

Abstract
ALS is a multisystem disorder affecting cognitive and motor functions. Bulbar-onset ALS (bALS) may be preferentially associated with language/cognitive impairments, compared with spinal-onset ALS (sALS), stemming from a potentially unique neuropathology. The objective of this systematic review was to compare neuropathology reported for bALS and sALS subtypes in studies of cadaveric brains. Using Cochrane guidelines, we reviewed articles in MEDLINE, Embase, and PsycINFO databases using standardized search terms for ALS and neuropathology, from inception until July 16th 2016. 17 studies were accepted. In summary, both subtypes presented with involvement in motor and frontotemporal cortices, deep cortical structures, and cerebellum, characterized by neuronal loss, spongiosis, myelin pallor, and ubiquitin+ and TDP43+ inclusion bodies. Changes in Broca and Wernicke areas, regions associated with speech and language processing, were noted exclusively in bALS. Further, some bALS cases presented with atypical pathology, neurofibrillary tangles and basophilic inclusions, which were not found in any sALS cases. Given the few studies, all with methodological biases, further work is required to better understand neuropathology of ALS subtypes.
Keywords: bulbar ALS, systematic review, neuropathology, histopathology, cognitive/language impairments, ALS subtypes
1. INTRODUCTION
Amyotrophic Lateral Sclerosis (ALS) is a progressive neurodegenerative disease that affects upper and lower motor neurons in the brain, brainstem, and spinal cord, but has also been associated with extra-motor (i.e., cognitive and language) impairments, similar to those found in frontotemporal dementia (FTD) (1, 2). ALS has two typical presentations at disease onset – approximately 70% of patients present initially with the spinal form of the disease, characterized by muscle weakness and atrophy in the limbs and trunk, when the remaining patients present with bulbar changes, affecting speech and swallowing musculature (3). Nearly 85% of patients with spinal-onset ALS, however, exhibit bulbar changes with disease progression (4, 5). Approximately 50% of all patients diagnosed with ALS show cognitive and language impairments, while 10% of the patients present with clear signs of FTD (6, 7). ALS is a complex disorder with considerable heterogeneity across affected individuals (8, 9). This heterogeneity is not well understood, however. Addressing the heterogeneity by developing accurate means of patient subtyping (10) is essential for providing more targeted approaches to treatment development, recruitment into clinical trials, and disease management in a clinic (e.g., early identification of bulbar disease in order to plan supportive interventions and predict disease progression).
Bulbar ALS is arguably the most devastating variant of the disease as it is characterized by the fastest decline, the shortest survival (<2 years post diagnosis), and a significantly reduced quality of life (11, 12). In addition to the rapid motor decline, some neuroimaging and behavioural studies have observed that bulbar ALS may present with an increased burden of cognitive/language impairments (1, 6, 13–17). This latter finding remains disputed, however (18, 19). Two hypotheses have been proposed regarding the association between motor and extramotor abnormalities in ALS, in relation to the disease subtype: 1) it has been suggested that the site of symptom onset may be related to the burden of extra motor impairments, with bulbar-onset ALS showing a unique neurodegenerative profile associated with specific and concomitant extramotor impairments (1, 6, 13–15, 20–22); and 2) the presence of bulbar motor dysfunction, regardless of site of onset, may be associated with extramotor impairments (6, 7, 16, 17). Neither of the two hypotheses has been investigated neuropathologically in cadaveric brain tissue.
Studies that examined the underlying neuropathology in cases with cognitive and language impairments showed that ALS cases typically present with frontotemporal lobar degeneration (FTLD) (23, 24). The pathology in the frontotemporal regions consisted of neuronal loss, marked gliosis, and intraneuronal inclusion bodies that were positive for ubiquitin and TAR DNA-binding protein 43 (TDP-43) (24–26). The severity and distribution of TDP-43 in the brain has been shown to be well-correlated with antemortem cognitive profiles, often giving insight into the phenotypic presentations of the disease and representing a clinicopathologic spectrum (27–30) that ranges from pure motor neuron disease to frontotemporal dementia. The underlying neuropathology, however, has not been well-characterized in the context of bulbar- versus spinal-onset subtypes in the existing literature. An examination of the neuropathological findings from the subtype perspective might shed light into the underlying similarities and/or differences in clinical disease presentations.
This study aimed to contribute to our understanding of ALS subtypes through neuropathological examinations of cadaveric autopsy brains, and elucidate whether these subtypes are neuropathologically distinct or lie within a spectrum of the same disease. To do this, we conducted a systematic review investigating similarities and differences between neuropathological profiles of bulbar-onset ALS (bALS) and spinal-onset ALS (sALS) by regional distribution and types of pathology.
2. METHODS
2.1 Operational Definitions
Our search was guided by the following operational definitions, determined a priori: Amyotrophic Lateral Sclerosis, defined as a progressive neurological disease with upper and lower motor neuron involvement determined by clinical, electrophysiological or neuropathologic examination; and cadaveric neuropathological examination, defined as the post-mortem study of disease on the brain and brainstem by gross or microscopic examination.
2.2 Search Methodology
Studies were identified by searching the Medline (1946 to July 12th, 2016), Embase (1980 to July 12th, 2016), and PsycInfo (2002 to July 12th, 2016) databases. Main search terms included Amyotrophic Lateral Sclerosis, ALS, Lou Gehrig’s disease, or motor neuron disease combined with neuropathology, histology, immunohistochemistry, or immunocytochemistry, limited to humans. The search strategy for Amyotrophic Lateral Sclerosis was adapted and modified from a previous Cochrane Review (31). The search terms were adapted for each database to accommodate for differences in subject headings (see Appendix A for full search strategies). Citation lists of included articles were hand-searched for articles relevant to the systematic review.
2.3 Study Selection
Articles were excluded for this review if they: 1) had no abstract; 2) included no human participants (i.e. animal study); 3) were classified as a tutorial, educational report, or narrative review; 4) were a neuroimaging study (i.e., fMRI, DTI, PET, EEG, MEG, etc.); 5) were a genetic study (i.e., focussing on cases with SOD1/C9ORF72/FUS mutations only); 6) involved a population where >90% of subjects were not diagnosed with ALS (i.e., all forms of motor neuropathies, Alzheimer’s Disease, etc.); 7) involved a population where >90% of subjects were diagnosed with an atypical subtype of ALS (i.e., Parkinson-Dementia-ALS of the Guam complex, or juvenile ALS of the Madras subtype); or, 8) did not involve a neuropathological examination, either microscopic or at a gross macroscopic level, of cadaveric brains (i.e., blood serum analysis, DNA fragmentation, or skin microscopy). Two independent raters (authors SS and KV) reviewed all unique abstracts identified from the primary search. Discrepant ratings were resolved by consensus. All accepted abstracts were brought to full review.
During the full article review, articles were excluded for the same reasons as above and also if they: 1) did not investigate the brain or brainstem (i.e., investigated muscle or spinal cord only), 2) involved a population where the site of onset were not specified, 3) did not have any bALS cases (i.e., sALS or FTD-onset cases only), or, 4) did not allow for data extraction of bALS cases (i.e., only aggregate data across all subjects of varying onsets). A full review of each article was conducted by the same two independent raters (Authors SS and KV). Discrepant ratings were again resolved by consensus.
2.4 Quality Assessment
The methodological quality of each included full article was critically appraised using a combination of guidelines designed to improve the quality of reporting non-randomized studies: The Effective Public Health Practice Project (EPHPP) Quality Assessment Tool for Quantitative studies (32), and the STROBE (Strengthening the Reporting of Observational Studies in Epidemiology) statement (33). Also, relevant quality assessment questions were adapted from the Cochrane Risk of Bias (34) for domains pertaining to selective enrolment, validity of bALS and sALS group comparisons (i.e., a high risk of bias (ROB) rating for dissimilar sites of tissue sampling, unequal number of patients using artificial respiratory support (ARS) between the two groups, etc.), incomplete clinical information provided (i.e., a high ROB rating for missing data on the cognitive and/or bulbar status of the patients, inadequate genetic testing, etc.), and poor methodological descriptions thereby limiting reproducibility of the study. Risk of bias for each article was judged relative to the dates of gene discovery. For example, articles published after 2011 that did not screen for C9ORF72 mutations were given a high ROB rating, but articles predating 2011 were not (see (35) for a chronological timeline of each discovered gene in ALS).
2.5 Data Extraction
Only full articles that met the inclusion criteria outlined above underwent data extraction by a single rater (Author SS) and included: 1) study design; 2) diagnosis of the examined group and sample size; 3) demographic variables (i.e. mean age at onset/death, site of initial symptom onset, and sex; 4) disease characteristics (i.e. severity of disease, disease duration, and cognitive status); 5) pathological assessment methods (i.e. sampling sites, staining methods, and immunohistochemistry techniques); and, 6) neuropathological data (i.e. anatomical areas involved, types and extent of pathology). Data on cases with a mixed-motor onset (i.e., limb and bulbar symptoms at initial disease presentation) were excluded. Neuropathological data were extracted for all areas within the cerebral cortex, as well as the hippocampus, amygdala, basal ganglia, thalamus, and cerebellum. For the brainstem, only data pertaining to the selected motor nuclei that innervate bulbar structures (i.e., the trigeminal motor, facial, vagus, and hypoglossal nuclei) were extracted. Data on the spinal cord were beyond the scope of this study and thus not extracted. Partial data extraction (25%) was checked by a second rater (VK) and discrepancies were resolved by consensus.
3. RESULTS
3.1 Literature Retrieval
The literature search and study selection flow diagram is shown in Figure 1. In addition to the 1377 studies identified through database searching, 5 were found by searching citation lists of included articles. Duplicate abstracts were removed leaving 1208 unique abstracts, of which 880 articles were excluded leaving 328 articles for full review. At this level, an additional 311 were excluded leaving 17 full articles (17, 21, 36–51). At abstract screening, the inter-rater agreement was 84.5% for the accept/reject criteria, with a 79% agreement for the reason of rejection. At full text screening, the accept/reject percent agreement was 89.5%, with an 81% agreement for reason of rejection.
