

Abstract
Protein–protein interactions are important in many essential biological functions, such as transcription, translation, and signal transduction. Much progress has been made in understanding protein–protein association in dilute solution via experimentation and simulation. Cells, however, contain various macromolecules, such as DNA, RNA, proteins, among many others, and a myriad of non-specific interactions (usually weak) are present between these cellular constituents. In this review article, we describe the important developments in recent years that have furthered our understanding and even allowed prediction of the consequences of macromolecular crowding on protein–protein interactions. We outline the development of our crowding theory that can predict the change in binding free energy due to crowding quantitatively for both repulsive and attractive protein–crowder interactions. One of the most important findings from our recent work is that weak attractive interactions between crowders and proteins can actually destabilize protein complex formation as opposed to the commonly assumed stabilizing effect predicted based on traditional crowding theories that only account for the entropic-excluded volume effects. We also discuss the implications of macromolecular crowding on the population of encounter versus specific native complex.
Keywords: Protein–protein interactions, Macromolecular crowding, Protein–crowder interactions, Crowding theories, Cellular environment, Dilute solution
Introduction
Many biological processes rely on protein–protein interactions in a highly crowded cellular environment; up to 40 % of a cell’s volume is occupied by various macromolecules, such as DNA, RNA, proteins, sugars and other organelles. Consequently, it is necessary for a protein to move around crowding macromolecules to find its way to bind its interaction partner(s) to carry out a specific biological function. Therefore, it is important to understand the effects of macromolecular crowding on protein–protein interactions to better understand biological processes in a living cell.
In recent years, significant progress has been made in understanding protein–protein interactions at the molecular level by both experimentation and simulation (Clore and Iwahara 2009). However, most studies have been performed in dilute solutions—in vitro or in silico—which are not representative of in vivo conditions. This raises a number of important following questions: (1) how can we understand protein–protein interactions in a living cell given the difficulties in designing experiments and simulations that attempt to mimic the in vivo environment? (2) Can we utilize the enormous amount of information collected on protein–protein interactions in dilute solutions collected over the years and relate it to protein–protein interactions in a crowded cellular environment?
Studying protein–protein interactions via in vivo experiments is extremely challenging although in-cell spectroscopic techniques are enabling great progress to be made in terms of achieving this goal (Wang et al. 2011). Thus, to answer the above questions, many experimental studies have been performed using synthetic polymers or specific proteins as crowding agents. These studies have addressed both the thermodynamics and kinetics of protein–protein interactions in a crowded environment (Minton and Wilf 1981; Minton 1983; Jarvis and Ring 1990; van den Berg et al. 1999; Wenner and Bloomfield 1999; Morar et al. 2001; Patel et al. 2002; Kozer and Schreiber 2004; Zorrilla et al. 2004; Phillip et al. 2009; 2012; Wang et al. 2011; Fodeke and Minton 2011). Using “inert” crowding agents, the primary focus in most of these studies was to understand the excluded volume effects of crowding agents on the formation of protein complexes (Minton 1983; Kim et al. 2010). As expected based on simple theoretical models, these experiments have shown that the consequence of excluded volume effects (entropic in nature) is to force proteins to form a stable complex, thereby increasing the volume available to the crowding agents. However, a few experiments have shown an unexpected trend, i.e., destabilization of protein complexes in the presence of crowding agents (Phillip et al. 2009; 2012; Jiao et al. 2010). This observation can be explained by attractive interactions between proteins and crowding agents, which can inevitably arise from a combination of electrostatic interactions, hydrogen bonding, hydrophobic interactions, and van der Waals interactions. As opposed to the stabilizing effect of entropic-excluded volume interactions, the attractive protein–crowder interactions will actually increase the binding free energy due to the enthalpic penalty in breaking favorable protein–crowder contacts to form a protein complex. The separation of these competing effects in an experimental setup is quite challenging due to unknown interaction parameters and presents a major barrier in developing a theoretical model to interpret (and even predict a priori) experimental observations.
Computational models can be helpful to form a basis of our understanding in separating these various effects as the interactions between proteins and crowding agents can be tuned precisely. Earlier computational studies based on purely repulsive protein–crowder interactions (such as hard-sphere as well as soft repulsive interactions) have shown that the repulsive interactions stabilize the formation of the protein complex by lowering the binding free energy. However, the extent of such stabilization has been found to be rather modest (Kim et al. 2010). Interestingly, the population of nonspecific encounter complexes, which are now believed to play an important role in forming the native functional complex, may well be decreased by the presence of repulsive crowders. Rosen et al. very recently considered attractive protein–crowder interactions in their protein binding simulations (2011). The work of these researchers has highlighted the importance of accounting for enthalpic effects arising from the attractive protein–crowder interactions (if present) in addition to the commonly invoked excluded volume effects (Douglas et al. 2009). Modest protein–crowder attractions can actually increase the binding free energy with respect to the crowder-free solution. Even if the binding free energy is decreased with respect to the crowder-free solution (weak protein–crowder attractions), the extent of this decrease is overestimated by theoretical models based solely on repulsive protein–crowder interactions.
Most analytical theories of macromolecular crowding are based on the scaled particle theory (SPT) of hard-sphere fluids developed almost half a century ago (Lebowitz and Rowlinson 1964). The SPT provides an analytical expression for the free energy cost of creating a spherical cavity in a bath of hard-sphere particles. By approximating proteins as spherical particles, these theories have been applied to interpret experimental (Patel et al. 2002; Snoussi and Halle 2008) and computational data with varying degrees of success (Zhou et al. 2008; Kim et al. 2010; Mittal and Best 2010). However, these theories fail to account for any kind of attractive protein–crowder interactions and need to be modified before they can be applied to more realistic circumstances.
A modification to account for protein–crowder attractions was recently proposed by Jiao et al. (2010) and Rosen et al. (2011). These researchers added a phenomenological mean-field term (proportional to the protein surface area) to the respective SPT-based crowding theories to interpret experimental and simulation data successfully.
