

Abstract
The cystic fibrosis transmembrane conductance regulator (CFTR) is abundantly expressed in the kidney. CFTR mRNA is detected in all nephron segments of rats and humans and its expression is higher in the renal cortex and outer medulla than in the inner medulla. CFTR protein is detected at the apical surface of both proximal and distal tubules of rat kidney but not in the outer medullary collecting ducts. The localization of CFTR in the proximal tubules is compatible with that of endosomes, suggesting that CFTR might regulate pH in endocytic vesicles by equilibrating H+ accumulation due to H+-ATPase activity. Many studies have also demonstrated that CFTR also regulates channel pore opening and the transport of sodium, chloride and potassium. The kidneys also express a CFTR splicing variant, called TNR-CFTR, in a tissue-specific manner, primarily in the renal medulla. This splicing variant conserves the functional characteristics of wild-type CFTR. The functional significance of TNR-CFTR remains to be elucidated, but our group proposes that TNR-CFTR may have a basic function in intracellular organelles, rather than in the plasma membrane. Also, this splicing variant is able to partially substitute CFTR functions in the renal medulla of Cftr-/- mice and CF patients. In this review we discuss the major functions that have been proposed for CFTR and TNR-CFTR in the kidney.
Keywords: CFTR, TNR-CFTR, Endocytosis, Kidney, Potassium channel, Sodium channel
Introduction
The major role of the kidneys is to maintain the extracellular sodium chloride (NaCl) concentration that regulates extracellular fluid volume and blood pressure (Morales et al. 2000). Na+ and Cl– are reabsorbed along the nephron, reaching over 99 % of the filtered load under a low-salt diet. Cl–, the predominant anion in the glomerular ultrafiltrate, is reabsorbed along the nephron either by transcellular or paracellular pathways (Taal et al. 2012). Transcellular transport of the Cl– involves several membrane proteins, including specialized channels (Berend et al. 2012; Morales et al. 2000).
Cl– is the major anion in the blood and is responsible for approximately one-third of plasma tonicity, 97–98 % of all anionic charges, and two-thirds of all negative charges in the plasma (Yunos et al. 2010). The main source of Cl– is dietary NaCl; the recommended intake is 133–202 mmol/day for adult men and 99–133 mmol/day for adult women in the USA. The concentration of Cl− in plasma is approximately 100 mmol/L, of which about 10 mmol/L is in the intracellular space; the range varies widely from 2 mmol/L in skeletal muscle to 90 mmol/L in erythrocytes. The concentration of Cl− in the interstitial fluid is usually 5–10 % greater than that in plasma (Yunos et al. 2010). Numerous chloride channels have been discovered in a variety of animal and plant cells, and their modulation and involvement in physiological processes are widely described in the literature.
Several human inherited diseases are caused by mutations in the large ClC superfamily of chloride channels or transporters which includes the CLC gene family. The CLC family is found in all phyla, from bacteria to man, and ClC proteins function as chloride channels or Cl−/H+ exchangers. ClC channels and exchangers operate as dimers with two largely independent permeation pathways. There are nine different CLC genes in mammals (Planells-Cases and Jentsch 2009), and these encode either plasma membrane chloride channels or transporters that are mainly localized to intracellular compartments such as endosomes, lysosomes, or synaptic vesicles. It seems very likely that all of these primarily intracellular ClC proteins are Cl−/H+ exchangers rather than chloride channels (Planells-Cases and Jentsch 2009).
Human mutations in CLCN genes (CLCN1, CLCN2, among others, encode the proteins ClC-1, ClC-2, respectively) cause genetic diseases, among which some of these are directly associated with changes in renal function, producing symptoms such as kidney stones, renal salt loss, low-molecular-weight proteinuria, glycosuria, and more (Planells-Cases and Jentsch 2009). Other genetic disease related to chloride channel dysfunction is cystic fibrosis (CF), a common lethal autosomal recessive disorder caused by mutations in the cystic fibrosis transmembrane conductance regulator gene (CFTR). CFTR is not a member of the CLC gene family; rather, it is a member of the ABC transporter family expressed in a variety of epithelia, including the renal tubules (Li and Naren 2010; Riordan 1993). Although CFTR is widely expressed in the kidney, minor changes in renal function are observed in patients with CF that differ from the changes observed for renal genetic disease associated with mutations in the CLC gene family. The expression of TNR-CFTR, a splicing variant of CFTR, in the kidney is one possible explanation for this paradox found in patients with CF (Morales et al. 1996). The main purpose of this review was to discuss the role of CFTR and TNR-CFTR in renal function. Clinical and experimental data suggest that CFTR and TNR-CFTR have important roles in the regulation of a number of the reabsorption processes along the nephron.
Structure and function of CFTR
The CFTR is one of the few members of the ABC protein family that does not function as an active transporter, but as a chloride channel (Bear et al. 1992; Rogan et al. 2011). CFTR is composed of an intracellular N-terminus followed by six transmembrane-spanning domains (TMD1) that in turn is followed by a first nucleotide-binding domain (NBD1) containing Walker A and B consensus sequences that bind ATP (Collins et al. 1990, 1992; Morales et al. 1996, 1999; Rogan et al. 2011). Flanking this site is a large regulatory domain (R) [rich in cAMP-dependent kinase (PKA) phosphorylation sites] which is followed by a second set of six transmembrane-spanning domains (TMD2) and a second nucleotide-binding domain (NBD2) (Fig. 1).
Fig. 1.

Schematic representation of the cystic fibrosis transmembrane conductance regulator (CFTR) protein in the plasma membrane. C Carboxy-terminus domain, N N-terminus domain, NBD1 nucleotide-binding domain 1, NBD2 nucleotide-binding domain 2, R regulatory domain, TMD1 transmembrane-spanning domain 1, TMD2 transmembrane-spanning domain 2
There is strong evidence that CFTR’s two NBDs form a head-to-tail dimer similar to those found in other ABC transporters (Vergani et al. 2005). The two ATP-binding pockets (ABP) for CFTR are defined as follows: ABP1, formed by Walker A and B motifs of NBD1 and the signature sequence of NBD2; ABP2, formed by Walker A and B motifs of NBD2 and the signature sequence of NBD1. The amino acids sequences of CFTR’s two NBDs show significant differences—even in the conserved motifs (Chen and Hwang 2008). For example, the glutamate residue adjacent to the Walker B motif, found in most ABC members, is replaced by a serine in NBD1. A histidine residue that has been shown to play an important role in ATP hydrolysis in other ABC proteins (Zaitseva et al. 2005) is also replaced by a serine in NBD1. In addition, the signature sequence in CFTR’s NBD2 is degenerated (LSHGH instead of LSGGQ). This structural asymmetry of these two NBDs in CFTR likely accounts for the observation that only ABP2, but not ABP1, hydrolyzes ATP (Aleksandrov et al. 2002; Basso et al. 2003; Stratford et al. 2007).
