

Abstract
Background
Most ischemic strokes in humans are caused by ruptured arterial atheroma, which activate platelets and produce thrombi that occlude cerebral vessels.
Methods
To simulate these events, we threaded a catheter through the internal carotid artery toward the middle cerebral artery (MCA) orifice and injected collagen directly into the cerebral circulation of male C57Bl/6 mice and Wistar rats.
Results
Laser-Doppler flowmetry demonstrated reductions in cerebral blood flow (CBF) of ~80% in mice and ~60% in rats. CBF spontaneously increased but remained depressed after catheter withdrawal. Magnetic resonance imaging showed that ipsilateral CBF was reduced at 3 h after collagen injection and markedly improved at 48 h. Micro-computed tomography revealed reduced blood vessel density in the ipsilateral MCA territory at 3 h. Gross examination of excised brains revealed thrombi within ipsilateral cerebral arteries at 3 h, but not 24 h, after collagen injection. Immunofluorescence microscopy confirmed that platelets and fibrinogen/fibrin were major components of these thrombi at both macrovascular and microvascular levels. Cerebral infarcts comprising ~30% of hemispheric volume and neurobehavioral deficits were observed 48 h after ischemic injury in both mice and rats.
Comparison with existing methods
Collagen injection caused brain injury that was similar in magnitude and variability to mechanical MCA occlusion or injection of a pre-formed clot; however, alterations in CBF and the mechanism of vascular occlusion were more consistent with clinical ischemic stroke.
Conclusion
This novel rodent model of ischemic stroke has pathophysiologic characteristics consistent with clinical atherothrombotic stroke, is technically feasible, and creates reproducible brain injury.
Keywords: Atherothrombosis, Collagen, Ischemic stroke, Platelet, Mouse, Rat
1. Introduction
Stroke is now the second leading worldwide cause of death and the third leading cause of disability-adjusted life-years (Murray and Lopez, 2013). More than 85% of human strokes are ischemic in nature, and of these, the majority are atherothrombotic (non-cardioembolic) in origin (Roger et al., 2012). Anti-platelet agents, such as aspirin and clopidogrel, are standard of care for primary and secondary prevention of atherothrombotic stroke, whereas anticoagulant drugs, such as warfarin, provide protection against cardioembolic stroke (Warden et al., 2012; Lansberg et al., 2012). Thrombolytic therapy is standard of care for treatment of acute ischemic stroke that has either an atherothrombotic or cardioembolic origin, but it requires administration within a few hours of symptom onset (Lansberg et al., 2012). Unfortunately, none of the currently available therapies is completely effective at reducing brain injury. Despite decades of intensive research and successful intervention in animal stroke models, novel laboratory therapies have not translated to the clinical environment. This failure can be attributed in part to shortcomings in animal stroke models, which do not faithfully recapitulate the pathophysiologic events that occur in most human strokes (Macrae, 2011; Fisher et al., 2009).
Most ischemic strokes in humans are caused by the rupture of unstable atherosclerotic plaque present in large extracranial and intracranial arterial vessels, with the carotid artery being a particularly common site (Nieswandt et al., 2011; Badimon and Vilahur, 2007; Warden et al., 2012; Hennerici, 2004). Components of plaques that remain on the arterial surface and that embolize distally activate platelets and produce thrombi that occlude large cerebral vessels and distal microvessels, causing ischemic injury, the magnitude of which depends on the size of the vessel and duration of occlusion (Davi and Patrono, 2007; Hennerici, 2004). The spectrum of clinical events, from transient ischemic attack to hemispheric stroke, reflects these pathologic events, which commonly include spontaneous or therapeutic recanalization of the occluded vessel and subsequent reperfusion injury. Much of the experimental stroke literature has focused on searching for neuroprotective strategies using models that include mechanical occlusion of the middle cerebral artery (MCA) and intra-arterial injection of a preformed blood clot, or on repair strategies with a permanent photothrombotic occlusion model of the distal MCA or its branches. However, these models do not lend themselves well for the study of atherothrombotic mechanisms, which involve platelet activation and thrombus formation in situ in cerebral macro- and microvessels. To more closely mimic human events, we modified the standard transient filament occlusion model of ischemia/reperfusion injury. Rather than threading a filament to mechanically occlude the MCA or injecting a preformed clot, we thread a narrow catheter near the MCA origin but do not occlude blood flow. Instead, we perform a series of intra-arterial injections of collagen, a potent platelet activator and component of ruptured atherosclerotic plaque (Fuster et al., 1990). The collagen induces platelet-rich clot formation and consequent vascular occlusion.
2. Materials and methods
2.1. Rodent model of ischemic stroke
The investigational protocol was approved by the Johns Hopkins University Animal Care and Use Committee. Male C57Bl/6 mice (26–28 g; 3–4 months) were anesthetized with 1–2% isoflurane/30% O2 in a temperature-controlled environment that maintained body temperature at 37.0 ± 0.5 °C. The right neck and carotid bifurcation were dissected. The external carotid artery was ligated and used as a stump for passage of a PE8 catheter (SAI Infusion Technologies, Lake Villa, IL), and the right common carotid artery was temporarily ligated. The PE8 catheter was advanced into the internal carotid artery (ICA) approximately 5 mm past its bifurcation with the external carotid. A 10-μl Hamilton syringe was used to inject 1 μg of collagen (1 μg/μl, Chrono-Log Corp., Havertown, PA) through the catheter six times at 5-min intervals (N = 10). In additional experiments, a single injection of collagen (5 μg or 1 μg) was used (N = 5 each dose). Cerebral blood flow (CBF) was measured with a laser-Doppler flow (LDF) (Moor Instruments Ltd., Wilmington, DE) apparatus fixed on a cranial window that was lateral and slightly posterior to the bregma and devoid of large vessels, as previously described (Alkayed et al., 1998). Carotid artery reperfusion was initiated 60 min after the first collagen injection by withdrawing the catheter and releasing the temporary carotid ligature. After necks were sutured, animals were placed into a humidity- and temperature-controlled chamber for the first 2 h of recovery.
The model was modified for use in male Wistar rats (300–400 g) (N = 12). We catheterized the right femoral artery to monitor blood pressure. In addition, with rats in the lateral position, we temporarily occluded both the left and right common carotid arteries with microvascular clips. The PE8 catheter was threaded through the ICA to the aperture of the MCA, as determined by a slight decrement (~10%) of LDF signal. Collagen was injected six times (10 μl per injection) at 5-min intervals. Carotid reperfusion was achieved 60 min after the first injection by removal of the catheter and removal of the microvascular clips placed on the common carotid arteries.