More recently, we introduced a microscopic theory based on the statistical mechanics of simple liquids to describe the protein binding simulation data in the presence of attractive protein–crowder interactions (Kim and Mittal 2012). Without any adjustable parameter in the model, this theory was able to predict the change in binding free energy from the molecular simulation remarkably well over a wide range of parameters, such as crowder size, packing fraction and the protein–crowder attraction strength. The aim of the review presented here is to outline a brief summary of our efforts to understand the thermodynamics of protein–protein interactions in a crowded environment using computational methods. This improved understanding is utilized in the development of a quantitative analytical theory that can be applied in future studies to predict macromolecular crowding effects on protein–protein interactions in an environment closely resembling living cells.
Thermodynamics of protein–protein interactions in a crowder solution
How can we characterize thermodynamics of protein–protein interactions in the presence of crowding molecules? This is illustrated in Fig. 1 for the case of a simple dimerization reaction, 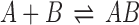 , in the presence of spherical crowders. The thermodynamic quantity that is mostly sought in both experiments and simulations is the change in the binding free energy relative to that of the crowder-free solution (ϕ = 0),
, in the presence of spherical crowders. The thermodynamic quantity that is mostly sought in both experiments and simulations is the change in the binding free energy relative to that of the crowder-free solution (ϕ = 0), 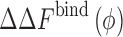 , in a crowded solution with crowder packing fraction ϕ (bottom horizontal reaction in Fig. 1). Theoretically, given the various solvation (or crowding) free energies,
, in a crowded solution with crowder packing fraction ϕ (bottom horizontal reaction in Fig. 1). Theoretically, given the various solvation (or crowding) free energies, 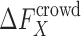 (X = A, B, AB) − the free energy of inserting X in a crowded solution,
(X = A, B, AB) − the free energy of inserting X in a crowded solution, 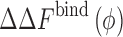 is then simply given by (using a thermodynamic cycle),
is then simply given by (using a thermodynamic cycle),
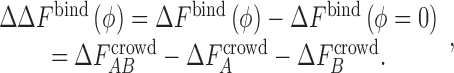 |
1 |
Fig. 1.
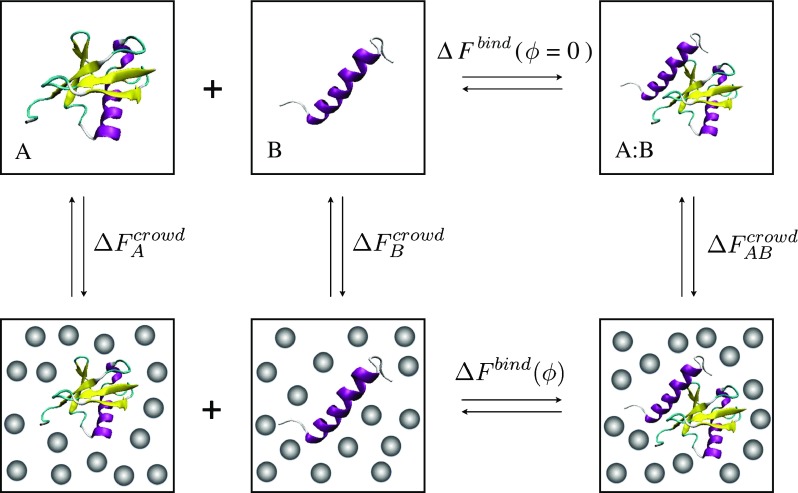
Thermodynamic cycle for the formation of a protein complex (between ubiquitin and UIM1) in bulk and in a crowded solution
A major challenge is thus to calculate the crowding free energy 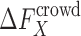 for a protein or a complex X.
for a protein or a complex X.
Next, we describe our procedure for calculating this crowding free energy. Without loss of generality, let 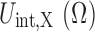 be the overall interaction energy between a protein X and a crowder, where Ω denotes the collective variables of protein atoms and the crowders. Thus, the crowding free energy,
be the overall interaction energy between a protein X and a crowder, where Ω denotes the collective variables of protein atoms and the crowders. Thus, the crowding free energy, 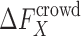 , can be obtained by
, can be obtained by
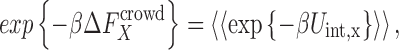 |
2 |
where 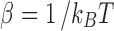 and
and  refers to the canonical ensemble average over protein and crowder configurations. For weakly-to-moderately interacting proteins, structural changes are minimal during binding events. Thus, the double ensemble average in Eq. 2 can be further approximated to,
refers to the canonical ensemble average over protein and crowder configurations. For weakly-to-moderately interacting proteins, structural changes are minimal during binding events. Thus, the double ensemble average in Eq. 2 can be further approximated to,
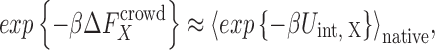 |
3 |
where 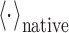 is the ensemble average over crowder configurations using the native structures of individual proteins and complexes.
is the ensemble average over crowder configurations using the native structures of individual proteins and complexes.
Following the Weeks–Chandler–Andersen (WCA) theory (Weeks 1971), we decompose 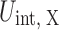 into the repulsive and attractive parts. The crowding free energy,
into the repulsive and attractive parts. The crowding free energy, 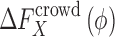 , can thus be divided into two separable contributions as,
, can thus be divided into two separable contributions as,
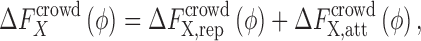 |
4 |
where 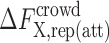 is the contribution to the crowding free energy from the repulsive (attractive) protein–crowder interactions.
is the contribution to the crowding free energy from the repulsive (attractive) protein–crowder interactions.
Now, for any generic type of interaction between proteins and crowders, the task of calculating crowding free energy is divided into calculating the repulsive and attractive contributions for isolated proteins and the bound complex.
We note that most of the currently used theoretical models aiming to describe experimental data ignore the attractive contribution to the change in the binding free energy, assuming that the interactions between inert crowding agents and proteins can be approximated effectively by repulsive potentials.
Next, we outline our recently developed theory (Kim and Mittal 2012) that can account for repulsive as well as attractive protein–crowder interactions.