Figure 1.

Selection of included studies.
3.2 Study Characteristics
Of all 17 included articles, none directly investigated the neuropathological differences between sALS and bALS. Out of the included studies, however, 11 reported individual neuropathology results for a mixed group of patients—those with bALS or sALS (17, 21, 36, 40–42, 44–46, 48, 49)—allowing for a comparison between the two groups (Table 1). The other 6 studies investigated the clinical and neuropathological abnormalities in bALS patients only (37–39, 47, 50, 51) (Table 3). Below, we present these two groups of findings separately as the former set of studies allows the comparison of the two disease subtypes, and the latter set of studies provides a more in-depth clinicopathologic analysis of bulbar-onset cases only.
Table 1.
Patient demographics and clinical characteristics for studies with bulbar-onset and spinal-onset ALS subjects.
| Bulbar-Onset Group | Spinal-Onset Group | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Included Studies | N (M/F) | Age at onset; Age at death (years, SD) | Disease Duration (months, SD) | Bulbar symptomology | Number of cases with clinical signs of cognitive/language impairments (N, % of group, Type) | Number of cases with artificial respiratory support (N, % of group) | N | Age at onset; Age at death (years, SD) | Disease Duration (months, SD) | Bulbar symptomology | Number of cases with clinical signs of cognitive/ language impairments (N, % of group, Type) | Number of cases with artificial respiratory support (N, % of group) |
| Kamo et al., 1987 (36) | 1M/3F | NR, 66.5 (9.67) | 21 (3.83) | NR | NR | NR | 3M/4F | NR, 58.43 (12.57) | 55 (35.78) | NR | NR | NR |
| Troost et al., 1992 (45) | 3M/6F | NR, 62.88 (9.16) | 24.88 (9.51) | NR | NR | NR | 16M/8F | NR, 64.04 (10.25) | 31.5 (13.19) | NR | NR | NR |
| Kato et al., 1993 (48) | 1M/2F | NR, 62.33 (5.03) | 15 (6.56) | NR | 3 (100%), NR | None | 7M/5F | NR, 65.58 (8.56) | 97.33 (85.74) | NR | None | 5 (42%) |
| Kato et al., 1994 (21) | 4M | 66.75 (8.88), NR | 26.5 (4.79) | Moderate to severe dysarthria/Dysphagia (via CNE) | 3 (75%), Eventual ALS-FTD diagnosis; Impaired emotion perception, memory, judgment, and language perseveration | None | 1M/1F | 50 (16.97), NR | 53.5 (30.4) | Moderate to severe dysarthria/Dysphagia (via CNE) | None | None |
| Nagy et al., 1994 (46) | 1F | NR, 55 | NR | NR | None | NR | 12M/2F | NR, 58.65 (10.49) | NR | NR | None | NR |
| Mochizuki et al., 1995 (41) | 1M | 52, 53 | 12 | Dysarthria, dysphagia | None | NR | 1M | 42, 44 | 18 | Dysarthria, dysphagia, tongue atrophy and fasciculation | None | NR |
| Averbuch-Heller et al., 1998 (40) | 1F | 54, 56 | 60 | Complete anarthria, atrophic tongue with fasciculation | 1 (100%), inappropriate behaviour/pseudobul bar affect | None | 1M | 50, 56 | 72 | Dysarthria, dysphagia, no atrophy or fasciculation of tongue | 1 (100%), inappropriate affect/pseudobul bar affect | None |
| Nakano et al., 2004 (49) | 1M | 40, NR | 137 | NR | None | 1 (100%) | 2M/2F | 62 (11.53),NR | 28.33 (15.94) | NR | 4 (100%), NR | None |
| Ota et al., 2005 (17) | 2F | 70.5 (2.12) NR | 17 (9.89) | Dysarthria, dysphagia, tongue atrophy and fasciculation | None | None | 1M/1F | 56.5 (6.36) NR | 29.5 (13.43) | Dysarthria, dysphagia, tongue atrophy and fasciculation | None | None |
| Bodansky et al., 2010 (42) | 2M/3F | NR, 77.8 (3.96) | 23.6 (10.4) | NR | NR | NR | 11M/4F | NR, 72.33 (8.34) | 30.36 (18.45) | NR | NR | NR |
| Sugiyama et al., 2013 (44) | 1M | 72, 74 | 36 | NR | NR | 1 (100%), non invasive positive pressure ventilation | 3M/4F | 58.42 (14.28), 62.42 (12.17) | 42.9 (29.6) | NR | NR | 2 (29%), Artificial ventilator |
NR= Not Reported. CNE= Cranial Nerve Exam
Table 3.
Patient demographics and clinical characteristics in studies with only a bulbar-onset population.
| Included Studies | N (M/F) | Age at onset; Age at death (years, SD) | Disease Duration (months, SD) | Bulbar symptomology | Number of cases with clinical signs of cognitive/ language impairments (N, % of group, Type) | Number of cases with artificial respiratory support (N, % of group) |
|---|---|---|---|---|---|---|
| Tsuchiya et al., 2000 (47) | 1F | 74; 75 | 12 | Severe; dysphagia (tube feeding) and dysarthria; atrophy and fasciculation of the tongue | 1 (100%), Primary Progressive Aphasia – non-fluent variant | NR, (but died of respiratory disturbances) |
| Bak et al., 2001 (51) | 4M/1 F | 58.5 (9.56); NR | 26 (4.89) | Severe dysarthria | 5 (100%), Personality and behaviour change with aphasia (66%); purely aphasic (33%) | NR |
| Tsuchiya et al., 2002 (39) | 1F | 69; 71 | 24 | Dysarthria, dysphagia | None | None |
| Ishihara et al., 2006 (37) | 1F | 52; NR | 90 | Severe dysarthria-hoarse-breathy voice, short speech duration, slow speech movements, incomprehensible at time of death | 1 (100%), ALS-FTD with progressive anterior opercular syndrome | NR |
| Kuwahara et al., 2010 (38) | 1M | 63; 65 | 20 | Severe dysarthria, severe dysphagia, feeding tube | 1 (100%); Behavioura l changes (bFTD), no cognitive disturbance s | None |
| Miki et al., 2010 (50) | 1M | 51; 60 | 108 | Severe; unable to vocalize | None | 1 (100%) |
3.3 Studies allowing direct comparison between bALS and sALS
3.3.1 Study Characteristics
All 11 included articles had a case series study design. The 11 studies included a total of 123 subjects, with a median of 8 subjects per study and range of 1 to 24. A total of 32 bALS cases and 91 sALS cases were examined. None of the studies reported any genetic mutations for any of the included subjects as the studies predated the discovery of most ALS genetic mutations. Four (17, 21, 40, 41) studies reported patients’ bulbar symptomology prior to death, which included severe dysphagia and dysarthria, along with tongue atrophy and fasciculation. The cognitive status of the patients was described in 7 studies (17, 21, 40, 41, 46, 48, 49), specifically in 41% of the bALS cases and 40% of the sALS cases, and mostly reported symptomatically as the presence or absence of dementia.
3.3.2 Comparison of clinical outcomes between bALS and sALS
Patient demographics and clinical characteristics of the cases are presented in Table 1. The average disease duration was significantly shorter (p<.05) for bALS cases (M = 29.04 months, SD = 25.98) than sALS cases (M = 51.74 months, SD = 51.59). The two groups were similar in age of onset, except for three studies: two (17, 21) studies reported bALS patients that were older (>15 years) than the sALS patients at disease onset, and one study (49) reported sALS older than bALS patients.
Across all studies assessing cognitive abnormalities, it was reported in 54% of bALS cases and 14% of the sALS cases. Two patients with bALS (disease duration: M=86.5 months, SD=71.42) and 7 patients with sALS cases (disease duration: M=78.57 months, SD=39.25) received artificial respiratory support (ARS) for more than 1 year prior to death, while 10 bALS patients (disease duration: M = 24.75 months, SD= 18.31) and 21 sALS patients (disease duration: M = 98.93 months, SD = 90.85) did not. ARS status for the remaining 81 cases was not reported.
Patients from both groups, bALS and sALS, presented with spinal symptoms at time of death, including weakness and paralysis of their extremities and/or respiratory difficulties.
3.3.3 Critical Appraisal
Methodological quality of the studies with bulbar-onset and spinal-onset patients is addressed in Table 2. Almost all studies recruited ALS patients using appropriate eligibility criteria. There was no evidence of attrition bias, in that all cases that were tested were reported, and very little evidence of reporting bias, where all pre-specified outcomes, such as all brain regions and stains, were reported.
Table 2.
