

Abstract
Background
Pyrethroids are potentially harmful to human health and ecosystems. It is necessary to develop some efficient strategies to degrade pyrethroid residues. Biodegradation is generally considered as a safe, efficient, and inexpensive way to eliminate environmental contaminants. To date, although several pyrethroid-hydrolyzing esterases have been cloned, there has been no report about a pyrethroid hydrolase with high hydrolytic activity, good stability, and high productivity, indispensable enzymatic properties in practical biodegradation. Almost all pyrethroid hydrolases are intracellular enzymes, which require complex extraction protocols and present issues in terms of easy inactivation and low production.
Results
In this study, random mutagenesis was performed on one pyrethroid-hydrolyzing esterase, Sys410, to enhance its activity and thermostability. Two beneficial mutations, A171V and D256N, were obtained by random mutagenesis and gave rise to the mutant M2. The mutant displayed ~1.5-fold improvement in the kcat/Km value and 2.46-fold higher catalytic activity. The optimal temperature was 10 °C higher than that of the wild-type enzyme (55 °C). The half-life at 40–65 °C was 3.3–310 times longer. It was surprising that M2 has a half-life of 12 h at 70 °C while Sys410 was completely inactivated at 70 °C. In addition, the desired gene was extracellularly expressed in a Pichia pastoris host system. The soluble expression level reached up to 689.7 mg/L. Remarkably, the enzyme could efficiently degrade various pyrethroids at moderate temperature for 15 min, exceeding a hydrolysis rate of 98%, which is the highest value ever reported.
Conclusions
This is the first report about random mutagenesis and secretory expression of pyrethroid-hydrolyzing esterase with high-level productivity and purity in P. pastoris. Broad substrate specificity, enhanced activity and thermostability make M2 an ideal candidate for the biodegradation of pyrethroid residues.
Electronic supplementary material
The online version of this article (doi:10.1186/s12934-017-0698-5) contains supplementary material, which is available to authorized users.
Keywords: Pyrethroids, Random mutagenesis, Pichia pastoris, Secretory expression
Background
Pyrethroids have been most widely used as insecticides throughout the world in agriculture, forestry, public health, and houses, as well as for protection of textiles and buildings [1, 2]. Previously considered to be of low toxicity and great selective lethality, pyrethroids have been used for more than 30 years and have accounted for almost 25% of the global pesticide market [3]. As a replacement for the more toxic and environmentally persistent organochlorine and organophosphorus pesticides, the demand for pyrethroids continues to grow [4, 5]. However, non-judicious use has raised great concerns as it has caused many problems, such as pest resistance, soil and water contamination, high residue in agricultural products, and human exposure [6, 7]. Therefore, the development of some efficient strategies to solve these problems is urgently required.
Biodegradation is generally considered as a safe, efficient, and inexpensive way of eliminating environmental contaminants [8]. The major metabolic pathway of pyrethroids involves cytochrome P450 oxidation and ester-bond hydrolysis by esterases that results in nontoxic acid and alcohol production [9, 10]. To date, many pyrethroid-degrading microorganisms have been isolated and studied [11–15]. Compared with living microorganisms, recombinant enzymes have greater potential in eliminating pyrethroid residuals, especially with mass production and safety [16]. So far, quite a few pyrethroid-degrading genes have been cloned and characterized, such as pytY, estP, pytH, pye3, pytZ, sys410, EstSt7 gene, and CMO gene [16–23]. However, no one enzyme has all the properties, such as high hydrolytic activity [22, 23], good stability [16, 23], and high productivity [17–21], which are indispensable for practical biodegradation. And almost all of them are intracellular enzymes, which require complex extraction protocols and present issues in terms of easy deactivation and low production [24]. To facilitate downstream processing in large-scale biotechnological applications, secretion of the overexpressed enzymes into the culture medium is desirable [25, 26].
Increasing interest in applying enzymes to industry has spurred the search for biocatalysts with new or improved properties [27]. Directed evolution has emerged as a ubiquitous technique to enhance the stability, activity, and selectivity of enzymes [28]. Notably, it has not relied on a priori structural and mechanistic information about the proteins [29], more suitable for modifying novel enzymes. Over the past decade, a large number of molecular biological methods for gene mutagenesis has been developed [29]. Random mutagenesis, as a powerful tool, has been advocated for modifying various enzyme functions [28, 30]. Jang et al. created a mutant β-agarase from Zobellia galactanivorans that had enhanced thermostability by random mutagenesis. The melting temperature (Tm) was increased by 5.2 °C over that of the wild-type enzyme (54.6 °C) [31]. Yu et al. enhanced the thermostability of a Rhizopus chinensis lipase by two rounds of error-prone polymerase chain reaction (PCR) and two rounds of DNA shuffling. The S4-3 variant was the most thermostable lipase; Tm was 22° higher and the half-lives at 60 and 65 °C were 46 and 23 times longer [32]. For practical use in clinical diagnosis, directed evolution was applied to improve the thermostability of fructosyl peptide oxidases. The sextuple mutant enzyme, R94K/G184D/F265L/N272D/H302R/H388Y, had a half-life of thermal inactivation at 50 °C that was 79.8-fold longer than that of the parental fructosyl peptide oxidase [33].
In our previous work [21], we identified a novel pyrethroid-hydrolyzing enzyme, Sys410, with broad substrate specificity and relatively high activity. High catalytic activity and thermostability are very attractive properties for practical applications. Herein, we chose Sys410 as a starting point for further improvement. Two beneficial mutations were obtained by random mutagenesis and gave rise to the mutant M2. The enzymatic properties and pyrethroid degradation were well characterized. Meanwhile, the desirable mutant was extracellularly expressed with high-level productivity and purity in Pichia pastoris.
Results and discussion
Directed evolution and screening for mutants with enhanced activity and stability
In the study, random mutations were introduced into the Sys410 gene by error-prone PCR. A two-step screening strategy was designed for screening. For the first round of error-prone PCR, plasmid pET28a-Sys410 was used as the template. Several clones displayed bigger hydrolysis zones on tributyrin plates (Fig. 1), and were then evaluated for thermostability. Only one positive clone named M1 turned deep brown after being treated at 60 °C for 55 min (Fig. 2). For the second round of error-prone PCR, plasmid pTV118N-M1 was used as the template. One positive clone harboring plasmid pTV118N-M2 was obtained (Additional file 1: Figure S1).
Fig. 1.
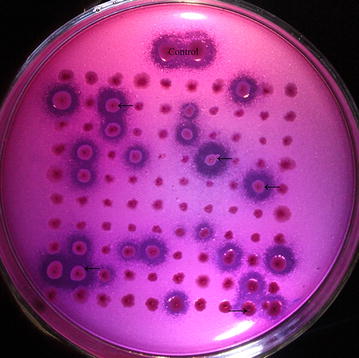
Screening of mutants with better activity (→). The transformants were replicated on LB agar plates supplemented with 0.1% (v/v) tributyrin. A transparent zone due to tributyrin hydrolysis appeared at 37 °C in 1–2 days. Clones with enhanced activity displayed bigger hydrolysis zones on tributyrin plates
Fig. 2.