In additional experiments, we induced ischemia in mice by mechanical occlusion of the MCA (N = 7), and in rats by intra-arterial injection of a clot (N = 12). Briefly, we performed the filament occlusion model in the mouse as previously described (Eliasson et al., 1997) using a 6-0 nylon monofilament to mechanically occlude the MCA for 60 min. Occlusion was followed by 60 min of monitored reperfusion. We performed the embolic clot model in the rat by injecting a single preformed 25-mm-long blood clot into the MCA, as previously described by us and others (Zhang et al., 1997; Papangelou et al., 2013).
2.2. Measurement of cerebral perfusion in vivo by magnetic resonance imaging (MRI)
Three hours after collagen injection, we assessed cerebral perfusion and brain injury in mice by MRI using T2, diffusion, and perfusion-weighted images (N = 3). MRI was carried out on a horizontal bore 11.7 Tesla Bruker AVANCE 3 system equipped with a triple-axis gradient unit (maximum strength 740 mT/m) with modifications of protocols previously described by us and others (Wu et al., 2013; Duhamel et al., 2012). Briefly, the animal’s respiration rate was kept at ~60 breaths/min by adjusting the dose of anesthesia (approximately 1–1.5% isoflurane with air and oxygen mixed at a 3:1 ratio). Body temperature was kept at approximately 35–37 °C by circulating warm water through the animal holder and was monitored constantly via a thermocouple placed under the body. Imaging acquisition was synchronized by respiratory gating signals. We used a fast spin echo sequence for T2 MRI and a diffusion-weighted echo planar imaging (EPI) sequence for diffusion tensor imaging (DTI). We acquired T2-weighted images with echo times (TEs) of 40 ms and a repetition time (TR) of 3000 ms, two signal averages, 0.4 mm slice thickness, and an in-plane resolution of 0.08 mm × 0.08 mm. For DTI, we used TE/TR = 30/3000 ms, 4 shots with navigator correction, two signal averages, 30 diffusion-weighted images with a maximum b value of 1000 s/mm2, 0.4 mm slice thickness, and an in-plane resolution of 0.16 mm × 0.16 mm. Un-inverted flow-sensitive alternating inversion recovery (UNFAIR) arterial spin labeling was used with 1 mm slice thickness, 5 slices, and an in-plane resolution of 0.25 mm × 0.25 mm. Diffusion tensor was calculated using a log-linear fitting method implemented in DTIstudio (www.mristudio.org), and maps of apparent diffusion coefficient (ADC) and fractional anisotropy (FA) were also obtained (Basser et al., 1994; Basser and Pierpaoli, 1996). Maps of cerebral blood flow (CBF) were calculated as previously described (Tanabe et al., 1999).
2.3. Assessment of cerebrovascular architecture ex vivo by micro-computerized tomography (CT)
In mice, we assessed gross cerebrovascular architecture and cerebral blood vessel density by micro-CT ex vivo using methods previously described (Pathak et al., 2011) (N = 3). Briefly, 3 h after collagen injection, mice were deeply anesthetized with isoflurane and then perfused through the left ventricle with phosphate-buffered saline (PBS) and 10% buffered formalin, followed by silicone rubber Microfil (FlowTech Inc., Carver, MA). Samples were sent to Numira Biosciences (Salt Lake City, UT) for imaging on a high-resolution, volumetric micro-CT scanner (μCT40, ScanCo Medical, Brüttisellen, Switzerland). All images were acquired with the following parameters: 8 μm isotropic resolution at 55 kVp, 300 ms exposure time, 2000 views, and 5 frames per view. We segmented and characterized the neurovasculature as we have described previously (Cebulla et al., 2014).
2.4. Histologic assessment of thrombus formation in the cerebral vasculature
Three or 24 h after collagen injection, animals were deeply anesthetized with 5% isoflurane and transcardially perfused with heparinized saline and 4% paraformaldehyde. Brains were removed, and gross images were taken of the dorsal and ventral aspects for macrovascular clot visualization. Whole brains were then coronally cryosectioned (10- and 80-μm slices), or the ICA/MCA segment was removed, embedded in paraffin or gelatin, and sectioned transversely (10- and 80-μm slices) for further processing (N = 6). For microvascular assessment, brain sections were incubated in blocking buffer (PBS, 10% horse serum, 0.1% Triton X) for 2 h at room temperature followed by primary antibodies diluted in incubation buffer (PBS, 1% horse serum, 0.1% Triton X). The antibodies used were anti-CD41 (a platelet marker, cat# ab11024 for mouse and rat, 1:400; Abcam, Cambridge, MA), anti-fibrinogen (fibrinogen/fibrin, cat# ab119948 for mouse, cat# ab74057 for rat, 1:200; Abcam), and anti-laminin (for architecture of blood vessels, cat# L9393 for rat, 1:25; Sigma, St. Louis, MO; anti-laminin for mouse tissue was a generous gift from Dr. Sorokin, 1:500; (Sorokin et al., 1997)). Sections were incubated overnight at 4 °C, rinsed with PBS, and incubated with Alexa Fluorochrome (−488 or −555, 1:200; Invitrogen, Eugene, OR)-linked secondary antibodies in incubation buffer for 2 h at room temperature. The slices were mounted by using Prolong Anti-Fade Gold with DAPI (Invitrogen). Images were acquired on an inverted Zeiss LSM780 confocal microscope with Zen 2009 Light Edition software.
2.5. Assessment of platelet activation in vivo by neutrophil-platelet conjugate formation
Quantification of neutrophil-platelet conjugates in the blood is a sensitive measure of platelet activation in vivo because activated platelets bind rapidly (via P-selectin) to neutrophils (via P-selectin glycoprotein ligand-1) (Faraday et al., 2004; Michelson et al., 2001). We assessed neutrophil-platelet conjugate formation by flow cytometry in mouse blood as previously described with modification (Faraday et al., 2013) (N = 4). Blood was obtained from the ipsilateral jugular vein of anesthetized mice before and 90 min after collagen injection. Collected blood was diluted in Tyrode’s buffer (137 mM NaCl, 2.7 mM KCl, 1 mM MgCl2, 1.8 mM CaCl2, 0.2 mM Na2HPO4, 12 mM NaHCO3, 5.5 mM d-glucose; pH to 7.40) and incubated with anti-neutrophil-FITC (clone 7/4, 1 μg/ml; Abcam), anti-CD41-APC (clone MWReg30, 1 μg/ml; eBioscience, San Diego, CA;), IgG-APC (1 μg/ml; eBioscience), and IgG-FITC (1 μg/ml; eBio-science) isotype control antibodies. Neutrophils were identified by their characteristic scatter and FITC-fluorescence on a FACScan flow cytometer. Neutrophil-platelet conjugate formation is expressed as the proportion of neutrophils displaying platelet CD41-APC-fluorescence compared to the total neutrophil population.