Phosphorylation of many of the consensus serine residues in the R domain is a prerequisite for CFTR function. The phosphorylation of consensus sites by PKA is one mechanism by which CFTR activity is regulated. However, some findings also suggest that protein kinase C can also regulate CFTR activity (Chen and Hwang 2008).
Once ATP binds to the homologous nucleotide-interacting motifs in the two NBDs, these domains are thought to approach each other closely, sandwiching two ATPs in the NBD1–NBD2 interface. When this intramolecular heterodimer-like interaction occurs, a signal is transmitted through the cytoplasmic-linking domains, resulting in opening of the gate in the transmembrane domain. This channel-opening signal is sustained until hydrolysis of one of the ATPs leads to disruption of the NBD1–NBD2 interface and separation of the NBDs. Loss of the signal allows the channel gate to close, terminating anion flow until ATP once again binds to the NBDs (Gadsby et al. 2006).
CFTR function is not only important for chloride transport through its structure; this channel can interact with other transporters to inhibit or increase their respective ion transportation. These interactions could have important effects on epithelia intensively involved in the transport of ions and fluid, such as those found in the lungs and kidneys. For example, CFTR function leads to stimulation of outward rectifying chloride channels (ORCC) (Fulmer et al. 1995; Schwiebert et al. 1998) and inhibition of epithelial sodium channels (ENaC) (Kunzelmann 2003; Kunzelmann and Schreiber 1999; Kunzelmann et al. 2000, 2001).
Renal epithelia contain an enormous quantity of different membrane transporters, and the expression of these transporters differs along different nephron segments. CFTR is abundantly expressed in the kidney (de Andrade Pinto et al. 2007; Morales et al. 1996, 2000, 2001; Souza-Menezes et al. 2008), suggesting the possibility of an important function in renal physiology. Also the question of whether there are interaction between CFTR with other transporters expressed in renal epithelia remains as yet unanswered. In the following sections, we discuss some important findings that suggest a role for CFTR in the regulation of many of the reabsorption processes along the nephron. We also present some experimental evidence that supports a function for TNR-CFTR (a splicing variant of CFTR found only in renal tissues) in the kidney.
CFTR and the kidney
Since the discovery of the gene encoding CFTR, an impressive number of studies have been performed to elucidate the role of this protein in the organs affected by CF. These studies have converged toward the conclusion that CFTR is a small linear Cl– channel regulated by cAMP and that the ΔF508 mutation (present in 70 % of patients with CF) is associated with a loss of this cAMP sensitivity. However, although the vital role of CFTR in secretory epithelia is now widely accepted, its role in reabsorbing epithelia, which includes the kidney, remains incompletely understood (Jouret and Devuyst 2008; Wang 1999).
It is well known that CFTR is abundantly expressed in the kidney. CFTR mRNA is detected in all nephron segments in rats and humans, and this abundance is more prominent in the renal cortex and outer medulla renal areas than in the inner medulla (Morales et al. 1996). In agreement with the mRNA findings, CFTR protein has been detected by immunostaining at the apical surface of both proximal and distal tubules of the rat kidney but not in the outer medullary collecting ducts (Crawford et al. 1991; Jouret and Devuyst 2008). Studies in the mouse kidney revealed that CFTR is mainly expressed in the apical area of the proximal tubules (PTs) (pars recta, S3 segment), with a subcellular distribution compatible with that of endosomes. This distribution resembles that reported for the ClC-5 transporter and vacuolar H+-ATPase (V-ATPase) in Rab5a (a common component of the apical and basolateral endocytic machinery in polarized epithelial cells) enriched fractions (Jouret and Devuyst 2008). In the human kidney, CFTR protein expression has been detected in the PTs, thin limbs of the loop of Henle, distal tubules, and collecting ducts (Crawford et al. 1991; Devuyst et al. 1996; Morales et al. 1996). CFTR is also expressed in the branching ureteric bud during early nephrogenesis (Devuyst et al. 1996). In addition to being expressed in the plasma membrane, CFTR is also located in intracellular organelles along the endocytic and secretory pathways in the PTs where it might act as a pH regulator by importing Cl− into endocytic vesicles, equilibrating H+ accumulation caused by transport via H+-ATPase (Bradbury 1999; Carraro-Lacroix et al. 2010; Jouret and Devuyst 2008). For this reason, CFTR, a well-known regulator of other membrane transporters, could also work in the kidney as a regulator of intracellular pathways.
CFTR and PT endocytosis
The distribution of CFTR in the apical endosomes of mouse PT cells has revealed its possible involvement in renal endocytosis (Fig. 2). This hypothesis is supported by evidence obtained using CFTR knockout mouse models to characterize the role of CFTR in the kidney. Plasma and urine analyses revealed that the baseline renal function was normal in Cftr−/− mice. However, the urinary excretion of low-molecular-weight (LMW) proteins, such as the Clara cell secretory protein 16 (CC16; 16-kDa protein which leaks from the respiratory tract and is known to be filtrated by glomeruli), was significantly increased in Cftr−/− mice compared with controls, reflecting a possible defect in PT cell apical endocytosis. This finding was supported by the demonstration of a significant decrease in renal uptake of 125I-labeled β2-microglobulin and aminoglycosides in Cftr−/− mice compared with wild-type mice, both known to be endocytosed by proximal tubules (Jouret et al. 2007).
Fig. 2.