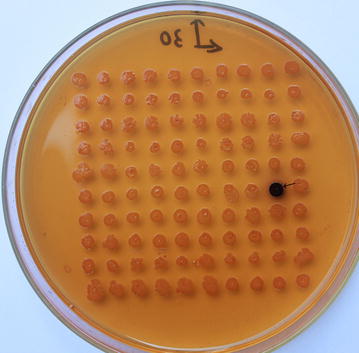
Screening of mutants with better thermostability (→). After being treated at 60 °C for 55 min, the clones were overlayed with 0.3 mg/mL α-naphthyl acetate and 1.3 mg/mL Fast Blue B. Clones with enhanced thermostability (M1) rapidly turned deep brown around the colony
A good method is crucial to screen the target gene from the enormous number of clones in a mutagenesis library. Hydrolysis areas were measured semi-quantitatively [34]. Clones with enhanced activity showed a bigger hydrolysis zone, and were selected for further study. The second screening was also based on esterase activity. In that step, it was very important to determine the critical inactivation temperature and time of the control clone harboring the template plasmid. The critical inactivation conditions were 60 °C for 55 min in the first round, and 80 °C for 60 min in the second round. After being heated, clones with enhanced thermostability still had esterase activity, and could react with α-naphthyl acetate and Fast Blue B, which resulted in a deep brown color around the colony [35], while clones with the same or reduced thermostability had no color change. In the end, two positive clones were obtained through two rounds of random mutagenesis. The results showed that this two-step screening strategy was efficient in screening esterases with enhanced activity and thermostability.
Sequence and structure analysis
Sequence analysis identified one point mutation (C512T) in M1, and two point mutations (C512T and G766A) in M2. Nucleotide mutations at C512T and G766A caused amino acid mutations from alanine to valine at position 171 and from aspartic acid to asparagine at position 256, respectively. M3 with one point mutation (G766A) was generated by site-directed mutagenesis. Homology models of Sys410 and variant M2 were built by SWISS-MODEL (https://www.swissmodel.expasy.org) and the models were evaluated with MolProbity [36, 37] and SAVES (The Structure Analysis and Verification Server, version 4). The overall structures of the two enzymes are extremely similar (Fig. 3). The catalytic triads S102-H260-D228 are well conserved, suggesting that these enzymes share a common catalytic mechanism. Mutation A171V is inside the molecule, near to the catalytic center, and D256N is on the surface of the protein.
Fig. 3.
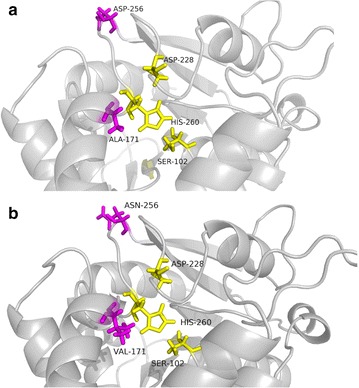
Homology models of Sys410 (a) and variant M2 (b). The catalytic triads S102-H260-D228 are colored yellow. Mutations are colored carmine
Heterologous expression and purification of the mutants
The full-length mutant genes were amplified and cloned into the expression vector pET-28a (+) with a C-terminal 6 × His tag, expressed in E. coli BL21 (DE3) with 0.6 mM IPTG induction at 37 °C for 8 h, then purified by Ni–NTA-agarose chromatography. The sodium dodecyl sulfate–polyacrylamide gel electrophoresis analysis showed that the target recombinant protein appeared as a single band on SDS-PAGE with molecular weight 36.7 kDa (Additional file 2: Figure S2), consist of the 280 amino acids with a fusion of 54 amino acids corresponding to polyhistidine tag (His-tag), a unique thrombin cleavage site (Thrombin). The molecular weight and expression quantity of the mutants were the same as the wild-type enzyme. The fractions containing the recombinant protein were stored at −20 °C for further study.
Determination of substrate specificity and kinetic parameters
To determine the substrate specificity of the mutants, we tested their activity on various ρ-nitrophenyl esters with acyl chain lengths of C2, C4, C6, C8, C10, and C12 under assay conditions of pH 6.5 and 55 °C. Substrate specificity for ρ-nitrophenyl esters of various fatty acids are shown in Fig. 4. The bar charts are very similar. All of the wild-type and mutant enzymes showed a specific preference for ρ-nitrophenyl acetate over other substrates. ρ-Nitrophenyl acetate was then used to test the activity and kinetic parameters of Sys410 and M2 (Table 1). The Km and kcat values were calculated by fitting the data to the Michaelis–Menten equation. The mutations A171V and D256N improved both the kcat value and the affinity. Combination of the two mutations resulted in ~1.5-fold improvement in the kcat/Km value. Generally, for most of esterases, there is a negative correlation between Km and kcat values for one enzyme toward different substrates. A low Km value for a substrate indicates positive affinity for the enzyme, followed with higher catalytic activity and consequently a higher kcat value. The catalytic activity of M2 was 2.46-fold higher than that of Sys410. The activity against ρ-nitrophenyl acetate was much better than other pyrethroid-hydrolyzing esterases reported [16–19, 21–23], except PytZ with no data shown [20]. Efficient catalytic activity is a very attractive property of enzymes for practical applications.
Fig. 4.
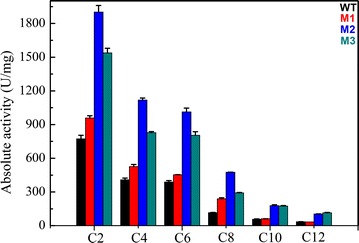
Substrate specificity of the wild-type and mutant enzymes against various ρ-nitrophenyl esters. Substrate specificity against various ρ-nitrophenyl esters with acyl chain lengths of C2, C4, C6, C8, C10, and C12 was determined under assay conditions of pH 6.5 and 55 °C. The activity of the wild-type enzyme, M1, M2, and M3 are colored black, red, blue, and dark cyan, respectively. Data points are the average of triplicate measurements, and error bars represent the standard deviation
Table 1.
Kinetic characterization and enzyme activity of the wild-type and mutant enzymes
| Enzyme | Km (μM) | k cat (s−1) | k cat/Km (s−1 M−1) | Maximum enzyme activity (U/mg) | Fold |
|---|---|---|---|---|---|
| Sys410 | 14.11 ± 3.22 | 289.12 ± 3.03 | 20.50 | 772.92 ± 3.41 | 1.00 |
| M1 | 13.70 ± 2.02 | 293.31 ± 1.27 | 21.41 | 958.43 ± 1.27 | 1.24 |
| M2 | 11.61 ± 2.74 | 350.15 ± 6.05 | 30.16 | 1901.31 ± 4.20 | 2.46 |
| M3 | 12.77 ± 1.92 | 307.32 ± 4.89 | 24.07 | 1538.14 ± 2.21 | 1.99 |
Effect of pH and temperature on enzyme activity
Enzyme activity can be significantly influenced by pH value and temperature. In this work, the effect of pH on the enzyme activity was determined between pH 4.5 and pH 9.0. The optimal pH was 0.5 higher than that of the wild-type enzyme (pH 7.0) (Fig. 5). Mutants have higher activity than WT over a range of pH values. Similar results were obtained in most other pyrethroid hydrolases [16–21, 23], very different from alkaliphilic EstSt7 [22]. Good pH adaptability is indispensable when dealing with the frequently changeable conditions during bioremediation.
Fig. 5.