Summary of methodological quality assessment for studies with bulbar-onset and spinal-onset patients.
| Kamo et al., 1987 (36) | Troost et al., 1992 (45) | Kato et al, 1993 (48) | Kato et al 1994 (21) | Nagy et al., 1994 (46) | Mochizuki et al, 1995 (41) | |
|---|---|---|---|---|---|---|
| Selection Bias | ||||||
| 1. Did the study develop and apply appropriate eligibility criteria for the ALS patients and a control population? | + | + | + | + | + | + |
| 2. Does the analysis account for important confounding and modifying variables through matching, stratification, or other approaches? | ||||||
| a) Were the number of patients with long-term artificial respiratory support equal for both groups? | ? | ? | − | + | ? | ? |
| b) Were antemortem cognitive/language signs of the patients reported? | − | − | + | + | + | + |
| c) Was appropriate genetic testing reported for all cases? | NA | NA | − | − | − | − |
| d) Was the presence of bulbar signs during the disease course reported? | − | − | − | + | − | + |
|
| ||||||
| Attrition Bias | ||||||
| 1. Were all the cases that were tested, reported? | + | + | + | + | + | + |
|
| ||||||
| Detection Bias | ||||||
| 1. Were the histopathological outcomes assessed using valid and reliable measures? | − | − | + | ? | − | − |
| 2. Was there more than one rater for the qualitative outcome measures? | ? | ? | NA | ? | ? | ? |
| 3. Was the histopathology methodology, such as sites of tissue sampling and staining, similar for all cases? | + | + | − | ? | − | + |
| 4. Did the study screen for, and document co-existing FTLD? | − | + | − | + | − | − |
| 5. Did the study screen for, document, and stage coexisting neurodegenerative phenomena (i.e., AD, mesial temporal sclerosis)? | + | + | + | + | + | + |
|
| ||||||
| Reporting Bias | ||||||
| 1. Were all pre-specified outcomes, such as areas of the brain and stains, reported? | − | + | − | + | + | + |
|
| ||||||
| Reproducibility | ||||||
| 1. Did the study clearly define each region of interest anatomically? | + | + | + | − | + | + |
| 2. Was the site of onset well-defined? | − | + | + | + | − | + |
| Averbuch-Heller et al., 1998 (40) | Nakano et al., 2004 (49) | Ota et al., 2005 (17) | Bodansky et al, 2010 (42) | Sugiyama et al., 2013 (44) | |
|---|---|---|---|---|---|
| Selection Bias | |||||
| 1. Did the study develop and apply appropriate eligibility criteria for the ALS patients and a control population? | ? | + | + | + | + |
| 2. Does the analysis account for important confounding and modifying variables through matching, stratification, or other approaches? | |||||
| a) Were the number of patients with long-term artificial respiratory support equal for both groups? | + | − | + | − | − |
| b) Were antemortem cognitive/language signs of the patients reported? | ? | + | + | − | − |
| c) Was appropriate genetic testing reported for all cases? | − | − | − | − | − |
| d) Was the presence of bulbar signs during the disease course reported? | + | − | + | − | − |
|
| |||||
| Attrition Bias | |||||
| 1. Were all the cases that were tested, reported? | + | + | + | + | + |
|
| |||||
| Detection Bias | |||||
| 1. Were the histopathological outcomes assessed using valid and reliable measures? | + | + | ? | + | − |
| 2. Was there more than one rater for the qualitative outcome measures? | NA | NA | ? | NA | ? |
| 3. Was the histopathology methodology, such as sites of tissue sampling and staining, similar for all cases? | ? | + | − | + | + |
| 4. Did the study screen for, and document co-existing FTLD? | − | + | + | + | + |
| 5. Did the study screen for, document, and stage coexisting neurodegenerative phenomena (i.e., AD, mesial temporal sclerosis)? | + | + | + | − | + |
|
| |||||
| Reporting Bias | |||||
| 1. Were all prespecified outcomes, such as areas of the brain, reported? | + | + | − | + | + |
|
| |||||
| Reproducibility | |||||
| 1. Did the study clearly define each region of interest anatomically? | − | − | − | + | + |
| 2. Was the site of onset well-defined? | + | + | − | + | + |
+=Yes, −=No; ? = not reported/ unknown/ unclear.
Of the 11 “mixed-group” studies, only 3 (17, 21, 40) had an equal number of patients with long-term artificial respiratory support (ARS) in the bulbar- and spinal-onset groups. Four studies (36, 41, 45, 46) failed to specify the prevalence and duration of ARS, and the remaining 4 (42, 44, 48, 49) introduced bias in the neuropathology data by having an unequal proportion of ARS users for the two groups.
Seven of the 11 “mixed-group” studies (17, 21, 40, 41, 46, 48, 49) reported antemortem cognitive signs of the patients; five of these studies only reported the presence or absence of dementia; another study (40) reported “inappropriate behaviour”, however, it was unclear whether these were signs of cognitive behavioural dysfunction or pseudobulbar affect; only one study reported specific cognitive changes on an extensive neuropsychological assessment battery (21). The remaining four studies (36, 42, 44, 45) did not mention the cognitive status of their patient population.
Most studies (36, 42, 44–46, 48, 49) did not report the presence or absence of bulbar signs (i.e., dysarthria, dysphagia) during the disease course for their patient population.
Five of the 11 “mixed-group” studies (17, 21, 40, 46, 49) had methodological biases concerning their neuropathology protocols; the studies either did not uniformly sample the brain regions across cases, or did not anatomically define the regions of interest. Furthermore, most studies used a semi-quantitative rating scale to evaluate the severity of neuronal loss and gliosis. This poses another limitation as subjective rating scales were not standardized across studies. The studies also did not indicate whether more than one rater was used to obtain these measures, suggesting a potential detection bias.
Only one of the studies screened for and documented the presence of coexisting FTLD through TDP-43 immunostaining (42). Most studies, albeit, were much older and predated the discovery of TDP-43 as the major component of the NCIs in ALS-FTD subtypes (25, 26). Four (36%) of the mixed-group studies (17, 21, 45, 49) stained for ubiquitin, which was the only identifiable component of the inclusions found in ALS-FTD pathology at the time. 10 studies (91%) screened for and documented coexisting neurodegenerative phenomena, such as Alzheimer’s Disease (AD) pathology, using various silver staining methods and/or anti-tau sera. None of the studies staged the AD-related changes using the Braak staging method (52).
None of the studies conducted the appropriate genetic testing to validate the observed neuropathological abnormalities, even though two studies (42, 46) discussed SOD1 and TARDBP mutations as potential explanations to the observed pathological findings.
3.3.4 Comparison of neuropathology between bALS and sALS
Regional analyses: The results of the regional analysis between bALS and sALS cases are summarized in Figure 2. Four studies suggested differences in the regional profiles of involvement for bALS as compared to sALS patients (21, 44, 48, 49) (Figure 2A). These studies suggested an increased prevalence of coexisting FTLD in bALS cases, compared to sALS cases. The unique regional distribution of pathological findings for bALS patients involved, primarily, the extramotor cortical regions, including the frontal, temporal, cingulate, and insular cortices, and frontotemporal white matter (WM). One study also reported pathology in the hippocampus and amygdala in bALS but not in sALS cases (48). Most of these cortical and subcortical areas, however, may not be differentially involved in bALS as 4 other studies in this subgroup of articles reported pathology in these areas for both subtypes (17, 44, 46, 49). Of particular interest is the one study that reported differential involvement of the Broca and Wernicke areas – regions that are highly associated with speech and language processing – in bALS cases, which were not involved in the sALS cases (21). While lobular assessment of the frontal and temporal cortices was conducted in other studies, these smaller regions were not individually examined in any of the other included 11 mixed-group studies.
Figure 2.
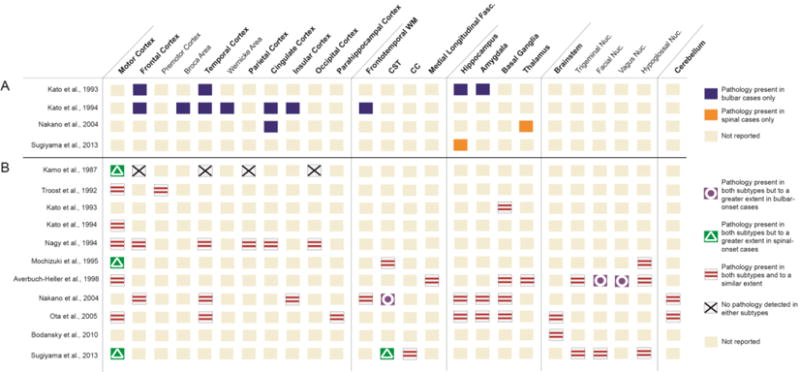
Summary of anatomic regions of involvement between bALS and sALS cases. Summary in panel A (above the solid line) lists studies reporting differences in the regional involvement between ALS variants. Summary in panel B (below the solid line) shows studies reporting shared regions of the brain for both variants. Some studies are shown in both panels as they report results of both types (i.e., differences in some regions but similarities in other regions). CST = corticospinal tract; CC = corpus callosum; Fasc. = fasciculus; Nuc. = nucleus.
In contrast, all the mixed-group studies (n=11) reported an overlap in the regional distribution of pathology between bALS and sALS cases —including the frontotemporal cortices and other extra motor cortical regions—and to a similar extent, suggesting that both subtypes were within the same spectrum of disease (see Figure 2B). Some studies that reported these overlapping regions of involvement, however, noted differences in the degree of pathological processes, defined as the number of inclusions, or severity of neuronal loss or gliosis ranked on a standardized scale. For example, the primary motor cortex was involved in both subtypes, but more notably affected in sALS than bALS in three studies (36, 41, 44), and another study (40) reported a greater degree of neuronal loss in the facial and vagus nuclei for the bALS cases compared to the sALS cases.
Type of pathology: Figure 3 displays differences in the types of pathology, cumulative across the whole brain, for the two subtypes. Panel A lists four studies that reported unique pathological features for the two disease variants. Specifically, one study reported gliosis, spongiosis, and ubiquitinated inclusions in the whole brain for bALS cases, but not sALS cases (21). An ALS diagnosis was confirmed for these sALS cases based on the loss of Betz cells in the primary motor cortex. Another study reported the presence of senile plaques in the motor cortex of bALS cases, that were absent from sALS cases (36). Two other studies reported skein-like inclusions and ubiquitinated inclusions that were immunoreactive to TDP-43 in sALS cases, which were absent in bALS cases (42, 44).
Figure 3.

Summary of pathology types compared between bALS and sALS cases. Summary in panel A (above the solid line) lists studies that report unique pathological characteristics for one subtype, but not the other. Summary in panel B (below the solid line) reports studies that indicate similar pathology types for both subtypes. Some studies are shown in both panels as they report results of both types (i.e., differences in some pathology types but similarities in other pathology types). Ubi+ = ubiquitin- positive; NCI = neuronal cytoplasmic inclusions. Incl. = inclusions.
All 11 studies, however, identified similar types of pathology for both subtypes (Figure 3B). The complied data suggested that, on a neurological examination, both bALS and sALS cases displayed an equal extent of neuronal loss, gliosis, and myelin pallor, as well as a comparable amount of bunina bodies, phosphorylated neurofilaments, senile plaques, and skein-like or round intraneuronal inclusions that were positive for ubiquitin and TDP-43.
