

Abstract
Alternate BRAF splicing is the most common mechanism of acquired resistance to BRAF inhibitor treatment in melanoma. Recently, alternate BRAF exon 4–8 splicing was shown to involve an intronic mutation, located 51 nucleotides upstream of BRAF exon 9 within a predicted splicing branch point. This intronic mutation was identified in a single cell line but has not been examined in vivo. Herein we demonstrate that in three melanomas biopsied from patients with acquired resistance to BRAF inhibitors, alternate BRAF exon 4–8 splicing is not associated with this intronic branch point mutation. We also confirm that melanoma cells expressing BRAF splicing variants retain exquisite sensitivity to existing FDA-approved MEK inhibitors.
Electronic supplementary material
The online version of this article (doi:10.1186/s40364-017-0098-3) contains supplementary material, which is available to authorized users.
Keywords: Melanoma, BRAF inhibitor, BRAF splicing, Dabrafenib, Resistance
Main text
The serine/threonine kinase BRAF is mutated and constitutively activated in 40–60% of cutaneous melanomas. Selective inhibitors of mutant BRAF, including dabrafenib and vemurafenib, improve the progression-free and overall survival of BRAF-mutant melanoma patients [1, 2]. Despite this activity, acquired resistance develops in most melanoma patients and multiple mechanisms of resistance have been described. Most of these resistance effectors, including alterations in MEK1, BRAF and N-RAS, promote the reactivation of the mitogen activated protein kinase pathway [3]. Alternate splicing of oncogenic BRAF is the most common driver of acquired resistance to BRAF inhibitors and is evident in approximately 30% of resistant melanomas [4–6]. Splice variants of BRAF encode an active kinase, but lack an intact RAS binding domain. These truncated proteins are prone to dimerization and have reduced affinity for the class I BRAF inhibitors, vemurafenib and dabrafenib [4]. Predictably, melanomas expressing BRAF splice variants display MAPK re-activation in the presence of BRAF inhibitors, and retain sensitivity to the inhibition of the downstream kinases MEK and ERK [5, 7].
One mechanism of altered BRAF splicing was recently shown to involve a C-to-G mutation in intron 8 within a predicted splicing branch point, 51 nucleotides upstream of BRAF exon 9 (i.e C > G − 51). This intron 8, variant was sufficient to promote the expression of a BRAF transcript lacking exons 4–8 (BRAF exon 4–8∆; also referred to as BRAF3–9) in a single vemurafenib resistant melanoma cell line [8]. The significance of this intronic mutation was not clinically validated, although we are aware of five patients with acquired BRAF inhibitor resistance driven by the BRAF exon 4–8∆ alternate transcript [4–6].
We selected three of these five BRAF inhibitor resistant patients (Patients 7, 10 and 28 [5]). These BRAFV600-mutant patients progressed within 1 year of receiving BRAF inhibitor monotherapy and analysis of the matched pre-treatment and progressing tumours revealed that only the progressing biopsies expressed the BRAF exon 4–8∆ splice variant (Additional file 1: Figure S1). We analysed the intron 8 splicing branch site in these matched pre-treatment and progressing melanomas and in all melanomas the -51– -45 branch point sequence and the exon 9 acceptor splice site were wild type (Fig. 1).
Fig. 1.
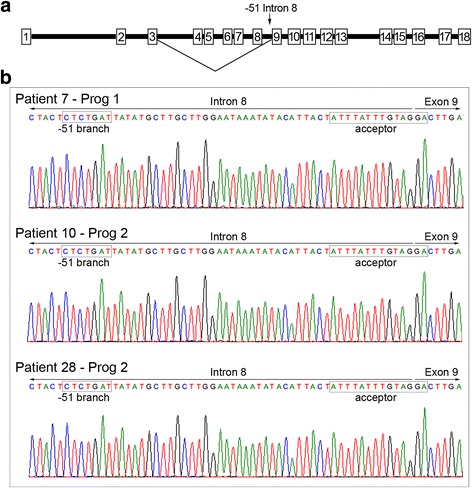
BRAF intronic splice and branch point sequences in melanomas expressing the BRAF exon 4–8∆ transcript. a Schematic diagram of the BRAF gene, showing exons (numbered boxes) and introns (black bars) and the alternate splicing event leading to the BRAF exon 4–8∆ transcript. b Sanger sequencing traces showing the sequence encompassing the junction between BRAF intron 8 and exon 9 in BRAF inhibitor resistant melanomas (Progs) expressing the BRAF exon 4–8∆ transcripts and derived from patient 7, 10 and 28. The branch point motifs, including the −51 nucleotide associated with BRAF exon 4–8∆ splicing, and splice sites are boxed
We analysed the impact of the C > G − 51 intron 8 mutation using several algorithms, and it was not predicted to break the branch point nor alter splice factor binding (Table 1). The −51 G mutation potentially activates an intronic cryptic donor site (Table 1), but usage of this splice site would not generate the BRAF exon 4–8∆ transcript. Moreover, an alignment of 66 intronic branch point disease mutations revealed that mutations altering splicing occur predominantly at the highly conserved adenine residue (equivalent to −48 in the intron 8 branch site), with no reported disease causing mutation at the position equivalent to −51 [9]. It is also worth noting that unlike the branch point adenine, which is highly conserved in mammalian branch point sequences, the −5 branch point nucleotide (equivalent to the −51 BRAF intron 8 branch site) is degenerate in mammals [10], and the BRAF intron 8 sequence shows a cytosine to thymine bias in many non-human primates (data not shown).