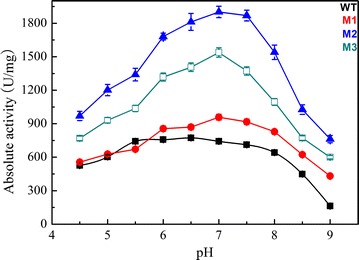
Effect of pH on the activity of the wild-type enzyme (filled square), M1 (filled circle), M2 (filled triangle), and M3 (open square). The optimum pH was measured using ρ-nitrophenyl acetate as a substrate at 55 °C. pH stability was tested after incubation of the purified enzyme for 24 h at 30 °C in the above different buffers. Data points are the average of triplicate measurements, and error bars represent the standard deviation
In the present study, the activity of Sys410 and mutants was determined at temperatures of 35–80 °C (Fig. 6), with the consideration of environmental and industrial complexity. The mutants M1 and M2 had an increased optimal temperature (Topt) of 65 °C, 10° higher than that of the parental enzyme. The mutant M3 had an increased Topt of 70 °C, 15° higher than that of Sys410. Interestingly, the relative activity of M2 was more than 50% in a wide temperature range between 35 and 80 °C, which proved that this pyrethroid-hydrolyzing enzyme possesses remarkable adaptability over a broad range of temperatures.
Fig. 6.
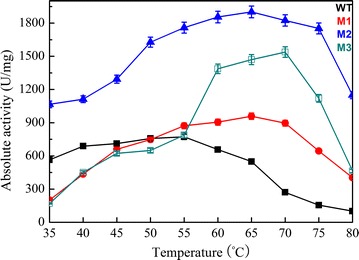
Effect of temperature on the activity of the wild-type enzyme (filled square), M1 (filled circle), M2 (filled triangle), and M3 (open square). The optimum temperature was determined analogously by measuring esterase activity in the temperature range of 35–80 °C in a phosphate buffer (50 mM, pH 6.5). Data points are the average of triplicate measurements, and error bars represent the standard deviation
Thermostability was determined by analysis of residual activity after preincubation at 45–65 °C for 12 h (Fig. 7). Mutants exhibited excellent thermostability (Table 2; Fig. 7). After preincubation at 65 °C for 12 h, M2 still retained as much as 76.4% of its total activity. Therefore, the thermostability of M2 was further analyzed by measuring the half-lives (T1/2) at 40–80 °C. Table 2 lists the T1/2 values of wild-type (WT) and mutant M2 at different temperatures. The T1/2 at 40–65 °C were 3.3–310 times longer than the WT. For practical applications, enhancement of the thermostability of the targeted enzyme is generally favorable, as it will extend the shelf life of the enzyme in reagents. It was surprising that M2 had a T1/2 of 12 h at 70 °C whereas the WT was immediately deactivated. These results proved that M2 is highly thermostable, much better than most of the pyrethroid hydrolases aforementioned, slightly less than the thermophilic pyrethroid-hydrolyzing enzyme EstSt7. PytZ lost enzymatic activity when it was incubated at 60 °C for 2 h [20]. When the temperature was higher than 45 °C, the stability of PytY began to decrease [16]. EstP and Pye3 were both fairly stable up to 45 °C and had 54% of their activity at 50 °C, but were completely inactivated at 65 °C [17, 19]. The enzyme PytH was fairly stable up to 50 °C, had 55% residual activity at 60 °C, and was completely inactivated at 70 °C [18]. CMO was highly stable below 10 °C, but at temperatures above 10 °C, catalytic activity decreased slowly along with increased incubation time [23]. EstSt7 had an optimum temperature of 80 °C [22]. Nevertheless, it showed worse adaptability to low and moderate temperatures, which are frequently encountered during degradation of pyrethroid residues.
Fig. 7.
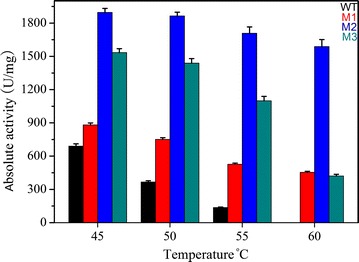
Effect of temperature on stability of the wild-type enzyme (black), M1 (red), M2 (blue) and M3 (dark cyan). Thermostability was measured by preincubation of the purified enzymes in 50 mM potassium phosphate buffer (pH 6.5) at 45–65 °C for 12 h. Data points are the average of triplicate measurements, and error bars represent the standard deviation
Table 2.
The half-lives (T1/2) of Sys410 and M2 at different temperatures
| Temperature (°C) | T1/2 of Sys410 and residual relative activity | T1/2 of M2 and residual relative activity | Fold improvement |
|---|---|---|---|
| 40 | 72 h (52.6%) | 236 h (50.7%) | 3.3 |
| 45 | 38 h (50.4%) | 154 h (53.8%) | 4.1 |
| 50 | 10 h (49.8%) | 133 h (54.1%) | 13.3 |
| 55 | 6 h (56.0%) | 77 h (48.3%) | 12.8 |
| 60 | 84 min (51.4%) | 53 h (54.6%) | 37.9 |
| 65 | 6 min (48.9%) | 31 h (52.8%) | 310 |
| 70 | – | 12 h (56.1%) | – |
| 80 | – | 4 h (45.3%) | – |
Enzymatic properties showed that the two amino acid substitutions, A171V and D256N, were beneficial for the enhanced activity and thermostability of Sys410. V171 is one of the two positions that underwent beneficial mutations obtained from our random screen, and the original residue was an alanine. This suggests that a bulkier residue like valine may be preferred at position 171, possibly to increase the hydrophobic interactions with substrates. D256 is on the surface of the protein and it is unlikely that this residue will interact with other residues before or after the mutation, as judged from the structure. We currently do not understand the reason for the 99% activity and thermostability enhancement caused by the D256N mutation. More details regarding the mechanisms involved in the enhanced properties will be elucidated by resolving the 3D structure and rational protein design.
Gas chromatography analysis of degradation of different pyrethroids
To assess the application potential of Sys410 and the mutants, their ability to degrade various pyrethroids was determined. Assays for pyrethroid hydrolysis were performed in 50 mM potassium phosphate buffer (pH 6.5 instead of 7.5) at 37 °C instead of high temperature for a number of reasons. Firstly, pyrethroids are unstable under alkaline conditions. Next, our aim was to evaluate the effectiveness of the enzyme at a temperature common in an application such as reducing pesticide residues in agro-products. And lastly, the hydrolysis parameters obtained could be directly compared with those of the WT Sys410 reported in the previous study. As shown in Fig. 8, the mutant M2 was able to efficiently degrade all the pyrethroids tested within a short time, indicating that M2 possessed broad substrate specificity. This feature was similar to most pyrethroid hydrolases [16–22], probably because most pyrethroid pesticides share a similar ester bond in their molecular structure. Remarkably, M2 hydrolyzed pyrethroids much more efficiently than the WT and other pyrethroid hydrolases reported [16–22], reaching over 98% conversion in 15 min. In contrast, the WT required 30 min, reaching about 80%. Unfortunately, in mid-to-low temperature conditions, both M1 and M3 had much lower activity and, as expected, the hydrolysis rates were both worse than Sys410. This demonstrated that it is very important for enzymes to possess remarkable adaptability over a broad range of temperatures to have practical application.
Fig. 8.