3.4 Neuropathology in bALS-only studies
3.4.1 Study Characteristics
All 6 included articles had a case series study design. Across studies, the total number of subjects was 11. All studies reported bulbar symptomology and cognitive status for the patients.
3.4.2 Clinical Characteristics
Nine out of 11 cases were diagnosed with ALS-FTD as disease progressed (See Table 3). All the bALS patients presented with spinal symptoms at time of death, characterized by weakness and paralysis of their extremities and/or respiratory difficulties. Only one case received ARS for more than one year prior to death. None of the studies reported genetic mutations.
3.4.3 Critical Appraisal
Methodological quality of the articles with only bALS cases is depicted in Table 4. Studies were evaluated using similar quality assessment criteria as the studies with mixed patients, but when appropriate, questions were modified for studies with only bALS patients.
Table 4.
Summary of methodological quality assessment for studies with bulbar-onset patients only.
| Tsuchiya et al., 2000 (47) | Bak et al., 2001 (43) | Tsuchiya et al., 2002 (39) | Ishihara et al., 2006 (37) | Kuwahara et al., 2010 (38) | Miki et al., 2010 (50) | |
|---|---|---|---|---|---|---|
| Selection Bias | ||||||
| 1. Did the study develop and apply appropriate eligibility criteria for the ALS patients and a control population? | + | + | + | + | + | + |
| 2. Does the analysis account for important confounding and modifying variables through matching, stratification, or other approaches? | ||||||
| a) Was long-term artificial respiratory support reported for all cases? | − | − | + | − | + | + |
| b) Were antemortem cognitive/language signs of the patients reported? | + | + | + | + | + | + |
| c) Was appropriate genetic testing reported for all cases? | − | − | − | − | − | − |
| d) Was the progression of bulbar signs during the disease course reported? | − | + | + | + | + | + |
|
| ||||||
| Attrition Bias | ||||||
| 1. Were all the cases that were tested, reported? | NA | + | NA | NA | NA | NA |
|
| ||||||
| Detection Bias | ||||||
| 1. Were the histopathological outcomes assessed using valid and reliable measures? | ? | ? | ? | ? | ? | ? |
| 2. Was there more than one rater for the qualitative outcome measures? | ? | ? | ? | ? | ? | ? |
| 3. Was the histopathology methodology, such as sites of tissue sampling and staining, similar for all cases? | NA | ? | NA | NA | NA | NA |
|
| ||||||
| 4. Did the study screen for, and documen co-existing FTLD? | + | + | + | + | + | + |
|
| ||||||
| 5. Did the study screen for, document, and stage coexisting neurodegenerative phenomena (i.e., AD, mesial lateral sclerosis)? | + | + | + | + | + | + |
|
| ||||||
| Reporting Bias | ||||||
| 1. Were all prespecified outcomes, such as areas of the brain and stains, reported? | NA | + | NA | NA | NA | NA |
|
| ||||||
| Reproducibility | ||||||
| 1. Did the study clearly define each region of interest anatomically? | − | − | − | − | − | − |
| 2. Was the site of onset well-defined? | + | + | + | + | + | + |
+=Yes, −=No; ? =not reported/ unknown/ unclear.
Three out of six studies (37, 47, 51) did not specify the presence and duration of ARS, and none of the studies characterized the genetic makeup of the patients or operationally defined the regions of interest using anatomical landmarks.
All of the bALS studies reported bulbar symptomology at some point of the disease course (37–39, 47, 50, 51), and all studies reported antemortem cognitive and language signs of the patients. All the bALS studies screened for and documented both co-existing FTLD, by staining against ubiquitin and/or TDP-43, as well as co-existing neurodegenerative phenomena, by staining for AD pathology. Three out of the 6 studies (38, 39, 51) staged the observed AD-related neurofibrillary changes in the brain using Braak’s staging methodology.
3.4.4 Neuropathology of bALS
Regional Analysis and Type of Pathology: The neuropathological findings for the studies that investigated exclusively the bALS cases (n=6) are summarized by the regions of involvement (Figure 4), and pathology types (Figure 5). Overall, these studies reported similar regions of involvement and types of pathology to the bALS subjects from the mixed-group studies (see Figures 2 and 3), including involvement in the Broca area. The findings form this group, however, also noted the presence of neurofibrillary tangles (NFTs) in the hippocampus and amgydala of bALS brains that were reported by 5 studies in which most patients developed FTD with disease progression (see Table 3). NFTs were not detected in any of the bALS cases within the mixed-group studies in Figure 3. Furthermore, one of the studies (37) identified basophilic inclusions that did not stain against ubiquitin, tau, or alpha-synuclein antibodies in the frontotemporal regions in the bALS cases. P62, neurofilament or FUS staining was not performed on these inclusions.
Figure 4.
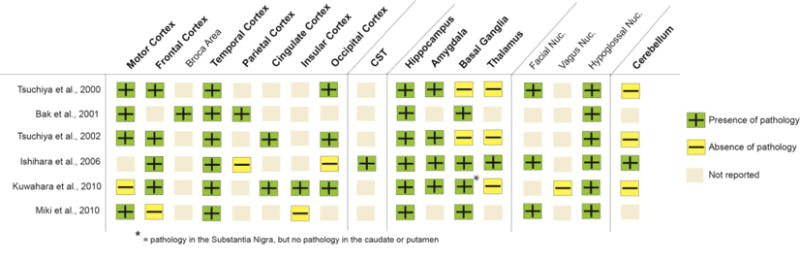
Summary of neuropathology by anatomical regions in bALS-only studies. CST = corticospinal tract; Nuc. = nucleus.
Figure 5.
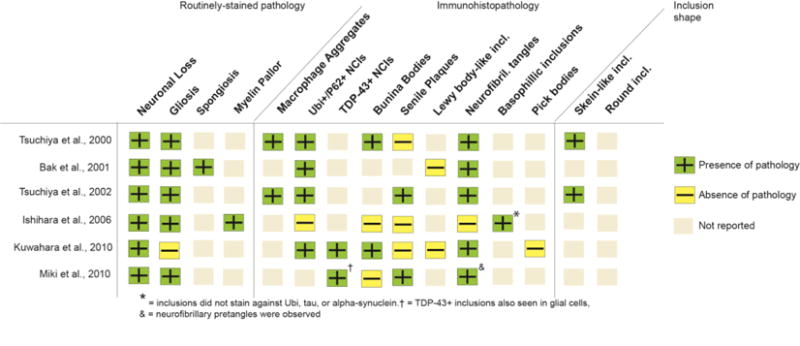
Summary of pathology types in bALS-only studies. Ubi+ = ubiquitin- positive; NCI = neuronal cytoplasmic inclusions. Incl. = inclusions.
4. DISCUSSION
The overall goal of this study was to compare and contrast the neuropathological features - both morphology and anatomic distribution - of bALS to sALS by synthesizing the findings from existing literature. This systematic review summarized clinical and neuropathological data from 17 studies. The studies were analysed with respect to two contrasting hypotheses (1, 53, 54). The first hypothesis proposed categorical pathological differences between bALS and sALS, with the former presenting with substantial extramotor (FTLD) signs as compared to the latter. The second hypothesis proposed that the two subtypes lay within the same spectrum of pathology. The results suggested that both subtypes, defined by the site of symptom onset, presented with comparable FTLD as well as the involvement of the brainstem and deep cortical structures. However, the involvement of regions that have been highly associated with speech and language functions (i.e., Broca and Wernicke areas) may be unique to bALS. When assessing the morphological properties of the pathology, both subtypes presented with neuronal loss and myelin pallor in the cortex and brainstem, along with P62/ubiquitin and/or TDP-43 positive inclusion bodies. In addition, there was evidence of secondary reactive changes, such as gliosis, spongiosis, and microglial activation in both subtypes. Yet, some bALS cases presented with atypical pathology, neurofibrillary tangles and basophilic inclusions, which were not found in any sALS cases.
4.1 Differential Involvement of Speech/Language Regions in bALS
Two studies reported differential involvement of specific regions of interest within the frontal and temporal cortices in bALS patients that were spared in sALS patients. Specifically, small regions related to speech and language processing— Broca and Wernicke areas— were differentially affected in bALS. The antemortem assessment of these patients indicated that these bALS cases initially presented with bulbar motor decline, which later evolved into primary progressive aphasia – a subtype of FTD with language changes. None of the sALS cases across the remaining studies were reported to have changes in these regions. Recent genetic and subsequent neuropathological studies have linked the co-existence of motor neuron disease and FTLD to C9ORF72 repeat expansions (55, 56). Extensive cortical and subcortical frontotemporal involvement may be associated with the C9ORF72 mutation, including reports of involvement of the Broca area (57). Genetic testing for C9ORF72 mutations for the subjects in the two included studies reporting Broca area involvement was not possible as the studies predated the discovery of the gene (58). The data, however, suggest a potential linkage between site of symptom onset, specifically bALS, and particular genotypes, specifically C9ORF72, reinforcing the concept that the pathogenesis of ALS may involve multiple distinct pathways that mediate disease onset and progression (59). Linking neuropathology with genetic information remains high priority for future studies.
The involvement of speech and language processing areas in bulbar ALS has been previously suggested by studies that reported language impairments, in ALS that were related to either bulbar-onset disease (15, 60, 61), or to the degree of bulbar motor decline (2, 6, 16, 54, 62), independent of dementia and motor disabilities. These observations, along with our analysis, suggest a potential co-development of aphasia and bulbar motor symptoms, which, from a pathophysiological perspective, may be directly related to the pattern of disease propagation within the brain. Currently, the factors that govern dissemination of pathology between segregated regions of the brain are unknown. A predominant theory postulates that propagation occurs within neuronal networks that form functional and structural connectomes, along the axonal wiring structure of the brain (63–67). A recent large-scale brain connectivity study in ALS has shown that abnormal proteins propagate to anatomic regions that are more closely interconnected by WM tracts than regions that are proximally closer, but with lesser connectivity (67). A co-development of aphasia following bulbar changes seems supportive of the connectome hypothesis, in which the pathways connecting the speech motor areas (i.e., the ventral region of the primary motor cortex) to speech/language processing areas may be degenerating in ALS. The speech-language connectome, however, has never been investigated in functional and structural connectivity studies in ALS. This may partially be due to the fact that tracts related to bulbar motor (i.e., corticobulbar tract) and language (i.e., arcuate fasciculus) functions were, until recently, difficult to isolate with existing imaging techniques (68) and were not anatomically well-defined (69, 70). Recent methodological advances have opened opportunities for this type of investigation.