Repulsive contribution to the crowding free energy
To calculate the repulsive contribution to the crowding free energy, we adopt the scaled particle theory (SPT) (Reiss et al. 1959).
The SPT provides an analytical expression for the free energy of creating a spherical cavity of radius R in a hard-sphere fluid (particle radius R c) at packing fraction ϕ which is given by,
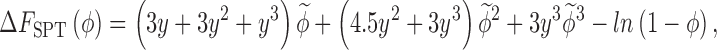 |
5 |
where 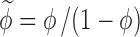 and y = R/R
c. The most important question that may arise from the use of SPT to predict the first term in Eq. 4 is: can anisometric proteins and commonly used crowding agents be represented as hard spheres to form the physical basis of excluded volume interactions?
and y = R/R
c. The most important question that may arise from the use of SPT to predict the first term in Eq. 4 is: can anisometric proteins and commonly used crowding agents be represented as hard spheres to form the physical basis of excluded volume interactions?
One may argue that for the cases in which long-range electrostatic interactions are negligible, the interactions between proteins and crowders and between crowders themselves can be approximated by effective hard-sphere potentials with contact distances adjusted to fit the experimental/simulation data. Previous successes in this direction indicate that such an approximation is acceptable in several cases (Minton 1983; Ellis 2001; Hatters et al. 2002; Ellis and Minton 2006; Stagg et al. 2007), and in fact one can even come up with rational ways to define effective hard-sphere diameter for proteins (Kim et al. 2010; Mittal and Best 2010). On the other hand, the failure of the SPT to explain the crowding data does not necessarily invalidate the SPT or the underlying spherical approximation. In fact, it highlights the importance of attractive interactions that must be present between proteins and crowders.
In most simulations crowders are represented by hard spheres or spheres with purely repulsive potentials. In addition, the details of pairwise interactions between protein atoms (or collection of atoms in a coarse-grained protein model) and crowders are given explicitly. Thus, to validate the SPT for the effects of repulsive protein–crowder interactions on protein binding thermodynamics, it is essential to provide an unambiguous way to determine the effective sphere radius, R X, for a protein or complex X. One approach suggested by us is to use the Boltzmann factor criteria (Ben-Amotz and Stell 2004) and define the effective radius for a crowder size R c as
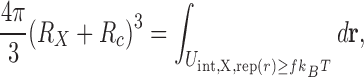 |
6 |
where 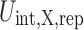 is the repulsive part of the interaction energy between protein X and a spherical crowder, while r is the three-dimensional vector pointing from the center of mass of the protein to the center of the crowder. We use f = 2, which has been successful in describing the thermodynamic and dynamic behavior of Lennard-Jones (LJ) fluid based on hard-sphere fluid (Mittal et al. 2007). Further justification of this f value can be found in the work of Speedy et al. (1989). Moreover, a slightly different value of f (e.g., f = 1) does not result in significantly different results. Note that R
X will then depend weakly on R
c as well as on a given protein conformation X.
is the repulsive part of the interaction energy between protein X and a spherical crowder, while r is the three-dimensional vector pointing from the center of mass of the protein to the center of the crowder. We use f = 2, which has been successful in describing the thermodynamic and dynamic behavior of Lennard-Jones (LJ) fluid based on hard-sphere fluid (Mittal et al. 2007). Further justification of this f value can be found in the work of Speedy et al. (1989). Moreover, a slightly different value of f (e.g., f = 1) does not result in significantly different results. Note that R
X will then depend weakly on R
c as well as on a given protein conformation X.
The success of the SPT Eq. 5 using the above prescription for the spherical approximation for anisometric proteins has been demonstrated in simulation studies of two distinct protein complexes, ubiquitin/UIM1 (Ubq/UIM1) and cytochrome c/cytochrome c peroxidase (Cc/CcP). Here, Ubq, Cc, and CcP are globular proteins close to spherical shape with 76, 108 and 294 residues, respectively. On the other hand, UIM1 is a 24-residue rod-like protein. It was observed that 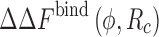 from the SPT-based theory for repulsive protein–crowder interactions agreed remarkably well with simulation data for a wide range of ϕ and R
c even for the Ubq/UIM1 complex (Kim et al. 2010). Note, however, that whether the theory will work for other highly anisometric proteins/complexes needs further investigation.
from the SPT-based theory for repulsive protein–crowder interactions agreed remarkably well with simulation data for a wide range of ϕ and R
c even for the Ubq/UIM1 complex (Kim et al. 2010). Note, however, that whether the theory will work for other highly anisometric proteins/complexes needs further investigation.
Attractive contribution to the crowding free energy
Calculating the attractive contribution, 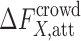 , to the crowding free energy is more challenging than calculating the repulsive contribution even for a simple spherical solute. In principle, one can use the thermodynamic perturbation theory approach to obtain an approximate analytical expression for
, to the crowding free energy is more challenging than calculating the repulsive contribution even for a simple spherical solute. In principle, one can use the thermodynamic perturbation theory approach to obtain an approximate analytical expression for 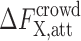 . The validity of this approach will then depend on the strength of the attractive interactions between proteins and crowders and if the reference repulsive interactions can describe the structure (protein–crowder, crowder–crowder) well. Thus, up to the first order in the attractive part of the interaction energy,
. The validity of this approach will then depend on the strength of the attractive interactions between proteins and crowders and if the reference repulsive interactions can describe the structure (protein–crowder, crowder–crowder) well. Thus, up to the first order in the attractive part of the interaction energy, 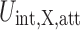 ,
, 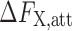 can be expressed as (Garde et al. 1999),
can be expressed as (Garde et al. 1999),
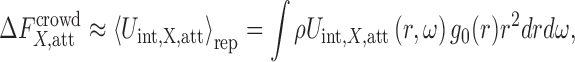 |
7 |
where ρ is the crowder number density related to ϕ via 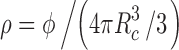 , and g0(r) is the radial distribution function between a protein and a crowder in the reference repulsive ensemble
, and g0(r) is the radial distribution function between a protein and a crowder in the reference repulsive ensemble 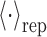 , while ω denotes the angular degree of freedom. We approximate g0(r) to be a stepwise function, i.e., g0(r) = 0 for r < r
0, g0(r) = gmax for r
0 ≤ r < r
1, and g0(r) = 1 for r > r
1. Employing the accurate Carnahan–Starling equation of state for a hard sphere fluid for gmax, we thereby obtain crowding free energy up to linear order in ϕ as
, while ω denotes the angular degree of freedom. We approximate g0(r) to be a stepwise function, i.e., g0(r) = 0 for r < r
0, g0(r) = gmax for r
0 ≤ r < r
1, and g0(r) = 1 for r > r
1. Employing the accurate Carnahan–Starling equation of state for a hard sphere fluid for gmax, we thereby obtain crowding free energy up to linear order in ϕ as
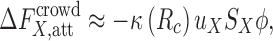 |
8 |
where S X is the surface area of a protein or complex encompassed by the center of a crowder, u X is the average strength of the attractive protein–crowder interaction on the surface S X, while κ depends only on R c.
