

Abstract
The intermolecular hydroamination of unactivated alkenes with simple dialkyl amines remains an unsolved problem in organic synthesis. Here we report a catalytic protocol for efficient additions of cyclic and acyclic secondary alkyl amines to a wide range of alkyl olefins with complete anti-Markovnikov regioselectivity. In this process, C–N bond formation proceeds through a key aminium radical cation intermediate that is generated via electron transfer between an excited state iridium photocatalyst and an amine substrate. These reactions are redox neutral, completely atom economical, exhibit broad functional group tolerance, and occur readily at room temperature under visible light irradiation. Certain tertiary amine products are formally endergonic relative to their constituent olefin and amine starting materials and thus are not accessible via direct coupling with conventional ground state catalysts.
Alkyl amines are common structural features in natural products, pharmaceutical agents, and other small-molecule probes of biological function. As such, methods for the construction of complex alkyl amines are critical synthetic technologies. In this regard, the addition of amines to olefins is a particularly powerful approach to C(sp3)–N bond formation, combining two abundant and structurally diverse feedstocks in a redox-neutral and atom-economical fashion. In light of these benefits, metal-catalyzed alkene hydroamination has been extensively studied for decades (1–3). Although tremendous advances have been realized (4–12), general methods for the intermolecular anti-Markovnikov coupling of common dialkyl amines and unactivated olefins are currently unknown (Figure 1A). This deficit is often attributed to the fact that weakly coordinating alkenes cannot compete effectively for metal coordination sites with strongly Lewis basic amines. But more fundamentally, the addition of dialkyl amine N–H bonds to substituted alkenes typically lacks a significant thermodynamic driving force (2,13). For example, the addition of diethylamine to the simplest trisubstituted olefin, 2-methyl-2-butene, is approximately thermoneutral (calc. ΔG° = −0.1 kcal/mol, CBS-QB3), and analogous aminations become increasingly unfavorable as the steric profiles of the reaction partners increase (Figure 1B) (14). These observations suggest that many desirable hydroamination adducts are energetically uphill relative to their constituent amine and alkene precursors, and thus cannot be synthesized efficiently via their direct catalytic union. To overcome this limitation, many of the most general olefin amination methods couple C–N bond formation to a secondary reaction that provides a more favorable driving force. Several powerful protocols have been reported recently that make use of higher oxidation state amine donors coupled with the use of stoichiometric reductants to furnish formal hydroamination products (15–17). Although these reports have dramatically advanced the state of the art, this approach precludes the direct use of inexpensive and abundant secondary amine partners and necessarily results in the formation of stoichiometric byproducts.
Figure 1.

Reaction development A. Intermolecular anti-Markovnikov hydroaminations of simple alkenes with 2° alkyl amines are currently unknown. B. Thermodynamic challenges associated with hydroaminations of substituted alkenes. C. Kinetically facile C-N bond formation between aminium radical cations and unactivated alkenes. D. ARC-based protocol for intermolecular hydroamination of unactivated alkenes.
Seeking to address these challenges, we became interested in the olefin amination chemistry of aminium radical cations (ARCs) (18–20). In 2014, we reported that aniline-derived ARCs, generated via one-electron oxidation of an arylamine starting material by an excited-state photoredox catalyst, could undergo efficient intramolecular additions to aryl olefin acceptors (21–23). However, intermolecular aminations proved unsuccessful in this system, presumably as the reported rates of bimolecular C–N bond formation with anilinium ions (k < 106 M−1s−1) are too slow to compete with back electron transfer between the ARC and the reduced state of the iridium photocatalyst (24). To expand these protocols to intermolecular variants, we were drawn to work from Lusztyk and coworkers noting that dialkyl ARCs undergo addition to a variety of olefin acceptors, including unactivated internal olefins, with second order rate constants greater than 108 M−1s−1 at room temperature (Figure 1C) (24). We reasoned that for ARCs generated via excited-state electron transfer, such rapid C–N bond formation would be kinetically competitive with both unproductive charge recombination between the ARC and the reduced state of the photocatalyst, and thermodynamically favorable deprotonation of the aminium by the secondary amine starting material or the tertiary amine product. Moreover, the use of an excited-state redox event to generate the ARC would provide the additional driving force necessary to offset the unfavorable reaction energetics associated with the direct coupling of hindered olefins and amines, enabling access to formally endergonic hydroamination adducts that cannot be synthesized efficiently using ground state catalysts (25). Lastly, as is characteristic of electrophilic radical additions to alkenes, aminiums would be expected to exhibit high levels of anti-Markovnikov regioselectivity in the C–N bond-forming step – an outcome that has proven difficult to achieve using traditional catalyst platforms. Here we report the successful realization of these goals and describe a general photocatalytic protocol for intermolecular alkene hydroamination with secondary alkyl amines and a wide range of unactivated olefins that proceed via ARC intermediates (Figure 1D).
