

Abstract
Objective(s):
Cognitive defects such as learning and memory impairment are amongst the most repetitious sequelae after sever and moderate traumatic brain injury (TBI). It was suggested that ellagic acid (EA), an innate phenol product, display neuroprotective properties against oxidative and inflammatory damages after brain injury. The object of the current study was therapeutic properties of EA on blood-brain barrier (BBB) interruption and elevated content of TNF-α in brain tissue followed by neurologic aftereffects, cognitive and brain electrophysiology deficits as outcomes of diffuse TBI in rat.
Materials and Methods:
TBI was induced by a 200 g weight falling by a 2-m height through a free-falling tube onto the head of anesthetized rat. TBI rats treated immediately after trauma with EA (100 mg/kg, IP) once every 8 hr until 48 hr later. Neurologic outcomes, passive avoidance task (PAT), hippocampal long-term potentiation (LTP), BBB permeability and content of TNF-α in brain tissue were evaluated.
Results:
TBI induced significant impairments in neurological score, BBB function, PAT and hippocampal LTP in TBI+Veh group in compare with Sham+Veh (P<0.001). EA treatment decreased neurologic severity score (NSS), restored increased BBB permeability, cognitive and hippocampal LTP abnormalities, and elevated brain content of TNF-α due to TBI significantly (P<0.001).
Conclusion:
Our findings propose that EA can restore NSS, cognitive and LTP deficits and prevent brain inflammation may by restore BBB permeability as well as lowering brain content of TNF-α following TBI.
Keywords: Blood-brain barrier, Ellagic acid, LTP, Memory, TNF-α, Traumatic brain injury
Introduction
Traumatic brain injury (TBI) is a most important reason of neurological disability and fatality among young individuals worldwide (1), and is foresee to be as a major cause of death and disability by the year 2020 (2). Apart from an initial mechanical injury distinguished by interruption of the brain parenchyma and blood vessels, the extracellular and intracellular changes following a TBI have been termed ‘secondary injury’ including molecular events and physiological proceedings that may occur hours and days after the early injury (1-3). The immediate cell death and interruption of blood brain barrier (BBB) integrity leads to an inflammatory response which is distin-guished by excitotoxicity, changes in concentrations of ions and neurotransmitters, up-regulation and liberate of inflammatory molecules such as complement factors, chemokines, cytokines, and infiltration of blood derived immune cells into the brain tissue (1, 3-5). Previous studies have shown that inflammation has a key role in secondary injury, not only immediately after the trauma, but perhaps even after months and years (6). Cognitive deficiencies such as thought, memory, and reasoning difficulties and behavior or intellectual health disorders are amongst the most frequent consequences after severe and moderate TBI (7).
Natural yields have been believed as an exceptional and reputable source for the development of new drugs (8). Epidemiologic investigations reported a relationship among the intake of phenolic foods and the protection against several illnesses (9). Polyphenols are yields of plant metabolism and have been found to possess antioxidant properties (10, 11). Ellagic acid (EA, 2,3,7,8- tetrahydroxybenzo pyrano [5,4,3-cde]benzopyran-5-10-dione) is a naturally occurring polyphenolic compound which is originate in pomegranate, grapes, raspberries, blackberries, strawberries and walnuts (8, 9, 12, 13). EA exhibits a variety of actions such as anti-bacterial, anti-inflammatory, immune regulatory and anti-tumor effects and also it is regarded as a potent antioxidant (14-16). Some studies suggested that EA exhibits neuroprotective effects against oxidative damage and anti-proliferative effects in diabetic rats, but the mechanisms of its action at the cellular and molecular levels are still unclear (17, 18).
Long-term potentiation (LTP) in brain hippocampal synapses has considered as a model for the cellular changes that underlie learning and memory (19) and changes in LTP may contribute to a number of neurological diseases. Spatial cognitive deficit following brain trauma in adult rodents has been linked to loss of neurons in the dentate gyrus area (DG) of the brain hippocampus (20, 21). Previous reports have reported that the DG area is sensitive to brain traumatic insults regardless of age, injury type, or injury severity (22, 23).
One of the important cellular factors synthesized by microglia cells following activation within the brain is tumor necrosis-alpha (TNF-α) as a pro-inflammatory cytokine. TNF-α is a potent activator of the immune system and can provoke the activation of additional resting microglia cells or astrocytes located in the adjacent brain microenvironment. If this occurs it can result in an unregulated inflammatory response by central nervous system (CNS) (24).
The pro-inflammatory cytokine TNF-α receptors are expressed on neurons and glial cells following the CNS. Levels of TNF-α areelevated in several CNS disorders that are correlated with cognitive discrepancies, leading to the search for a possible role in plasticity, especially in the hippocampus (25). Astrocytes, microglia and some neurons can synthesize and release large amounts of TNF-α in the brain which leads to neuro-inflammatory response that is associated with several neurological disorders (26, 27).
In previous study we reported that7 days pretreatment of TBI rats with EA significantly prevented lowering neurological score, elevation of lipid peroxidation (by measuring MDA), increased the anti-oxidative enzymes activity (GPx and SOD). EA prohibited pro-inflammatory cytokines creation such as IL-1, IL-6 and TNF-α induced by TBI while elevated IL-4 and IL-10. These effects thereby prevented significant BBB disruption, cognitive and LTP impairment, with positive effects on antioxidant capacity and anti-inflammatory cytokines (28).
There is no available scientific statement on the role of EA against cognitive, learning and memory and brain electrophysiology deficits induced after TBI. With these findings we proposed EA may also have positive effects on behavioral and electrophysiological impairments after TBI. So, the present study aimed to examine the effects of EA on passive avoidance memory and hippocampal LTP destructions induced by closed head injury and established whether these effects are adjusted through anti-inflammatory mechanisms in the brain.
Materials and Methods
Animals
Adult male Wistar albino rats (250±20 g) were obtained from animal breeding center of Jundishapur University of Medical Sciences (Ahvaz, Iran). Animals were kept in room with constant conditions with temperature-controlled (22±2 °C) and permitted to adapt to the laboratory situations for at least 7 days prior to the study and exposed to a 12-hr light–dark cycle, lights on 07:00 AM. All experiments were done during the light phase of the cycle (8:00 AM to 5:00 PM). Access to food and water were ad libitum except during the experiments. Animal handling and experimental procedures were performed under observance of the University and Institutional legislation, controlled by the Local Ethics Committee for the Purpose of Control and Supervision of Experiments on Laboratory Animals (Ethics code: ajums.REC.1392.362). All attempts were made to minimize animal distress and to reduce the number of animals used. Prior to the onset of behavioral testing, all rats were handled for 5 days (10 min daily).