2.6. Histologic assessment of anatomical brain injury ex vivo
As previously described (Takahashi et al., 1996), we anesthetized animals deeply at 48 h after stroke induction and collected the brains (N = 8 for mice; N = 12 for rats). Brains were perfused, fixed, sliced coronally, and stained with 1% triphenyltetrazolium chloride (TTC; Sigma) in saline at 37 °C for 30 min. The area of infarcted brain, identified by the lack of TTC staining, was measured on the rostral and caudal surfaces of each slice and numerically integrated across the thickness of the slice to obtain an estimate of infarct volume in each slice (SigmaScan Pro, SPSS Inc., San Jose, CA). The infarct volumes were derived for the cortex, caudoputamenal complex, and entire hemisphere and expressed as a percent of the contralateral uninjured structure. Infarct volume was corrected for swelling by comparing the volumes in the ipsilateral and contralateral hemispheres. The corrected infarct volume was calculated as: volume of contralateral hemisphere – (volume of ipsilateral hemisphere – volume of infarct) (Swanson et al., 1990; Lin et al., 1993).
2.7. Neurobehavioral deficit score
At 48 h after stroke induction, neurologic deficit score in mice was graded as follows: 0 = no deficit; 1 = forelimb weakness and torso turning to the ipsilateral side when held by tail; 2 = circling to affected side; 3 = unable to bear weight on affected side; and 4 = no spontaneous locomotor activity or barrel rolling (Faraday et al., 2013) (N = 8). In rats, a modified system published by Bederson et al. (1986) was used at 48 h. Briefly, animals received a score of 0–4 as follows: 0 = no deficit, 1 = forelimb flexion, 2 = forelimb flexion plus decreased resistance to lateral push, 3 = unidirectional circling, 4 = unidirectional circling plus decreased level of consciousness (N = 12).
2.8. Statistics
Data are expressed as mean ± SD and were analyzed by Student’s t-test or Mann–Whitney U (for comparison of behavioral data). Leukocyte-platelet conjugate formation was analyzed by paired t-test. All testing was two-tailed, and results were considered significant at P < 0.05.
3. Results
3.1. Mouse model
3.1.1. Collagen injection into the distal ICA disrupts ipsilateral CBF
LDF was used to monitor CBF during collagen injection and for 90 min thereafter. LDF decreased to ~40% of baseline after the 4th injection of collagen and remained depressed to ~20–30% of baseline after the 6th injection (Fig. 1). Blood flow slowly increased during the carotid reperfusion phase, but it remained depressed (~40% of baseline) at 60 min. In control experiments, injection of saline instead of collagen into the cerebral circulation had no impact on LDF (data not shown). The pattern of CBF disruption observed after transient occlusion of the MCA differed from that observed after collagen injection (Fig. 1). Both models achieved a reduction in CBF to approximately 20% of baseline; however, filament occlusion of the MCA was characterized by an abrupt cessation of blood flow upon insertion of the filament and abrupt return to baseline upon its withdrawal. In contrast, interruption of blood flow was more gradual with collagen injection, and CBF slowly increased, but remained depressed, for at least 60 min after catheter withdrawal.
Fig. 1.
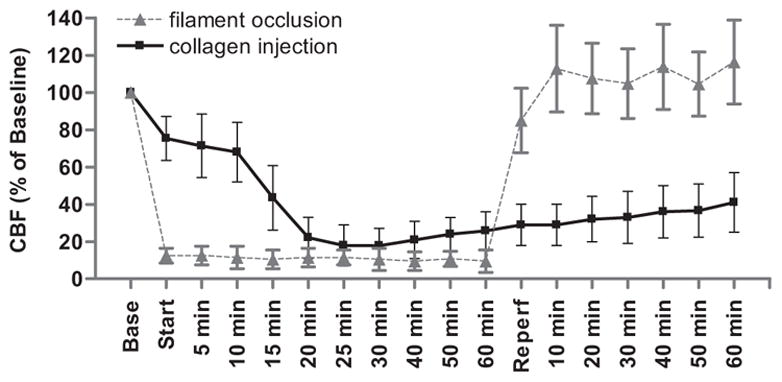
Injection of collagen into the cerebral vasculature reduces cerebral blood flow (CBF) in the mouse. Laser-Doppler flowmetry was used to monitor CBF in the territory of the middle cerebral artery before, during, and after transient occlusion of the MCA with a nylon filament or injection of collagen through a catheter threaded into the distal internal carotid artery. “Base” represents stabilized CBF at baseline expressed as percent; “start” represents insertion of the filament or first collagen injection (total of 6 injections every 5 min); “reperfusion” represents removal of the filament or catheter and release of the ipsilateral common carotid ligature. N = 7 filament occlusion; N = 10 collagen injection.
Using in vivo MRI, we examined the effects of collagen injection on microvascular CBF and brain injury after 3 and 48 h (Fig. 2). T2-weighted images and ADC maps demonstrated ischemic lesions in the cortex, thalamus, and hippocampus, and maps of CBF showed marked reductions in microvascular flow ipsilateral to collagen injection at the 3 h time point. MRI-visible lesions remained in the thalamus and hippocampus at 48 h, but were smaller; hemispheric differences in blood flow were much less, although some regional differences remained (Fig. 2).
Fig. 2.

Cerebral perfusion and architecture are disrupted after injection of collagen in the mouse. Focal cerebral ischemia was induced by injecting six 1-μg boluses of collagen into the distal internal carotid artery of the mouse. (A) An 11.7 T magnetic resonance scanner was used to acquire T2-weighted (T2), apparent diffusion coefficient (ADC), and perfusion images on live mice 3 and 48 h after collagen injection. Regions with hyperintense T2 signals and hypointense ADC values were detected in the ipsilateral cortex (red arrow), hippocampus (yellow arrow), and thalamus (white arrow) at 3 h, accompanied by marked reductions in regional cerebral blood flow (CBF). At 48 h, T2 signals, ADC hypointensities, and CBF were improved, but abnormalities remained. (B) Micro-CT was performed on brains excised from mice 3 h after collagen injection. A coronal brain section shows disruption of vascular network in the middle cerebral artery territory and reduced vascular density ipsilateral to collagen injection (red arrow) compared to that in the contralateral hemisphere (blue arrow). (C) Micro-CT was used to construct cerebral heat maps that display average vessel density in the ischemic (red square) and contralateral brain (yellow square). Lighter colors represent higher vessel densities. (D) Right sagittal view from a micro-CT scan of the neurovasculature shows a poorly defined vascular network ipsilateral to collagen injection. (E) Left sagittal view of the neurovasculature contralateral to collagen injection. Images are representative of three separate experiments. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
We also examined the effect of intra-arterial collagen injection on cerebrovascular architecture by micro-CT of brain tissue ex vivo at the 3 h time point. Three-dimensional reconstruction of micro-CT images revealed disruption of the vascular network in the MCA territory ipsilateral to collagen injection and markedly reduced vascular density in that territory compared to that seen in the contralateral hemisphere (Fig. 2).