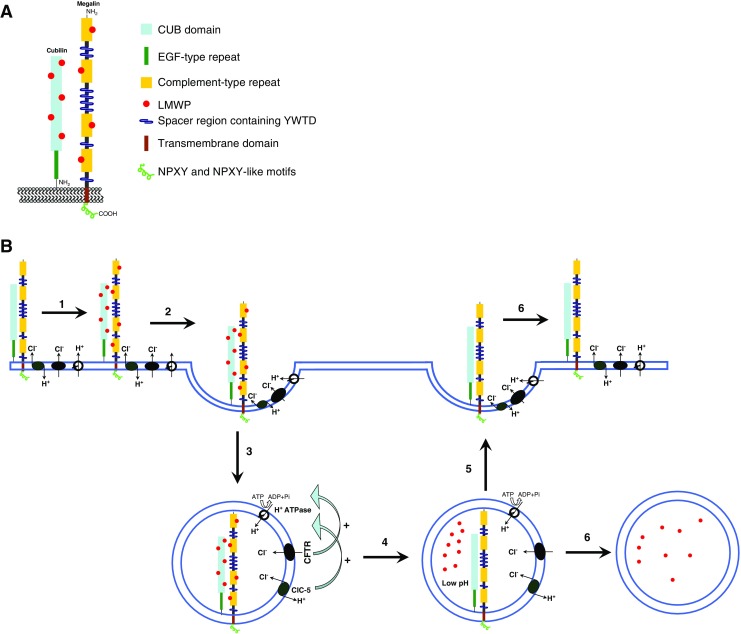
Schematic representation of the mechanism of megalin, cubilin, and endocytosis in the proximal tubules. a Megalin is a transmembrane protein of approximately 4,600 amino acids (aa) and a nonglycosylated molecular weight of approximately 517 kDa. The extracellular domain (approx. 4,400 aa) contains four cysteine-rich clusters of low-density-lipoprotein receptor type A repeats, which constitute the ligand-binding regions, separated and followed by 17 epidermal growth factor (EGF)-type repeats and eight spacer regions that contain YWTD repeats. A single transmembrane domain (22 aa) is followed by the cytoplasmic tail (213 aa), which contains two NPXY sequences and one NPXY-like sequence in addition to several Src-homology-3 (SH3) and one Src-homology-2 (SH2) recognition sites. Cubilin (left) is an approximately 3,600-aa protein with no transmembrane domain and a nonglycosylated molecular weight of approximately 400 kDa. The extracellular domain contains 27 CUB domains, which constitute the molecular basis for interaction with several proteins. The CUB domains are preceded by a stretch of 110 aa followed by eight EGF-type repeats. The amino-terminal region contains a potential palmitoylation site and an amphipathic α-helical structure with some similarity to the lipid-binding regions of apolipoproteins. Both are potential contributors to the anchoring of the receptor in the membrane. b Schematic view of the endocytosis process described in renal proximal tubules. Observe the important role of ClC-5 and CFTR in the steps involved in endosomal acidification: 1 binding of low-molecular-weight proteins (LMWP) to megalin/cubilin complex, 2 internalization of LMWP bound to megalin/cubilin complex, 3 formation of primary endosome, 4 endosomal acidification by H+-ATPase type V activity and its regulation by CFTR and ClC-5; in this step, the LMWPs unbind from the megalin/cubilin complex, 5 recycling of megalin/cubilin and other transporters to the luminal membrane, 6 LMWPs follow the lysosomal or transcytosis pathway. Here, only H+-ATPase type V, megalin/cubilin, ClC-5, and CFTR are represented; for simplicity, other membrane proteins and transporters are not shown
The endocytic uptake of aminoglycosides and LMW proteins, such as CC16 and β2-microglobulin, is mediated by the multiligand receptors, megalin and cubilin (Christensen and Birn 2002). Cubilin is a highly conserved membrane glycoprotein with little structural homology to other well-known endocytic receptors, and it is characterized by the absence of a transmembrane domain (Fig. 2). High-affinity binding of purified megalin to the N-terminal region of cubilin has been shown in vitro, suggesting that megalin participates not only in the endocytosis and intracellular trafficking of cubilin but also in the anchoring of cubilin to plasma membranes (Fig. 2). Christensen and Birn (2002) observed a selective decrease of cubilin expression in the straight (S3) segment of the PTs of Cftr−/− mice and an increase in urinary excretion of cubilin ligands, such as transferrin and CC16. Further investigations demonstrated that the lack of CFTR in the kidney is not associated with changes in the biosynthesis of cubilin but, rather, with a significant increase in the excretion of cubilin in the urine of Cftr−/− mice (Jouret and Devuyst 2008; Jouret et al. 2007). In contrast to findings reported for ClC-5 knockout mice (Christensen et al. 2003; Souza-Menezes et al. 2007), these authors observed no significant changes in the abundance of megalin in the kidney and urine of Cftr−/− mice and no changes in ClC-5 protein expression. These data suggest that the lack of CFTR in renal PT cells could induce cubilin instability at the brush border, leading to accelerated shedding into urine (Jouret and Devuyst 2008; Jouret et al. 2007). As such, these data are very intriguing because a similar mechanism is observed in ClC-5-/- mice and in Dent disease—but with a lower protein expression of both megalin and cubilin. Only cubilin shows a lower expression in Cftr-/- mice (Christensen et al. 2003; Jouret and Devuyst 2008; Jouret et al. 2007; Souza-Menezes et al. 2007).
Although the entire process remains unclear, the possibility that CFTR can work together with other proteins in the acidification of endosomal vesicles in PT cells cannot be rejected (Fig. 2). Working within the framework of this hypothesis, in 2010 our group showed that once the rat PT cell line (IRPTC) was cultured in medium containing no Cl–, the activity of H+-ATPase was significantly reduced (Carraro-Lacroix et al. 2010). In the same study, our group applied the siRNA technique and showed that a reduction in CFTR mRNA content resulted in a reduction in both H+-ATPase activity and the expression of the B2 subunit of type V H+-ATPase. Corroborating the results observed in Cftr-/- mice, we did not observe any changes in megalin protein expression when the CFTR mRNA content was reduced (Carraro-Lacroix et al. 2010).
These data (from cell culture experiments and Cftr-/- mice) are strong evidence that at least one of the functions of CFTR in PTs is to regulate the activity of type V H+-ATPase and endosomal acidification, both of which play a very important role in the endocytosis of LMW proteins (Fig. 2).
CFTR and regulation of potassium channel activity
In the kidney, the renal outer medullary potassium (K+) channel (ROMK) plays an important role in K+ recycling in the lumen of the thick ascending limbs of the loop of Henle (TAL) and in K+ secretion in the cortical collecting duct (CCD) based on its expression in the luminal membrane of both nephron segments (Wang et al. 1997). K+ recycling across the apical membrane of the TAL is important for NaCl reabsorption. This mechanism is directly associated with solute concentration in the renal medulla (Greger 1985; Hebert and Andreoli 1984). Two important renal phenomena are connected with K+ recycling across the apical membrane of TAL (Giebisch 1998). First, K+ recycling is essential for the positive potential of the lumen, which is the driving force for transepithelial Na+ reabsorption. Second, K+ recycling provides an adequate supply of K+ for the Na+/K+/2Cl- cotransporter. Mutations in the ROMK channel are associated with Bartter syndrome with reduced urine concentrating capacity and renal medulla activity.
In the CCD, K+ enters the cell across the basolateral membrane via Na+/K+-ATPase and is then secreted into the lumen through the apical K+ channels. It is believed that the ROMK channel provides the major route for K+ movement across the apical membrane of the CCD (Wang 1999).