Animals divided randomly into three main groups with 24 rats in each group: 1) Sham + Veh; animals underwent same operation procedure of inducing brain trauma except trauma induction, received intraperitoneal injection (IP) of vehicle (10% DMSO in normal saline in a total volume of 10 ml/kg) was started immediately after the injury and continued until 48 hr later (three times daily, once every 8 hr). 2) TBI+Veh; TBI animals that received IP injection of vehicle similar to the first group. 3) TBI+EA; TBI animals that obtained IP injection of 100 mg/kg EA immediately after trauma three times daily until 48 hr later. Each main experimental group were divided into 3 sub-groups with 8 in each in order to; 1) passive avoidance task and electrophysiological recording, 2) measuring the BBB permeability, 3) determination of TNF-α content in brain tissue respectively.
Drug administration and chemicals
EA (purity≥98%), Evans blue, dimethyl sulfoxide (DMSO), Triton-X100 and protease inhibitor cocktail were purchased from Sigma-Aldrich Co (St Louis, MO, USA). Tris base, sodium phosphate, sodium chloride, potassium phosphate, potassium chloride and all other chemicals purchased from Merck Company (Darmstadt, Germany). EA dissolved in 10% DMSO solution. Its concentration was freshly prepared in such a way that the required dose could be injected in a volume of 10 ml/kg, IP.
Dose and drug administration timetables were selected based on our previous works (28, 29) and on pilot tests in our laboratory. The mean serum elimination half-life of EA is about 8.4±1.4 hr (30). It was administered 3 times daily (once every 8 hr), considering the bioavailability of EA starting immediately after induction of TBI continued for two consecutive days. To habituate the animals to the IP injections, all rats were administered saline (10 ml/kg) 3 times daily for three days prior to the experiment.
Induction of experimental diffuse TBI
Rats were anesthetized with ketamine hydro-chloride/xylazine (50/4 mg/kg, IP). Tracheal intubation was performed in all anesthetized rats followed by diffuse traumatic brain trauma using a device prepared in Physiology Research Center of Ahvaz Jundishapur University of Medical Sciences according to Marmarou’s technique (31). Briefly, a 200 g weight was fallen from a 2-m height through a free-falling tube onto the head of anesthetized animal while a steel disc (with 10 mm diameter and 2 mm thickness) was attached to dorsal surface of animal cranium. Following brain trauma generation, the animal was urgently connected to the small animal ventilator (Ugo Basile, Italy) and as soon as starting the spontaneous breathing, it was detached from ventilator and returned to the cage to be cared (32).
Passive avoidance task
The effects of EA on memory evaluated using a two-way shuttle box for test the step-through passive avoidance task (Borj Sanat Azma, Tehran). The device consisted two compartments with similar in dimensions (200 × 250 × 200 mm) includes a lighted and a dark partitions with a grid floor. These two partitions were separated by a sliding gate. In the adapting session, the animals were placed into the lighted partition while the sliding gate was opened and permitted to explore into both partitions for 5 min. After 10 min the rat was re-located into the lighted division again facing away from the closed sliding gate and 10 sec later the door was raised and the incoming delay of rat into the dark compartment was recorded as initial latency (IL). As soon as the animals entered the dark compartment, the door was closed; an inescapable foot-shock (50 Hz square wave, 1.2 mA for 3 sec) was delivered through the grid floor with a constant current shock generator. All animals examined entered the dark compartment within 300 sec as cut-off latency in the training session and received a foot-shock. The memory evaluated 24 hr after the training session using the same pattern, but without the foot-shock, and the step-through latency (STL) for animals to enter the dark compartment was measured. When an animal did not enter the dark room within 300 sec, the step-through latency was recorded as 300 sec (33).
Electrophysiological studies
Forty eight hours after TBI induction, animals that have received EA or vehicle 3 times daily for 2 consecutive days were prepared for electrophysio-logy surgery and recording. Animals were anesthe-tized with ketamine and xylazine (90/10 mg/kg, IP) and their heads mounted in a stereotaxic device for surgery (electrode implantation and fEPSP recording). Supplementary injections of ketamine/xylazine (1/3 main dose) were given when necessary to ensure full anesthesia. A non-electric heating pad was used to maintain the animals’ body temperature at 36.5 ± 0.5°C. Small holes were drilled in the skull at the positions of the stimulating and recording electrodes. A couple of stimulating metal wire microelectrode (stainless steel, 100 µm in diameter, tip separation 500 µm, CFW, USA) and a pair of recording metal wire microelectrodes (tungsten, 50µm in diameter, tip separation 1 mm, CFW, USA) were implanted into the perforant pathway (PP) at AP =−7.5 mm from bregma; ML = -4 mm; DV = -3.9 mm from dura and granular cells of hippocampus dentate gyrus (DG) with stereotaxic coordination of AP =−3.8 mm from bregma; ML = -2.3 mm; DV = –3.5 mm from dura consistent with the atlas of Paxinos and Watson, respectively (34). The electrodes were lowered gradually (0.1mm/30 sec) from dura to the DG and PP, in order to minimize trauma to brain tissue (35).
LTP induction
The field potential recordings were obtained in the granular cells of the DG following stimulation of the PP. Test stimuli were delivered to the PP every 30 sec. Electrodes were positioned gently to elicit a maximal field excitatory postsynaptic potential (fEPSP). The amplitude of post-tetanic stimulation population spike (PS) was calculated as the difference in voltage between the peak of the first positive wave and the peak of the first negative deflection. The EPSP slope was quantified as the maximum slope between initial point of fEPSP and the first positive peak of wave in order to measure synaptic efficacy. Extracellular field potentials were amplified (1000×); band pass filtered between 0.1 Hz to 3 KHz, digitized, recorded and analyzed using homemade software (Biochart software version 1.53, Science Beam Co, Iran). After ensuring a steady state baseline response, LTP was induced using high-frequency stimulation (HFS) protocols of 400 Hz (10 bursts of 20 stimuli, 0.2 ms stimulus duration, 10 sec interburst interval) at a stimulus intensity that evoked a PS amplitude and field EPSP slope of approximately 80% of maximum response. All potentials used as baseline before and after HFS were evoked at a stimulus intensity which produced 40% of this maximum by input/output (I/O) curve with different intensities for LTP recording. LTP was recorded for the periods of -1, 0.25, 0.5, 1, 3 and 24 hr following the HFS in order to conclude any changes in the synaptic response of DG neurons. For each time-point, 10 consecutive evoked responses were averaged at 10 s stimulus interval (33, 36, 37).