3.1.2. Collagen injection induces thrombus formation and platelet activation within the cerebral vasculature
Three hours after collagen injection, thrombus was grossly visualized in the ipsilateral MCA (and/or branches) in 100% (6/6) of mice and in the ipsilateral ICA in 83% (5/6) of mice (Fig. 3); thrombus was visualized in the posterior cerebral artery (and/or branches) in 33% (2/6) of mice and in the anterior cerebral artery (and/or branches) in 17% (1/6) of mice. Immunofluorescent images obtained through cross sections of these macrovessels confirmed that platelets and fibrinogen/fibrin were major components of these thrombi. Images of coronal brain slices also revealed platelet and fibrin deposition within microvessels of the cortex and striatum ipsilateral to collagen injection. These thrombi were present in vessels ranging in size from 30 μm down to the capillary level (Fig. 3). Microvascular thrombi were not observed in the contralateral hemisphere.
Fig. 3.

Collagen injection causes formation of platelet- and fibrin-rich thrombi in cerebral macro- and microvessels. Focal cerebral ischemia was induced by injecting six 1-μg boluses of collagen into the distal right internal carotid artery (ICA) of the mouse. Three hours after collagen injection, brains were excised for histologic and immunofluorescent examination of cerebral vasculature. (A) Ventral (top panel) and dorsal (bottom panel) gross images show thrombus in internal carotid, middle cerebral, and anterior cerebral arteries and their branches. Black box in ventral view is magnified in (B) top panel. (B) Middle and lower panels show platelet- and fibrin-rich thrombi in cross sections of the ICA. Middle panel is labeled with anti-laminin antibody (red), anti-CD41 platelet antibody (green), and anti-nuclear stain (blue). Bottom panel is labeled with anti-laminin antibody (red), anti-fibrinogen/fibrin antibody (Fbn; green), and anti-nuclear stain (blue). (C) Top panel: triphenyltetrazolium chloride-stained coronal brain section of brain excised 3 h after collagen injection. Black box denotes area from which the images in the two lower panels were taken. Middle panel labeled with anti-laminin antibody (red), anti-CD41 platelet antibody (green), and anti-nuclear stain (blue). Bottom panel labeled with anti-laminin antibody (red), anti-fibrinogen antibody (green), and anti-nuclear stain (blue). Images are representative of six separate experiments. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
The accumulation of platelets within cerebral vessels ipsilateral to collagen injection was consistent with collagen-dependent platelet activation in vivo. To confirm platelet activation, we examined leukocyte-platelet conjugate formation, a sensitive measure of platelet activation in vivo (Michelson et al., 2001; Faraday et al., 2004), in jugular venous blood samples obtained before and after intra-arterial collagen injection. The proportion of leukocyte-platelet conjugates in jugular blood was significantly greater after collagen injection than in samples collected before injection (35.4 ± 16% vs. 17.6 ± 12%; P = 0.018; n = 4).
3.1.3. Infarct size and behavioral deficits after ischemic stroke
We collected mouse brains 48 h after collagen injection or filament occlusion of the MCA to assess infarct size. Corrected infarct volumes after collagen injection were 41 ± 11%, 49 ± 10%, and 33 ± 9% in the cortex, striatum, and hemisphere, respectively. These volumes were similar in magnitude to those of infarcts produced by mechanical occlusion of the MCA (Fig. 4). The coefficients of variation of infarct volume were as follows: 27% in the cortex, 20% in the striatum, and 27% in the hemisphere. These values represent precision that was at least as good as the corresponding values of 54%, 26%, and 50%, respectively, observed in the filament model. Behavioral deficits of mice 48 h after stroke were also similar in collagen injection and filament occlusion models (Fig. 4).
Fig. 4.

Injection of collagen into the cerebral vasculature causes anatomic brain injury and behavioral deficits in the mouse. Focal cerebral ischemia was induced by transient filament-occlusion of the middle cerebral artery or by injecting six 1-μg boluses of collagen into the distal right internal carotid artery of the mouse. After 48 h, cerebral infarct volume was quantified by staining brain slices with triphenyltetrazolium chloride (TTC) and neurobehavioral deficit score was assessed. (A) Representative TTC-stained coronal brain sections from the mouse filament-occlusion model (top) and collagen injection model (bottom). (B) Cerebral infarct volumes for the filament-occlusion (solid bar) and collagen injection (open bar) models. (C) Neurobehavioral deficit scores for the filament-occlusion (solid bar) and collagen injection (open bar) models. N = 5 filament occlusion; N = 8 collagen injection.
In additional experiments, we examined the impact of a single injection of 5 μg of collagen on neurologic outcomes and compared them to those we observed after six serial injections of 1 μg. Results from both experimental sets were very similar, with similar but more rapid reductions in CBF during the first 1.5 h after collagen injection and histologic and behavioral injuries that were not significantly different at 48 h (Fig. 5). A smaller single dose of collagen (1 μg) caused significantly less hemispheric injury (21 ± 4% vs. 34 ± 6%, P < 0.05) and a trend toward lesser behavioral deficit (0.6 ± 0.5 vs. 1.4 ± 0.5, P = 0.12) compared to the 5 μg dose.
Fig. 5.

Effect of serial vs. single injection of collagen on cerebral blood flow (CBF), anatomic brain injury and behavioral deficit in the mouse. (A) Laser-Doppler flowmetry was used to monitor CBF in the territory of the MCA before, during, and after serial injections of collagen (6× 1 μg collagen) or a single injection of collagen (5 μg collagen) through a catheter threaded into the distal internal carotid artery. “Base” represents stabilized CBF at baseline expressed as percent; “start” represents first collagen injection; “reperfusion” represents removal of the catheter and release of the the ipsilateral common carotid ligature. After 48 h, cerebral infarct volume was quantified by staining brain slices with triphenyltetrazolium chloride (TTC) and neurobehavioral deficit score was assessed. (B) Cerebral infarct volumes after serial injections of collagen (6× 1 μg; solid bar) or single injection of collagen (1× 5 μg; open bar). (C) Neurobehavioral deficit scores after serial collagen injection (6× 1 μg; solid bar) or single collagen injection (1 × 5 μg; open bar). N = 5 for 5 μg group; N = 10 for 6× 1 μg group.
