Roth CB, Jaakola VP, Chien EY, Velasquez J, Kuhn P, Stevens RC. A specific cholesterol binding site is established by the 2.8 A structure of the human beta2-adrenergic receptor. Structure. 2008;16:897–905. doi: 10.1016/j.str.2008.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding PJ, Attrill H, Boehringer J, Ross S, Wadhams GH, Smith E, Armitage JP, Watts A. Constitutive dimerization of the G-protein coupled receptor, neurotensin receptor 1, reconstituted into phospholipid bilayers. Biophys J. 2009;96:964–973. doi: 10.1016/j.bpj.2008.09.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harikumar KG, Miller LJ. Monitoring the state of cholecystokinin receptor oligomerization after ligand binding using decay of time-resolved fluorescence anisotropy. Ann N Y Acad Sci. 2008;1144:21–27. doi: 10.1196/annals.1418.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hern JA, Baig AH, Mashanov GI, Birdsall B, Corrie JE, Lazareno S, Molloy JE, Birdsall NJ. Formation and dissociation of M1 muscarinic receptor dimers seen by total internal reflection fluorescence imaging of single molecules. Proc Natl Acad Sci USA. 2010;107:2693–2698. doi: 10.1073/pnas.0907915107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrick-Davis K, Grinde E, Weaver BA. Serotonin 5-HT(2C) receptor homodimerization is not regulated by agonist or inverse agonist treatment. Eur J Pharmacol. 2007;568:45–53. doi: 10.1016/j.ejphar.2007.04.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaakola VP, Griffith MT, Hanson MA, Cherezov V, Chien EY, Lane JR, Ijzerman AP, Stevens RC. The 2.6 angstrom crystal structure of a human A2A adenosine receptor bound to an antagonist. Science. 2008;322:1211–1217. doi: 10.1126/science.1164772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacoby E, Bouhelal R, Gerspacher M, Seuwen K. The 7 TM G-protein-coupled receptor target family. ChemMedChem. 2006;1:761–782. doi: 10.1002/cmdc.200600134. [DOI] [PubMed] [Google Scholar]
- James JR, Oliveira MI, Carmo AM, Iaboni A, Davis SJ. A rigorous experimental framework for detecting protein oligomerization using bioluminescence resonance energy transfer. Nat Methods. 2006;3:1001–1006. doi: 10.1038/nmeth978. [DOI] [PubMed] [Google Scholar]
- Kasai RS, Suzuki KG, Prossnitz ER, Koyama-Honda I, Nakada C, Fujiwara TK, Kusumi A. Full characterization of GPCR monomer-dimer dynamic equilibrium by single molecule imaging. J Cell Biol. 2011;192:463–480. doi: 10.1083/jcb.201009128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuder K, Kiec-Kononowicz K. Fluorescent GPCR ligands as new tools in pharmacology. Curr Med Chem. 2008;15:2132–2143. doi: 10.2174/092986708785747599. [DOI] [PubMed] [Google Scholar]
- Latif R, Graves P, Davies TF. Ligand-dependent inhibition of oligomerization at the human thyrotropin receptor. J Biol Chem. 2002;277:45059–45067. doi: 10.1074/jbc.M206693200. [DOI] [PubMed] [Google Scholar]
- Lebon G, Warne T, Edwards PC, Bennett K, Langmead CJ, Leslie AG, Tate CG. Agonist-bound adenosine A2A receptor structures reveal common features of GPCR activation. Nature. 2011;474:521–525. doi: 10.1038/nature10136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leitz AJ, Bayburt TH, Barnakov AN, Springer BA, Sligar SG. Functional reconstitution of Beta2-adrenergic receptors utilizing self-assembling nanodisc technology. Biotechniques. 2006;40:601–602. doi: 10.2144/000112169. [DOI] [PubMed] [Google Scholar]