Previous studies have demonstrated that blocking of the sulfonylurea receptor (SUR) by glybenclamide inhibits ROMK2 activity, probably by increasing the local ATP concentration or by direct interaction between SUR and ROMK2 (Inagaki et al. 1995a, b, c). Consequently, an accessory protein is required for the complete activity of ROMK2. The expression of SUR1 and SUR2, involved in the interaction with ROMK2 was not detected in the kidney (Aguilar-Bryan et al. 1998). Because CFTR expression is abundant in renal epithelia, several groups have explored the possibility that CFTR may couple to the ROMK channels to form a functional renal low-conductance ATP-sensitive K+ channel (Fuller and Benos 1992). Previous studies (McNicholas et al. 1996a, b, 1997) have reported that when CFTR is coexpressed with ROMK2, application of sulfonylurea agents, such as glybenclamide, blocks the activity of the ROMK2 channel. This same group has further suggested that a functional CFTR is required for CFTR/ROMK2 interaction (McNicholas et al. 1997). The observation that coupling of the ROMK channel with CFTR is essential for restoring the response to sulfonylurea agents has also been reported (Ruknudin et al. 1998). In addition, using the voltage clamp technique in Xenopus oocytes, these authors found that millimolar concentrations of Mg-ATP had no significant effect on ROMK1 channel activity. However, when ROMK1 was coexpressed with CFTR, the K+ channel became sensitive to ATP (Ruknudin et al. 1998), suggesting that CFTR may be required to increase this sensitivity.
In CFTR knockout mice and CFTR-ΔF508 transgenic mice, the sensitivity of ROMK to ATP is absent (Lu et al. 2010).
These data suggest that CFTR could play an important role in the kidney by regulating the activity of ROMK channels in organs where both channels are expressed.
CFTR and regulation of sodium channel activity
A large body of evidence indicates that CFTR is co-localized with ENaC in airway, colonic, and other epithelial tissues (Kunzelmann et al. 2000; Schwiebert et al. 1999). It has been proposed that ENaC is inhibited when cAMP activates CFTR; this inhibitory effect of CFTR has been used as an argument to explain the pathophysiology of CF in airway epithelia (Mall et al. 1998; Stutts et al. 1995). In Xenopus oocytes expressing ENaC, co-expression of CFTR reduces the ENaC-mediated Na+ current (Briel et al. 1998), an effect that might be attributed to a direct effect of CFTR on ENaC activity. In addition, Kunzelman (2003) reported that the inhibitory effect of CFTR can be mimicked by the coexpression of other anion channels (ClC-0 and ClC-2) or treatment with amphotericin B and that this effect can be explained by an increase in Cl− currents or intracellular Cl− concentration ([Cl−]i). Some authors have shown that an increase in [Cl−]i inhibits ENaC activity in FL-MDCK cells (MDCK cells that have been transfected with rat ENaC subunits containing the FLAG epitope in their extracellular loops) (Morris and Schafer 2002); however, this effect cannot explain the effect of Cl− secretion on Na+ absorption with cAMP treatment because the stimulation of Cl− secretion by cAMP results in a decrease—rather than an increase—in [Cl−]i (Xie and Schafer 2004). Moreover, in the Xenopus oocyte expression system, CFTR activation inhibits ENaC at a continuous holding potential at which CFTR mediates inward currents corresponding to Cl− efflux (Konstas et al. 2003). Under these conditions, activation of CFTR does not result in an increase in [Cl−]i. Therefore, a change in [Cl−]i is unlikely to fully explain the observed reciprocal regulation of ENaC and CFTR. In addition, extracellular ATP has been shown to attenuate amiloride-sensitive Na+ absorption in a variety of tissues, including airways (Devor and Pilewski 1999; Inglis et al. 1999; Iwase et al. 1997) and renal epithelia (Cuffe et al. 2000; Inglis et al. 1999; McCoy et al. 1999).
Similarly, activation of CFTR not only inhibits ENaC but also increases Cl− secretion in native epithelial tissues. The influence of CFTR on the activity of other transporters could be explained by the possible release of ATP through CFTR, and this phenomenon may happen along the nephron segments where both ENaC and CFTR are coexpressed (Kunzelmann and Schreiber 1999; Stutts et al. 1995; Sugita et al. 1998). Schwiebert and Kishore (2001) demonstrated the endogenous release of ATP by the FL-MDCK monolayers into the apical but not into the basolateral medium under basal conditions, consistent with measurements in the CCD. This release increased by more than threefold with cAMP treatment (Xie and Schafer 2008).
These data suggest a role of CFTR in the regulation of ENaC, with an interaction between these two channels in renal epithelia where they are coexpressed.
TNR-CFTR expression and function in the kidney
The kidney of patients with CF does not display major changes in renal function. This absence of an obvious renal phenotype is a priori paradoxical because CFTR protein is expressed in the kidney (de Andrade Pinto et al. 2007; Morales et al. 1996, 2000, 2001; Souza-Menezes et al. 2008), and it is involved in the regulation of many reabsorption and secretion mechanisms. In the absence of functional CFTR in the kidney, as observed in CF, other Cl− channels can compensate for the CFTR function, but this does not seem to happen because CFTR is not only involved with Cl− transport but also with the regulation of other channels by mechanisms other than Cl– transport.
Morales et al. (1996) showed that CFTR pre-mRNA in the renal medulla can form a splice variant called TNR-CFTR which is highly abundant in the renal medulla compared with wild-type CFTR expression. In their study, the authors showed that TNR-CFTR mRNA lacks a 145-bp fragment corresponding to segments of exons 13 and 14, which encode the last part (7 %) of the regulatory (R) domain of the CFTR molecule (Fig. 3). This deletion causes a shift in the reading frame, leading to creation of a premature termination codon at exon 14. This alternative structure contains only the first transmembrane domain, the first NBD, and the R domain.
Fig. 3.

Schematic representation of the protein structure of TNR-CFTR. C Carboxy-terminus domain, N N-terminus domain, NBD1 nucleotide-binding domain 1, R regulatory domain, TMD1 transmembrane-spanning domain 1
The hypothesis proposed by Morales et al. (1996) that TNR-CFTR is a splicing variant of CFTR, based on the homology of cDNA molecules forming TNR-CFTR and CFTR, is supported by the findings of Souza-Menezes et al. (2008) who showed that the small nuclear RNAs U11 and U12 (small nuclear RNAs present in the composition of spliceosomes that participate in alternative splicing mechanisms) are probably involved in the production of TNR-CFTR mRNA using a primary transcript from the CFTR gene. These authors showed that the pattern of U11 and U12 RNA expression in rat kidneys is similar to that observed for TNR-CFTR in rat kidneys, i.e., high levels of expression in the renal medulla and low levels of expression in the renal cortex (de Andrade Pinto et al. 2007; Souza-Menezes et al. 2008). Souza-Menezes et al. (2008) also showed that it was possible to observe a significant reduction in TNR-CFTR mRNA expression when U11 and U12 RNA expression was suppressed in a PT cell line.