TNF-α assay
Rats were anesthetized (as described above) 48 hr after trauma and were perfused intracardially with phosphate buffered saline (pH=7.4) for 1 min to remove vesicular blood. Then, brain was take out from skull quickly and immediately frozen in a freezer (-80 °C) until assay. The brain tissue homogenized in tissue protein extraction reagent with 0.5% Triton-x100, 150 mM NaCl, 50 mM Tris, and a protease inhibitor cocktail (500 mg tissue per 2ml of the reagent). Following homogenization, samples were shaken on a shaker for 90 min and then centrifuged at 4 °C and 4000’ g for 15 min, the supernatant was collected as a homogenate. The protein content of the supernatant was estimated using a protein assay reagent kit to ensure an equal amount of protein from each sample was used for the assay (38).
ELISA kit for TNF-α obtained from eBioscience (San Diego, USA) and the assay carried out according to the manufacturer’s instructions. The concentration of TNF-α measured as picograms of antigen per milliliter of the supernatant.
Evaluation of BBB permeability
BBB permeability was determined by measuring extravascular Evans blue dye and using spectrophotometer device. Forty eight hours after trauma, 20 mg/kg Evans blue (Eb) dye 2% (1 ml/kg) was injected through femoral vein of anesthetized rats and brain vascular permeability was determined one hour after injection of Eb by opening thorax and clip the descending aorta. Then, isotonic saline solution (200-300 ml) was infused through the heart left ventricle for 20 min in order to remove intravascular Eb dye. For this purpose, jugular vein was cut bilaterally and infusion was continued until complete removal of Eb dye. Rats were then decapitated under deep anesthetize, the brains take out and homogenized in phosphate buffered saline. Trichloroacetic acid was then added to precipitate protein, and the samples were cooled and centrifuged. The resulting supernatant was gotten and the absorbance of Eb was evaluated at 620 nm using a spectrophotometer. The amount of color based on µg/mg brain tissue was calculated by the following formula:
Evans blue dye (µg) in brain tissue (g)= (13.24×20×absorbance)/tissue weight
Higher amount of dye in brain tissue represents more vascular permeability and more severe blood-brain barrier disruption (39, 40).
Assessment of neurologic outcomes
Neurologic severity score (NSS) was evaluated based on Veterinary Coma Scale (VCS). In this scale a total score (3-15) obtains by adding the scores of motor (1-8), visual (1-4) and respiratory (1-4) responses respectively. Higher score represents better neurological outcome. In current study neurological outcomes were also evaluated at -1, 1, 4, 24, and 48 hr after TBI. Derived from the VCS score, the NSS can be classified as following; mild (13–15), moderate (9–12), and severe (8 or less) (38, 41, 42).
Statistical analysis
Data were presented as mean ± SEM and analyzed by one-way ANOVA followed by Tukey’s post hoc test, while LTP data and neurological scores were analyzed by repeated measures ANOVA followed by Tukey’s post hoc test. P- Value lower than 0.05 were considered to be statistically significant. Statistical analysis was performed using Graph Pad Prism 6 software (Graph Pad Software Inc, San Diego, USA).
Results
Passive avoidance task
Figure 2 shows the effects of ellagic acid (EA) on the passive avoidance task, IL, and STL in TBI rats. Data was analyzed using one-way ANOVA analysis followed by Tukey’s post hoc test. Results showed that after administration of EA (100 mg/kg, IP), 3 times a day (once every 8 hr) for two consecutive days after TBI, IL was not different in groups [F(2,21)=1.004, P<0.49]. STL increased significantly in TBI rats treated with EA [F (2, 21)=115.9, P<0.001, for Sham+Veh; 61.48±14.78 sec compared to TBI+Veh; 29.43±2.25 sec, and F (2, 21) =97.42, P<0.001, for TBI+EA: 239.50±15.40 sec compared to TBI+Veh)].
Figure 1.

A design of several stages of experiments
Figure 2.
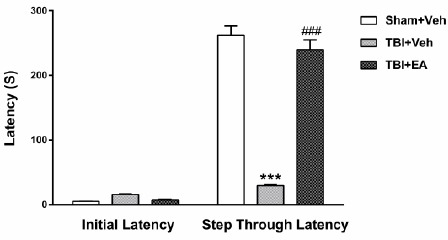
Initial latency and memory retention (STL) 48 hr after TBI. Memory retention was damaged in TBI+Veh group (***P<0.001 compared to Sham+Veh). Administration of EA (100 mg/kg, IP) reversed memory 48 hr after TBI (# # # P<0.001 compared to TBI+Veh). Data is shown as mean±SEM and analyzed by one-way ANOVA, followed by Tukey’s post hoc test, n=8
Electrophysiology
Sample PS recorded from the hippocampus DG area, induced following 400 Hz tetanic stimulation is displayed in Figure 3.
Figure 3.
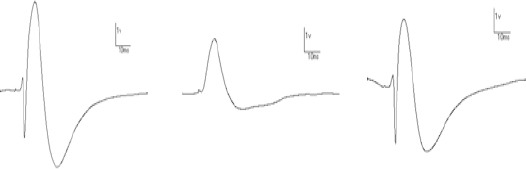
Sample population spikes recorded from the hippocampus DG area induced following 400 Hz tetanic stimulation, Sham+Veh (A), TBI+Veh (B), and TBI+EA (C) 48 hr after TBI
PS amplitude: As shown in Figure 4A, PS amplitude significantly decreased in TBI+Veh group in all LTP recording times (P<0.001). Analysis of data with repeated measures ANOVA and then by Tukey’s post hoc test showed that PS amplitude in TBI+Veh group was decreased significantly during 0.25, 0.5, 1, 3, and 24 hr after HFS compared to Sham+Veh [F(10,105)=28.27, P<0.001]. Administration of EA (100 mg/kg, IP), 3 times daily (once every 8 hr) for two consecutive days after TBI, restored PS amplitude significantly at all of the same recording times compared to TBI+Veh[F (2,21)=41.87, P<0.001].
Figure 4.
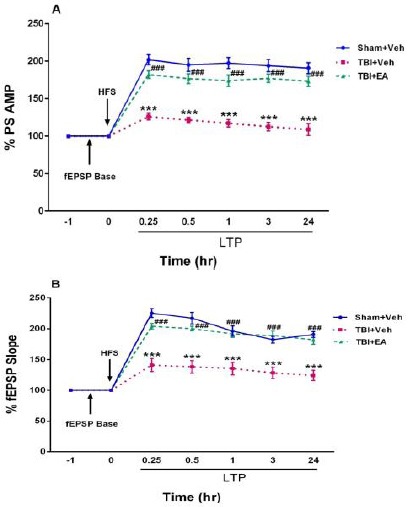
Amplitude of population spike (A) and slope of fEPSP (B) recorded from the hippocampal dentate gyrus area before and after high-frequency stimulation. PS amplitude and fEPSP incline were decreased in TBI+Veh group (***P<0.001 compared to Sham+Veh). Administration of EA (100 mg/kg, IP) restored amplitude and slope 48 hr after TBI (# # # P<0.001 in compared with TBI+Veh). Data represented as mean±SEM and analyzed using RM- ANOVA and then by Tukey’s post hoc test, n=8
fEPSP slope: Figure 4B shows the effects of EA (100 mg/kg, IP), 3 times daily (once every 8 hr) for two consecutive days after TBI on EPSP incline in TBI rats. Analysis of data using repeated measures ANOVA and then by Tukey’s post hoc test showed that in all LTP recording times PS slope significantly decreased in TBI+Veh group compared to Sham+Veh [(F (10,105)=21.13, P<0.001)], while in TBI+EA group it was increased significantly compared with TBI+Veh [(F (2,21)=24.27, P<0.001)].