- Lukasiewicz S, Blasiak E, Faron-Gorecka A, Polit A, Tworzydlo M, Gorecki A, Wasylewski Z, Dziedzicka-Wasylewska M. Fluorescence studies of homooligomerization of adenosine A2A and serotonin 5-HT1A receptors reveal the specificity of receptor interactions in the plasma membrane. Pharmacol Rep. 2007;59:379–392. [PubMed] [Google Scholar]
- Luttrell LM. Reviews in molecular biology and biotechnology: transmembrane signaling by G protein-coupled receptors. Mol Biotechnol. 2008;39:239–264. doi: 10.1007/s12033-008-9031-1. [DOI] [PubMed] [Google Scholar]
- Mattheyses AL, Simon SM, Rappoport JZ. Imaging with total internal reflection fluorescence microscopy for the cell biologist. J Cell Sci. 2010;123:3621–3628. doi: 10.1242/jcs.056218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurel D, Kniazeff J, Mathis G, Trinquet E, Pin JP, Ansanay H. Cell surface detection of membrane protein interaction with homogeneous time-resolved fluorescence resonance energy transfer technology. Anal Biochem. 2004;329:253–262. doi: 10.1016/j.ab.2004.02.013. [DOI] [PubMed] [Google Scholar]
- Maurel D, Comps-Agrar L, Brock C, Rives ML, Bourrier E, Ayoub MA, Bazin H, Tinel N, Durroux T, Prezeau L, Trinquet E, Pin JP. Cell-surface protein-protein interaction analysis with time-resolved FRET and snap-tag technologies: application to GPCR oligomerization. Nat Methods. 2008;5:561–567. doi: 10.1038/nmeth.1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- May LT, Bridge LJ, Stoddart LA, Briddon SJ, Hill SJ. Allosteric interactions across native adenosine-A3 receptor homodimers: quantification using single-cell ligand-binding kinetics. FASEB J. 2011;25:3465–3476. doi: 10.1096/fj.11-186296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McVey M, Ramsay D, Kellett E, Rees S, Wilson S, Pope AJ, Milligan G. Monitoring receptor oligomerization using time-resolved fluorescence resonance energy transfer and bioluminescence resonance energy transfer. The human delta-opioid receptor displays constitutive oligomerization at the cell surface, which is not regulated by receptor occupancy. J Biol Chem. 2001;276:14092–14099. doi: 10.1074/jbc.M008902200. [DOI] [PubMed] [Google Scholar]
- Milligan G, Ramsay D, Pascal G, Carrillo JJ. GPCR dimerisation. Life Sci. 2003;74:181–188. doi: 10.1016/j.lfs.2003.09.005. [DOI] [PubMed] [Google Scholar]
- Oldham WM, Hamm HE. Heterotrimeric G protein activation by G-protein-coupled receptors. Nat Rev Mol Cell Biol. 2008;9:60–71. doi: 10.1038/nrm2299. [DOI] [PubMed] [Google Scholar]
- Overton MC, Blumer KJ. G-protein-coupled receptors function as oligomers in vivo. Curr Biol. 2000;10:341–344. doi: 10.1016/S0960-9822(00)00386-9. [DOI] [PubMed] [Google Scholar]
- Paila YD, Kombrabail M, Krishnamoorthy G, Chattopadhyay A. Oligomerization of the serotonin(1A) receptor in live cells: a time-resolved fluorescence anisotropy approach. J Phys Chem B. 2011;115:11439–11447. doi: 10.1021/jp201458h. [DOI] [PubMed] [Google Scholar]
- Palczewski K. Oligomeric forms of G protein-coupled receptors (GPCRs) Trends Biochem Sci. 2010;35:595–600. doi: 10.1016/j.tibs.2010.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piehler J. GPCRs: caught in a spectroscopic trap. Nat Chem Biol. 2011;7:578–579. doi: 10.1038/nchembio.641. [DOI] [PubMed] [Google Scholar]