While the bALS cases in the included studies presented with obvious bulbar changes during the disease course, all sALS cases also presented with bulbar changes towards the end stage of the disease, characterized by dysarthria and dysphagia. This may explain why some extra-motor cortical regions that are involved in the speech-language connectome, such as the insular and cingulate cortices (71), were pathologically involved in both bALS and sALS cases (46, 49). Future studies would need to examine the pathological profiles of those with fully preserved bulbar function at the time of death (72), and compare them with those with bulbar disease, in order to understand the link between bulbar disease, irrespective of onset site, and extramotor involvement.
The comparison is further complicated by the fact that ALS, by definition, is a disease with upper motor neuron (UMN) and lower motor neuron (LMN) pathology. The associated bulbar changes are often characterised clinically by mixed spastic and flaccid dysarthria. The examined studies did not define bulbar signs from the perspective of this distinction. Existing cognitive behavioural studies reported a more prominent cognitive impairment in patients with UMN involvement as compared to those with primarily LMN involvement (73–76), suggesting that abnormalities of cognitive/language function may be indicators of a subgroup of patients with corticobulbar (UMN) neuronal damage. However, such abnormalities do not seem to be exclusive to the pseudobulbar palsy subtype (76) and the relation between extramotor dysfunction and LMN bulbar symptoms should be addressed in future studies.
4.2 Atypical Pathological Features in some bALS cases
The diagnostic tools and classification for ALS have evolved rapidly in recent years. The classic descriptions of ALS focused on the degeneration of the motor neurons, suggesting a motor neuron selective disorder (77, 78). However, with the recent discovery of TDP-43 protein (26) and TARDBP mutation (79, 80) linking ALS and FTD, a wave of new clinical, genetic, neuropathological and epidemiological studies have suggested that ALS and FTD represent a continuum of disease with shared clinical and pathological features (7, 81, 82) including the presence of ubiquitin-positive, TDP-43 positive, tau- and α -synuclein-negative inclusions throughout the central nervous system (26, 83). For the cases included in this review, both bALS and sALS equally presented with typical ALS pathology, including TDP-43 proteinopathy.
Interestingly, however, 5 out of 16 studies that stained for co-existing AD pathology reported the presence of tau-positive neurofibrillary tangles in the hippocampus (38, 39, 43, 47, 50) and amygdala (39) for bALS cases, which were not reported for any of the sALS cases. One study also reported basophilic inclusions in the frontotemporal cortices and cerebellum (37) for bALS patients. FUS or neurofilament staining was not performed on these inclusions. Both NFTs and basophilic inclusions are not typically seen in classic ALS/FTD subtypes, but more common in other neurodegenerative phenomena (e.g. AD) and atypical forms of ALS, such as Guamanian ALS (84, 85) and Juvenile ALS (86, 87). These atypical forms of the disease do not show a greater prevalence of bulbar-onset cases, however (86, 88). Furthermore, the anatomical distribution of pathology for the bALS patients with these atypical features was not distinctively different when compared with bALS cases with typical pathology; they all presented with a multi-system degeneration, including involvement of the amygdala, basal ganglia, thalamus, and cerebellum (17, 40, 48, 49). Although the medical history of these cases indicated a seemingly typical ALS, the clinical disease profiles were not sufficiently defined (e.g., cognitive evaluation was not reported), making it difficult to understand the clinical significance of these atypical pathological features. Noteworthy, these atypical inclusions were not observed in any of the sALS patients across all studies with similar staining protocols, even for the cases with similar disease durations, suggesting that these bALS patients may represent a unique subtype that is neuropathologically distinct from typical bALS and sALS.
4.3 Quality Assessment: Limitations of Existing Studies
Critical appraisal of the individual studies identified a number of methodological challenges of the published studies. They include: 1) a lack of matching between patient groups for their disease characteristics (i.e., use of ARS) and demographics, resulting in large variability in disease durations and severities; 2) insufficient clinical (i.e., both motor and cognitive) description of cases; 3) insufficient documentation of co-existing FTLD and other neurodegenerative phenomena; 4) large variability in the neuropathology methodology across cases within and between studies, and 5) a lack of genetic information. Each of these methodological violations can place a study at substantial risk of bias, affecting the external validity of the findings. For example, the use and duration of ARS may be a confounding factor to the development of extra-motor impairments in ALS (6, 89, 90), as it can lead to a more widespread pattern of cortical atrophy due to longer disease durations, and consequently, greater disease progression (91). Secondly, antemortem bulbar signs were rarely reported and were limited to the mention of dysarthria (92). The perceptual judgement of the presence of speech changes may not be sensitive to detect more subtle abnormalities in bulbar physiology (9). As a result, a clear distinction between cases with bulbar disease and those with pure spinal symptoms may be challenging. Thirdly, studies under documented co-existing FTLD in the ALS cases. This may be because most studies predated the discovery of the TDP-43 protein (26), and the encoding TARDBP gene (79, 80) that were first to link ALS and FTD. Furthermore, the large variability in neuropathology protocols between subjects may have introduced a detection bias, where the observed pathological differences may be a result of differences in staining methods and sampling sites between subjects, and not a consequence of the disease. Lastly, some pathological abnormalities, especially the atypical features that were reported for some bALS cases, may be directly related to a known genetic mutation, which would better account for the pathological differences between groups.
4.4 Suggestions for Future Studies
Synthesizing findings across studies for the purposes of this review was difficult, primarily due to a lack of standardization across the neuropathology protocols. Overall, the existing studies offer limited information for determining if bALS has a unique neurodegenerative profile relative to sALS. In order to adequately distinguish the neuropathology for subtypes in ALS, future studies need to:
Expand and standardize clinical assessments to include antemortem clinical signs and symptoms, with an emphasis on bulbar changes and cognitive testing;
Include genetic testing in order to validate the neuropathological findings;
Expand and standardize neuropathology protocols regionally to include whole-brain analyses—with regions related to both motor and extramotor functions within the cortex, brainstem, deep cortical structures, and cerebellum—as well as include smaller, more specific, regions of interest within the cortex and WM tracts;
Expand and standardize staining methodology to include standardized screening and staging of co-existing neurodegenerative pathology such as Alzheimer’s disease, FTLD, and mesial temporal sclerosis;
Match patient groups or control for confounding factors, such as specific disease characteristics (i.e., duration of bulbar disease, and duration of ARS) and demographics.
4.5 Conclusions
The distinction between bulbar- versus spinal-onset patient groups is common in ALS literature and has implications for clinical management (i.e., predicting disease course and planning symptom management), and allocation to clinical trials. Yet, there is limited knowledge regarding the neuropathological differences between these subtypes. Neuroimaging and behavioural studies have suggested that the two subtypes may be distinct, with greater extramotor involvement in bALS (13–15). However the literature remains inconclusive. This systematic review approached this knowledge gap by comparing neuropathology of the two subtypes across a number of existing studies. The findings revealed a great deal of overlap in the regions of involvement and types of pathology between the two subtypes. However a handful of studies suggested unique distribution and nature of pathology in bALS with a subsequent progression to primary progressive aphasia. Specifically, smaller cortical regions of interest related to speech and language processing seemed differentially involved in this subtype of bulbar-onset ALS. Critical appraisal of the literature gleaned that further work is needed as existing studies revealed multiple methodological limitations. In summary, determining if and how subtypes of ALS differ will require future studies designed to have standardized neuropathology protocols with clinical and genetic patient profiles.
Highlights.
-
-
Neuropathology of brain tissue is compared between bulbar-onset (bALS) and spinal-onset ALS (sALS)
-
-
Both bALS and sALS present with similar anatomical regions of involvement and similar types of pathology
-
-
Specific regions of interest associated to speech/language functions may be differentially involved in bALS
-
-
Some bALS cases may present with pathological features atypical to ALS - Majority of existing studies have methodological biases, therefore, further work is required
Acknowledgments
This work was supported by the National Institutes of Health [R01 DC009890] and ALS Society of Canada Bernese Ramsey Discovery Grant.
APPENDIX A
Electronic Search Strategies. Original search was conducted Sept. 1, 2015 (shown) and updated July 12, 2016.