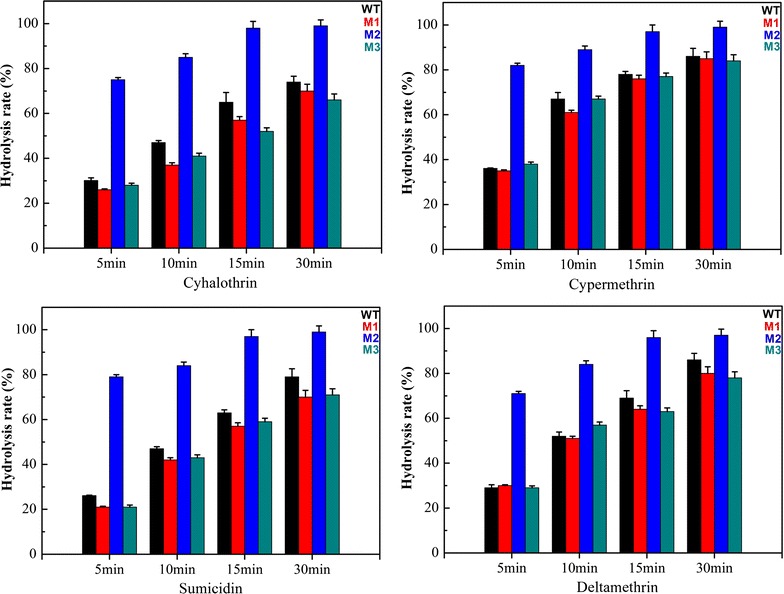
Hydrolysis rate of different pyrethroids degraded by the wild-type enzyme (black), M1 (red), M2 (blue), and M3 (dark cyan)
Extracellular expression and characterization of M2 in P. pastoris X-33
Because of its remarkable enzymatic properties, the mutant M2 was selected as the candidate for further study. Secretion is preferred for heterologous protein production due to the ease of recovery [38]. The expression system of the yeast P. pastoris is widely used to express various proteins due to its high production and secretion efficiency [39]. Furthermore, the secreted recombinant protein in P. pastoris constitutes the vast majority of total proteins in the medium because the host secretes low levels of endogenous proteins [40]. In the present study, the M2 coding sequence was cloned into the pPICZα B vector at the EcoRI/KpnI restriction sites as described previously. This construction allowed the M2 gene to be theoretically in-frame with the α-factor secretion signal in pPICZα B. Following a sequence check, the construct denoted as pPICZα B-M2 was SacI-linearized and electroporated into P. pastoris X-33. The transformant (pPICZα B-M2) with the highest esterase activity was selected for further experiments through activity screening and gene integration analysis.
After methanol induction for 7 days, the recombinant enzyme was secreted into culture supernatants using α-factor signal sequence and showed a single band at approximately 31 kDa by SDS-PAGE analysis (Additional file 3: Figure S3). This was in good agreement with the molecular mass deduced from the amino acid sequence with a fusion 6 × His tag, smaller than the recombinant protein in Escherichia coli BL21 (DE3) with a molecular weight of 36.7 kDa, consisting of 280 amino acids with a fusion of 54 amino acids corresponding to a polyhistidine tag (His tag), a unique thrombin cleavage site. It was presumed that there is little or no glycosylation in the protein. More importantly, solubility analysis revealed that the recombinant enzyme was soluble and accounted for more than 94% of the total protein in the supernatant calculated according to Quantity One software (Bio-Rad Laboratories Inc., Hercules, USA) for protein band visualization, which facilitated its purification in the application. The purity of the recombinant esterase could meet further enzyme analysis. The optimal induction time was 5 days determined by SDS-PAGE and activity analysis. The recombinant enzyme expression hit a highest level of approximately 689.7 mg/L, which was 2.87 times that expressed in E. coli BL21.
The expression system of the yeast P. pastoris is widely used for the expression of various proteins due to its high production, secretion efficiency, and few endogenous proteins in the medium [24]. However, to our knowledge, there has only been one study about a pyrethroid-metabolizing esterase expressed in P. pastoris. The CzEst9-pPICZα A vector transformed into P. pastoris GS115 produced a methanol-inducible intracellular recombinant protein, and the protein yield seemed low [41]. So, our study is the first report about extracellular and high-yield expression of a pyrethroid-hydrolyzing esterase gene in a P. pastoris host system.
We also determined the degrading ability of M2 from the P. pastoris host system towards various pyrethroids. Enzyme samples (2.4 μg) with 5 mg/mL substrate in 50 mM potassium phosphate buffer (pH 6.5) were incubated at 37 °C for 15 min. As shown in Fig. 9, the hydrolysis rates of cyhalothrin, cypermethrin, sumicidin, and deltamethrin were 98.6, 99.53, 98.1, and 98.9%, respectively. The results demonstrated that the ability of the enzyme from P. pastoris to degrade various pyrethroids was in good agreement with that expressed in E. coli BL21 (DE3). Generally, pesticide residues are mixtures in the environment. A broad-spectrum pyrethroid-degrading enzyme with higher hydrolytic activity will play a better role in practical biodegradation.
Fig. 9.
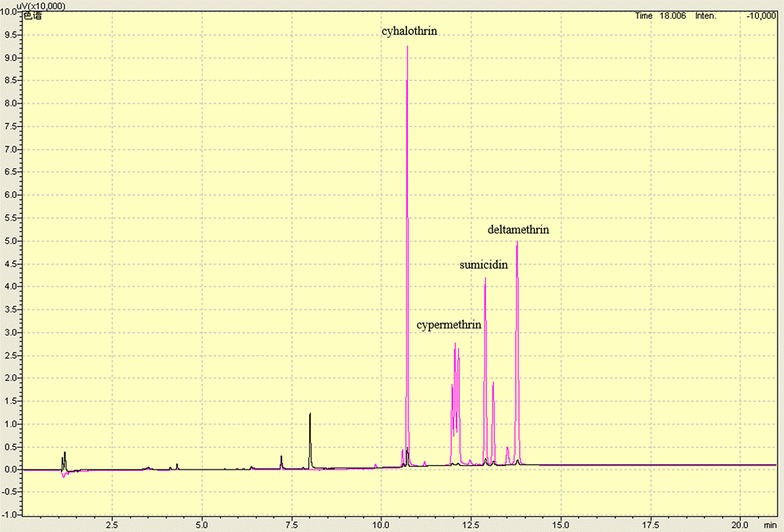
Gas chromatography analysis of different pyrethroids hydrolyzed by M2. The red and black lines denote hydrolysis chromatography by inactive enzyme (control) and active enzyme, respectively. The enzyme solution were boiled for 20 min, and then centrifugated at 13,000g for 10 min. The supernatants were taken as inactive enzymes
Conclusions
Pyrethroids are potentially harmful to human health and ecosystems. Thus, great concerns have been raised about pyrethroid residues and their persistence in the environment. In this study, we enhanced the activity and thermostability of one novel pyrethroid-hydrolyzing esterase by random and site-directed mutagenesis, and obtained three positive mutants. Remarkably, combination of two mutations A171V and D256N resulted in ~1.5-fold improvement in the kcat/Km value and 2.46-fold higher catalytic activity compared to the WT Sys410. In addition, the desired gene M2 was extracellularly expressed with high yield and good solubility in a P. pastoris host system for the first time. More interestingly, the enzyme could efficiently degrade cyhalothrin, cypermethrin, sumicidin, and deltamethrin under assay conditions of 37 °C for 15 min, exceeding a hydrolysis rate of 98%, higher than other pyrethroid-hydrolyzing enzymes reported so far. These favorable features make M2 an ideal candidate for the biodegradation of pyrethroids. More details regarding the mechanisms involved in the enhanced properties of M2 will be elucidated by resolving the 3D structure and rational protein design; these studies are currently in progress.