In the kidney, TNR-CFTR is expressed in a tissue-specific manner, primarily in the renal medulla, and conserves the functional characteristics of wild-type CFTR. This CFTR form is also found in the human kidney medulla, but not in the human kidney cortex (Morales et al. 1996). Huber et al. (1998), using primary monolayer cultures of rat ureteric buds and CCDs in different embryonic and postnatal developmental stages, showed that TNR-CFTR does not seem to have a specific embryonic–morphogenetic function but that around birth, TNR-CFTR is upregulated.
After TNR-CFTR identification and characterization, it was hypothesized that all of the factors able to regulate CFTR mRNA and protein expression were also able to regulate TNR-CFTR mRNA and protein expression in renal tissues. This hypothesis is supported by the findings of de Andrade Pinto et al. (2007) who demonstrated the modulation of CFTR and TNR-CFTR mRNA and protein expression by thyroid hormone. Comparing hypothyroid rats and hyperthyroid rats with a control group, these authors observed a decrease in both CFTR and TNR-CFTR mRNA and protein expression in hypothyroid rats and a significant increase in both CFTR and TRN-CFTR mRNA and protein expression in hyperthyroid rats.
Lassance-Soares et al. (2010) showed that the hypertonic environment of the renal medulla regulates the expression of CFTR and TNR-CFTR. Stable Madin-Darby canine kidney (MDCK) cell lines treated with hypertonic medium at 480 mOsm/kg or 560 mOsm/kg using NaCl, urea, or sucrose were used in this study. Compared with the control group, CFTR total protein was increased when the cells were treated with NaCl and sucrose but, in contrast, when the medium was hypertonic using urea, CFTR total protein expression did not change. TNR-CFTR total protein expression was increased in cells subjected to hypertonic medium using NaCl, urea, or sucrose. In this work, the authors showed that CFTR was located mainly at the cell surface but that TNR-CFTR was localized primarily within the cells (Lassance-Soares et al. 2010). In addition, urea was able to increase TNR-CFTR protein expression but not CFTR protein expression. This was the first study demonstrating that the expressions of the CFTR and TNR-CFTR proteins were differentially regulated in a stimulus-dependent manner.
The functional significance of TNR-CFTR remains to be elucidated, but Morales et al. (1996) brought forward two hypotheses. Because several related members of the ATP-binding cassette family, to which CFTR belongs, function in intracellular organelles as half molecules with TMD1-NBD1 (Mosser et al. 1993; Valle and Gartner 1993), there is a possibility that TNR-CFTR may have a basic function in intracellular organelles rather than in the plasma membrane. Another hypothesis is based on the fact that this unusual form of mRNA processing occurs only in the renal medulla, a portion of the kidney with high osmolality. Once this half molecule of CFTR works as a Cl– channel and regulates other conductances (such as ORCC), its high expression, especially in the renal medulla, could protect the kidney from the functional defects observed in CF.
Conclusion
In the kidney, CFTR protein and mRNA are detected in different nephron segments. CFTR in the kidney has been proposed to function as a regulator of the conductance of other channels (e.g., ENaC and ROMK) and to be involved in the acidification of endosomal vesicles present in the endocytotic pathway of PTs.
No expressive changes in renal function have been observed in patients with CF or in CFTR knockout mice. One possibility to show that renal function in patients with CF or in CFTR knockout mice does undergo minor changes is the replacement of CFTR function by other transporters or proteins where CFTR is absent or not functioning correctly. One candidate for this is TNR-CFTR, an alternative splice of CFTR, which, in vitro, functions as a wild-type CFTR. However, further studies are necessary to completely understand the role of TNR-CFTR in renal physiology.
Acknowledgments
This study was supported by FAPERJ, CNPq, CAPES, and INCT.
Compliance with Ethical Guidelines
ᅟ
Conflict of Interest
Jackson Souza-Menezes, Geórgia da Silva Feltran and Marcelo M. Morales declare that they have no conflict of interest.
Human and Animal Studies
This article does not contain any studies with human or animal subjects performed by the any of the authors.
Contributor Information
Jackson Souza-Menezes, Phone: +55-22-33993905, Email: jacksonmenezes@gmail.com, Email: jacksonmenezes@macae.ufrj.br.
Geórgia da Silva Feltran, Email: georgiafeltran@yahoo.com.br.