3.2. Rat model
3.2.1. Collagen injection into the MCA disrupts CBF
LDF fell to ~40% of baseline after the 4th injection of collagen and remained depressed at this level for the next 30 min (Fig. 6). During carotid reperfusion, LDF returned to ~60% of baseline by 30 min. By way of comparison, CBF reduction was much more abrupt and severe (~20% of baseline) after injection of a pre-formed clot, but CBF returned to similar levels by 30 min of carotid reperfusion in both models (Fig. 6).
Fig. 6.
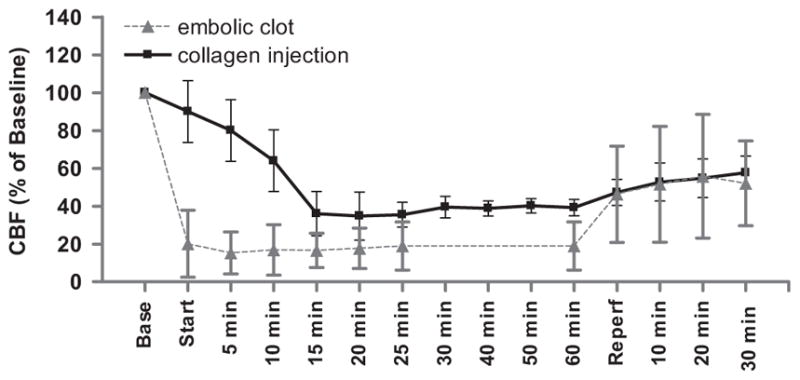
Injection of collagen into the middle cerebral artery (MCA) reduces cerebral blood flow (CBF) in the rat. Laser-Doppler flowmetry was used to monitor CBF in the territory of the MCA before, during, and after occlusion of the MCA by injection of collagen or a preformed clot through a catheter threaded to the MCA orifice. “Base” represents stabilized CBF at baseline expressed as percent; “start” represents insertion of the clot or first collagen injection (total of 6 injections every 5 min); “reperfusion” represents removal of the catheter and release of the bilateral common carotid ligatures. N = 12 each group.
3.2.2. Collagen injection induces thrombus formation in the cerebral vasculature
Three hours after collagen injection, rat brains were perfusion-fixed and inspected for thrombus formation. Gross examination of the ventral surface revealed thrombus within the MCA and its branches in all rats (Fig. 7). Immunofluorescent images of cross sections through the MCA revealed intra-luminal thrombi that were rich in platelets and fibrin/fibrinogen. Platelet- and fibrin-rich thrombi were also present within the ipsilateral microvasculature 3 h after collagen injection (Fig. 7), whereas thrombi were not present on the contralateral side. Brains were also harvested at 24 h after collagen injection. Gross examination and cross sectional immunofluorescent images of the MCA showed that thrombus was no longer present in the macrovasculature. However, microvascular platelet thrombi remained within the cortex and striatum ipsilateral to collagen injection at the 24 h time point (Fig. 7)
Fig. 7.

Injection of collagen into the middle cerebral artery (MCA) causes formation of platelet- and fibrin-rich thrombi in cerebral macro- and microvessels. Focal cerebral ischemia was induced by administering six 10-μl boluses of collagen near the right MCA orifice of the rat. At 3 and 24 h after collagen injection, brains were excised for histologic and immunofluorescent examinations of cerebral vasculature. (A) Ventral (upper panel) and dorsal (lower panel) gross images show thrombus in the MCA and its branches at 3 h (left panel) but not at 24 h (right panel) after collagen injection. (B) Immunofluorescent images show platelet- and fibrin-rich thrombi in cross sections of the MCA at 3 h (left panel) but not 24 h (right panel) after collagen injection. Upper panels are labeled with anti-laminin antibody (red), anti-CD41 platelet antibody (green), and anti-nuclear stain (blue). Lower panels are labeled with anti-fibrinogen/fibrin antibody (Fbn; red), anti-CD41 platelet antibody (green), and anti-nuclear stain (blue). (C) Coronal brain slices display platelet and fibrin deposition in cortical cerebral microvessels at 3 h (left panel) and 24 h (right panel) after collagen injection. Upper panels are labeled with anti-laminin antibody (red), anti-CD41 platelet antibody (green), and anti-nuclear stain (blue). Lower panels are labeled with anti-fibrinogen/fibrin antibody (red), anti-CD41 platelet antibody (green), and anti-nuclear stain (blue). Images are representative of four separate experiments. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
3.2.3. Infarct size and behavioral deficits after stroke
We collected rat brains at 48 h after collagen injection or clot injection to assess infarct size. Corrected infarct volumes after collagen injection were 27 ± 16%, 50 ± 30%, and 27 ± 17% in the cortex, striatum, and hemisphere, respectively (Fig. 8). These volumes were comparable in size to brain injury after clot injection. The coefficients of variation for infarct volume were 59% in the cortex, 60% in the striatum, and 63% in the hemisphere, indicating that precision was at least as good as that after clot injection, as the corresponding values were 77%, 63%, and 73%, respectively. Behavioral deficit scores 48 h after stroke were similar in the two stroke models (Fig. 8).
Fig. 8.

Injection of collagen into the middle cerebral artery (MCA) causes anatomic brain injury and behavioral deficits in the rat. Focal cerebral ischemia was induced by injection of a preformed clot into the MCA or by injection of six 10-μl boluses of collagen near the MCA orifice of the rat. After 48 h, cerebral infarct volume was quantified by staining brain slices with triphenyltetrazolium chloride and neurobehavioral deficit score was assessed. (A) Cerebral infarct volumes for the embolic clot (solid bar) and collagen injection (open bar) models. (B) Neurobehavioral deficit scores for the embolic clot (solid bar) and collagen injection (open bar) models. N = 12 each group.