It was observed that u X depends weakly on the type of protein X (Kim and Mittal 2012). We then obtain the attractive contribution to the change in the binding free energy using Eqs. (1), (4), and (8) as
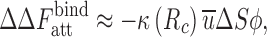 |
9 |
where 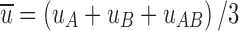 and
and 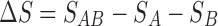 are the change in the surface area upon binding. Note that our theory provides a microscopic foundation for the phenomenological expressions proposed earlier by Jiao et al. (2010) and Rosen et al. (2011).
are the change in the surface area upon binding. Note that our theory provides a microscopic foundation for the phenomenological expressions proposed earlier by Jiao et al. (2010) and Rosen et al. (2011).
From Eq. 9 it is evident that the attractive protein–crowder interactions destabilize complex formation or stabilize isolated proteins in a crowded solution (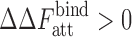 ) because
) because 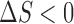 in general. This enthalpic effect (dependent on protein surface areas) is in contrast to the entropic effect caused by repulsive interactions (dependent on protein volumes) that favors compact structures that occupy lesser volume. This competition can then give rise to numerous different scenarios in experiments or simulations depending on their individual contributions.
in general. This enthalpic effect (dependent on protein surface areas) is in contrast to the entropic effect caused by repulsive interactions (dependent on protein volumes) that favors compact structures that occupy lesser volume. This competition can then give rise to numerous different scenarios in experiments or simulations depending on their individual contributions.
Computational model of protein–protein interactions in a crowded solution
It is quite challenging to test the theory presented above experimentally since the exact nature of the microscopic interactions between proteins and crowders is unknown in most cases. In the case of experimental data (if available), one can consider the physical parameters in our crowding theory as fitting parameters to describe the experimental data. The resulting fit parameters will have a sound physical basis and can therefore provide information on unknown microscopic interactions. However, such an approach is still ambiguous, as pointed out by Elcock (2010); for dextran, a commonly used crowding agent, three different spherical approximations have been used to fit the SPT equation in three different experiments (Batra et al. 2009a, b).
Computational models can provide a concrete platform on which to test a theory without much difficulty and can also provide new physical insight. Although there have been computational studies mimicking the cellular environment by employing various molecules measured experimentally inside the cytoplasm of Escherichia coli (McGuffee and Elcock 2010), such an approach can be computationally prohibitive to the sampling of binding thermodynamics and kinetics accurately. Thus, most simulations have been performed using spherical crowders and such models still continue to provide valuable insights (Wang and Cheung 2012). Although atomistic description is desirable for protein simulations, computational cost is again prohibitive in sampling protein–protein interactions. Consequently, a coarse-grained protein model is often used in simulating protein–protein interactions (Kim and Hummer 2008). In particular, the residue-level coarse-grained model developed by one of us has been quite successful in yielding binding free energies for weakly-to-moderately binding protein complexes and in describing the non-specific complexes in good agreement with nuclear magnetic resonance experiments (Kim et al. 2008).
For the interaction between a protein residue and a crowder, modified LJ potential has been found to be suitable and is given by
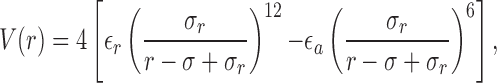 |
10 |
where σ is the contact radius [i.e., V(r = σ) = 0] between a protein residue and a crowder, while σ r is the interaction range (set equal to 6 Å). The potential acts from r = σ − σ r to an arbitrary r defined by the simulation cutoff distance. Note that unlike the standard LJ form, this potential ensures that the interaction range is independent of the crowder size R c.
Effects of crowding on the thermodynamic stability of a protein complex
Our crowding theory described here and its variants proposed earlier by others predict that excluded volume interactions due to repulsive protein–crowder interactions will lower the binding free energy, thereby favoring complex formation in a crowded solution. The extent of this binding free energy change with respect to crowder-free solution will depend on various factors, such as the crowder packing fraction, crowder size, and size of the protein molecules. Simulation studies employing repulsive protein–crowder interactions have essentially validated this qualitative expectation and shown strengthened protein complex formation (Kim et al. 2010). In our recent work (Kim et al. 2010) in which we used LJ-type repulsive interaction potential, a similar trend was observed as shown in Fig. 2a for the Cc/CcP complexes. Again, these results were anticipated, since these protein complexes occupy less volume as compared to the total volume occupied by the isolated proteins. With increasing crowder packing fraction, the binding free energy is lowered in a non-linear fashion. Also, for a given crowder packing fraction, smaller crowders have a more stabilizing influence on the complex formation—a prediction borne out by the SPT.
Fig. 2.