TNF-α level in brain tissue
Figure 5 shows the effects of EA administration (100 mg/kg, IP), 3 times a day (once every 8 hr) for two consecutive days after TBI, on TNF-α level in whole brain tissue. Analysis of data by one-way ANOVA analysis and then by Tukey’s post hoc test revealed a significant enhancement of TNF-αin in TBI+Veh group (1300±62.91 pg/ml) compared with Sham+Veh (754.2± 16.8 pg/ml) [(F (2, 21) =54.00, P<0.001)], whereas in TBI+EA group (820±24.03 pg/ml) it was elevated significantly [(F (2, 21) =54.00, P<0.001)] when compared with TBI+Veh.
Figure 5.
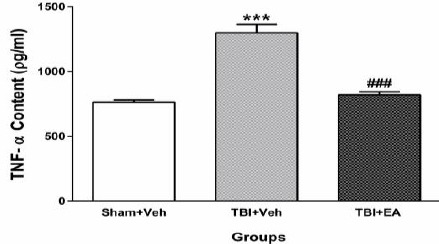
Brain tissue content of TNF-α 48 hr after TBI. TNF-α increased significantly after induction of TBI (***P<0.001 compared to Sham+Veh). Administration of EA (100 mg/kg, IP) reversed TNF-α 48 hr after TBI (# # # P<0.001 compared to TBI+Veh). Data represented as mean ± SEM and analyzed using one-way ANOVA followed by Tukey’s post hoc test, n=8
Evans blue dye content in brain
Figure 6 shows the Evans blue dye content in brains of different groups. Analysis of data using one-way ANOVA and then Tukey’s post hoc test showed that the amount of Evans blue dye in TBI+Veh group (29.21±0.76 µg/g tissue) increased significantly compared with Sham+Veh (22.12±0.47 µg/g tissue) [(F (2, 21) = 36.57, P<0.001)], whereas it decreased significantly in TBI+EA group (23.23±0.63 µg/g tissue) when compared with TBI+Veh [(F (2, 21) =36.57, P<0.001)].
Figure 6.
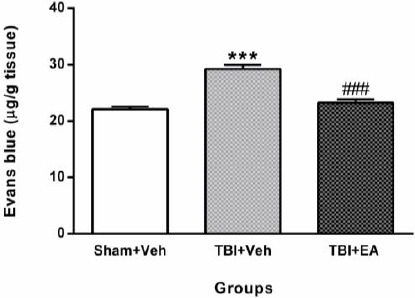
Content of Evans blue dye in brain tissue 48 hr after TBI. Content of Evans blue dye increased significantly in TBI+Veh (***P<0.001 compared with Sham+Veh). Administration of EA (100 mg/kg, IP) decreased it in TBI+EA (# # # P<0.001 when compared with TBI+Veh). Data shown as mean±SEM and analyzed using one-way ANOVA followed by Tukey’s post hoc test, n=8
Evaluation of neurologic severity score (NSS)
As seen in Figure 7, regarding results of analyzing data using repeated measures ANOVA and then Tukey’s post hoc test, NSS in TBI+Veh group decreased significantly [(F (10,105)=81.22, P<0.001)] compared with Sham+Veh in all hours following TBI. Nurological severity score increased significantly in TBI+EA group at 24 and 48 hr after TBI [(F (2,21)=208.5, P<0.001)] when compared with TBI+Veh.
Figure 7.
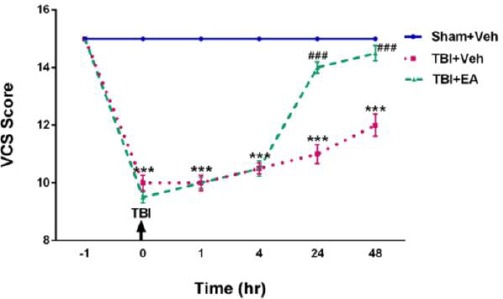
Veterinary coma scale (VCS) as NSS of TBI rats during one hour before, just after TBI, 1, 4, 24, and 48 hr following TBI induction. VCS in TBI+Veh was decreased significantly (***P<0.001 compared with Sham+Veh). Administration of EA (100 mg/kg, IP) reversed it 24 and 48 hr following TBI (# # # P<0.001 when compared with TBI+Veh). Data shown as mean±SEM and analyzed using RM- ANOVA followed by Tukey’s post hoc test, n=8
Discussion
The main goal of the current work was to examine the effects of intraperitoneal injection of EA on some brain functions such as cognition, sensory-motor signs, and generation and perseverance of LTP in hippocampal DG area following TBI. These findings indicated: 1) passive avoidance task was impaired after TBI and treatment with EA restored it significantly; 2) hippocampal LTP specifications were impaired and EA treatment restored the PS amplitude and slope of fEPSP significantly; 3) disrupted BBB permeability after TBI was improved with EA; 4) increased brain content of TNF-α due to TBI was recovered to normal levels and 5) NSS brought back to normal level in treated animals with EA.
A constant failure to induce LTP following brain injury has been shown in rats with several different brain injury models (43, 44). Our data indicated that TBI caused enhanced brain content of TNF-α and administration of EA decreased it, which may lead to restoration of cognitive behavior, the hippocampal LTP, and neurologic behaviors after repairing the BBB. Current findings are in agreement with the results of Chao et al study in rats which shows that the expression of TNF-α is efficiently decreased by EA, indicating EA anti-inflammatory activity (45).
It was demonstrated that TNF-α has both homeostatic and pathophysiological roles in the CNS (27, 46). The homeostatic functions of TNF-α regulate indispensable physiological processes such as synaptic plasticity (47, 48), learning and memory (49, 50), and astrocyte-induced synaptic strengthening (51). In pathological states, large amounts of TNF-α are released by astrocytes and microglia which leads to the neuro-inflammatory response that is associated with several neurological disorders (52-54). Like other studies as shown in our results, the brain content of TNF-α was increased after TBI (Figure 5) (55).