| Ovid MEDLINE(R) 1946 to August Week 4 2015 | |||
|---|---|---|---|
| # | Searches | Results | Search Type |
| 1 | Amyotrophic Lateral Sclerosis/ | 14231 | Advanced |
| 2 | Motor Neuron Disease/ | 3738 | Advanced |
| 3 | (Lou Gehrig* adj5 disease).mp,kw. | 124 | Advanced |
| 4 | immunohistochemistry/ or histological techniques/ | 275474 | Advanced |
| 5 | Immunocytochemistry/ | 258980 | Advanced |
| 6 | exp Histology/ | 346909 | Advanced |
| 7 | or/1–3 | 17475 | Advanced |
| 8 | or/4–6 | 275474 | Advanced |
| 9 | 7 and 8 | 708 | Advanced |
| 10 | exp animals/ not (exp animals/ and exp humans/) | 4096239 | Advanced |
| 11 | 9 not 10 | 557 | Advanced |
| Embase 1980 to 2015 Week 34 | |||
|---|---|---|---|
| # | Searches | Results | Search Type |
| 1 | (Lou Gehrig* adj5 disease).mp,kw. | 162 | Advanced |
| 2 | Amyotrophic Lateral Sclerosis/ | 25232 | Advanced |
| 3 | Motor Neuron Disease/ | 7679 | Advanced |
| 4 | 1 or 2 or 3 | 30460 | Advanced |
| 5 | histochemistry/ | 45029 | Advanced |
| 6 | histopathology/ | 392734 | Advanced |
| 7 | 5 or 6 | 436223 | Advanced |
| 8 | 4 and 7 | 765 | Advanced |
| 9 | exp animals/ not (exp animals/ and exp humans/) | 4083328 | Advanced |
| 10 | 8 not 9 | 747 | Advanced |
| 11 | limit 10 to embase | 747 | Advanced |
| PsycINFO 2002 to August Week 4 2015 | |||
|---|---|---|---|
| # | Searches | Results | Search Type |
| 1 | Amyotrophic Lateral Sclerosis/ | 2395 | Advanced |
| 2 | Amyotrophic Lateral Sclerosis.mp. | 3328 | Advanced |
| 3 | Motor Neuron* Disease.mp. | 748 | Advanced |
| 4 | 1 or 2 or 3 | 3715 | Advanced |
| 5 | histology/ | 413 | Advanced |
| 6 | immunocytochemistry/ | 1103 | Advanced |
| 7 | 5 or 6 | 1507 | Advanced |
| 8 | 4 and 7 | 28 | Advanced |
| 9 | animals/ not (animals/ and humans/) | 1618 | Advanced |
| 10 | 8 not 9 | 28 | Advanced |
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Schreiber H, Gaigalat T, Wiedemuth-Catrinescu U, Graf M, Uttner I, Muche R, et al. Cognitive function in bulbar- and spinal-onset amyotrophic lateral sclerosis. A longitudinal study in 52 patients. Journal of neurology. 2005;252(7):772–81. doi: 10.1007/s00415-005-0739-6. [DOI] [PubMed] [Google Scholar]
- 2.Phukan J, Elamin M, Bede P, Jordan N, Gallagher L, Byrne S, et al. The syndrome of cognitive impairment in amyotrophic lateral sclerosis: a population-based study. Journal of Neurology, Neurosurgery and Psychiatry. 2012;83(1):102–8. doi: 10.1136/jnnp-2011-300188. [DOI] [PubMed] [Google Scholar]
- 3.Bonduelle VM. The familial forms of amyotrophic lateral sclerosis. Wiener medizinische Wochenschrift (1946) 1975;125(21):330–1. [PubMed] [Google Scholar]
- 4.Armon C, Moses D. Linear estimates of rates of disease progression as predictors of survival in patients with ALS entering clinical trials. J Neurol Sci. 1998;160(Suppl 1):S37–41. doi: 10.1016/s0022-510x(98)00196-8. [DOI] [PubMed] [Google Scholar]
- 5.Haverkamp LJ, Appel V, Appel SH. Natural history of amyotrophic lateral sclerosis in a database population. Validation of a scoring system and a model for survival prediction. Brain : a journal of neurology. 1995;118(Pt 3):707–19. doi: 10.1093/brain/118.3.707. [DOI] [PubMed] [Google Scholar]
- 6.Massman PJ, Sims J, Cooke N, Haverkamp LJ, Appel V, Appel SH. Prevalence and correlates of neuropsychological deficits in amyotrophic lateral sclerosis. Journal of Neurology, Neurosurgey and Psychiatry. 1996;61(5):450–5. doi: 10.1136/jnnp.61.5.450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ringholz GM, Appel SH, Bradshaw M, Cooke NA, Mosnik DM, Schulz PE. Prevalence and patterns of cognitive impairment in sporadic ALS. Neurology. 2005;65(4):586–90. doi: 10.1212/01.wnl.0000172911.39167.b6. [DOI] [PubMed] [Google Scholar]
- 8.Robert D, Pouget J, Giovanni A, Azulay JP, Triglia JM. Quantitative voice analysis in the assessment of bulbar involvement in amyotrophic lateral sclerosis. Acta Otolaryngol. 1999;119(6):724–31. doi: 10.1080/00016489950180702. [DOI] [PubMed] [Google Scholar]
- 9.Green J, Yunusova Y, Kuruvilla MS, Wang J, Pattee GL, Synhorst L, et al. Bulbar and speech motor assessment in ALS: challenges and future directions. Amyotrophic lateral sclerosis & frontotemporal degeneration. 2013;14(7–8):494–500. doi: 10.3109/21678421.2013.817585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brooks BR, Sufit RL, DePaul R, Tan YD, Sanjak M, Robbins J. Design of clinical therapeutic trials in amyotrophic lateral sclerosis. Adv Neurol. 1991;56:521–46. [PubMed] [Google Scholar]
- 11.Goldstein LH, Atkins L, Leigh PN. Correlates of Quality of Life in people with motor neuron disease (MND) Amyotroph Lateral Scler Other Motor Neuron Disord. 2002;3(3):123–9. doi: 10.1080/146608202760834120. [DOI] [PubMed] [Google Scholar]
- 12.Mitsumoto H, Del Bene M. Improving the quality of life for people with ALS: the challenge ahead. Amyotroph Lateral Scler Other Motor Neuron Disord. 2000;1(5):329–36. doi: 10.1080/146608200300079464. [DOI] [PubMed] [Google Scholar]
- 13.Strong MJ, Grace GM, Orange JB, Leeper HA, Menon RS, Aere C. A prospective study of cognitive impairment in ALS. Neurology. 1999;53(8):1665–70. doi: 10.1212/wnl.53.8.1665. [DOI] [PubMed] [Google Scholar]
- 14.Ogawa T, Tanaka H, Hirata K. Cognitive deficits in amyotrophic lateral sclerosis evaluated by event-related potentials. Clinical Neurophysiology. 2009;120(4):659–64. doi: 10.1016/j.clinph.2009.01.013. [DOI] [PubMed] [Google Scholar]
- 15.Lomen-Hoerth C, Murphy J, Langmore S, Kramer JH, Olney RK, Miller B. Are amyotrophic lateral sclerosis patients cognitively normal? Neurology. 2003;60(7):1094–7. doi: 10.1212/01.wnl.0000055861.95202.8d. [DOI] [PubMed] [Google Scholar]
- 16.Sterling LE, Jawaid A, Salamone AR, Murthy SB, Mosnik DM, McDowell E, et al. Association between dysarthria and cognitive impairment in ALS: A prospective study. Amyotrophic Lateral Sclerosis. 2010;11(1–2):46–51. doi: 10.3109/17482960903207997. [DOI] [PubMed] [Google Scholar]
- 17.Ota S, Tsuchiya K, Akiyama H. “Forme fruste” of amyotrophic lateral sclerosis with dementia: A report of five autopsy cases without dementia and with ubiquitinated intraneuronal inclusions. Neuropathology: official journal of the Japanese Society of Neuropathology. 2005;25(4):326–35. doi: 10.1111/j.1440-1789.2005.00646.x. [DOI] [PubMed] [Google Scholar]
- 18.Taylor LJ, Brown RG, Tsermentseli S, Al-Chalabi A, Shaw CE, Ellis CM, et al. Is language impairment more common than executive dysfunction in amyotrophic lateral sclerosis? Journal of Neurology, Neurosurgery and Psychiatry. 2013;84(5):494–8. doi: 10.1136/jnnp-2012-303526. [DOI] [PubMed] [Google Scholar]
- 19.Gordon PH, Goetz RR, Rabkin JG, Dalton K, Mcelhiney M, Hays AP, et al. A prospective cohort study of neuropsychological test performance in ALS. Amyotrophic Lateral Sclerosis. 2010;11(3):312–20. doi: 10.3109/17482961003622585. [DOI] [PubMed] [Google Scholar]
- 20.Ichikawa H, Koyama S, Ohno H, Ishihara K, Nagumo K, Kawamura M. Writing errors and anosognosia in amyotrophic lateral sclerosis with dementia. Behavioural Neurology. 2008;19(3):107–16. doi: 10.1155/2008/814846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kato S, Oda M, Hayashi H, Kawata A, Shimizu T. Participation of the limbic system and its associated areas in the dementia of amyotrophic lateral sclerosis. Journal of the neurological sciences. 1994;126(1):62–9. doi: 10.1016/0022-510x(94)90095-7. [DOI] [PubMed] [Google Scholar]
- 22.Portet F, Cadilhac C, Touchon J, Camu W. Cognitive impairment in motor neuron disease with bulbar onset. Amyotrophic Lateral Sclerosis and Other Motor Neuron Disorders. 2001;2(1):23–9. doi: 10.1080/146608201300079382. [DOI] [PubMed] [Google Scholar]
- 23.Geser F, Lee VMY, Trojanowski JQ. Amyotrophic lateral sclerosis and frontotemporal lobar degeneration: A spectrum of TDP-43 proteinopathies. Neuropathology: official journal of the Japanese Society of Neuropathology. 2010;30(2):103–12. doi: 10.1111/j.1440-1789.2009.01091.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liscic R, Grinberg LT, Zidar J, Gitcho MA, Cairns NJ. ALS and FTLD: two faces of TDP-43 proteinopathy. European Journal of Neurology. 2008;15(8):772–80. doi: 10.1111/j.1468-1331.2008.02195.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Arai T, Hasegawa M, Akiyama H, Ikeda K, Nonaka T, Mori H, et al. TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem Biophys Res Commun. 2006;351(3):602–11. doi: 10.1016/j.bbrc.2006.10.093. [DOI] [PubMed] [Google Scholar]
- 26.Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006;314(5796):130–3. doi: 10.1126/science.1134108. [DOI] [PubMed] [Google Scholar]
- 27.Mackenzie IR, Feldman H. The relationship between extramotor ubiquitin-immunoreactive neuronal inclusions and dementia in motor neuron disease. Acta neuropathologica. 2003;105(2):98–102. doi: 10.1007/s00401-002-0620-y. [DOI] [PubMed] [Google Scholar]