The change in the binding free energy (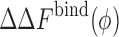 ) data obtained from replica exchange Monte Carlo (REMC) simulations (symbols) are shown as a function of the crowder packing fraction (ϕ) for different protein–crowder interaction strengths ranging from a purely repulsive interaction εr = 1.69k
B
T (a) to attractive interactions for the cytochrome c/cytochrome c peroxidase (Cc/CcP) complex(b). R
c Effective particle radius for a crowder size, εr crowder–protein repulsive strength, solid lines the predictions of our crowding theory as described in the text
) data obtained from replica exchange Monte Carlo (REMC) simulations (symbols) are shown as a function of the crowder packing fraction (ϕ) for different protein–crowder interaction strengths ranging from a purely repulsive interaction εr = 1.69k
B
T (a) to attractive interactions for the cytochrome c/cytochrome c peroxidase (Cc/CcP) complex(b). R
c Effective particle radius for a crowder size, εr crowder–protein repulsive strength, solid lines the predictions of our crowding theory as described in the text
The predictions of our crowding theory presented in the previous section, for which proteins are mapped onto spheres, agree remarkably well with the simulation data over a wide range of crowder sizes and crowder packing fractions. This theory can provide quantitative predictions for the change in binding free energy in the presence of repulsive spherical crowders with minimal information (protein structures).
Note, however, that whether the theory can be still valid for highly anisometric proteins, in general, requires further tests in future work.
Thus, thermodynamically, the excluded volume effect due to crowding favors the association of macromolecules. Various experimental studies that are consistent with this expectation are reviewed by Zimmerman and Minton (1993; Table 2) and Zhou et al. (2008; Table 1). Adding to the repertoire, a recent study (Aguilar et al. 2011) probed the effect of crowding (using Ficoll 70) on a heptameric protein (human cpn10 or GroES in E. coli) consisting of seven identical β-barrel subunits assembling into a ring. Using tyrosine fluorescence, it was observed that the monomer–heptamer dissociation constant value is lower in the Ficoll 70 solution than in the buffer, thereby suggesting a stabilization of the heptameric complex due to crowding.
Although crowding effects by hemoglobin, serum albumin and dextran can be quantitatively accounted for (Rivas et al. 2001), excluded volume-based models fail to account for the crowding effects exerted by another commonly used crowding agent, i.e., polyethylene glycol (PEG). Phillip et al. (2009) found a negligible impact on the binding affinity of TEM1-β-lactamase with its inhibitor β-lactamase and barnase with barstar due to increased PEG 1000 crowding that varied up to 30 % packing fraction. Results from experimental studies (Crowley et al. 2008; Phillip et al. 2009) suggest the presence of an attractive interaction between PEG molecules and proteins. Based on our earlier discussion, attractive protein–crowder interactions will actually counteract the stabilizing effect of excluded volume on complex formation and can help explain this trend.
To probe the effect of attractive interactions in addition to the excluded volume effects, we conducted simulations over a wide range of parameters (Rosen et al. 2011). As shown in Fig. 2b, after a critical threshold, increasing the protein–crowder attraction strength results in the destabilization of the protein complex relative to the crowder-free solution (i.e., 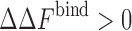 ). This effect is found to be more pronounced for crowders of smaller sizes. At low protein–crowder attraction strengths, we find that the stabilizing entropic effect is dominant over the destabilizing enthalpic effect. In fact, the critical attraction strength for which the binding free energy exhibits no change in a crowded solution as compared to the crowder-free solution (i.e.,
). This effect is found to be more pronounced for crowders of smaller sizes. At low protein–crowder attraction strengths, we find that the stabilizing entropic effect is dominant over the destabilizing enthalpic effect. In fact, the critical attraction strength for which the binding free energy exhibits no change in a crowded solution as compared to the crowder-free solution (i.e., 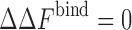 ) is approximately independent of the crowder packing fraction ϕ as shown in Fig. 3. This observation is a reflection of the approximate linear dependence of the binding free energy near the critical attraction strength with fraction up to modest packing fractions. Importantly, the agreement between our crowding theory (solid curves in Fig. 3) and simulation data is quite remarkable.
) is approximately independent of the crowder packing fraction ϕ as shown in Fig. 3. This observation is a reflection of the approximate linear dependence of the binding free energy near the critical attraction strength with fraction up to modest packing fractions. Importantly, the agreement between our crowding theory (solid curves in Fig. 3) and simulation data is quite remarkable.
Fig. 3.

The change in binding free energy data calculated from REMC simulations (symbols) are shown as a function of crowder–protein attraction strengths (εa) for various crowder packing fractions ϕ for the Cc/CcP complex. The fit curves for different ϕ converge around the point where 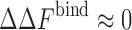
Probing the effects of attractive interactions experimentally also have begun only quite recently. Jiao et al. (2010) have recently studied catalase and superoxide dismutase interactions in the presence of dextran 70, ficoll 70, and PEG 2000 at various concentrations and temperatures. These researchers found that above a particular temperature, denoted by T θ, the primary effect of addition of crowders was the enhancement of protein association, whereas at a temperature below T θ, attractive interactions between the proteins and polymers predominate, inhibiting protein association. At a temperature T approximately equal to T θ the two effects cancel each other, thereby showing no effect on protein association (similar to our critical protein–crowder interaction strength) upon increasing the concentration of the crowding polymers.
Effects of mixed macromolecular crowding
So far we have discussed the development of a theory and associated simulation results for a single type of crowder in a crowded solution only. However, macromolecules present in a cell are quite diverse in terms of their sizes and interactions. Therefore, mixed crowding with different types of crowder particles will be a more realistic description of cellular crowding. One then may ask if there is any difference in treating the effects of mixed macromolecular crowding compared to the single-component crowders and if one may observe qualitatively different results?