Furthermore, TNF-α may lead to glutamate-mediated cytotoxicity by two mechanisms; 1) indirectly, by inhibiting glutamate transport on astrocytes, 2) directly, by increasing the localization of ionotropic glutamate receptors to synapses (25). Neuro-inflammation and excitotoxicity are among the major causes of the neurodegenerative process (56) and thus, the levels of TNF-α in brain tissue are typically enhanced in a wide range of CNS disorders, such as TBI, cerebral ischemia, multiple sclerosis (MS), Alzheimer’s disease (AD), Parkinson’s disease (PD) and amyotrophic lateral sclerosis (ALS) (55, 57-59). The production and release of TNF-α accelerate under pathological conditions, about 1 hr following brain insult and well before neuronal death (57, 60, 61). The TNF-R1 (p55) and TNF-R2 (p75), two different types of receptors for TNF-α, are present in both neurons and glia (62). Although both receptors can lead to the activation of transcription factors, only TNF-R1 activation can initiate activation of the caspase pathways leading to apoptosis (25).
It has been revealed that TNF-α adjusts the growth of the hippocampus (63), perhaps via activation of TNF-R2, which does not lead to caspase-3 activation but is known to transduce the trophic effect of TNF-α (64). It has been shown that TNF-α inhibits LTP induction in CA1 and DG areas of the rat hippocampus at pathophysiological levels (65, 66). Our results showed that TBI-induced hippocampal LTP inhibition probably due to elevated levels of TNF-α in the brain tissue.
There are varieties in the mechanisms by which TNF-α inhibits early phase (less than 2 hr) and late phase LTP (more than 2 hr) post induction and it was established that p38 MAP kinase plays a major role in the inhibitory effect of TNF-α on early LTP. It has been shown that using the p38 inhibitor inversed the effect of TNF-α on early LTP without affecting late LTP (66). On the other hand, TNF-α has an important role in the regulation of AMPA-type glutamate receptors (AMPARs) trafficking being a serious component of the homeostatic regulatory system controlling synaptic plasticity (27). Considering the previous studies, TNF-α causes increased expression of AMPARs which may be more calcium permeable, therefore in the presence of TNF-α, the elevation of intracellular concentrations of calcium may perturb the LTP (25, 67). The effects of TNF-α on N-methyl-D-aspartate receptors (NMDARs) trafficking are less lessened; however, it was indicated that TNF-α potentiates NMDAR-mediated excitotoxicity in cortical neurons (54, 68). The results of several lines of studies showed that mGLuRs may also participate in the inhibitory effects of TNF-α on early LTP (25). Blockage of mGluR1 and mGluR5 receptors can stop the TNF-α-dependent inhibition of early and late LTPs (69). The results of the current study have revealed that administration of EA (100 mg/kg) restores the hippocampal LTP in both early and late phases as a result of a significant decrease in brain content of TNF-α. As shown in Figure 4, treatment with EA causes reduction of brain content of TNF-α to normal levels. Together with other parts of our results, the recovery effect of EA on LTP may directly depend on its anti-inflammatory effects through decreasing the elevated levels of TNF-α.
It seems this is the first study using EA to re-establish synaptic plasticity following TBI. These findings indicated that EA treatment restores significantly the TBI-induced behavioral abnormalities (impaired passive avoidance memory) and reduced fEPSP slope and PS amplitude appeared in TBI rats. In accordance with the current results, administration of EA leads to significant reduction of TNF-α content in the brain following induction of TBI in rats. Furthermore, the BBB function was restored in treated animals with EA which may explain the effects of EA on astrocytes improvement because of its anti-inflammatory actions. Although numerous reports have shown different effects of EA such as neuroprotective, anti-inflammatory, anti-oxidant, and free-radical scavenging (18, 70, 71), the precise mechanisms by which EA provokes these effects have not been fully revealed and require more investigations.
Neurological severity score in treated animals with EA reversed to normal level within 24 and 48 hr after TBI. It is shown that EA administration to TBI rats could improve neurologic outcomes via restoring the BBB function followed by brain tissue TNF-α level as well as cognitive and electrophysiological activity of hippocampus.
Conclusion
Current findings demonstrated that EA is capable of restoring cognitive behavior and hippocampal electrophysiological deficits induced by TBI and improve brain inflammation after TBI, followed by diminishing the pro-inflammatory factors such as TNF-α. According to our findings in previous and current research, these effects could be attributed to the anti-oxidative and anti-inflammatory nature of EA. These results suggest that EA may have a potential therapeutic effect on subjects with TBI.
Acknowledgment
The results reported in this paper were part of Dr Shahram Mashhadizadeh’s PhD thesis. This study was accomplished in Physiology Research Centers, Ahvaz, Iran. It was supported by the Vice-Chancellor for Research, Ahvaz Jundishapur University of Medical Sciences (Grant No: PRC-147), Ahvaz, Iran. Funding sources had no involvement in the conduct of the research and/or preparation of the article.
Conflict of interest
The authors have no conflict of interest to declare.