4. Discussion
We describe a new model of atherothrombotic stroke in rodents in which regional CBF is interrupted by injection of collagen directly into the cerebral vasculature. Disruption of the cerebral circulation was confirmed by LDF, MRI diffusion-perfusion imaging, and micro-CT. Histologic studies showed that thrombi were present within cerebral macro- and microvessels up until at least 3 h after collagen injection (and perhaps until 24 h in microvessels) and that they were composed largely of platelets and fibrinogen/fibrin. Improvements in CBF at 48 h (in mice) and the absence of thrombi in the macrovasculature at 24 h (in rats) suggested that these thrombi resolve spontaneously over time. The magnitude of anatomic brain injury and behavioral damage from collagen injection was comparable to that created after transient filament occlusion of the MCA in mice or intra-arterial injection of a preformed clot in rats. However, the pattern of disruption in CBF and the pathophysiologic mechanisms underlying vascular occlusion after collagen injection more closely simulate human atherothrombotic stroke, which constitutes the majority of human stroke events.
Interruption of brain blood flow by atheroemboli in cerebral blood vessels is the predominant cause of ischemic stroke in humans (Nieswandt et al., 2011; Badimon and Vilahur, 2007). We simulated atheroembolism by direct intra-arterial injection of collagen, a potent platelet activator and component of ruptured atherosclerotic plaque (Fuster et al., 1990). Using LDF, we observed successive decreases in CBF after serial collagen (1 μg) injections. CBF became maximally depressed by the 4th or 5th injection. Depression of CBF persisted throughout the monitoring period, for at least 1.5 h after the first collagen injection in our protocols, and gradually increased to 50–60% of baseline within 30–60 min after withdrawal of the catheter. A single 5 μg injection of collagen caused a rapid reduction in CBF to levels similar to those observed after serial 1 μg injections, followed by a similar pattern of blood flow recovery. This pattern of initial disruption followed by gradual restoration of CBF, whether spontaneous or therapeutic, resembles clinical ischemic stroke (Hossmann, 2012). Progressive reduction in CBF after serial injections of collagen presented a pattern that was different from the abrupt decrease in flow seen with filament occlusion or clot injection models and was reminiscent of repeated transient ischemic attacks or “stuttering stroke” in humans. Indeed, the early presence and partial resolution of ADC abnormalities that we observed on MRI by 48 h in mice was reported to occur in a human case of stuttering stroke (Peters et al., 2010). Gradual restoration of CBF during reperfusion was common to the collagen injection and clot injection models but differed from the abrupt increase in CBF seen after withdrawal of an occluding filament. The time-dependent return of CBF to baseline after initial vascular occlusion is thought to play an important role in expansion of ischemic injury into the penumbra in both human stroke and animal stroke models, and the nonphysiologic restoration of CBF in the filament occlusion model is understood to present limitations for its use in preclinical stroke studies (Hossmann, 2012).
The rupture of unstable atherosclerotic plaques, present primarily in the aorta and large extracranial vessels (e.g. carotid artery), causes cerebrovascular occlusion in humans by inducing platelet activation and thrombus formation in situ (Warden et al., 2012; Badimon and Vilahur, 2007; Nieswandt et al., 2011; Hennerici, 2004). Plaque instability can persist for days to weeks, during which time an individual is at high risk for repeated thromboembolic ischemic events, which may manifest as crescendo TIAs or separate strokes (Redgrave et al., 2006; Lovett et al., 2004; Marnane et al., 2014). We attempted to simulate these pathophysiologic events in rodents by administering repeated injections of collagen distal to the ICA. Gross anatomic studies confirmed the presence of thrombus in the MCA territory of all rodents. Immunohistochemistry confirmed that these thrombi were largely composed of platelets and fibrinogen/fibrin. These thrombi were present in vessels as large as the ICA and as small as capillaries 3 h after collagen injection, but they were absent from the macrovasculature by 24 h, consistent with the improvements in CBF seen by diffusion-perfusion MRI at 48 h. Intra-arterial activation of platelets in vivo was confirmed after collagen injection by increased numbers of leukocyte-platelet conjugates in jugular venous blood. Increased numbers of these conjugates have also been observed in blood obtained from patients suffering from atherothrombotic stroke (Ishikawa et al., 2012). Although platelet activation is known to occur in filament occlusion, clot injection, and photothrombotic models (Nieswandt et al., 2011; Zhang et al., 2001; Kleinschnitz et al., 2008), it is not a primary mechanism for cerebrovascular occlusion in any of these models. Thus, these models are limited in their utility for investigating the cellular and molecular events that incite cerebrovascular occlusion in most human strokes. Intravascular injection of thrombin has also been used to induce cerebrovascular thrombus formation in situ. Although thrombin is a platelet activator, this methodology produces cerebral thrombi that contain few platelets; instead, thrombi are composed primarily of polymerized fibrin (Orset et al., 2007).
Despite producing vascular occlusion by a different mechanism, collagen injection produced anatomic brain injury and behavioral deficits that were comparable in magnitude to those produced by filament occlusion in mice and clot-injection in rats. In experiments using a single collagen injection in mice, we found that the magnitude of anatomic and behavioral injury could be modified by altering the dose of collagen administered. From a catheter position just caudal to the MCA, collagen injection consistently produced thrombi in the MCA and/or its branches in all mice and in the ICA in the majority; a minority of mice also developed thrombi in the anterior or posterior cerebral arterial territories. The magnitudes of cerebral injury and behavioral deficit in mice were consistent, with coefficients of variation for infarct volumes suggesting that precision was at least as good as that obtained by filament occlusion. Injection of collagen into the cerebrovasculature of rats produced thrombi that were localized to the MCA and its branches, ischemic brain injury that was consistent in size and location, and consistent behavioral deficits. Historically, reliable reproduction of infarct size has been challenging in the embolic clot model owing to obstacles in clot delivery and placement (Marinescu et al., 2014). We note that the model of stroke is multifocal and its utility in the mouse may be limited in experiments where variability in the anatomic location of stroke cannot be tolerated. Nevertheless, the coefficient of variation of infarct volume in the mouse and rat models will permit investigators to perform hypothesis testing experiments with reasonable sample sizes and statistical power.
We believe that the described ischemic stroke model has potential for use in preclinical studies that test the effectiveness of therapeutic interventions. Similar to the clot injection model, the collagen injection model could be used to examine the impact of therapies designed to accelerate recovery of CBF by dissolution of a vascular thrombus (e.g. tPA). The model could also be used to evaluate possible mechanisms for the failure of such therapies when thrombosis is caused by atheroembolism instead of cardioembolism. Furthermore, because this is the only model to simulate ischemic brain injury from unstable atherosclerotic plaque, the model provides investigators a unique opportunity to test the effectiveness of therapies designed to prevent the formation of cerebrovascular thrombi caused by plaque rupture – the putative mechanism for the clinical benefits derived from aspirin and clopidogrel (Warden et al., 2012). In addition, we demonstrate that the model can be used in both mice and rats, which should allow investigators the flexibility to adapt the model to address a variety of research questions in multiple laboratory environments.