Marcelo M. Morales, Email: mmorales@biof.ufrj.br
References
- Aguilar-Bryan L, Clement JP, Gonzalez G, Kunjilwar K, Babenko A, Bryan J. Toward understanding the assembly and structure of KATP channels. Physiol Rev. 1998;78:227–245. doi: 10.1152/physrev.1998.78.1.227. [DOI] [PubMed] [Google Scholar]
- Aleksandrov L, Aleksandrov AA, Chang XB, Riordan JR. The first nucleotide binding domain of cystic fibrosis transmembrane conductance regulator is a site of stable nucleotide interaction, whereas the second is a site of rapid turnover. J Biol Chem. 2002;277:15419–15425. doi: 10.1074/jbc.M111713200. [DOI] [PubMed] [Google Scholar]
- Basso C, Vergani P, Nairn AC, Gadsby DC. Prolonged nonhydrolytic interaction of nucleotide with CFTR's NH2-terminal nucleotide binding domain and its role in channel gating. J Gen Physiol. 2003;122:333–348. doi: 10.1085/jgp.200308798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bear CE, Li CH, Kartner N, Bridges RJ, Jensen TJ, Ramjeesingh M, Riordan JR. Purification and functional reconstitution of the cystic fibrosis transmembrane conductance regulator (CFTR) Cell. 1992;68:809–818. doi: 10.1016/0092-8674(92)90155-6. [DOI] [PubMed] [Google Scholar]
- Berend K, van Hulsteijn LH, Gans RO. Chloride: the queen of electrolytes? Eur J Intern Med. 2012;23:203–211. doi: 10.1016/j.ejim.2011.11.013. [DOI] [PubMed] [Google Scholar]
- Bradbury NA. Intracellular CFTR: localization and function. Physiol Rev. 1999;79:S175–S191. doi: 10.1152/physrev.1999.79.1.S175. [DOI] [PubMed] [Google Scholar]
- Briel M, Greger R, Kunzelmann K. Cl– transport by cystic fibrosis transmembrane conductance regulator (CFTR) contributes to the inhibition of epithelial Na+ channels (ENaCs) in Xenopus oocytes co-expressing CFTR and ENaC. J Physiol. 1998;508(Pt 3):825–836. doi: 10.1111/j.1469-7793.1998.825bp.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carraro-Lacroix LR, Lessa LM, Bezerra CN, Pessoa TD, Souza-Menezes J, Morales MM, Girardi AC, Malnic G. Role of CFTR and ClC-5 in modulating vacuolar H+-ATPase activity in kidney proximal tubule. Cell Physiol Biochem. 2010;26:563–576. doi: 10.1159/000322324. [DOI] [PubMed] [Google Scholar]
- Chen TY, Hwang TC. CLC-0 and CFTR: chloride channels evolved from transporters. Physiol Rev. 2008;88:351–387. doi: 10.1152/physrev.00058.2006. [DOI] [PubMed] [Google Scholar]
- Christensen EI, Birn H. Megalin and cubilin: multifunctional endocytic receptors. Nat Rev Mol Cell Biol. 2002;3:256–266. doi: 10.1038/nrm778. [DOI] [PubMed] [Google Scholar]
- Christensen EI, Devuyst O, Dom G, Nielsen R, Van der Smissen P, Verroust P, Leruth M, Guggino WB, Courtoy PJ. Loss of chloride channel ClC-5 impairs endocytosis by defective trafficking of megalin and cubilin in kidney proximal tubules. Proc Natl Acad Sci USA. 2003;100:8472–8477. doi: 10.1073/pnas.1432873100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins FS. Cystic fibrosis: molecular biology and therapeutic implications. Science. 1992;256:774–779. doi: 10.1126/science.1375392. [DOI] [PubMed] [Google Scholar]
- Collins FS, Riordan JR, Tsui LC. The cystic fibrosis gene: isolation and significance. Hosp Pract (Off Ed) 1990;25:47–57. doi: 10.1080/21548331.1990.11704019. [DOI] [PubMed] [Google Scholar]
- Crawford I, Maloney PC, Zeitlin PL, Guggino WB, Hyde SC, Turley H, Gatter KC, Harris A, Higgins CF. Immunocytochemical localization of the cystic fibrosis gene product CFTR. Proc Natl Acad Sci USA. 1991;88:9262–9266. doi: 10.1073/pnas.88.20.9262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuffe JE, Bielfeld-Ackermann A, Thomas J, Leipziger J, Korbmacher C. ATP stimulates Cl– secretion and reduces amiloride-sensitive Na+ absorption in M-1 mouse cortical collecting duct cells. J Physiol. 2000;524(Pt 1):77–90. doi: 10.1111/j.1469-7793.2000.00077.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Andrade Pinto AC, Barbosa CM, Ornellas DS, Novaira HJ, de Souza-Menezes J, Ortiga-Carvalho TM, Fong P, Morales MM. Thyroid hormones stimulate renal expression of CFTR. Cell Physiol Biochem. 2007;20:83–90. doi: 10.1159/000104156. [DOI] [PubMed] [Google Scholar]
- Devor DC, Pilewski JM. UTP inhibits Na+ absorption in wild-type and DeltaF508 CFTR-expressing human bronchial epithelia. Am J Physiol. 1999;276:C827–C837. doi: 10.1152/ajpcell.1999.276.4.C827. [DOI] [PubMed] [Google Scholar]
- Devuyst O, Burrow CR, Schwiebert EM, Guggino WB, Wilson PD. Developmental regulation of CFTR expression during human nephrogenesis. Am J Physiol. 1996;271:F723–F735. doi: 10.1152/ajprenal.1996.271.3.F723. [DOI] [PubMed] [Google Scholar]
- Fuller CM, Benos DJ. CFTR! Am J Physiol. 1992;263:C267–C286. doi: 10.1152/ajpcell.1992.263.2.C267. [DOI] [PubMed] [Google Scholar]
- Fulmer SB, Schwiebert EM, Morales MM, Guggino WB, Cutting GR. Two cystic fibrosis transmembrane conductance regulator mutations have different effects on both pulmonary phenotype and regulation of outwardly rectified chloride currents. Proc Natl Acad Sci USA. 1995;92:6832–6836. doi: 10.1073/pnas.92.15.6832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gadsby DC, Vergani P, Csanady L. The ABC protein turned chloride channel whose failure causes cystic fibrosis. Nature. 2006;440:477–483. doi: 10.1038/nature04712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giebisch G. Renal potassium transport: mechanisms and regulation. Am J Physiol. 1998;274:F817–F833. doi: 10.1152/ajprenal.1998.274.5.F817. [DOI] [PubMed] [Google Scholar]
- Greger R. Ion transport mechanisms in thick ascending limb of Henle's loop of mammalian nephron. Physiol Rev. 1985;65:760–797. doi: 10.1152/physrev.1985.65.3.760. [DOI] [PubMed] [Google Scholar]
- Hebert SC, Andreoli TE. Control of NaCl transport in the thick ascending limb. Am J Physiol. 1984;246:F745–F756. doi: 10.1152/ajprenal.1984.246.6.F745. [DOI] [PubMed] [Google Scholar]