References
- 1.Al Nimer F, Lindblom R, Ström M, Guerreiro-Cacais AO, Parsa R, Aeinehband S, et al. Strain influences on inflammatory pathway activation, cell infiltration and complement cascade after traumatic brain injury in the rat. Brain Behav Immun. 2013;27:109–122. doi: 10.1016/j.bbi.2012.10.002. [DOI] [PubMed] [Google Scholar]
- 2.Mathers CD, Loncar D. Projections of global mortality and burden of disease from 2002 to 2030. PLoS Med. 2006;3:1–20. doi: 10.1371/journal.pmed.0030442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kalsotra A, Zhao J, Anakk S, Dash PK, Strobel HW. Brain trauma leads to enhanced lung inflammation and injury: evidence for role of P4504Fs in resolution. J Cereb Blood Flow Metab. 2007;27:963–974. doi: 10.1038/sj.jcbfm.9600396. [DOI] [PubMed] [Google Scholar]
- 4.Kumar A, Loane D. Neuroinflammation after traumatic brain injury: opportunities for therapeutic intervention. Brain Behav Immun. 2012;26:1191–1201. doi: 10.1016/j.bbi.2012.06.008. [DOI] [PubMed] [Google Scholar]
- 5.Shlosberg D, Benifla M, Kaufer D, Friedman A. Blood–brain barrier breakdown as a therapeutic target in traumatic brain injury. Nat Rev Neurol. 2010;6:393–403. doi: 10.1038/nrneurol.2010.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ramlackhansingh AF, Brooks DJ, Greenwood RJ, Bose SK, Turkheimer FE, Kinnunen KM, et al. Inflammation after trauma: microglial activation and traumatic brain injury. Ann Neurol. 2011;70:374–383. doi: 10.1002/ana.22455. [DOI] [PubMed] [Google Scholar]
- 7.Ribbers GM. Stone JH, Blouin M, editors. Brain Injury: Long term outcome after traumatic brain injury. Int Encyclopedia of Rehabilitation [serial online] pp. 1–9. Available from: http://cirrie.buffalo.edu/encyclopedia/en/article/338/
- 8.Newman DJ, Cragg GM. Natural Products as Sources of New Drugs over the Last 25 Years. J Nat Prod. 2007;70:461–477. doi: 10.1021/np068054v. [DOI] [PubMed] [Google Scholar]
- 9.Morton LW, Abu-Amsha Caccetta R, Puddey IB, Croft KD. Chemistry and biological effects of dietary phenolic compounds: relevance to cardiovascular disease. Clin Exp Pharmacol Physiol. 2000;27:152–159. doi: 10.1046/j.1440-1681.2000.03214.x. [DOI] [PubMed] [Google Scholar]
- 10.Ben Nasr C, Ayed N, Metche M. Quantitative determination of the polyphenolic content of pomegranate peel. Z Lebensm Unters Forsch. 1996;203:374–378. doi: 10.1007/BF01231077. [DOI] [PubMed] [Google Scholar]
- 11.Kostrzewa RM, Segura-Aguilar J. Novel mechanisms and approaches in the study of neurodegeneration and neuroprotection. A review. Neurotox Res. 2003;5:375–383. doi: 10.1007/BF03033166. [DOI] [PubMed] [Google Scholar]
- 12.Clifford MN, Scalbert A. Ellagitannins–nature, occurrence and dietary burden. J Sci Food Agric. 2000;80:1118–1125. [Google Scholar]
- 13.Wada L, Ou B. Antioxidant activity and phenolic content of Oregon caneberries. J Agric Food Chem. 2002;50:3495–500. doi: 10.1021/jf011405l. [DOI] [PubMed] [Google Scholar]
- 14.tta-Ur-Rahman F, Ngounou M, Choudhary M, Malik S, Makhmoor T, Nur-E-Alam M, et al. New antioxidant and antimicrobial ellagic acid derivatives from Pteleopsis hylodendron. Planta Med. 2001;67:335–339. doi: 10.1055/s-2001-14306. [DOI] [PubMed] [Google Scholar]
- 15.Chen HS, Liu M, Shi LJ, Zhao JL, Zhang CP, Lin LQ, et al. Effects of raspberry phytochemical extract on cell proliferation, apoptosis, andserum proteomics in a rat model. J Food Sci. 2011;76:T192–T198. doi: 10.1111/j.1750-3841.2011.02373.x. [DOI] [PubMed] [Google Scholar]
- 16.Festa F, Aglitti T, Duranti G, Ricordy R, Perticone P, Cozzi R. Strong antioxidant activity of ellagic acid in mammalian cells in vitro revealed by the comet assay. Anticancer Res. 2001;21:3903–3908. [PubMed] [Google Scholar]
- 17.Uzar E, Alp H, Cevik MU, Fırat U, Evliyaoglu O, Tufek A, et al. Ellagic acid attenuates oxidative stress on brain and sciatic nerve and improves histopathology of brain in streptozotocin-induced diabetic rats. Neurol Sci. 2012;33:567–574. doi: 10.1007/s10072-011-0775-1. [DOI] [PubMed] [Google Scholar]
- 18.Narayanan BA, Geoffroy O, Willingham MC, Re GG, Nixon DW. p53/p21 (WAF1/CIP1) expression and its possible role in G1 arrest and apoptosis in ellagic acid treated cancer cells. Cancer Lett. 1999;136:215–221. doi: 10.1016/s0304-3835(98)00323-1. [DOI] [PubMed] [Google Scholar]
- 19.Selig DK, Segal MR, Liao D, Malenka RC, Malinow R, Nicoll RA, et al. Examination of the role of cGMP in long-term potentiation in the CA1 region of the hippocampus. Learn Mem. 1996;3:42–48. doi: 10.1101/lm.3.1.42. [DOI] [PubMed] [Google Scholar]
- 20.Baldwin SA, Gibson T, Callihan CT, Sullivan PG, Palmer E, Scheff SW. Neuronal cell loss inthe CA3 subfield of the hippocampus following cortical contusion utilizing the optical disector method for cell counting. J Neurotrauma. 1997;14:385–398. doi: 10.1089/neu.1997.14.385. [DOI] [PubMed] [Google Scholar]
- 21.Colicos MA, Dixon CE, Dash PK. Delayed, selective neuronal death following experimental cortical impact injury in rats: possible role in memory deficits. Brain Res. 1996;739:111–119. doi: 10.1016/s0006-8993(96)00819-0. [DOI] [PubMed] [Google Scholar]
- 22.Huh JW, Widing AG, Raghupathi R. Differential effects of injury severity on cognition and cellular pathology after contusive brain trauma in the immature rat. J Neurotrauma. 2011;28:245–257. doi: 10.1089/neu.2010.1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Huh JW, Widing AG, Raghupathi R. Midline brain injury in the immature rat induces sustainedcognitive deficits, bihemispheric axonal injury and neuro-degeneration. Exp Neurol. 2008;213:84–92. doi: 10.1016/j.expneurol.2008.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Frankola KA, Greig NH, Luo W, Tweedie D. Targeting TNF-alpha to elucidate and ameliorate neuroinflammation in neurodegenerative diseases. CNS Neurol Disord DrugTargets. 2011;10:391–403. doi: 10.2174/187152711794653751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pickering M, Cumiskey D, O’Connor JJ. Actions of TNF-α on glutamatergic synaptic transmission in the central nervous system. Exp. Physiol. 2005;90:663–670. doi: 10.1113/expphysiol.2005.030734. [DOI] [PubMed] [Google Scholar]