In summary, we describe a novel rodent model of ischemic stroke induced by direct injection of collagen distal to the ICA. The model is compatible with the pathophysiology of clinical atherothrombotic stroke, is technically feasible, and creates reproducible injury. This model may be useful in determining the pathophysiologic mechanisms underlying ischemic brain injury from atherothrombic causes, which may provide greater opportunities for the development of therapies to prevent and treat human stroke.
HIGHLIGHTS.
We developed a stroke model similar in pathophysiology to atherothrombotic stroke.
Collagen was injected directly into the cerebral circulation of mice and rats.
Cerebral blood flow remained depressed for at least 1 h after collagen injection.
Platelet and fibrin rich thrombi formed in macro- and microvascular cerebral arteries.
Cerebral infarcts and neurobehavioral deficits were observed after 48 h.
Acknowledgments
This work was supported by RC1 HL099677-01 (NF), NS038684 (RCK), and NS0677525 (RCK). We thank Claire Levine, MS, ELS, for editing this manuscript during its preparation.
Footnotes
Disclosures
The funding sources had no involvement in study design, collection or analysis of data, writing of the manuscript, or in the decision to submit the article for publication. The authors have no conflicts of interest to disclose.
References
- Alkayed NJ, Harukuni I, Kimes AS, London ED, Traystman RJ, Hurn PD. Gender-linked brain injury in experimental stroke. Stroke. 1998;29:159–65. doi: 10.1161/01.str.29.1.159. discussion 166. [DOI] [PubMed] [Google Scholar]
- Badimon L, Vilahur G. Platelets, arterial thrombosis and cerebral ischemia. Cerebrovasc Dis. 2007;24(Suppl 1):30–9. doi: 10.1159/000107377. [DOI] [PubMed] [Google Scholar]
- Basser PJ, Mattiello J, LeBihan D. Estimation of the effective self-diffusion tensor from the NMR spin echo. J Magn Reson B. 1994;103:247–54. doi: 10.1006/jmrb.1994.1037. [DOI] [PubMed] [Google Scholar]
- Basser PJ, Pierpaoli C. Microstructural and physiological features of tissues elucidated by quantitative-diffusion-tensor MRI. J Magn Reson B. 1996;111:209–19. doi: 10.1006/jmrb.1996.0086. [DOI] [PubMed] [Google Scholar]
- Bederson JB, Pitts LH, Tsuji M, Nishimura MC, Davis RL, Bartkowski H. Rat middle cerebral artery occlusion: evaluation of the model and development of a neurologic examination. Stroke. 1986;17:472–6. doi: 10.1161/01.str.17.3.472. [DOI] [PubMed] [Google Scholar]
- Cebulla J, Kim E, Rhie K, Zhang J, Pathak AP. Multiscale and multi-modality visualization of angiogenesis in a human breast cancer model. Angiogenesis. 2014 doi: 10.1007/s10456-014-9429-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davi G, Patrono C. Platelet activation and atherothrombosis. N Engl J Med. 2007;357:2482–94. doi: 10.1056/NEJMra071014. [DOI] [PubMed] [Google Scholar]
- Duhamel G, Callot V, Tachrount M, Alsop DC, Cozzone PJ. Pseudo-continuous arterial spin labeling at very high magnetic field (11. 75 T) for high-resolution mouse brain perfusion imaging. Magn Reson Med. 2012;67:1225–36. doi: 10.1002/mrm.23096. [DOI] [PubMed] [Google Scholar]
- Eliasson MJ, Sampei K, Mandir AS, Hurn PD, Traystman RJ, Bao J, et al. Poly(ADP-ribose) polymerase gene disruption renders mice resistant to cerebral ischemia. Nat Med. 1997;3:1089–95. doi: 10.1038/nm1097-1089. [DOI] [PubMed] [Google Scholar]
- Faraday N, Braunstein JB, Heldman AW, Bolton ED, Chiles KA, Gerstenblith G, et al. Prospective evaluation of the relationship between platelet-leukocyte conjugate formation and recurrent myocardial ischemia in patients with acute coronary syndromes. Platelets. 2004;15:9–14. doi: 10.1080/09537100310001644006. [DOI] [PubMed] [Google Scholar]
- Faraday N, Schunke K, Saleem S, Fu J, Wang B, Zhang J, et al. Cathepsin G-dependent modulation of platelet thrombus formation in vivo by blood neutrophils. PLoS ONE. 2013;8:e71447. doi: 10.1371/journal.pone.0071447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher M, Feuerstein G, Howells DW, Hurn PD, Kent TA, Savitz SI, et al. Update of the stroke therapy academic industry roundtable preclinical recommendations. Stroke. 2009;40:2244–50. doi: 10.1161/STROKEAHA.108.541128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuster V, Stein B, Ambrose JA, Badimon L, Badimon JJ, Chesebro JH. Atherosclerotic plaque rupture and thrombosis. Evolving concepts. Circulation. 1990;82:II47–59. [PubMed] [Google Scholar]
- Hennerici MG. The unstable plaque. Cerebrovasc Dis. 2004;17(Suppl 3):17–22. doi: 10.1159/000075300. [DOI] [PubMed] [Google Scholar]
- Hossmann KA. The two pathophysiologies of focal brain ischemia: implications for translational stroke research. J Cereb Blood Flow Metab. 2012;32:1310–6. doi: 10.1038/jcbfm.2011.186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikawa T, Shimizu M, Kohara S, Takizawa S, Kitagawa Y, Takagi S. Appearance of WBC-platelet complex in acute ischemic stroke, predominantly in atherothrombotic infarction. J Atheroscler Thromb. 2012;19:494–501. doi: 10.5551/jat.10637. [DOI] [PubMed] [Google Scholar]
- Kleinschnitz C, Braeuninger S, Pham M, Austinat M, Nolte I, Renne T, et al. Blocking of platelets or intrinsic coagulation pathway-driven thrombosis does not prevent cerebral infarctions induced by photothrombosis. Stroke. 2008;39:1262–8. doi: 10.1161/STROKEAHA.107.496448. [DOI] [PubMed] [Google Scholar]