- Huber S, Braun G, Burger-Kentischer A, Reinhart B, Luckow B, Horster M. CFTR mRNA and its truncated splice variant (TRN-CFTR) are differentially expressed during collecting duct ontogeny. FEBS Lett. 1998;423:362–366. doi: 10.1016/S0014-5793(98)00112-4. [DOI] [PubMed] [Google Scholar]
- Inagaki N, Gonoi T, Clement JP, Namba N, Inazawa J, Gonzalez G, Aguilar-Bryan L, Seino S, Bryan J. Reconstitution of IKATP: an inward rectifier subunit plus the sulfonylurea receptor. Science. 1995;270:1166–1170. doi: 10.1126/science.270.5239.1166. [DOI] [PubMed] [Google Scholar]
- Inagaki N, Inazawa J, Seino S. cDNA sequence, gene structure, and chromosomal localization of the human ATP-sensitive potassium channel, uKATP-1, gene (KCNJ8) Genomics. 1995;30:102–104. doi: 10.1006/geno.1995.0018. [DOI] [PubMed] [Google Scholar]
- Inagaki N, Tsuura Y, Namba N, Masuda K, Gonoi T, Horie M, Seino Y, Mizuta M, Seino S. Cloning and functional characterization of a novel ATP-sensitive potassium channel ubiquitously expressed in rat tissues, including pancreatic islets, pituitary, skeletal muscle, and heart. J Biol Chem. 1995;270:5691–5694. doi: 10.1074/jbc.270.11.5691. [DOI] [PubMed] [Google Scholar]
- Inglis SK, Collett A, McAlroy HL, Wilson SM, Olver RE. Effect of luminal nucleotides on Cl– secretion and Na+ absorption in distal bronchi. Pflugers Arch. 1999;438:621–627. doi: 10.1007/s004240051085. [DOI] [PubMed] [Google Scholar]
- Iwase N, Sasaki T, Shimura S, Yamamoto M, Suzuki S, Shirato K. ATP-induced Cl– secretion with suppressed Na+ absorption in rabbit tracheal epithelium. Respir Physiol. 1997;107:173–180. doi: 10.1016/S0034-5687(96)02516-9. [DOI] [PubMed] [Google Scholar]
- Jouret F, Devuyst O. CFTR and defective endocytosis: new insights in the renal phenotype of cystic fibrosis. Pflugers Arch. 2008;457:1227–1236. doi: 10.1007/s00424-008-0594-2. [DOI] [PubMed] [Google Scholar]
- Jouret F, Bernard A, Hermans C, Dom G, Terryn S, Leal T, Lebecque P, Cassiman JJ, Scholte BJ, de Jonge HR, Courtoy PJ, Devuyst O. Cystic fibrosis is associated with a defect in apical receptor-mediated endocytosis in mouse and human kidney. J Am Soc Nephrol. 2007;18:707–718. doi: 10.1681/ASN.2006030269. [DOI] [PubMed] [Google Scholar]
- Konstas AA, Koch JP, Korbmacher C. cAMP-dependent activation of CFTR inhibits the epithelial sodium channel (ENaC) without affecting its surface expression. Pflugers Arch. 2003;445:513–521. [Google Scholar]
- Kunzelmann K. ENaC is inhibited by an increase in the intracellular Cl(–) concentration mediated through activation of Cl(–) channels. Pflugers Arch. 2003;445:504–512. [Google Scholar]
- Kunzelmann K, Schreiber R. CFTR, a regulator of channels. J Membr Biol. 1999;168:1–8. doi: 10.1007/s002329900492. [DOI] [PubMed] [Google Scholar]
- Kunzelmann K, Schreiber R, Nitschke R, Mall M. Control of epithelial Na+ conductance by the cystic fibrosis transmembrane conductance regulator. Pflugers Arch. 2000;440:193–201. doi: 10.1007/s004240000255. [DOI] [PubMed] [Google Scholar]
- Kunzelmann K, Schreiber R, Boucherot A. Mechanisms of the inhibition of epithelial Na(+) channels by CFTR and purinergic stimulation. Kidney Int. 2001;60:455–461. doi: 10.1046/j.1523-1755.2001.060002455.x. [DOI] [PubMed] [Google Scholar]
- Lassance-Soares RM, Cheng J, Krasnov K, Cebotaru L, Cutting GR, Souza-Menezes J, Morales MM, Guggino WB. The hypertonic environment differentially regulates wild-type CFTR and TNR-CFTR chloride channels. Cell Physiol Biochem. 2010;26:577–586. doi: 10.1159/000322325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C, Naren AP. CFTR chloride channel in the apical compartments: spatiotemporal coupling to its interacting partners. Integr Biol (Camb) 2010;2:161–177. doi: 10.1039/b924455g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu M, Dong K, Egan ME, Giebisch GH, Boulpaep EL, Hebert SC. Mouse cystic fibrosis transmembrane conductance regulator forms cAMP-PKA-regulated apical chloride channels in cortical collecting duct. Proc Natl Acad Sci USA. 2010;107:6082–6087. doi: 10.1073/pnas.0902661107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mall M, Bleich M, Greger R, Schreiber R, Kunzelmann K. The amiloride-inhibitable Na+ conductance is reduced by the cystic fibrosis transmembrane conductance regulator in normal but not in cystic fibrosis airways. J Clin Invest. 1998;102:15–21. doi: 10.1172/JCI2729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy DE, Taylor AL, Kudlow BA, Karlson K, Slattery MJ, Schwiebert LM, Schwiebert EM, Stanton BA. Nucleotides regulate NaCl transport in mIMCD-K2 cells via P2X and P2Y purinergic receptors. Am J Physiol. 1999;277:F552–F559. doi: 10.1152/ajprenal.1999.277.4.F552. [DOI] [PubMed] [Google Scholar]
- McNicholas CM, Guggino WB, Schwiebert EM, Hebert SC, Giebisch G, Egan ME. Sensitivity of a renal K+ channel (ROMK2) to the inhibitory sulfonylurea compound glibenclamide is enhanced by coexpression with the ATP-binding cassette transporter cystic fibrosis transmembrane regulator. Proc Natl Acad Sci USA. 1996;93:8083–8088. doi: 10.1073/pnas.93.15.8083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNicholas CM, Yang Y, Giebisch G, Hebert SC. Molecular site for nucleotide binding on an ATP-sensitive renal K+ channel (ROMK2) Am J Physiol. 1996;271:F275–F285. doi: 10.1152/ajprenal.1996.271.2.F275. [DOI] [PubMed] [Google Scholar]
- McNicholas CM, Nason MW, Jr, Guggino WB, Schwiebert EM, Hebert SC, Giebisch G, Egan ME. A functional CFTR-NBF1 is required for ROMK2-CFTR interaction. Am J Physiol. 1997;273:F843–F848. doi: 10.1152/ajprenal.1997.273.5.F843. [DOI] [PubMed] [Google Scholar]
- Morales MM, Carroll TP, Morita T, Schwiebert EM, Devuyst O, Wilson PD, Lopes AG, Stanton BA, Dietz HC, Cutting GR, Guggino WB. Both the wild type and a functional isoform of CFTR are expressed in kidney. Am J Physiol. 1996;270:F1038–F1048. doi: 10.1152/ajprenal.1996.270.6.F1038. [DOI] [PubMed] [Google Scholar]