Methods
Chemicals and materials
Technical-grade pyrethroids were provided by Jiangsu Yangnong Chemical Group Co., Ltd., Jiangsu, China. All ρ-nitrophenyl esters were purchased from Sigma. Restriction endonuclease, TaKaRa MutanBEST Kit, T4 DNA ligase and PrimeSTAR® HS DNA Polymerase were purchased from TaKaRa (Dalian, China) and used according to the recommendations of the manufacturer. E.Z.N.A. Plasmid Mini Kit, E.Z.N.A. Gel Extraction Kit and E.Z.N.A. Yeast DNA Kit were purchased from OMEGA (Norcross, USA). GeneMorph® II Random Mutagenesis Kit was purchased from Stratagene (La Jolla, CA, USA). Zeocin™ and EasySelect™ Pichia Expression Kit were purchased from Invitrogen (Carlsbad, CA, USA). All other chemicals and reagents were of analytical grade and were purchased from commercial sources, unless otherwise stated.
Bacterial strains and plasmids
Escherichia coli DH5α (Novagen, Madison, WI, USA), E. coli BL21 (DE3) (Novagen, Madison, WI, USA) and P. pastoris strain X-33 (Invitrogen, Carlsbad, CA, USA) were used as the hosts for gene cloning and protein expression, respectively. pTV118N (TaKaRa, Dalian, China) and pPICZα B (Invitrogen, Carlsbad, CA, USA) were used to construct mutant libraries and express the target protein, respectively.
Directed evolution and screening for mutants with enhanced activity and thermostability
Random mutagenesis of the Sys410 gene (GenBank accession number: JQ272178) was carried out by error-prone PCR using a GeneMorph® II Random Mutagenesis Kit at low frequency (0–4.5 mutations/kb) according to the manufacturer’s protocol, performed with pET28a-Sys410 as a template. The following primers were used: fw (5′-TTATTGGATCCATGTTCGCTCAGCCCCCGAA-3′; the BamHI cutting site is underlined) and rv (5′-CCGGAATTCTCACTCCGCCAAGAACCGATCCACG-3′; the EcoRI cutting site is underlined). The PCR product digested with BamHI/EcoRI was ligated into BamHI/EcoRI-digested cloning vector pTV118N, and then transformed into E. coli DH5α cells. The transformants were plated onto LB agar containing 100 μg/mL ampicillin and further incubated at 37 °C overnight.
The mutant library was screened by a two-step screening strategy. Initially, the transformants were replicated on LB agar plates supplemented with 0.1% (v/v) tributyrin, 100 μg/mL ampicillin and 0.1 mM IPTG (isopropyl-β-d-thiogalactoside) [42]. A transparent zone due to tributyrin hydrolysis appeared at 37 °C in 1–2 days. Positive clones would develop bigger transparent zones around the colony than WT clones. Then the positive clones obtained by the initial screening were transferred to LB agar plates containing 100 μg/mL ampicillin and 0.1 mM IPTG, and further incubated at 37 °C for 12 h. After heat treatment in critical inactivation conditions, the clones were overlayed with 0.3 mg/mL α-naphthyl acetate and 1.3 mg/mL Fast Blue B [35]. The positive clones rapidly turned deep brown around the colony. The positive clones from the first round of random mutagenesis were used as the template for the second round of mutagenesis. The critical inactivation conditions were 60 °C for 55 min in the first round, and 80 °C for 60 min in the second round. The detailed methods were performed as described above.
Site-directed mutagenesis
We created mutant M3 with a single mutation by site-directed mutagenesis for determining the effect of each mutation. In vitro site-directed mutagenesis was performed by using a TaKaRa MutanBEST Kit (TaKaRa, Dalian, China) following the instructions of the manufacturer. The plasmid pET28a-Sys410 was used as the template. The following primers were used: fw (5′-TCTGGAGCGTGTCAACGACAGTCGC-3′; the mutation site is underlined) and rv (5′-TGGACGCCCGGCAAGGTGGCGTATT-3′). The correctness of the mutant was confirmed by DNA sequencing.
Cloning, expression, and purification of the mutants
The mutant genes were amplified by PCR with pTV118N-gene as a template using the primers mentioned above which contained restriction enzyme sites BamHI and EcoRI. Amplified DNA was digested by BamHI/EcoRI, ligated into pET-28a (+) which was linearized by BamHI/EcoRI, then transformed into E. coli BL21 (DE3) cells. E. coli cell transformants were plated onto LB agar containing 50 μg/mL kanamycin. Transformed cells were grown in a 250 mL flask containing 50 mL of LB (50 μg/mL kanamycin) at 37 °C until the cell concentration reached an OD600 of 1.0, then induced with 0.6 mM IPTG. After incubation at 37 °C for 8 h with shaking at 220 rpm, cells were harvested by centrifugation (6000g, 10 min) at 4 °C and suspended in binding buffer (0.5 M NaCl, 5 mM imidazole, 20 mM Tris–HCl, pH 7.9). The cells were disrupted by sonication, and the supernatant was collected by centrifugation (13,000g, 10 min) at 4 °C. The sample was loaded onto an Ni–NTA His·Bind column pre-equilibrated with binding buffer. Then the column was washed with binding buffer and washing buffer (0.5 M NaCl, 60 mM imidazole, 20 mM Tris–HCl, pH 7.9). Finally, the bound protein was eluted with eluting buffer (1 M imidazole, 0.5 M NaCl, 20 mM Tris–HCl, pH 7.9). The fractions containing the recombinant protein were collected and stored at −20 °C.
Determination of substrate specificity and kinetic parameters
ρ-Nitrophenyl esters are general substrates of esterases. Substrate specificity against various ρ-nitrophenyl esters with acyl chain lengths of C2, C4, C6, C8, C10, and C12 was determined under assay conditions of pH 6.5 and 55 °C, according to the method of Fan et al. [21]. The activity was tested under conditions of pH 6.5 and 55 °C (the best conditions of Sys410), pH 6.5 and 65 °C (the best conditions of M1 and M2), and pH 6.5 and 70 °C (the best conditions of M3), respectively. One unit of enzyme activity was defined as the amount of enzyme that produced 1 μmol of ρ-nitrophenol per minute under these conditions. The purified enzyme was incubated with various concentrations of ρ-nitrophenyl acetate. The final concentration ranged from 1.0 to 10.0 mM in potassium phosphate buffer (pH to 6.5). The kinetic constants were calculated by fitting the initial rate data into the Michaelis–Menten equation using GraFit software version 6 (Erithacus Software Ltd., Horley, UK).
Effect of temperature and pH on enzyme activity
The effect of temperature and pH on the initial reaction rates of Sys410 and the mutants was determined by using ρ-nitrophenyl acetate as a substrate. The optimum pH was measured using ρ-nitrophenyl acetate as a substrate at 55 °C. The pH buffers included citric acid-NaOH buffer (pH 3.5–5.5), potassium phosphate buffer (pH 5.0–7.0), and Tris–HCl buffer (pH 6.5–9.0). pH stability was tested after incubation of the purified enzyme for 24 h at 30 °C in the above different buffers. The optimum temperature was determined analogously by measuring esterase activity in the temperature range of 35–80 °C in a phosphate buffer (50 mM, pH 6.5). Thermostability was measured by preincubation of the purified enzyme in 50 mM potassium phosphate buffer (pH 6.5) at 45–65 °C for 12 h. The residual activity was tested under conditions of pH 6.5 and 55 °C (the best conditions of Sys410), pH 6.5 and 65 °C (the best conditions of M1 and M2), and pH 6.5 and 70 °C (the best conditions of M3), respectively. In addition, the T1/2 of Sys410 and M2 at 40–80 °C were measured. T1/2 is defined as the incubation time inactivating 50% of the initial enzyme activity. Samples containing 0.1 mg/mL purified enzyme (50 mM phosphate buffer, pH 6.5) were treated by incubating for various time intervals at different temperatures. Then the residual activities were quantified by using ρ-nitrophenyl acetate as a substrate based on the method of Fan et al. [21].