- Pioszak AA, Harikumar KG, Parker NR, Miller LJ, Xu HE. Dimeric arrangement of the parathyroid hormone receptor and a structural mechanism for ligand-induced dissociation. J Biol Chem. 2010;285:12435–12444. doi: 10.1074/jbc.M109.093138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen SG, Choi HJ, Rosenbaum DM, Kobilka TS, Thian FS, Edwards PC, Burghammer M, Ratnala VR, Sanishvili R, Fischetti RF, Schertler GF, Weis WI, Kobilka BK. Crystal structure of the human beta2 adrenergic G-protein-coupled receptor. Nature. 2007;450:383–387. doi: 10.1038/nature06325. [DOI] [PubMed] [Google Scholar]
- Rasmussen SG, Choi HJ, Fung JJ, Pardon E, Casarosa P, Chae PS, Devree BT, Rosenbaum DM, Thian FS, Kobilka TS, Schnapp A, Konetzki I, Sunahara RK, Gellman SH, Pautsch A, Steyaert J, Weis WI, Kobilka BK. Structure of a nanobody-stabilized active state of the beta(2) adrenoceptor. Nature. 2011;469:175–180. doi: 10.1038/nature09648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen SG, DeVree BT, Zou Y, Kruse AC, Chung KY, Kobilka TS, Thian FS, Chae PS, Pardon E, Calinski D, Mathiesen JM, Shah ST, Lyons JA, Caffrey M, Gellman SH, Steyaert J, Skiniotis G, Weis WI, Sunahara RK, Kobilka BK. Crystal structure of the beta2 adrenergic receptor-Gs protein complex. Nature. 2011;477:549–555. doi: 10.1038/nature10361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenbaum DM, Zhang C, Lyons JA, Holl R, Aragao D, Arlow DH, Rasmussen SG, Choi HJ, Devree BT, Sunahara RK, Chae PS, Gellman SH, Dror RO, Shaw DE, Weis WI, Caffrey M, Gmeiner P, Kobilka BK. Structure and function of an irreversible agonist-beta(2) adrenoceptor complex. Nature. 2011;469:236–240. doi: 10.1038/nature09665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stryer L. Fluorescence energy transfer as a spectroscopic ruler. Annu Rev Biochem. 1978;47:819–846. doi: 10.1146/annurev.bi.47.070178.004131. [DOI] [PubMed] [Google Scholar]
- Stryer L, Haugland RP. Energy transfer: a spectroscopic ruler. Proc Natl Acad Sci USA. 1967;58:719–726. doi: 10.1073/pnas.58.2.719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urizar E, Yano H, Kolster R, Gales C, Lambert N, Javitch JA. CODA-RET reveals functional selectivity as a result of GPCR heteromerization. Nat Chem Biol. 2011;7:624–630. doi: 10.1038/nchembio.623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warne T, Moukhametzianov R, Baker JG, Nehme R, Edwards PC, Leslie AG, Schertler GF, Tate CG. The structural basis for agonist and partial agonist action on a beta(1)-adrenergic receptor. Nature. 2011;469:241–244. doi: 10.1038/nature09746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whorton MR, Bokoch MP, Rasmussen SG, Huang B, Zare RN, Kobilka B, Sunahara RK. A monomeric G protein-coupled receptor isolated in a high-density lipoprotein particle efficiently activates its G protein. Proc Natl Acad Sci USA. 2007;104:7682–7687. doi: 10.1073/pnas.0611448104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wurch T, Matsumoto A, Pauwels PJ. Agonist-independent and -dependent oligomerization of dopamine D(2) receptors by fusion to fluorescent proteins. FEBS Lett. 2001;507:109–113. doi: 10.1016/S0014-5793(01)02969-6. [DOI] [PubMed] [Google Scholar]
- Xu F, Wu H, Katritch V, Han GW, Jacobson KA, Gao ZG, Cherezov V, Stevens RC. Structure of an agonist-bound human A2A adenosine receptor. Science. 2011;332:322–327. doi: 10.1126/science.1202793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanagawa M, Yamashita T, Shichida Y. Comparative fluorescence resonance energy transfer analysis of metabotropic glutamate receptors: implications about the dimeric arrangement and rearrangement upon ligand bindings. J Biol Chem. 2011;286:22971–22981. doi: 10.1074/jbc.M110.206870. [DOI] [PMC free article] [PubMed] [Google Scholar]