A few recent studies, experimental as well as theoretical, have proposed that the mixed crowding solution may actually enhance the effect of crowding and may result in non-monotonic effects unlike single-component results (Batra et al. 2009b; Du et al. 2006). However, our simulation data for a binary crowder mixture (repulsive protein–crowder interactions) did not show anything qualitatively different from a single-component crowder solution (Kim et al. 2010). Moreover, within the SPT-like approach the effects of mixed repulsive crowding on the binding free energy are actually additive and we had proposed the following “ansatz,”
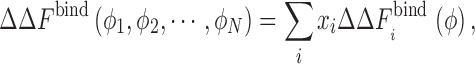 |
11 |
where i = 1 to N, ϕ
i is the packing fraction of component i in a crowding mixture, x
i = ϕ
i/ϕ is the relative fraction of component i, 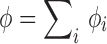 , and
, and 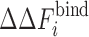 is the change in the binding free energy of a pure component i at total crowder packing fraction ϕ. Simulations for repulsive protein–crowder interactions with a binary crowder mixture for the sizes between 12 and 20 Å showed an excellent agreement with the prediction of Eq. 11 (Kim et al. 2010). In the case of attractive protein–crowder interactions, the additivity ansatz Eq. 11 still holds for a range of crowder sizes (12–20 Å) and for weak attractive interactions. However, mutual attraction between different crowder components can affect the behavior in a way that cannot be captured in an additive sense. More work is needed to identify the parameter range for which Eq. 11 is a good approximation to yield an accurate estimate of
is the change in the binding free energy of a pure component i at total crowder packing fraction ϕ. Simulations for repulsive protein–crowder interactions with a binary crowder mixture for the sizes between 12 and 20 Å showed an excellent agreement with the prediction of Eq. 11 (Kim et al. 2010). In the case of attractive protein–crowder interactions, the additivity ansatz Eq. 11 still holds for a range of crowder sizes (12–20 Å) and for weak attractive interactions. However, mutual attraction between different crowder components can affect the behavior in a way that cannot be captured in an additive sense. More work is needed to identify the parameter range for which Eq. 11 is a good approximation to yield an accurate estimate of 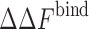 for a crowder mixture.
for a crowder mixture.
Effects of crowding on protein complex specificity: native vs encounter complex
Recent experiments and simulation have shown the existence of nonspecific complexes in solution, albeit in low population (<10 %), which may play an important role in the complex formation by reducing the degree of freedom during the binding target search process (Schreiber et al. 2009; Li et al. 2010; Wang et al. 2011). What is the effect of macromolecular crowding on the presence and stability of such nonspecific complexes?
Figure 4a shows the cumulative distributions of distance root mean square (dRMS) with increasing crowder packing fraction ϕ for the Cc/CcP complex in the presence of repulsive crowders. Here, dRMS is a measure of similarity between the experimental native complex and simulated structures. Structures with a dRMS of <5 Å are very similar to the native complex structure. It was observed that as the crowder packing fraction increases, the population of native-like structures with dRMS of <5 Å increases, while the populations of transient encounter complexes (dRMS >5 Å) decrease (Kim et al. 2010). This result suggests that proteins in the presence of repulsive crowders are more likely to form more compact native complexes (Fig. 4c) than metastable intermediate states (Fig. 4d).
Fig. 4.

a, b Cumulative distributions of bound complexes are shown as a function of dRMS calculated based on the experimental native structure for the Cc/CcP complex. c The specific bound complexes are shown where the red–blue combination is used for the experimental structure (PDB: 2pcc) and the red–green combination is used for the complex structure obtained from our simulation. d Several instances of the nonspecific bound complexes (red–yellow combination) are shown and the experimental native structure (red–blue) is also shown for the reference
On the contrary, as the attractive protein–crowder interactions tend to maximize contact between a protein and crowders, one would expect that the nonspecific complexes with larger surface area exposed to the crowders are stabilized in this case. This expectation is confirmed in our simulations as shown in Fig. 4b; the population of nonspecific complexes (dRMS >5 Å) is enhanced as the protein–crowder attraction strength increases. These findings are also consistent with recent experimental results (Phillip et al. 2012).
Conclusions
We have presented a review of the development of a general theory to describe the effects of macromolecular crowding on protein–protein interactions. The theory accounts for both repulsive and attractive protein–crowder interactions. The change in the binding free energy due to crowding can be separated into repulsive and attractive contributions. The repulsive contribution is well described by the scaled particle theory (SPT) by approximating proteins as spherical objects. Meanwhile, an approximate analytical expression is obtained for weak protein–crowder interactions by using the statistical mechanics of hard-sphere fluid and the first-order perturbation theory. To validate the theory, we performed simulations on two distinct protein complexes, Ubq/UIM1 and Cc/CcP, using a residue-level coarse-grained protein model and spherical crowders. The results of these simulations show that the theory can describe the simulation data remarkably well for both repulsive and attractive protein–crowder interactions over a wide range of parameters. In addition, the simulations show that the repulsive protein–crowder interactions increase the population of the native complex at the expense of transient encounter complexes, while the opposite trend was observed for the attractive protein–crowder interactions.
Most of the work to date, however, has been focused on the formation of complexes between well-structured proteins, thus ignoring any protein conformational change. In such cases, it was found that a single conformation of a protein or complex was sufficient to calculate the crowding free energy. However, there exist many eukaryotic proteins which are disordered in isolation under physiological conditions, but fold into their native conformations upon binding to target proteins (Wright and Dyson 2009). No theoretical and computational studies have focused on the effects of macromolecular crowding on such protein complexes. Since the folding of a protein is tightly coupled to the binding event, and crowding agents may exert a different level of effect on the stability of the folded state and the association equilibria, one may encounter complex scenarios in the presence of crowders. Although it can easily be predicted that the binding of these complexes would be enhanced by the excluded volume effects, it is still unclear whether the theory introduced here will be adequate to describe their behavior. In addition, one may ask whether the kinetics (Kim and Yethiraj 2009), and, in particular, the underlying mechanism remain the same in the presence of generic crowding agents. Thus, further theoretical as well as computational studies are warranted along these directions.
Acknowledgments
It is our great pleasure to contribute this article in a special issue to celebrate Allen Minton’s 70th birthday and his contributions to the field of macromolecular crowding.
Conflict of interest
None.
Footnotes
Special Issue: Protein–Protein and Protein–Ligand Interactions in Dilute and Crowded Solution Conditions. In Honor of Allen Minton’s 70th Birthday.