Gas chromatography analysis of the degradation of different pyrethroids
Sys410 and the mutants were tested for hydrolysis of cyhalothrin, cypermethrin, sumicidin, and deltamethrin. Hydrolytic activity of the enzyme was determined based on the method described previously [21], with slight modification. The enzyme samples (2.4 μg, a tenth of the amount in the previous study) with 5 mg/mL substrate in 50 mM potassium phosphate buffer (pH 6.5) were incubated at 37 °C at different time intervals. Then the residual pyrethroids were quantified by gas chromatography. Aliquots (1 μL) of the reaction mixtures were loaded onto a Gas chromatography system (GC-2010, Shimadzu Corporation, Japan) with ECD Detector using RestekRTX-5 column (30 m × 0.25 mm × 0.25 μm). The column flow was 2 mL/min. The column was set at 150 °C for 1 min, and heated to 270 °C at 30 °C/min−1. The temperature of ECD Detector was 300 °C. The electric current was 1 nA. In each measurement, the effect of nonenzymatic hydrolysis of substrates was taken into consideration and subtracted from the value measured when the enzyme was added.
Extracellular expression of the mutant in P. pastoris X-33
The mutant gene M2 was amplified by PCR with pTV118N-Sys410-M5 as a template using primers which contained restriction enzyme sites EcoRI and KpnI. The following primers were used: fw (5′-CCGGAATTCATGTTCGCTCAGCCCCCGAA-3′; the EcoRI cutting site is underlined) and rv (5′-ATATAGGTACCTCACTCCGCCAAGAACCGATCCACG-3′; the KpnI cutting site is underlined). The PCR product was cloned into EcoRI- and KpnI-digested pPICZα B. After being transformed into E. coli DH5a, some recombinant clones were selected on low salt LB agar plates containing 25 μg/mL Zeocin™. The proper insert orientation was checked by restriction analysis and sequencing. The recombinant plasmid was linearized with SacI and electroporated into P. pastoris X-33. The construction of recombinant transformants was based on the method described previously [43].
Transformants were induced on BMMY plates and screened on the basis of esterase activity. Positive clones displayed a blue color by hydrolysis with 100 μM 5-bromo-4-chloro-3-indolyl caprylate, and were further tested to confirm gene integration [44]. Some transformants were selected and cultivated in liquid media in Erlenmeyer flasks, to check their production levels [45]. The best transformant was subjected to high cell-density fermentation, according to the EasySelect™ Pichia Expression Kit manual. Methanol was added into the culture to a final concentration of 0.5% for maintaining induction every day. At different times (day1–day7), 1 mL of the culture was centrifuged and the amount of target protein in the supernatant was estimated by activity measurement assays and SDS-PAGE. The recombinant enzyme was also tested for hydrolysis of cyhalothrin, cypermethrin, sumicidin, and deltamethrin, based on the method described above.
Additional files
Additional file 1. Screening of mutants with better activity (A) and better thermostability (B) in the second round of random mutagenesis. The transformants were replicated on LB agar plates supplemented with 0.1% (v/v) tributyrin. A transparent zone due to tributyrin hydrolysis appeared at 37 °C in 1–2 days. Clones with enhanced activity displayed bigger hydrolysis zones on tributyrin plates. After being treated at 80 °C for 60 min, the clones were overlayed with 0.3 mg/mL α-naphthyl acetate and 1.3 mg/mL Fast Blue B. Clones with enhanced thermostability (M2) rapidly turned deep brown around the colony.
Additional file 2. SDS-PAGE of gene expression in E. coli BL21 (DE3). M, protein MW marker (low); lanes 1–4, supernatant of E. coli BL21 (DE3) (pET-28a (+)-WT), E. coli BL21 (DE3) (pET-28a (+)-M1), E. coli BL21 (DE3) (pET-28a (+)-M2), E. coli BL21 (DE3) (pET-28a (+)-M3); lanes 5–8, purified WT, M1, M2, M3.
Additional file 3. SDS-PAGE of gene expression in P. pastoris X-33 (pPICZα B-M2). M, protein MW marker (low); lanes 1–7, supernatant of P. pastoris X-33 (pPICZα B-M5) induced for 1–7 days; lane 8, supernatant of P. pastoris X-33(pPICZα B) at day 7; lane 9, cell lysis solution of E. coli BL21 (DE3) (pET-28a(+)-M2) (arrow). After after being treated at 60 °C for 55 min, the clones were overlayed with 0.3 mg/mL α-naphthyl acetate and 1.3 mg/mL Fast Blue B. Clones with enhanced thermostability rapidly turned deep brown around the colony.
Authors’ contributions
XLL: have performed construction of mutagenesis library, gene cloning and expression. MJL: have performed enzyme characterization. YHL: have conceived the study. FXJ: have designed and supervised the experiments and written the manuscript. All authors read and approved the final manuscript.
Acknowledgements
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Availability of data and materials
All data generated or analysed during this study are included in this published article.
Funding
This work was supported by the National Natural Science Foundation of China (31400680), Grants for Scientific Research of BSKY (XJ201322) from Anhui Medical University, Science and Technology Plan Project of Guangdong Province (2012B010300021, 2013B010404044) and Education Project of Guangdong Province (2013KJCX0107).
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Footnotes
Electronic supplementary material
The online version of this article (doi:10.1186/s12934-017-0698-5) contains supplementary material, which is available to authorized users.
Contributor Information
Xiaolong Liu, Email: liuxiaolong85@126.com.
Mingjun Liang, Email: m18063053941@126.com.
Yuhuan Liu, Email: lsslyh@mail.sysu.edu.cn.
Xinjiong Fan, Email: fanxinjiong@126.com.