Table 1.
Predicted effects of −51 mutation in intron 8
| Branch Site | Donor Splice | SRSF6 | |
|---|---|---|---|
| Position/Site/Score | Position/Site/Score | Sequence/Site/Score | |
| Wild Type | -51/CTCTGAT/88 | -54/ACTctcgat/40 | -134/TGTGTA/84 |
| Mutant | -51/GTCTGAT/86 | -138/ACTgtctga/67 | -134/TGTGTA/84 |
Data derived using the Human Splice Finder Tool [12]
Branch Site score above 67 is considered a potential break point, and score variation between wild type and mutant sequence of less than −10% is considered to break the branch point
Splice Site values above 65 are predicted splice sites, the −51 G mutation is underlined in Cryptic Donor Site
Splicing factor SRSF6 motif with highest predicted scores is shown [12]
Motif position is relative to first nucleotide of exon 9
Significantly, the expression of BRAF splice variants is insufficient to confer MEK inhibitor resistance in melanoma (Additional file 2: Figure S2). Consequently, BRAFV600-mutant metastatic melanoma patients treated with combination BRAF and MEK inhibitors, rarely develop resistance due to alternate BRAF splicing [11]. In this clinical setting, therefore, the therapeutic potential of splicing modulators, such as spliceostatin A, which inhibit the formation of the BRAF exon 4–8∆ via inhibition of the SF3B1 splicing factor [8], is diminished.
BRAF inhibitor resistance mediated by alternate BRAF splicing is likely a result of cis-acting alterations that disrupt the use of allele-specific constitutive splice sites. Trans-acting mutations can also affect basal and alternate splicing machinery, but these mutations would be predicted to alter the processing of both wild type and mutant BRAF pre-mRNA. Several studies have now confirmed that the wild type BRAF allele is transcribed and processed correctly in mutant BRAF melanoma, and the shorter, variant BRAF transcripts are derived only from the mutant BRAF allele [4, 5]. Our data show that alternate BRAF exon 4–8∆ splicing in melanomas derived from patients with resistance to BRAF inhibitor monotherapy is not associated with a mutation in the −51 position in intron 8. Regardless of the precise mechanisms altering the splicing of BRAF, downstream MEK and ERK inhibition effectively inhibit the proliferation of BRAF inhibitor resistant melanoma cells expressing BRAF splice variants [5, 7].
Methods
Patients
All patients included in this study had BRAFV600 mutant metastatic melanoma, and were treated with either dabrafenib or vemurafenib [5]. All patients had a pre-treatment melanoma tissue sample obtained before commencing BRAF inhibitor and a matched progressing melanoma metastasis (Prog). Informed consent was obtained for each patient under approved Human Research Ethics Committee protocols.
BRAF sequence analysis
Sequences were amplified using Taq polymerase (Fisher Biotech) using primers shown in Additional file 3: Table S1. PCR products were purified using QIAquick PCR purification kit (Qiagen, Limburg, Netherlands) followed by Sanger sequencing on the 3730xl DNA Analyser (AGRF, Westmead, NSW, Australia). The Human Splicing Finder system [12] was used to identify and analyse BRAF splice sites and branch point sites and auxiliary splicing enhancer sequences.
Melanoma cell lines
The short-term patient-derived dabrafenib-resistant melanoma cells, WMD009 and SMU027, as well as SKMel28 parental and the dabrafenib-resistant BR4 cell line [7] were grown in in Dulbecco’s Modified Eagle Medium (DMEM) with 10% FBS and glutamine (Gibco-BRL).