- 26.Rogerio AP, Fontanari C, Melo MC, Ambrosio SR, Souza GE, Pereira PS, et al. Anti-inflammatory, analgesic and anti-oedematouseffects of Lafoensia pacari extract and ellagic acid. J Pharm Pharmacol. 2006;58:1265–1273. doi: 10.1211/jpp.58.9.0014. [DOI] [PubMed] [Google Scholar]
- 27.Montgomery SL, Bowers WJ. Tumor necrosis factor-alpha and the roles it plays in homeostatic and degenerative processes within the central nervous system. J Neuroimmune Pharmacol. 2012;7:42–59. doi: 10.1007/s11481-011-9287-2. [DOI] [PubMed] [Google Scholar]
- 28.Farbood Y, Sarkaki A, Dianat M, Khodadadi A, Haddad MK, Mashhadizadeh S. Ellagic acid prevents cognitive and hippocampal long-term potentiation deficits and brain inflammation in rat with traumatic brain injury. Life Sci. 2015;124:120–127. doi: 10.1016/j.lfs.2015.01.013. [DOI] [PubMed] [Google Scholar]
- 29.Mansouri MT, Naghizadeh B, Ghorbanzadeh B, Farbood Y. Central and peripheral antinociceptive effects of ellagic acid in different animal models of pain. Eur J Pharmacol. 2013;707:46–53. doi: 10.1016/j.ejphar.2013.03.031. [DOI] [PubMed] [Google Scholar]
- 30.bdul-Wahab RH, Al-Momani WM, Janakat S, Oran SA. Bioavailability of ellagic acid after single dose administration using HPLC. Pak J Nut. 2009;8:1661–1664. [Google Scholar]
- 31.Marmarou A, Foda MA, van den Brink W, Campbell J, Kita H, Demetriadou K. A new model of diffuse brain injury in rats: Part I: Pathophysiology and biomechanics. J Neurosurg. 1994;80:291–300. doi: 10.3171/jns.1994.80.2.0291. [DOI] [PubMed] [Google Scholar]
- 32.Khaksari M, Soltani Z, Shahrokhi N, Moshtaghi G, Asadikaram G. The role of estrogen and progesterone, administered alone and in combination, in modulating cytokine concentration following traumatic brain injury. Can J Physiol Pharmacol. 2011;89:31–40. doi: 10.1139/y10-103. [DOI] [PubMed] [Google Scholar]
- 33.Lashgari R, Motamedi F, Asl SZ, Shahidi S, Komaki A. Behavioral and electrophysiological studies of chronic oral administration of L-type calcium channel blocker verapamil on learning and memory in rats. Behav Brain Res. 2006;171:324–328. doi: 10.1016/j.bbr.2006.04.013. [DOI] [PubMed] [Google Scholar]
- 34.Paxinos G, Watson C. The rat brain in stereotaxic coordinates: hard cover. 6th ed. Academic Press; 2006. [Google Scholar]
- 35.Gureviciene I, Ikonen S, Gurevicius K, Sarkaki A, van Groen T, Pussinen R, et al. Normal induction but accelerated decay of LTP in APP +PS1 transgenic mice. Neurobiol Dis. 2004;15:188–195. doi: 10.1016/j.nbd.2003.11.011. [DOI] [PubMed] [Google Scholar]
- 36.Sarkaki A, Fathimoghaddam H, Mansouri SMT, Shahrani Korrani M, Farbood Y. Gallic acid improves cognitive, hippocampal long-term potentiation deficits and brain damage induced by chronic cerebral hypoperfusion in rats. Pak J Biol Sci. 2014;17:978–990. doi: 10.3923/pjbs.2014.978.990. [DOI] [PubMed] [Google Scholar]
- 37.Sarkaki A, Assaei R, Motamedi F, Badavi M, Pajouhi N. Effect of parental morphine addiction on hippocampal long-term potentiation in rats offspring. Behav Brain Res. 2008;186:72–77. doi: 10.1016/j.bbr.2007.07.041. [DOI] [PubMed] [Google Scholar]
- 38.Sarkaki A, Khaksari Haddad M, Soltani Z, Shahrokhi N, Mahmoodi M. Time-and dose-dependent neuroprotective effects of sex steroid hormones on inflammatory cytokines after a traumatic brain injury. J Neurotrauma. 2013;30:47–54. doi: 10.1089/neu.2010.1686. [DOI] [PubMed] [Google Scholar]
- 39.O’Connor CA, Cernak I, Vink R. Both estrogen and progesterone attenuate edema formation following diffuse traumatic brain injury in rats. Brain Res. 2005;1062:171–174. doi: 10.1016/j.brainres.2005.09.011. [DOI] [PubMed] [Google Scholar]
- 40.Khaksari M, Mahmmodi R, Shahrokhi N, Shabani M, Joukar S, Aqapour M. The effects of shilajit on brain edema, intracranial pressure and neurologic outcomes following the traumatic brain injury in rat. Iran J Basic Med Sci. 2013;16:858–864. [PMC free article] [PubMed] [Google Scholar]
- 41.King DR, Cohn SM, Proctor KG. Changes in intracranial pressure, coagulation, and neurologic outcome after resuscitation from experimental traumatic brain injury with hetastarch. Surgery. 2004;136:355–363. doi: 10.1016/j.surg.2004.05.011. [DOI] [PubMed] [Google Scholar]
- 42.Sternbach GL. The Glasgow coma scale. J Emerg Med. 2000;19:67–71. doi: 10.1016/s0736-4679(00)00182-7. [DOI] [PubMed] [Google Scholar]
- 43.Reeves TM, Lyeth BG, Povlishock JT. Long-term potentiation deficits and excitability changes following traumatic brain injury. Exp Brain Res. 1995;106:248–256. doi: 10.1007/BF00241120. [DOI] [PubMed] [Google Scholar]
- 44.Sanders MJ, Sick TJ, Perez-Pinzon MA, Dietrich WD, Green EJ. Chronic failure in the maintenance of long-term potentiation following fluid percussion injury in the rat. Brain Res. 2000;861:69–76. doi: 10.1016/s0006-8993(00)01986-7. [DOI] [PubMed] [Google Scholar]
- 45.Chao PC, Hsu CC, Yin MC. Anti-inflammatory and anti-coagulatory activities of caffeic acid and ellagic acid in cardiac tissue of diabetic mice. Nutr Metab (Lond) 2009;6:1–8. doi: 10.1186/1743-7075-6-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Santello M, Volterra A. TNFα in synaptic function: switching gears. Trends Neurosci. 2012;35:638–647. doi: 10.1016/j.tins.2012.06.001. [DOI] [PubMed] [Google Scholar]
- 47.Beattie EC, Stellwagen D, Morishita W, Bresnahan JC, Ha BK, Von Zastrow M, et al. Control of synaptic strength by glial TNFα. Science. 2002;295:2282–2285. doi: 10.1126/science.1067859. [DOI] [PubMed] [Google Scholar]