References
- 1.Lin QS, Chen SH, Hu MY, Haq MU, Yang L, Li H. Biodegradation of cypermethrin by a newly isolated actinomycetes HU-S-01 from wastewater sludge. Int J Environ Sci Technol. 2011;8:45–56. doi: 10.1007/BF03326194. [DOI] [Google Scholar]
- 2.Zhang C, Wang SH, Yan YC. Isomerization and biodegradation of beta-cypermethrin by Pseudomonas aeruginosa CH7 with biosurfactant production. Bioresour Technol. 2011;102:7139–7146. doi: 10.1016/j.biortech.2011.03.086. [DOI] [PubMed] [Google Scholar]
- 3.Zhang C, Jia L, Wang SH, Qu J, Li K, Xu LL, Shi YH, Yan YC. Biodegradation of beta-cypermethrin by two Serratia spp. with different cell surface hydrophobicity. Bioresour Technol. 2010;101:3423–3429. doi: 10.1016/j.biortech.2009.12.083. [DOI] [PubMed] [Google Scholar]
- 4.Maund SJ, Campbell PJ, Giddings JM, Hamer MJ, Henry K, Pilling ED, Warinton JS, Wheeler JR. Ecotoxicology of synthetic pyrethroids. Top Curr Chem. 2012;314:137–166. doi: 10.1007/128_2011_260. [DOI] [PubMed] [Google Scholar]
- 5.Housset P, Dickmann R. A promise fulfilled-pyrethroid development and the benefits for agriculture and human health. Bayer Crop Sci. 2009;62:135–145. [Google Scholar]
- 6.Li QY, Li Y, Zhu XK, Cai BL. Isolation and characterization of atrazine-degrading Arthrobacter sp. AD26 and use of this strain in bioremediation of contaminated soil. J Environ Sci. 2008;20:1226–1230. doi: 10.1016/S1001-0742(08)62213-5. [DOI] [PubMed] [Google Scholar]
- 7.Saxena P, Saxena AK. Cypermethrin induced biochemical alterations in the blood of albino rats. Jordan J Biol Sci. 2010;3:111–114. [Google Scholar]
- 8.Alexander M. Biodegradation of chemicals of environmental concern. Science. 1981;211:132–138. doi: 10.1126/science.7444456. [DOI] [PubMed] [Google Scholar]
- 9.Kasai S. Role of cytochrome P450 in mechanism of pyrethroid resistance. J Pestic Sci. 2004;29:220–221. doi: 10.1584/jpestics.29.220. [DOI] [Google Scholar]
- 10.Sogorb MA, Vilanova E. Enzymes involved in the detoxification of organophosphorus, carbamate and pyrethroid insecticides through hydrolysis. Toxicol Lett. 2002;128:215–228. doi: 10.1016/S0378-4274(01)00543-4. [DOI] [PubMed] [Google Scholar]
- 11.Tallur PN, Megadi VB, Ninnekar HZ. Biodegradation of cypermethrin by Micrococcus sp. strain CPN 1. Biodegradation. 2008;19:77–82. doi: 10.1007/s10532-007-9116-8. [DOI] [PubMed] [Google Scholar]
- 12.Maloeny SE, Maule A, Smith AR. Purification and preliminary characterization of permethrinase from a pyrethroid-transforming strain of Bacillus cereus. Appl Environ Microbiol. 1993;59:2007–2013. doi: 10.1128/aem.59.7.2007-2013.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liang WQ, Wang ZY, Li H, Wu PC, Hu JM, Liu YH. Purification and characterization of a novel pyrethroid hydrolase from Aspergillus niger ZD11. J Agric Food Chem. 2005;53:7415–7420. doi: 10.1021/jf051460k. [DOI] [PubMed] [Google Scholar]
- 14.Guo P, Wang BZ, Hang BJ, Li L, Ali SW, He J, Li SP. Pyrethroid-degrading Sphingobium sp. JZ-2 and the purification and characterization of a novel pyrethroid hydrolase. Int Biodeterior Biodegrad. 2009;63:1107–1112. doi: 10.1016/j.ibiod.2009.09.008. [DOI] [Google Scholar]
- 15.Cycoń M, Żmijowska A, Piotrowska-Seget Z. Enhancement of deltamethrin degradation by soil bioaugmentation with two different strains of Serratia marcescens. Int J Environ Sci Technol. 2014;11:1305–1316. doi: 10.1007/s13762-013-0322-0. [DOI] [Google Scholar]
- 16.Ruan ZY, Zhai Y, Song JL, Shi YH, Li K, Zhao B, Yan YC. Molecular cloning and characterization of a newly isolated pyrethroid-degrading esterase gene from a genomic library of Ochrobactrum anthropi YZ-1. PLoS ONE. 2013;8:e77329. doi: 10.1371/journal.pone.0077329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wu PC, Liu YH, Wang ZY, Zhang XY, Li H, Liang WQ, Luo N, Hu JM, Lu JQ, Luan TG, Cao LX. Molecular cloning, purification, and biochemical characterization of a novel pyrethroid-hydrolyzing esterase from Klebsiella sp. strain ZD112. J Agric Food Chem. 2006;54:836–842. doi: 10.1021/jf052691u. [DOI] [PubMed] [Google Scholar]
- 18.Wang BZ, Guo P, Hang BJ, Li L, He J, Li SP. Cloning of a novel pyrethroid-hydrolyzing carboxylesterase gene from Sphingobium sp. JZ-1 and characterization of the gene product. Appl Environ Microbiol. 2009;75:5496–5500. doi: 10.1128/AEM.01298-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li G, Wang K, Liu YH. Molecular cloning and characterization of a novel pyrethroid-hydrolyzing esterase originating from the metagenome. Microb Cell Fact. 2008;7:38. doi: 10.1186/1475-2859-7-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhai Y, Li K, Song JL, Shi YH, Yan YC. Molecular cloning, purification and biochemical characterization of a novel pyrethroid-hydrolyzing carboxylesterase gene from Ochrobactrum anthropi YZ-1. J Hazard Mater. 2012;221:206–212. doi: 10.1016/j.jhazmat.2012.04.031. [DOI] [PubMed] [Google Scholar]
- 21.Fan XJ, Liu XL, Huang R, Liu YH. Identification and characterization of a novel thermostable pyrethroid-hydrolyzing enzyme isolated through metagenomic approach. Microb Cell Fact. 2012;11:33–43. doi: 10.1186/1475-2859-11-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wei T, Feng SX, Shen YL, He PX, Ma GL, Yu X, Zhang F, Mao DB. Characterization of a novel thermophilic pyrethroid-hydrolyzing carboxylesterase from Sulfolobus tokodaii into a new family. J Mol Catal B Enzym. 2013;97:225–232. doi: 10.1016/j.molcatb.2013.07.022. [DOI] [Google Scholar]
- 23.Chen SH, Lin QS, Xiao Y, Deng YY, Chang CQ, Zhong GH, Hu MY, Zhang LH. Monooxygenase, a novel beta-cypermethrin degrading enzyme from Streptomyces sp. PLoS ONE. 2013;8:e75450. doi: 10.1371/journal.pone.0075450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jafari R, Sundström BE, Holm P. Optimization of production of the anti-keratin 8 single-chain Fv TS1-218 in Pichia pastoris using design of experiments. Microb Cell Fact. 2011;10:34. doi: 10.1186/1475-2859-10-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Karlskas IL, Maudal K, Axelsson L, Rud I, Eijsink VG, Mathiesen G. Heterologous protein secretion in lactobacilli with modified pSIP vectors. PLoS ONE. 2014;9:e91125. doi: 10.1371/journal.pone.0091125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sak-Ubol S, Namvijitr P, Pechsrichuang P, Haltrich D, Nguyen TH, Mathiesen G, Eijsink VGH, Yamabhai M. Secretory production of a beta-mannanase and a chitosanase using a Lactobacillus plantarum expression system. Microb Cell Fact. 2016;15:81. doi: 10.1186/s12934-016-0481-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hasan FA, Hameed A. Industrial applications of microbial lipases. Enzyme Microb Technol. 2006;39:235–251. doi: 10.1016/j.enzmictec.2005.10.016. [DOI] [Google Scholar]