- 28.Mackenzie IR. The neuropathology of FTD associated with ALS. Alzheimer Disease & Associated Disorders. 2007;21(4):S44–S9. doi: 10.1097/WAD.0b013e31815c3486. [DOI] [PubMed] [Google Scholar]
- 29.Yoshida M. Amyotrophic lateral sclerosis with dementia: the clinicopathological spectrum. Neuropathology: official journal of the Japanese Society of Neuropathology. 2004;24(1):87–102. doi: 10.1111/j.1440-1789.2003.00544.x. [DOI] [PubMed] [Google Scholar]
- 30.Prudlo J, König J, Schuster C, Kasper E, Büttner A, Teipel S, et al. TDP-43 pathology and cognition in ALS A prospective clinicopathologic correlation study. Neurology. 2016;87(10):1019–23. doi: 10.1212/WNL.0000000000003062. [DOI] [PubMed] [Google Scholar]
- 31.Dal Bello-Haas V, Florence JM, Krivickas LS. Therapeutic exercise for people with amyotrophic lateral sclerosis or motor neuron disease. The Cochrane Library; 2008. [DOI] [PubMed] [Google Scholar]
- 32.Thomas BH, Ciliska D, Dobbins M, Micucci S. A process for systematically reviewing the literature: providing the research evidence for public health nursing interventions. Worldviews on evidence-based nursing/Sigma Theta Tau International, Honor Society of Nursing. 2004;1(3):176–84. doi: 10.1111/j.1524-475X.2004.04006.x. [DOI] [PubMed] [Google Scholar]
- 33.Von Elm E, Altman DG, Egger M, Pocock SJ, Gøtzsche PC, Vandenbroucke JP, et al. The Strengthening the Reporting of Observational Studies in Epidemiology (STROBE) statement: guidelines for reporting observational studies. Preventive medicine. 2007;45(4):247–51. doi: 10.1016/j.ypmed.2007.08.012. [DOI] [PubMed] [Google Scholar]
- 34.Higgins JP, Altman DG, Gøtzsche PC, Jüni P, Moher D, Oxman AD, et al. The Cochrane Collaboration’s tool for assessing risk of bias in randomised trials. Bmj. 2011;343:d5928. doi: 10.1136/bmj.d5928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Renton AE, Chio A, Traynor BJ. State of play in amyotrophic lateral sclerosis genetics. Nat Neurosci. 2014;17(1):17–23. doi: 10.1038/nn.3584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kamo H, Haebara H, Akiguchi I, Kameyama M, Kimura H, McGeer P. A distinctive distribution of reactive astroglia in the precentral cortex in amyotrophic lateral sclerosis. Acta neuropathologica. 1987;74(1):33–8. doi: 10.1007/BF00688335. [DOI] [PubMed] [Google Scholar]
- 37.Ishihara K, Araki S, Ihori N, Shiota JI, Kawamura M, Nakano I. An autopsy case of frontotemporal dementia with severe dysarthria and motor neuron disease showing numerous basophilic inclusions. Neuropathology: official journal of the Japanese Society of Neuropathology. 2006;26(5):447–54. doi: 10.1111/j.1440-1789.2006.00717.x. [DOI] [PubMed] [Google Scholar]
- 38.Kuwahara H, Tsuchiya K, Saito Y, Kobayashi Z, Miyazaki H, Izumiyama Y, et al. Frontotemporal lobar degeneration with motor neuron disease showing severe and circumscribed atrophy of anterior temporal lobes. Journal of the neurological sciences. 2010;297(1):92–6. doi: 10.1016/j.jns.2010.07.004. [DOI] [PubMed] [Google Scholar]
- 39.Tsuchiya K, Takahashi M, Shiotsu H, Akiyama H, Haga C, Watabiki S, et al. Sporadic amyotrophic lateral sclerosis with circumscribed temporal atrophy: a report of an autopsy case without dementia and with ubiquitinated intraneuronal inclusions. Neuropathology: official journal of the Japanese Society of Neuropathology. 2002;22(4):308–16. doi: 10.1046/j.1440-1789.2002.00451.x. [DOI] [PubMed] [Google Scholar]
- 40.Averbuch-Heller L, Helmchen C, Horn AK, Leigh RJ, Büttner-Ennever JA. Slow vertical saccades in motor neuron disease: correlation of structure and function. Annals of neurology. 1998;44(4):641–8. doi: 10.1002/ana.410440410. [DOI] [PubMed] [Google Scholar]
- 41.Mochizuki Y, Mizutani T, Takasu T. Amyotrophic lateral sclerosis with marked neurological asymmetry: clinicopathological study. Acta neuropathologica. 1995;90(1):44–50. doi: 10.1007/BF00294458. [DOI] [PubMed] [Google Scholar]
- 42.Bodansky A, Kim JMH, Tempest L, Velagapudi A, Libby R, Ravits J. TDP-43 and ubiquitinated cytoplasmic aggregates in sporadic ALS are low frequency and widely distributed in the lower motor neuron columns independent of disease spread. Amyotrophic Lateral Sclerosis. 2010;11(3):321–7. doi: 10.3109/17482961003602363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bak TH, O’Donovan DG, Xuereb JH, Boniface S, Hodges JR. Selective impairment of verb processing associated with pathological changes in Brodmann areas 44 and 45 in the motor neuron disease-dementia-aphasia syndrome. Brain: a journal of neurology. 2001;124:103–20. doi: 10.1093/brain/124.1.103. [DOI] [PubMed] [Google Scholar]
- 44.Sugiyama M, Takao M, Hatsuta H, Funabe S, Ito S, Obi T, et al. Increased number of astrocytes and macrophages/microglial cells in the corpus callosum in amyotrophic lateral sclerosis. Neuropathology: official journal of the Japanese Society of Neuropathology. 2013;33(6):591–9. doi: 10.1111/neup.12027. [DOI] [PubMed] [Google Scholar]
- 45.Troost D, Smitt PS, de Jong J, Swaab D. Neurofilament and glial alterations in the cerebral cortex in amyotrophic lateral sclerosis. Acta neuropathologica. 1992;84(6):664–73. doi: 10.1007/BF00227744. [DOI] [PubMed] [Google Scholar]
- 46.Nagy D, Kato T, Kushner P. Reactive astrocytes are widespread in the cortical gray matter of amyotrophic lateral sclerosis. Journal of neuroscience research. 1994;38(3):336–47. doi: 10.1002/jnr.490380312. [DOI] [PubMed] [Google Scholar]
- 47.Tsuchiya K, Ozawa E, Fukushima J, Yasui H, Kondo H, Nakano I, et al. Rapidly progressive aphasia and motor neuron disease: a clinical, radiological, and pathological study of an autopsy case with circumscribed lobar atrophy. Acta neuropathologica. 2000;99(1):81–7. doi: 10.1007/pl00007411. [DOI] [PubMed] [Google Scholar]
- 48.Kato S, Oda M, Tanabe H. Diminution of dopaminergic neurons in the substantia nigra of sporadic amyotrophic lateral sclerosis. Neuropathology and applied neurobiology. 1993;19(4):300–4. doi: 10.1111/j.1365-2990.1993.tb00444.x. [DOI] [PubMed] [Google Scholar]
- 49.Nakano T, Nakaso K, Nakashima K, Ohama E. Expression of ubiquitin-binding protein p62 in ubiquitin-immunoreactive intraneuronal inclusions in amyotrophic lateral sclerosis with dementia: analysis of five autopsy cases with broad clinicopathological spectrum. Acta neuropathologica. 2004;107(4):359–64. doi: 10.1007/s00401-004-0821-7. [DOI] [PubMed] [Google Scholar]
- 50.Miki Y, Mori F, Nunomura J, Ookawa K, Yajima N, Yagihashi S, et al. Sporadic amyotrophic lateral sclerosis with pallido-nigro-luysian degeneration: A TDP-43 immunohistochemical study. Neuropathology: official journal of the Japanese Society of Neuropathology. 2010;30(2):149–53. doi: 10.1111/j.1440-1789.2009.01046.x. [DOI] [PubMed] [Google Scholar]
- 51.Bak TH, O’Donovan DG, Xuereb JH, Boniface S, Hodges JR. Selective impairment of verb processing associated with pathological changes in Brodmann areas 44 and 45 in the motor neuron disease-dementia-aphasia syndrome. 2001 doi: 10.1093/brain/124.1.103. [DOI] [PubMed] [Google Scholar]
- 52.Braak H, Braak E. Staging of Alzheimer’s disease-related neurofibrillary changes. Neurobiology of aging. 1995;16(3):271–8. doi: 10.1016/0197-4580(95)00021-6. [DOI] [PubMed] [Google Scholar]
- 53.Abrahams S. Executive dysfunction in ALS is not the whole story. Journal of Neurology, Neurosurgey and Psychiatry. 2013;84(5):474–5. doi: 10.1136/jnnp-2012-303851. [DOI] [PubMed] [Google Scholar]
- 54.Ichikawa H, Hieda S, Ohno H, Ishihara K, Kawamura M. Language impairment in amyotrophic lateral sclerosis from an historical review: kana and kanji versus alphabetical languages. Amyotrophic Lateral Sclerosis and the Frontotemporal Dementias. 2012:93. [Google Scholar]
- 55.Bigio EH. C90RF72, the new gene on the block, causes C9FTD/ALS: New insights provided by neuropathology. Acta neuropathologica. 2011;122(6):653–5. doi: 10.1007/s00401-011-0919-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Murray ME, DeJesus-Hernandez M, Rutherford NJ, Baker M, Duara R, Graff-Radford NR, et al. Clinical and neuropathologic heterogeneity of c9FTD/ALS associated with hexanucleotide repeat expansion in C9ORF72. Acta neuropathologica. 2011;122(6):673–90. doi: 10.1007/s00401-011-0907-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bede P, Bokde ALW, Byrne S, Elamin M, McLaughlin RL, Kenna K, et al. Multiparametric MRI study of ALS stratified for the C9orf72 genotype. Neurology. 2013;81(4):361–9. doi: 10.1212/WNL.0b013e31829c5eee. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Renton AE, Majounie E, Waite A, Simon-Sanchez J, Rollinson S, Gibbs JR, et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron. 2011;72(2):257–68. doi: 10.1016/j.neuron.2011.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rothstein JD. Current hypotheses for the underlying biology of amyotrophic lateral sclerosis. Ann Neurol. 2009;65(S1):S3–S9. doi: 10.1002/ana.21543. [DOI] [PubMed] [Google Scholar]