- Lansberg MG, O’Donnell MJ, Khatri P, Lang ES, Nguyen-Huynh MN, Schwartz NE, et al. American College of Chest Physicians. Antithrombotic and thrombolytic therapy for ischemic stroke: antithrombotic therapy and prevention of thrombosis, 9th ed: American college of chest physicians evidence-based clinical practice guidelines. Chest. 2012;141:e601S–36S. doi: 10.1378/chest.11-2302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin TN, He YY, Wu G, Khan M, Hsu CY. Effect of brain edema on infarct volume in a focal cerebral ischemia model in rats. Stroke. 1993;24:117–21. doi: 10.1161/01.str.24.1.117. [DOI] [PubMed] [Google Scholar]
- Lovett JK, Coull AJ, Rothwell PM. Early risk of recurrence by subtype of ischemic stroke in population-based incidence studies. Neurology. 2004;62:569–73. doi: 10.1212/01.wnl.0000110311.09970.83. [DOI] [PubMed] [Google Scholar]
- Macrae IM. Preclinical stroke research – advantages and disadvantages of the most common rodent models of focal ischaemia. Br J Pharmacol. 2011;164:1062–78. doi: 10.1111/j.1476-5381.2011.01398.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marinescu M, Bouley J, Chueh J, Fisher M, Henninger N. Clot injection technique affects thrombolytic efficacy in a rat embolic stroke model: implications for translaboratory collaborations. J Cereb Blood Flow Metab. 2014;34:677–82. doi: 10.1038/jcbfm.2014.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marnane M, Prendeville S, McDonnell C, Noone I, Barry M, Crowe M, et al. Plaque inflammation and unstable morphology are associated with early stroke recurrence in symptomatic carotid stenosis. Stroke. 2014;45:801–6. doi: 10.1161/STROKEAHA.113.003657. [DOI] [PubMed] [Google Scholar]
- Michelson AD, Barnard MR, Krueger LA, Valeri CR, Furman MI. Circulating monocyteplatelet aggregates are a more sensitive marker of in vivo platelet activation than platelet surface P-selectin: studies in baboons, human coronary intervention, and human acute myocardial infarction. Circulation. 2001;104:1533–7. doi: 10.1161/hc3801.095588. [DOI] [PubMed] [Google Scholar]
- Murray CJ, Lopez AD. Measuring the global burden of disease. N Engl J Med. 2013;369:448–57. doi: 10.1056/NEJMra1201534. [DOI] [PubMed] [Google Scholar]
- Nieswandt B, Pleines I, Bender M. Platelet adhesion and activation mechanisms in arterial thrombosis and ischaemic stroke. J Thromb Haemost. 2011;9(Suppl 1):92–104. doi: 10.1111/j.1538-7836.2011.04361.x. [DOI] [PubMed] [Google Scholar]
- Orset C, Macrez R, Young AR, Panthou D, Angles-Cano E, Maubert E, et al. Mouse model of in situ thromboembolic stroke and reperfusion. Stroke. 2007;38:2771–8. doi: 10.1161/STROKEAHA.107.487520. [DOI] [PubMed] [Google Scholar]
- Papangelou A, Toung TJ, Gottschalk A, Mirski MA, Koehler RC. Infarct volume after hyperacute infusion of hypertonic saline in a rat model of acute embolic stroke. Neurocrit Care. 2013;18:106–14. doi: 10.1007/s12028-012-9768-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pathak AP, Kim E, Zhang J, Jones MV. Three-dimensional imaging of the mouse neurovasculature with magnetic resonance microscopy. PLoS ONE. 2011;6:e22643. doi: 10.1371/journal.pone.0022643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters JM, Maclean AV, Young GS. Rapid resolution of diffusion weighted MRI abnormality in a patient with a stuttering stroke. BMJ Case Rep. 2010;2010 doi: 10.1136/bcr.07.2009.2063. http://dx.doi.org/10.1136/bcr.07.2009.2063[Epub 2010 February 8] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redgrave JN, Lovett JK, Gallagher PJ, Rothwell PM. Histological assessment of 526 symptomatic carotid plaques in relation to the nature and timing of ischemic symptoms: the Oxford plaque study. Circulation. 2006;113:2320–8. doi: 10.1161/CIRCULATIONAHA.105.589044. [DOI] [PubMed] [Google Scholar]
- Roger VL, Go AS, Lloyd-Jones DM, Benjamin EJ, Berry JD, Borden WB, et al. American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Heart disease and stroke statistics – 2012 update: a report from the American Heart Association. Circulation. 2012;125:e2–220. doi: 10.1161/CIR.0b013e31823ac046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanson RA, Morton MT, Tsao-Wu G, Savalos RA, Davidson C, Sharp FR. A semiautomated method for measuring brain infarct volume. J Cereb Blood Flow Metab. 1990;10:290–3. doi: 10.1038/jcbfm.1990.47. [DOI] [PubMed] [Google Scholar]
- Takahashi H, Kirsch JR, Hashimoto K, London ED, Koehler RC, Traystman RJ. PPBP [4-phenyl-1-(4-phenylbutyl) piperidine] decreases brain injury after transient focal ischemia in rats. Stroke. 1996;27:2120–3. doi: 10.1161/01.str.27.11.2120. [DOI] [PubMed] [Google Scholar]
- Tanabe JL, Yongbi M, Branch C, Hrabe J, Johnson G, Helpern JA. MR perfusion imaging in human brain using the UNFAIR technique. Un-inverted flow-sensitive alternating inversion recovery. J Magn Reson Imaging. 1999;9:761–7. doi: 10.1002/(sici)1522-2586(199906)9:6<761::aid-jmri2>3.0.co;2-c. [DOI] [PubMed] [Google Scholar]
- Warden BA, Willman AM, Williams CD. Antithrombotics for secondary prevention of noncardioembolic ischaemic stroke. Nat Rev Neurol. 2012;8:223–35. doi: 10.1038/nrneurol.2012.33. [DOI] [PubMed] [Google Scholar]
- Wu D, Xu J, McMahon MT, van Zijl PC, Mori S, Northington FJ, et al. In vivo high-resolution diffusion tensor imaging of the mouse brain. Neuroimage. 2013;83:18–26. doi: 10.1016/j.neuroimage.2013.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang RL, Chopp M, Zhang ZG, Jiang Q, Ewing JR. A rat model of focal embolic cerebral ischemia. Brain Res. 1997;766:83–92. doi: 10.1016/s0006-8993(97)00580-5. [DOI] [PubMed] [Google Scholar]
- Zhang ZG, Zhang L, Tsang W, Goussev A, Powers C, Ho KL, et al. Dynamic platelet accumulation at the site of the occluded middle cerebral artery and in downstream microvessels is associated with loss of microvascular integrity after embolic middle cerebral artery occlusion. Brain Res. 2001;912:181–94. doi: 10.1016/s0006-8993(01)02735-4. [DOI] [PubMed] [Google Scholar]