Additional files
Sequence and expression of BRAF exon 4-8Δ splice variant in BRAF inhibitor resistant melanomas. A. PCR analysis of BRAF cDNA from pre-treatment (Pre) and matching melanomas progressing (Prog) on BRAF inhibitor monotheraphy. kb, kilobase; Lane 1 1 kb marker. B. Traces showing alternate BRAF splice junctions and the exon 15 Val600 codon (boxed) in BRAF inhibitor progressing melanomas expressing the BRAF exon 4-8Δ splice variant. (TIFF 676 kb)
BRAF inhibitor-resistant melanoma cells expressing BRAF splice variants retain sensitivity to the FDA-approved MEK inhibitor, trametinib. Viability curves of the parental SKMel28 and the dabrafenib-acquired resistant BR4 subline, and patient-derived dabrafenib-resistant WMD009 and SMU027 cells treated with the indicated doses of dabrafenib or trametinib for 72 h (relative to DMSO-treated controls; mean ± SD; n = 2). (TIFF 411 kb)
Amplification and sequencing primers. (PDF 91 kb)
Acknowledgements
Clinicians and Staff at Melanoma Institute Australia, Royal Prince Alfred Hospital Department of Pathology and Crown Princess Mary Cancer Centre, Westmead Hospital.
Funding
RAS, HR, GVL and NKH are supported by NHMRC Fellowship programs. This work is supported by Program Grant 1093017 and project grants of the National Health and Medical Research Council of Australia (NHMRC).
Availability of data and materials
Data sharing is not applicable to this article as no datasets were generated or analysed during the current study.
Authors’ contributions
GMP and HR designed the study. CF, SCB, BP, GMP, NKH and PJ performed the experiments. GVL, AMM, MSC, NKH, PJ, RAS, RFK and HR analysed the data. GVL, AMM, MSC, RAS and RFK recruited the patients and provided the reagents/tissues. All authors contributed to the final version of the paper. All authors read and approved the final manuscript.
Competing interests
A.M.M – Travel Support, GlaxoSmithKline; Honoraria GlaxoSmithKline; M.S.C - Honoraria GlaxoSmithKline and Novartis; J.F.T - Travel support, GlaxoSmithKline, Provectus; Consultant Advisor – Roche, GlaxoSmithKline, Provectus; Honoraria – GlaxoSmithKline, Provectus; R.F.K - Consultant Advisor Roche, GlaxoSmithKline and Novartis; R.A.S - Consultant Advisor Roche, GlaxoSmithKline and Novartis; G.V.L - Consultant Advisor Roche, GlaxoSmithKline and Novartis; Travel Support Roche; Honoraria Roche, GlaxoSmithKline.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Written consent was obtained from all patients under approved Human Research ethics committee protocols from Royal Prince Alfred Hospital (Protocol X15-0454 & HREC/11/RPAH/444).
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Footnotes
Electronic supplementary material
The online version of this article (doi:10.1186/s40364-017-0098-3) contains supplementary material, which is available to authorized users.
Contributor Information
Gulietta M. Pupo, Email: gulietta.pupo@sydney.edu.au
Suzanah C. Boyd, Email: suzanah.boyd@mq.edu.au
Carina Fung, Email: carina.fung@mq.edu.au.
Matteo S. Carlino, Email: matteo.carlino@sydney.edu.au
Alexander M. Menzies, Email: alexander.menzies@sydney.edu.au
Bernadette Pedersen, Email: bernadette.pedersen@mq.edu.au.
Peter Johansson, Email: Peter.Johansson@qimrberghofer.edu.au.
Nicholas K. Hayward, Email: Nicholas.Hayward@qimrberghofer.edu.au
Richard F. Kefford, Email: richard.kefford@mq.edu.au
Richard A. Scolyer, Email: richard.scolyer@sswahs.nsw.gov.au
Georgina V. Long, Email: georgina.long@sydney.edu.au
Helen Rizos, Phone: +61 2 9850 2762, Email: helen.rizos@mq.edu.au.