- 60.Ichikawa H, Ohno H, Murakami H, Ohnaka Y, Kawamura M. Writing error may be a predictive sign for impending brain atrophy progression in amyotrophic lateral sclerosis: A preliminary study using x-ray computed tomography. European Neurology. 2011;65(6):346–51. doi: 10.1159/000328216. [DOI] [PubMed] [Google Scholar]
- 61.Ichikawa H, Takahashi N, Hieda S, Hideki O, Kawamura M. Agraphia in bulbar-onset amyotrophic lateral sclerosis: Not merely a consequence of dementia or aphasia. Behavioural Neurology. 2008;20(3):91–9. doi: 10.3233/BEN-2008-0219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yoshizawa K, Yasuda N, Fukuda M, Yukimoto Y, Ogino M, Hata W, et al. Syntactic comprehension in patients with amyotrophic lateral sclerosis. Behavioural neurology. 2014;2014 doi: 10.1155/2014/230578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Agosta F, Weiler M, Filippi M. Propagation of pathology through brain networks in neurodegenerative diseases: from molecules to clinical phenotypes. CNS neuroscience & therapeutics. 2015;21(10):754–67. doi: 10.1111/cns.12410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Seeley WW, Crawford RK, Zhou J, Miller BL, Greicius MD. Neurodegenerative diseases target large-scale human brain networks. Neuron. 2009;62(1):42–52. doi: 10.1016/j.neuron.2009.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.van den Berg LH, van den Heuvel MP. Simulating disease propagation across white matter connectome reveals. doi: 10.1016/j.neuroimage.2015.04.005. [DOI] [PubMed] [Google Scholar]
- 66.Evans J, Olm C, McCluskey L, Elman L, Boller A, Moran E, et al. Impaired cognitive flexibility in amyotrophic lateral sclerosis. Cognitive and behavioral neurology: official journal of the Society for Behavioral and Cognitive Neurology. 2015;28(1):17. doi: 10.1097/WNN.0000000000000049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Schmidt R, de Reus MA, Scholtens LH, van den Berg LH, van den Heuvel MP. Simulating disease propagation across white matter connectome reveals anatomical substrate for neuropathology staging in amyotrophic lateral sclerosis. NeuroImage. 2016;124:762–9. doi: 10.1016/j.neuroimage.2015.04.005. [DOI] [PubMed] [Google Scholar]
- 68.Li Z, Peck KK, Brennan NP, Jenabi M, Hsu M, Zhang Z, et al. Diffusion tensor tractography of the arcuate fasciculus in patients with brain tumors: comparison between deterministic and probabilistic models. Journal of biomedical science and engineering. 2013;6(2):192. doi: 10.4236/jbise.2013.62023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.de Schotten MT, Bizzi A, Dell’Acqua F, Allin M, Walshe M, Murray R, et al. Atlasing location, asymmetry and inter-subject variability of white matter tracts in the human brain with MR diffusion tractography. Neuroimage. 2011;54(1):49–59. doi: 10.1016/j.neuroimage.2010.07.055. [DOI] [PubMed] [Google Scholar]
- 70.Catani M, de Schotten MT. A diffusion tensor imaging tractography atlas for virtual in vivo dissections. Cortex. 2008;44(8):1105–32. doi: 10.1016/j.cortex.2008.05.004. [DOI] [PubMed] [Google Scholar]
- 71.Fuertinger S, Horwitz B, Simonyan K. The functional connectome of speech control. PLoS Biol. 2015;13(7):e1002209. doi: 10.1371/journal.pbio.1002209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Fujimura-Kiyono C, Kimura F, Ishida S, Nakajima H, Hosokawa T, Sugino M, et al. Onset and spreading patterns of lower motor neuron involvements predict survival in sporadic amyotrophic lateral sclerosis. Journal of Neurology, Neurosurgery & Psychiatry. 2011;82(11):1244–9. doi: 10.1136/jnnp-2011-300141. [DOI] [PubMed] [Google Scholar]
- 73.David AS, Gillham RA. Neuropsychological study of motor neuron disease. Psychosomatics. 1986;27(6):441–5. doi: 10.1016/S0033-3182(86)72673-X. [DOI] [PubMed] [Google Scholar]
- 74.Gallassi R, Montagna P, Ciardulli C, Lorusso S, Mussuto V, Stracciari A. Cognitive impairment in motor neuron disease. Acta neurologica scandinavica. 1985;71(6):480–4. doi: 10.1111/j.1600-0404.1985.tb03231.x. [DOI] [PubMed] [Google Scholar]
- 75.Gallassi R, Montagna P, Morreale A, Lorusso S, Tinuper P, Daidone R, et al. Neuropsychological, electroencephalogram and brain computed tomography findings in motor neuron disease. European neurology. 1989;29(2):115–20. doi: 10.1159/000116391. [DOI] [PubMed] [Google Scholar]
- 76.Abrahams S, Goldstein L, Al-Chalabi A, Pickering A, Morris R, Passingham R, et al. Relation between cognitive dysfunction and pseudobulbar palsy in amyotrophic lateral sclerosis. Journal of Neurology, Neurosurgery & Psychiatry. 1997;62(5):464–72. doi: 10.1136/jnnp.62.5.464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lawyer T, Jr, Netsky MG. Amyotrophic lateral sclerosis. AMA Arch Neurol Psychiatry. 1953;69(2):171–92. doi: 10.1001/archneurpsyc.1953.02320260029002. [DOI] [PubMed] [Google Scholar]
- 78.Hirano A. Cytopathology of amyotrophic lateral sclerosis. Adv Neurol. 1991;56:91. [PubMed] [Google Scholar]
- 79.Kabashi E, Valdmanis PN, Dion P, Spiegelman D, McConkey BJ, Velde CV, et al. TARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nat Genet. 2008;40(5):572–4. doi: 10.1038/ng.132. [DOI] [PubMed] [Google Scholar]
- 80.Sreedharan J, Blair IP, Tripathi VB, Hu X, Vance C, Rogelj B, et al. TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science. 2008;319(5870):1668–72. doi: 10.1126/science.1154584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Lomen-Hoerth C, Anderson T, Miller B. The overlap of amyotrophic lateral sclerosis and frontotemporal dementia. Neurology. 2002;59(7):1077–9. doi: 10.1212/wnl.59.7.1077. [DOI] [PubMed] [Google Scholar]
- 82.Lillo P, Savage S, Mioshi E, Kiernan MC, Hodges JR. Amyotrophic lateral sclerosis and frontotemporal dementia: A behavioural and cognitive continuum. Amyotroph Lateral Scler. 2012;13(1):102–9. doi: 10.3109/17482968.2011.639376. [DOI] [PubMed] [Google Scholar]
- 83.Lillo P, Hodges JR. Frontotemporal dementia and motor neurone disease: overlapping clinic-pathological disorders. Journal of clinical neuroscience: official journal of the Neurosurgical Society of Australasia. 2009;16(9):1131–5. doi: 10.1016/j.jocn.2009.03.005. [DOI] [PubMed] [Google Scholar]
- 84.Kokubo Y, Kuzuhara S. Neurofibrillary tangles in ALS and parkinsonism–dementia complex focus in Kii, Japan. Neurology. 2004;63(12):2399–401. doi: 10.1212/01.wnl.0000147241.52694.6a. [DOI] [PubMed] [Google Scholar]
- 85.Shankar SK, Yanagihara R, Garruto RM, Grundke-Iqbal I, Kosik KS, Gajdusek DC. Immunocytochemical characterization of neurofibrillary tangles in amyotrophic lateral sclerosis and parkinsonism-dementia of guam. Ann Neurol. 1989;25(2):146–51. doi: 10.1002/ana.410250207. [DOI] [PubMed] [Google Scholar]
- 86.Bäumer D, Hilton D, Paine S, Turner M, Lowe J, Talbot K, et al. Juvenile ALS with basophilic inclusions is a FUS proteinopathy with FUS mutations. Neurology. 2010;75(7):611–8. doi: 10.1212/WNL.0b013e3181ed9cde. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Tateishi T, Hokonohara T, Yamasaki R, Miura S, Kikuchi H, Iwaki A, et al. Multiple system degeneration with basophilic inclusions in Japanese ALS patients with FUS mutation. Acta Neuropathol. 2010;119(3):355–64. doi: 10.1007/s00401-009-0621-1. [DOI] [PubMed] [Google Scholar]
- 88.Elizan TS, Hirano A, Abrams BM, Need RL, Van Nuis C, Kurland LT. Amyotrophic lateral sclerosis and parkinsonism-dementia complex of Guam: Neurological reevaluation. Archives of neurology. 1966;14(4):356–68. doi: 10.1001/archneur.1966.00470100012002. [DOI] [PubMed] [Google Scholar]
- 89.Gordon PH, Cheng B, Salachas F, Pradat PF, Bruneteau G, Corcia P, et al. Progression in ALS is not linear but is curvilinear. Journal of Neurology. 2010;257(10):1713–7. doi: 10.1007/s00415-010-5609-1. [DOI] [PubMed] [Google Scholar]
- 90.Kim S-M, Lee K-M, Hong Y-H, Park KS, Yang J-H, Nam H-W, et al. Relation between cognitive dysfunction and reduced vital capacity in amyotrophic lateral sclerosis. Journal of Neurology, Neurosurgery & Psychiatry. 2007;78(12):1387–9. doi: 10.1136/jnnp.2006.111195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Whitwell JL, Jack CR, Senjem ML, Josephs KA. Patterns of atrophy in pathologically confirmed FTLD with and without motor neuron degeneration. Neurology. 2006;66(1):102–4. doi: 10.1212/01.wnl.0000191395.69438.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Cedarbaum JM, Stambler N, Malta E, Fuller C, Hilt D, Thurmond B, et al. The ALSFRS-R: a revised ALS functional rating scale that incorporates assessments of respiratory function. BDNF ALS Study Group (Phase III) Journal of the neurological sciences. 1999;169(1–2):13–21. doi: 10.1016/s0022-510x(99)00210-5. [DOI] [PubMed] [Google Scholar]