- Morales MM, Capella MA, Lopes AG. Structure and function of the cystic fibrosis transmembrane conductance regulator. Braz J Med Biol Res. 1999;32:1021–1028. doi: 10.1590/S0100-879X1999000800013. [DOI] [PubMed] [Google Scholar]
- Morales MM, Falkenstein D, Lopes AG. The cystic fibrosis transmembrane regulator (CFTR) in the kidney. An Acad Bras Cienc. 2000;72:399–406. doi: 10.1590/S0001-37652000000300013. [DOI] [PubMed] [Google Scholar]
- Morales MM, Nascimento DS, Capella MA, Lopes AG, Guggino WB. Arginine vasopressin regulates CFTR and ClC-2 mRNA expression in rat kidney cortex and medulla. Pflugers Arch. 2001;443:202–211. doi: 10.1007/s004240100671. [DOI] [PubMed] [Google Scholar]
- Morris RG, Schafer JA. cAMP increases density of ENaC subunits in the apical membrane of MDCK cells in direct proportion to amiloride-sensitive Na(+) transport. J Gen Physiol. 2002;120:71–85. doi: 10.1085/jgp.20018547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosser J, Douar AM, Sarde CO, Kioschis P, Feil R, Moser H, Poustka AM, Mandel JL, Aubourg P. Putative X-linked adrenoleukodystrophy gene shares unexpected homology with ABC transporters. Nature. 1993;361:726–730. doi: 10.1038/361726a0. [DOI] [PubMed] [Google Scholar]
- Planells-Cases R, Jentsch TJ. Chloride channelopathies. Biochim Biophys Acta. 2009;1792:173–189. doi: 10.1016/j.bbadis.2009.02.002. [DOI] [PubMed] [Google Scholar]
- Riordan JR. The cystic fibrosis transmembrane conductance regulator. Annu Rev Physiol. 1993;55:609–630. doi: 10.1146/annurev.ph.55.030193.003141. [DOI] [PubMed] [Google Scholar]
- Rogan MP, Stoltz DA, Hornick DB. Cystic fibrosis transmembrane conductance regulator intracellular processing, trafficking, and opportunities for mutation-specific treatment. Chest. 2011;139:1480–1490. doi: 10.1378/chest.10-2077. [DOI] [PubMed] [Google Scholar]
- Ruknudin A, Schulze DH, Sullivan SK, Lederer WJ, Welling PA. Novel subunit composition of a renal epithelial KATP channel. J Biol Chem. 1998;273:14165–14171. doi: 10.1074/jbc.273.23.14165. [DOI] [PubMed] [Google Scholar]
- Schwiebert EM, Kishore BK. Extracellular nucleotide signaling along the renal epithelium. Am J Physiol Renal Physiol. 2001;280:F945–F963. doi: 10.1152/ajprenal.2001.280.6.F945. [DOI] [PubMed] [Google Scholar]
- Schwiebert EM, Morales MM, Devidas S, Egan ME, Guggino WB. Chloride channel and chloride conductance regulator domains of CFTR, the cystic fibrosis transmembrane conductance regulator. Proc Natl Acad Sci USA. 1998;95:2674–2679. doi: 10.1073/pnas.95.5.2674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwiebert EM, Benos DJ, Egan ME, Stutts MJ, Guggino WB. CFTR is a conductance regulator as well as a chloride channel. Physiol Rev. 1999;79:S145–S166. doi: 10.1152/physrev.1999.79.1.S145. [DOI] [PubMed] [Google Scholar]
- Souza-Menezes J, Morales MM, Tukaye DN, Guggino SE, Guggino WB. Absence of ClC5 in knockout mice leads to glycosuria, impaired renal glucose handling and low proximal tubule GLUT2 protein expression. Cell Physiol Biochem. 2007;20:455–464. doi: 10.1159/000107529. [DOI] [PubMed] [Google Scholar]
- Souza-Menezes J, Tukaye DN, Novaira HJ, Guggino WB, Morales MM. Small nuclear RNAs U11 and U12 modulate expression of TNR-CFTR mRNA in mammalian kidneys. Cell Physiol Biochem. 2008;22:93–100. doi: 10.1159/000149786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stratford FL, Ramjeesingh M, Cheung JC, Huan LJ, Bear CE. The Walker B motif of the second nucleotide-binding domain (NBD2) of CFTR plays a key role in ATPase activity by the NBD1-NBD2 heterodimer. Biochem J. 2007;401:581–586. doi: 10.1042/BJ20060968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stutts MJ, Canessa CM, Olsen JC, Hamrick M, Cohn JA, Rossier BC, Boucher RC. CFTR as a cAMP-dependent regulator of sodium channels. Science. 1995;269:847–850. doi: 10.1126/science.7543698. [DOI] [PubMed] [Google Scholar]
- Sugita M, Yue Y, Foskett JK. CFTR Cl– channel and CFTR-associated ATP channel: distinct pores regulated by common gates. EMBO J. 1998;17:898–908. doi: 10.1093/emboj/17.4.898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taal MW, Chertow GM, Marsden PA, Skorecki K, Yu ASL, Brenner BM, editors. Brenner & Rector's the kidney. 9. Philadelphia: Elsevier Saunders; 2012. [Google Scholar]
- Valle D, Gartner J. Human genetics. Penetrating the peroxisome. Nature. 1993;361:682–683. doi: 10.1038/361682a0. [DOI] [PubMed] [Google Scholar]
- Vergani P, Lockless SW, Nairn AC, Gadsby DC. CFTR channel opening by ATP-driven tight dimerization of its nucleotide-binding domains. Nature. 2005;433:876–880. doi: 10.1038/nature03313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W. Regulation of the ROMK channel: interaction of the ROMK with associate proteins. Am J Physiol. 1999;277:F826–F831. doi: 10.1152/ajprenal.1999.277.6.F826. [DOI] [PubMed] [Google Scholar]
- Wang W, Hebert SC, Giebisch G. Renal K+ channels: structure and function. Annu Rev Physiol. 1997;59:413–436. doi: 10.1146/annurev.physiol.59.1.413. [DOI] [PubMed] [Google Scholar]
- Xie Y, Schafer JA. Inhibition of ENaC by intracellular Cl– in an MDCK clone with high ENaC expression. Am J Physiol Renal Physiol. 2004;287:F722–F731. doi: 10.1152/ajprenal.00135.2004. [DOI] [PubMed] [Google Scholar]
- Xie Y, Schafer JA. Endogenous ATP release inhibits electrogenic Na(+) absorption and stimulates Cl(–) secretion in MDCK cells. Purinergic Signal. 2008;4:125–137. doi: 10.1007/s11302-007-9053-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yunos NM, Bellomo R, Story D, Kellum J. Bench-to-bedside review: chloride in critical illness. Crit Care. 2010;14:226. doi: 10.1186/cc9052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaitseva J, Jenewein S, Jumpertz T, Holland IB, Schmitt L. H662 is the linchpin of ATP hydrolysis in the nucleotide-binding domain of the ABC transporter HlyB. EMBO J. 2005;24:1901–1910. doi: 10.1038/sj.emboj.7600657. [DOI] [PMC free article] [PubMed] [Google Scholar]