References
- 1.Chapman PB, Hauschild A, Robert C, Haanen JB, Ascierto P, Larkin J, Dummer R, Garbe C, Testori A, Maio M, et al. Improved survival with Vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364:2507–2516. doi: 10.1056/NEJMoa1103782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hauschild A, Grob JJ, Demidov LV, Jouary T, Gutzmer R, Millward M, Rutkowski P, Blank CU, Miller WH, Jr, Kaempgen E, et al. Dabrafenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet. 2012;380:358–365. doi: 10.1016/S0140-6736(12)60868-X. [DOI] [PubMed] [Google Scholar]
- 3.Johnson DB, Menzies AM, Zimmer L, Eroglu Z, Ye F, Zhao S, Rizos H, Sucker A, Scolyer RA, Gutzmer R, et al. Acquired BRAF inhibitor resistance: a multicenter meta-analysis of the spectrum and frequencies, clinical behaviour, and phenotypic associations of resistance mechanisms. Eur J Cancer. 2015;51:2792–2799. doi: 10.1016/j.ejca.2015.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Poulikakos PI, Persaud Y, Janakiraman M, Kong X, Ng C, Moriceau G, Shi H, Atefi M, Titz B, Gabay MT, et al. RAF inhibitor resistance is mediated by dimerization of aberrantly spliced BRAF (V600E) Nature. 2011;480:387–390. doi: 10.1038/nature10662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rizos H, Menzies AM, Pupo GM, Carlino MS, Fung C, Hyman J, Haydu LE, Mijatov B, Becker TM, Boyd SC, et al. BRAF inhibitor resistance mechanisms in metastatic melanoma: Spectrum and clinical impact. Clin Cancer Res. 2014;20:1965–1977. doi: 10.1158/1078-0432.CCR-13-3122. [DOI] [PubMed] [Google Scholar]
- 6.Shi H, Hugo W, Kong X, Hong A, Koya RC, Moriceau G, Chodon T, Guo R, Johnson DB, Dahlman KB, et al. Acquired resistance and clonal evolution in melanoma during BRAF inhibitor therapy. Cancer Discov. 2014;4:80–93. doi: 10.1158/2159-8290.CD-13-0642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Carlino MS, Todd JR, Gowrishankar K, Mijatov B, Pupo GM, Fung C, Snoyman S, Hersey P, Long GV, Kefford RF, Rizos H. Differential activity of MEK and ERK inhibitors in BRAF inhibitor resistant melanoma. Mol Oncol. 2014;8:544–554. doi: 10.1016/j.molonc.2014.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Salton M, Kasprzak WK, Voss T, Shapiro BA, Poulikakos PI, Misteli T. Inhibition of vemurafenib-resistant melanoma by interference with pre-mRNA splicing. Nat Commun. 2015;6:7103. doi: 10.1038/ncomms8103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Taggart AJ, DeSimone AM, Shih JS, Filloux ME, Fairbrother WG. Large-scale mapping of branchpoints in human pre-mRNA transcripts in vivo. Nat Struct Mol Biol. 2012;19:719–721. doi: 10.1038/nsmb.2327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gao K, Masuda A, Matsuura T, Ohno K. Human branch point consensus sequence is yUnAy. Nucleic Acids Res. 2008;36:2257–2267. doi: 10.1093/nar/gkn073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Long GV, Fung C, Menzies AM, Pupo GM, Carlino MS, Hyman J, Shahheydari H, Tembe V, Thompson JF, Saw RP, et al. Increased MAPK reactivation in early resistance to dabrafenib/trametinib combination therapy of BRAF-mutant metastatic melanoma. Nat Commun. 2014;5:5694. doi: 10.1038/ncomms6694. [DOI] [PubMed] [Google Scholar]
- 12.Desmet FO, Hamroun D, Lalande M, Collod-Beroud G, Claustres M, Beroud C. Human splicing Finder: an online bioinformatics tool to predict splicing signals. Nucleic Acids Res. 2009;37:e67. doi: 10.1093/nar/gkp215. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Sequence and expression of BRAF exon 4-8Δ splice variant in BRAF inhibitor resistant melanomas. A. PCR analysis of BRAF cDNA from pre-treatment (Pre) and matching melanomas progressing (Prog) on BRAF inhibitor monotheraphy. kb, kilobase; Lane 1 1 kb marker. B. Traces showing alternate BRAF splice junctions and the exon 15 Val600 codon (boxed) in BRAF inhibitor progressing melanomas expressing the BRAF exon 4-8Δ splice variant. (TIFF 676 kb)
BRAF inhibitor-resistant melanoma cells expressing BRAF splice variants retain sensitivity to the FDA-approved MEK inhibitor, trametinib. Viability curves of the parental SKMel28 and the dabrafenib-acquired resistant BR4 subline, and patient-derived dabrafenib-resistant WMD009 and SMU027 cells treated with the indicated doses of dabrafenib or trametinib for 72 h (relative to DMSO-treated controls; mean ± SD; n = 2). (TIFF 411 kb)
Amplification and sequencing primers. (PDF 91 kb)
Data Availability Statement
Data sharing is not applicable to this article as no datasets were generated or analysed during the current study.