References
- Aguilar X, Weise CF, Sparrman T, Wolf-Watz M, Wittung-Stafshede P. Macromolecular crowding extended to a Heptameric system: the Co-chaperonin protein 10. Biochem. 2011;50:3034–3044. doi: 10.1021/bi2002086. [DOI] [PubMed] [Google Scholar]
- Batra J, Xu K, Qin S, Zhou H-X. Effect of macromolecular crowding on protein binding stability: modest stabilization and significant biological consequences. Biophys J. 2009a;970(3):906–11. doi: 10.1016/j.bpj.2009.05.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batra J, Xu K, Zhou H-X. Nonadditive effects of mixed crowding on protein stability. Proteins. 2009b;770(1):133. doi: 10.1002/prot.22425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Amotz D, Stell G. Reformulation of Weeks–Chandler–Andersen perturbation theory directly in terms of a hard-sphere reference system. J Phys Chem B. 2004;1080(21):6877–6882. [Google Scholar]
- Clore G, Iwahara J. Theory, practice, and applications of paramagnetic relaxation enhancement for the characterization of transient low-population states of biological macromolecules and their complexes. Chem Rev. 2009;109:4108–4139. doi: 10.1021/cr900033p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crowley PB, Brett K, Muldoon J (2008) NMR spectroscopy reveals cytochrome cpoly(ethylene glycol) interactions. ChemBioChem 90(5):685–688. ISSN 1439–7633. doi:10.1002/cbic.200700603 [DOI] [PubMed]
- Douglas J, Dudowicz J, Freed K. Crowding induced self-assembly and enthalpy-entropy compensation. Phys Rev Lett. 2009;1030(13):135701. doi: 10.1103/PhysRevLett.103.135701. [DOI] [PubMed] [Google Scholar]
- Du F, Zhou Z, Mo Z-Y, Shi J-Z, Chen J, Liang Y. Mixed macromolecular crowding accelerates the refolding of rabbit muscle creatine kinase: implications for protein folding in physiological environments. J Mol Biol. 2006;3640(3):469–82. doi: 10.1016/j.jmb.2006.09.018. [DOI] [PubMed] [Google Scholar]
- Elcock AH. Models of macromolecular crowding effects and the need for quantitative comparisons with experiment. Curr Opin Struct Biol. 2010;200(2):196–206. doi: 10.1016/j.sbi.2010.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis RJ. Macromolecular crowding: obvious but underappreciated. Trends Biochem Sci. 2001;260(10):597–604. doi: 10.1016/s0968-0004(01)01938-7. [DOI] [PubMed] [Google Scholar]
- Ellis RJ, Minton AP. Protein aggregation in crowded environments. Biol Chem. 2006;3870(5):485–97. doi: 10.1515/BC.2006.064. [DOI] [PubMed] [Google Scholar]
- Fodeke AA, Minton AP. Quantitative characterization of temperature-independent and temperature-dependent protein–protein interactions in highly nonideal solutions. J Phys Chem B. 2011;1150(38):11261. doi: 10.1021/jp2049266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garde S, Garcìa AE, Pratt LR, Hummer G. Temperature dependence of the solubility of non-polar gases in water. Biophys Chem. 1999;780:21–32. doi: 10.1016/s0301-4622(99)00018-6. [DOI] [PubMed] [Google Scholar]
- Hatters DM, Minton AP, Howlett GJ. Macromolecular crowding accelerates amyloid formation by human apolipoprotein C-II. J Biol Chem. 2002;2770(10):7824–30. doi: 10.1074/jbc.M110429200. [DOI] [PubMed] [Google Scholar]
- Jarvis T, Ring D. Macromolecular crowding: thermodynamic consequences for protein–protein interactions within the T4 DNA replication complex*. J Biol Chem. 1990;2650(25):15160–15167. [PubMed] [Google Scholar]
- Jiao M, Li H-T, Chen J, Minton AP, Liang Y. Attractive protein-polymer interactions markedly alter the effect of Macromolecular crowding on protein association equilibria. Biophys J. 2010;990(3):914–23. doi: 10.1016/j.bpj.2010.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JS, Yethiraj A. Effect of Macromolecular crowding on reaction rates: a computational and theoretical study. Biophys J. 2009;960(4):1333–40. doi: 10.1016/j.bpj.2008.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YC, Hummer G. Coarse-grained models for simulations of multiprotein complexes: application to ubiquitin binding. J Mol Biol. 2008;3750(5):1416–33. doi: 10.1016/j.jmb.2007.11.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YC, Mittal J (2012) Crowding induced entropy-enthalpy compensation in protein association equilibria. arXiv:1209.6379 [cond-mat.soft] (1):1–5 [DOI] [PubMed]
- Kim YC, Tang C, Clore GM, Hummer G. Replica exchange simulations of transient encounter complexes in protein–protein association. Proc Natl Acad Sci USA. 2008;105:12855–12860. doi: 10.1073/pnas.0802460105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YC, Best RB, Mittal J. Macromolecular crowding effects on protein–protein binding affinity and specificity. J Chem Phys. 2010;1330(20):205101. doi: 10.1063/1.3516589. [DOI] [PubMed] [Google Scholar]
- Kozer N, Schreiber G. Effect of crowding on protein–protein association rates: fundamental differences between low and high mass crowding agents. J Mol Biol. 2004;3360(3):763–74. doi: 10.1016/j.jmb.2003.12.008. [DOI] [PubMed] [Google Scholar]
- Lebowitz JL, Rowlinson JS. Thermodynamic properties of mixtures of hard spheres. J Chem Phys. 1964;410(1):133. [Google Scholar]
- Li X, Moal IH, Bates PA. Detection and refinement of encounter complexes for protein–protein docking: taking account of macromolecular crowding. Proteins. 2010;780(15):3189–96. doi: 10.1002/prot.22770. [DOI] [PubMed] [Google Scholar]