- 48.Kaneko M, Stellwagen D, Malenka RC, Stryker MP. Tumor necrosis factor-α mediates one component of competitive, experience-dependent plasticity in developing visual cortex. Neuron. 2008;58:673–680. doi: 10.1016/j.neuron.2008.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Baune BT, Wiede F, Braun A, Golledge J, Arolt V, Koerner H. Cognitive dysfunction in mice deficient for TNF-α and its receptors. Am J Med Genet Bneuro-psychiatr Genet. 2008;147B:1056–1064. doi: 10.1002/ajmg.b.30712. [DOI] [PubMed] [Google Scholar]
- 50.Beste C, Baune BT, Falkenstein M, Konrad C. variations in the TNF-Gene (TNF--308G¡A) affect attention and action selection mechanisms in a dissociated fashion. J Neurophysiol. 2010;104:2523–2531. doi: 10.1152/jn.00561.2010. [DOI] [PubMed] [Google Scholar]
- 51.Santello M, Bezzi P, Volterra A. TNF-α controls glutamatergic gliotransmission in the hippocampal dentate gyrus. Neuron. 2011;69:988–1001. doi: 10.1016/j.neuron.2011.02.003. [DOI] [PubMed] [Google Scholar]
- 52.McCoy MK, Tansey MG. TNF signaling inhibition in the CNS: implications for normal brain function and neurodegenerativedisease. J Neuroinflamm. 2008;5:1–13. doi: 10.1186/1742-2094-5-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Frankola KA, Greig NH, Luo W, Tweedie D. Targeting TNF-alpha to elucidate and ameliorate neuroinflammation in neurodegenerative diseases. CNS Neurol Disord Drug Targets. 2011;10:391–403. doi: 10.2174/187152711794653751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wyss-Coray T, Mucke L. Inflammation in neurodegenerative disease—a double-edged sword. Neuron. 2002;35:419–432. doi: 10.1016/s0896-6273(02)00794-8. [DOI] [PubMed] [Google Scholar]
- 55.Goodman JC, Robertson CS, Grossman RG, Narayan RK. Elevation of tumor necrosis factor in head injury. J Neuroimmunol. 1990;30:213–217. doi: 10.1016/0165-5728(90)90105-v. [DOI] [PubMed] [Google Scholar]
- 56.Olmos G, Lladó J. Tumor necrosis factor alpha: A link between neuroinflammation and excitotoxicity. Mediators Inflamm. 2014;2014:861231. doi: 10.1155/2014/861231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Liu T, Clark R, McDonnell P, Young P, White R, Barone F, et al. Tumor necrosis factor-alpha expression in ischemic neurons. Stroke. 1994;25:1481–1488. doi: 10.1161/01.str.25.7.1481. [DOI] [PubMed] [Google Scholar]
- 58.Álvarez A, Cacabelos R, Sanpedro C, García-Fantini M, Aleixandre M. Serum TNF-alpha levels are increased and correlate negatively with free IGF-I in Alzheimer disease. Neurobiol Aging. 2007;28:533–536. doi: 10.1016/j.neurobiolaging.2006.02.012. [DOI] [PubMed] [Google Scholar]
- 59.Babu GN, Kumar A, Chandra R, Puri S, Kalita J, Misra U. Elevated inflammatory markers in a group of amyotrophic lateral sclerosis patients from northern India. Neurochem Res. 2008;33:1145–1149. doi: 10.1007/s11064-007-9564-x. [DOI] [PubMed] [Google Scholar]
- 60.llan SM, Rothwell NJ. Cytokines and acute neurodegeneration. Nat Rev Neurosci. 2001;2:734–744. doi: 10.1038/35094583. [DOI] [PubMed] [Google Scholar]
- 61.Wang X, Yue TL, Barone FC, White RF, Gagnon RC, Feuerstein GZ. Concomitant cortical expression of TNF-α and IL-1βmRNAs follows early response gene expression in transient focal ischemia. Mol Chem Neuropathol. 1994;23:103–114. doi: 10.1007/BF02815404. [DOI] [PubMed] [Google Scholar]
- 62.Boka G, Anglade P, Wallach D, Javoy-Agid F, Agid Y, Hirsch E. Immunocytochemical analysis of tumor necrosis factor and its receptors in Parkinson’s disease. Neurosci Lett. 1994;172:151–154. doi: 10.1016/0304-3940(94)90684-x. [DOI] [PubMed] [Google Scholar]
- 63.Golan H, Levav T, Mendelsohn A, Huleihel M. Involvement of tumor necrosis factor alpha in hippocampal development and function. Cereb Cortex. 2004;14:97–105. doi: 10.1093/cercor/bhg108. [DOI] [PubMed] [Google Scholar]
- 64.Yang L, Lindholm K, Konishi Y, Li R, Shen Y. Target depletion of distinct tumor necrosis factor receptor subtypes reveals hippocampal neuron death and survival through different signal transduction pathways. J Neurosc. 2002;22:3025–3032. doi: 10.1523/JNEUROSCI.22-08-03025.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Tancredi V, D’Arcangelo G, Grassi F, Tarroni P, Palmieri G, Santoni A, et al. Tumor necrosis factor alters synaptic transmission in rat hippocampal slices. Neurosci Lett. 1992;146:176–178. doi: 10.1016/0304-3940(92)90071-e. [DOI] [PubMed] [Google Scholar]
- 66.Butler M, O’connor J, Moynagh P. Dissection of tumor-necrosis factor-α inhibition of long-term potentiation (LTP) reveals a p38 mitogen-activated protein kinase-dependent mechanism which maps to early—but not late—phase LTP. Neuroscience. 2004;124:319–326. doi: 10.1016/j.neuroscience.2003.11.040. [DOI] [PubMed] [Google Scholar]
- 67.Stellwagen D, Beattie EC, Seo JY, Malenka RC. Differential regulation of AMPA receptor and GABA receptor trafficking by tumor necrosis factor-α. J Neurosci. 2005;25:3219–3228. doi: 10.1523/JNEUROSCI.4486-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Floden AM, Li S, Combs CK. β-Amyloid-stimulated microglia induce neuron death via synergistic stimulation of tumor necrosis factor α and NMDA receptors. J Neurosci. 2005;25:2566–2575. doi: 10.1523/JNEUROSCI.4998-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.O’Connor JJ, Moynagh PN, Butler MP, Cumiskey D. The inhibitory effect of tumour necrosis factor-α on long-term potentiation is attenuated by type 1 metabotropic glutamate receptor blockade. Brain Res. 2005;1062:171–174. doi: 10.1016/j.brainres.2006.12.019. [DOI] [PubMed] [Google Scholar]
- 70.Umesalma S, Sudhandiran G. Differential inhibitory effects of the polyphenol ellagic acid on inflammatory mediators NF-κB, iNOS, COX-2, TNF-α, and IL-6 in 1, 2-dimethylhydrazine-induced rat colon carcinogenesis. Basic Clin Pharmacol Toxicol. 2010;107:650–655. doi: 10.1111/j.1742-7843.2010.00565.x. [DOI] [PubMed] [Google Scholar]
- 71.Yu YM, Chang WC, Wu CH, Chiang SY. Reduction of oxidative stress and apoptosis in hyperlipidemic rabbits by ellagic acid. J Nutr Biochem. 2005;16:675–681. doi: 10.1016/j.jnutbio.2005.03.013. [DOI] [PubMed] [Google Scholar]