- 28.Peng XQ. Improved thermostability of lipase B from Candida antarctica by directed evolution and display on yeast surface. Appl Biochem Biotechnol. 2013;169:351–358. doi: 10.1007/s12010-012-9954-7. [DOI] [PubMed] [Google Scholar]
- 29.Dong YP, Yan J, Du HQ, Chen M, Ma T, Feng L. Engineering of LadA for enhanced hexadecane oxidation using random- and site-directed mutagenesis. Appl Microbiol Biotechnol. 2012;94:1019–1029. doi: 10.1007/s00253-012-4035-y. [DOI] [PubMed] [Google Scholar]
- 30.Song LT, Laguerre S, Dumon C, Bozonnet S, O’Donohue MJ. A high-throughput screening system for the evaluation of biomass-hydrolyzing glycoside hydrolases. Bioresour Technol. 2010;101:8237–8243. doi: 10.1016/j.biortech.2010.05.097. [DOI] [PubMed] [Google Scholar]
- 31.Jang MK, Lee SW, Lee DG, Kim NY, Yu KH, Jang HJ, Kim S, Kim A, Lee SH. Enhancement of the thermostability of a recombinant β-agarase, AgaB, from Zobellia galactanivorans by random mutagenesis. Biotechnol Lett. 2010;32:943–949. doi: 10.1007/s10529-010-0237-5. [DOI] [PubMed] [Google Scholar]
- 32.Yu XW, Wang R, Zhang M, Xu Y, Xiao R. Enhanced thermostability of a Rhizopus chinensis lipase by in vivo recombination in Pichia pastoris. Microb Cell Fact. 2012;11:102. doi: 10.1186/1475-2859-11-102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hirokawa K, Ichiyanagi A, Kajiyama N. Enhancement of thermostability of fungal deglycating enzymes by directed evolution. Appl Microbiol Biotechnol. 2008;78:775–781. doi: 10.1007/s00253-008-1363-z. [DOI] [PubMed] [Google Scholar]
- 34.Lin XJ, Xu WT, Huang KL, Mei XH, Liang ZH, Li ZM, Guo JX, Luo YB. Cloning, expression and characterization of recombinant elastase from Pseudomonas aeruginosa in Pichia pastoris. Protein Expr Purif. 2009;63:69–74. doi: 10.1016/j.pep.2007.12.011. [DOI] [PubMed] [Google Scholar]
- 35.Kim JH, Choi GS, Kim SB, Kim WH, Lee JY, Ryu YW, Kim GJ. Enhanced thermostability and tolerance of high substrate concentration of an esterase by directed evolution. J Mol Catal B Enzym. 2004;27:169–175. doi: 10.1016/j.molcatb.2003.11.010. [DOI] [Google Scholar]
- 36.Chen VB, Arendall WB, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, Richardson DC. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Cryst. 2010;D66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Davis IW, Leaver-Fay A, Chen VB, Block JN, Kapral GJ, Wang X, Murray LW, Arendall WB, Snoeyink J, Richardson JS, Richardson DC. MolProbity: all-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res. 2007;35:W375–W383. doi: 10.1093/nar/gkm216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sreekrishna K, Brankamp RG, Kropp KE, Blankenship DT, Tsay JT, Smith PL, Wierschke JD, Subramaniam A, Birkenberger LA, Brankamp RG. Strategies for optimal synthesis and secretion of heterologous proteins in the methylotrophic yeast Pichia pastoris. Gene. 1997;190:55–62. doi: 10.1016/S0378-1119(96)00672-5. [DOI] [PubMed] [Google Scholar]
- 39.Romanos MA, Scorer CA, Clare JJ. Foreign gene expression in yeast: a review. Yeast. 1992;8:423–488. doi: 10.1002/yea.320080602. [DOI] [PubMed] [Google Scholar]
- 40.Cereghino JL, Cregg JM. Heterologous protein expression in the methylotrophic yeast Pichia pastoris. FEMS Microbiol Rev. 2000;24:45–66. doi: 10.1111/j.1574-6976.2000.tb00532.x. [DOI] [PubMed] [Google Scholar]
- 41.Guerrero FD, Nene VM. Gene structure and expression of a pyrethroid-metabolizing esterase, CzEst9, from a pyrethroid resistant mexican population of Rhipicephalus (Boophilus) microplus (Acari: Ixodidae) J Med Entomol. 2008;45(4):677–685. doi: 10.1603/0022-2585(2008)45[677:gsaeoa]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 42.Kok RG, Christoffels VM, Vosman B, Hellingwerf KJ. Growth-phase-dependent expression of the lipolytic system of Acinetobacter calcoaceticus BD413: cloning of a gene encoding one of the esterases. J Gen Microbiol. 1993;139:2329–2342. doi: 10.1099/00221287-139-10-2329. [DOI] [PubMed] [Google Scholar]
- 43.Yang CH, Huang YC, Chen CY, Wen CY. Expression of Thermobifida fusca thermostable raw starch digesting alpha-amylase in Pichia pastoris and its application in raw sago starch hydrolysis. J Ind Microbiol Biotechnol. 2010;37:401–406. doi: 10.1007/s10295-009-0686-9. [DOI] [PubMed] [Google Scholar]
- 44.Reddy ST, Kumar SN, Haas AL, Dahms NM. Biochemical and functional properties of the full-length cation-dependent mannose 6-phosphate receptor expressed in Pichia pastoris. Biochem Biophys Res Commun. 2003;309:643–651. doi: 10.1016/j.bbrc.2003.08.048. [DOI] [PubMed] [Google Scholar]
- 45.Cedillo VB, Plou FJ, Martínez MJ. Recombinant sterol esterase from Ophiostoma piceae: an improved biocatalyst expressed in Pichia pastoris. Microb Cell Fact. 2012;11:73. doi: 10.1186/1475-2859-11-73. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1. Screening of mutants with better activity (A) and better thermostability (B) in the second round of random mutagenesis. The transformants were replicated on LB agar plates supplemented with 0.1% (v/v) tributyrin. A transparent zone due to tributyrin hydrolysis appeared at 37 °C in 1–2 days. Clones with enhanced activity displayed bigger hydrolysis zones on tributyrin plates. After being treated at 80 °C for 60 min, the clones were overlayed with 0.3 mg/mL α-naphthyl acetate and 1.3 mg/mL Fast Blue B. Clones with enhanced thermostability (M2) rapidly turned deep brown around the colony.
Additional file 2. SDS-PAGE of gene expression in E. coli BL21 (DE3). M, protein MW marker (low); lanes 1–4, supernatant of E. coli BL21 (DE3) (pET-28a (+)-WT), E. coli BL21 (DE3) (pET-28a (+)-M1), E. coli BL21 (DE3) (pET-28a (+)-M2), E. coli BL21 (DE3) (pET-28a (+)-M3); lanes 5–8, purified WT, M1, M2, M3.
Additional file 3. SDS-PAGE of gene expression in P. pastoris X-33 (pPICZα B-M2). M, protein MW marker (low); lanes 1–7, supernatant of P. pastoris X-33 (pPICZα B-M5) induced for 1–7 days; lane 8, supernatant of P. pastoris X-33(pPICZα B) at day 7; lane 9, cell lysis solution of E. coli BL21 (DE3) (pET-28a(+)-M2) (arrow). After after being treated at 60 °C for 55 min, the clones were overlayed with 0.3 mg/mL α-naphthyl acetate and 1.3 mg/mL Fast Blue B. Clones with enhanced thermostability rapidly turned deep brown around the colony.
Data Availability Statement
All data generated or analysed during this study are included in this published article.