- McGuffee SR, Elcock AH. Diffusion, crowding & protein stability in a dynamic molecular model of the bacterial cytoplasm. PLoS Comput Biol. 2010;60(3):e1000694. doi: 10.1371/journal.pcbi.1000694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minton AP. The effect of volume occupancy upon the thermodynamic activity of proteins: some biochemical consequences. Mol Cell Biochem. 1983;55:119–140. doi: 10.1007/BF00673707. [DOI] [PubMed] [Google Scholar]
- Minton AP, Wilf J. Effect of Macromolecular crowding upon the structure and function of an enzyme: glyceraldehyde-3-phosphate dehydrogenase. Biochemistry. 1981;200(17):4821–6. doi: 10.1021/bi00520a003. [DOI] [PubMed] [Google Scholar]
- Mittal J, Best RB. Dependence of protein folding stability and dynamics on the density and composition of macromolecular crowders. Biophys J. 2010;980(2):315–20. doi: 10.1016/j.bpj.2009.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mittal J, Errington J, Truskett T. Relationships between self-diffusivity, packing fraction, and excess entropy in simple bulk and confined fluids. J Phys Chem B. 2007;111:10054–10063. doi: 10.1021/jp071369e. [DOI] [PubMed] [Google Scholar]
- Morar AS, Wang X, Pielak GJ. Effects of crowding by mono-, di-, and tetrasaccharides on cytochrome c-cytochrome c peroxidase binding: comparing experiment to theory. Biochem. 2001;400(1):281–5. doi: 10.1021/bi002296r. [DOI] [PubMed] [Google Scholar]
- Patel CN, Noble SM, Weatherly GT, Tripathy A, Winzor DJ, Pielak GJ. Effects of molecular crowding by saccharides on alpha-chymotrypsin dimerization. Protein Sci. 2002;11:997–1003. doi: 10.1110/ps.4450102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillip Y, Sherman E, Haran G, Schreiber G. Common crowding agents have only a small effect on protein–protein interactions. Biophys J. 2009;970(3):875–85. doi: 10.1016/j.bpj.2009.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillip Y, Kiss V, Schreiber G. Protein-binding dynamics imaged in a living cell. Proc Natl Acad Sci USA. 2012;1090(5):1461–6. doi: 10.1073/pnas.1112171109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiss H, Frisch HL, Lebowitz JL. Statistical mechanics of rigid spheres. J Chem Phys. 1959;310(2):369. [Google Scholar]
- Rivas G, Fernández JA, Minton (2001) Direct observation of the enhancement of noncooperative protein self-assembly by Macromolecular crowding: indefinite linear self-association of bacterial cell division protein FtsZ. Proc Natl Acad Sci USA 980(6):3150–5. ISSN 0027–8424. doi:10.1073/pnas.051634398 [DOI] [PMC free article] [PubMed]
- Rosen J, Kim YC, Mittal (2011) Modest protein–crowder attractive interactions can counteract enhancement of protein association by intermolecular excluded volume interactions. J Phys Chem B 1150(11):2683–9. ISSN 1520–5207. doi:10.1021/jp200625k [DOI] [PubMed]
- Schreiber G, Haran G, Zhou H-X. Fundamental aspects of protein–protein association kinetics. Chem Rev. 2009;1090(3):839–60. doi: 10.1021/cr800373w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snoussi K, Halle B. Protein self-association induced by macromolecular crowding: a quantitative analysis by magnetic relaxation dispersion. Biophys J. 2008;880(4):2855–2866. doi: 10.1529/biophysj.104.055871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speedy R, Prielmeier F, Vardag T, Lang E, Ludemann H. Diffusion in simple fluids. Mol Phys. 1989;660(3):577–590. [Google Scholar]
- Stagg L, Zhang S.-Q, Cheung MS, Wittung-Stafshede P. Molecular crowding enhances native structure and stability of alpha/beta protein flavodoxin. Proc Natl Acad Sci USA. 2007;1040(48):18976–81. doi: 10.1073/pnas.0705127104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Berg B, Ellis RJ, Dobson CM (1999) Effects of macromolecular crowding on protein folding and aggregation. EMBO J 180 (24):6927–33. ISSN 0261–4189. doi:10.1093/emboj/18.24.6927 [DOI] [PMC free article] [PubMed]
- Wang Q, Cheung MS (2012) A physics-based approach of coarse-graining the cytoplasm of Escherichia coli (CGCYTO). Biophys J 1020(10):2353–2361. ISSN 00063495. doi:10.1016/j.bpj.2012.04.010 [DOI] [PMC free article] [PubMed]
- Wang Q, Zhuravleva A, Gierasch LM. Exploring weak, transient protein–protein interactions in crowded in vivo environments by in-cell nuclear magnetic resonance spectroscopy. Biochemistry. 2011;500(43):9225–36. doi: 10.1021/bi201287e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weeks JD. Role of repulsive forces in determining the equilibrium structure of simple liquids. J Chem Phys. 1971;540(12):5237. [Google Scholar]
- Wenner JR, Bloomfield VA. Crowding effects on EcoRV kinetics and binding. Biophys J. 1999;770(6):3234–41. doi: 10.1016/S0006-3495(99)77154-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright PE, Dyson HJ. Linking folding and binding. Curr Opin Struct Biol. 2009;190(1):31. doi: 10.1016/j.sbi.2008.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou H.-X, Rivas G, Minton AP. Macromolecular crowding and confinement: biochemical, biophysical, and potential physiological consequences. Annu Rev Biophys. 2008;37:375–97. doi: 10.1146/annurev.biophys.37.032807.125817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmerman S, Minton AP. Macromolecular crowding: biochemical, biophysical, and physiological consequences. Annu Rev Biophys Biomol Struct. 1993;22:27–65. doi: 10.1146/annurev.bb.22.060193.000331. [DOI] [PubMed] [Google Scholar]
- Zorrilla S, Rivas G, Acuña A, Lillo M. Protein self-association in crowded protein solutions: a time-resolved fluorescence polarization study. Protein Sci. 2004;13:2960–2969. doi: 10.1110/ps.04809404. [DOI] [PMC free article] [PubMed] [Google Scholar]