We envisioned a catalytic cycle wherein an excited state redox catalyst would first oxidize a secondary alkyl amine to its corresponding aminium radical cation (Figure 2). This electrophilic N-centered radical would then undergo intermolecular addition to an olefin acceptor to furnish a new C–N bond and an adjacent carbon-centered radical. Based on previous radical olefin hydrofunctionalization work from Nicewicz, Studer, and our own group, we proposed that this alkyl radical could be reduced via H-atom transfer (HAT) from an aryl thiol co-catalyst to form a closed-shell ammonium ion intermediate and a transient thiyl radical (26–28). The thiyl could accept an electron from the reduced state of the photocatalyst and the resulting thiolate could deprotonate the closed-shell ammonium adduct to provide the desired tertiary amine product and close the catalytic cycle. Aminiums are potent H-atom abstractors, and are known to react rapidly with aryl thiols containing much weaker S-H bonds (20). However, we recently demonstrated that aryl thiols can function effectively as H-atom donor catalysts in the presence of even more reactive amidyl and alkoxy radical abstractors (28, 29), likely due to mismatched polar effects that are well known to impact the rates of organic HATs (30).
Figure 2.
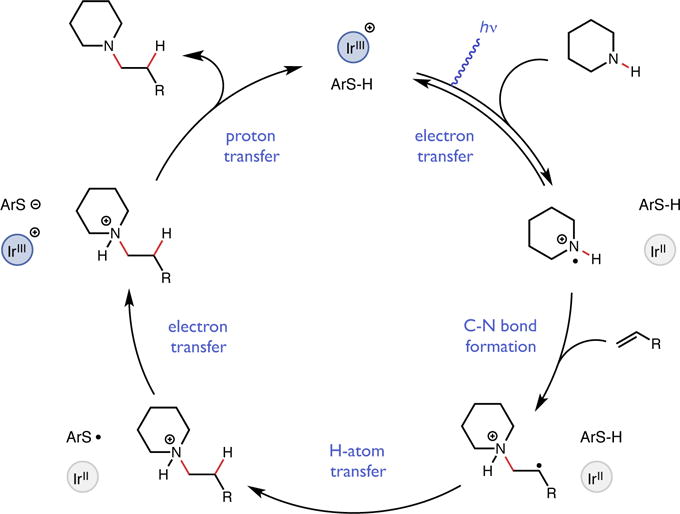
Proposed catalytic cycle for hydroamination.
Based on this proposal, our initial evaluation focused on the hydroamination of 2-methylhex-1-ene with piperidine as a model reaction. A promising lead result was observed using 2 mol% of [Ir(dF(CF3)ppy)2(bpy)]PF6 [dF(CF3)ppy = 2,4-dimethyl-2-[5-(trifluoromethyl)-2-pyridinyl; bpy = 2,2′-bipyridine] (A) and 50 mol% of 2,4,6-triisopropylbenzenethiol (TRIP thiol) in dioxane, providing 34% of the desired amination product upon irradiation with blue LEDs for 12 h at room temperature (for details of the optimization, see Supplementary Information). The related complex [Ir(dF(CF3)ppy)2(dtbbpy)]PF6 [dtbbpy = 4,4′-bis(tert-butyl) bipyridine] (B) proved even more efficient, furnishing the desired amination adduct in 86% yield. Further investigation revealed that hydroaminations using acyclic secondary amine substrates, such as diethylamine, were much less successful with B, providing the desired amination adduct in only trace yields, even when five equivalents of olefin were employed. Seeking to improve on this result, we found that an alternative complex, [Ir(dF(Me)ppy)2(dtbbpy)]PF6 [dF(Me)ppy = 2,4-dimethyl-2-[5-(methyl)-2-pyridinyl] phenyl] (C), was more effective, providing the diethylamine adduct in 17% yield (31). Further study revealed that toluene was the optimal solvent for this catalyst, and these conditions were used to evaluate the synthetic scope of the reaction. Control experiments lacking photocatalyst, thiol, or visible light provided none of the desired hydroamination adduct. The use of lower thiol loadings (10 mol %) also proved effective in the model reaction with piperidine, providing the expected adduct in 95% yield. However, this finding did not prove to be general for all alkene substrate classes surveyed and we elected to use the higher thiol loading to evaluate the full scope of the reaction.
With these optimized conditions, we were pleased to find that a wide range of olefins could be successfully hydroaminated (Figure 3). With piperidine we observed that every olefin substitution pattern could be accommodated, including terminal α-olefins, internal 1,2-disubstituted, 1,1-disubstituted, trisubstituted, and tetrasubstituted olefins (Figure 3, 1–6). As electrophilic radicals, we expected that aminiums should react readily with nucleophilic olefins. Indeed, we observed that aminations of cyclic and acyclic enol ethers proceeded smoothly to form 7, 8, and 9 in good yields. In addition, we found that silyl enol ethers were viable substrates for hydroamination, providing access to useful 1,2-amino alcohol products 10 and 11 following an acidic desilylative workup. Similarly, both cyclic and acyclic enamides could be aminated to provide differentiated 1,2-diamines 12 and 13. We were pleased to find that various allylic alcohols and protected allylic amines could be aminated successfully to provide access to both 1,2- and 1,3-amino alcohol and diamine motifs with high efficiency and complete anti-Markovnikov regioselectivity (14–17). An analogous homoallylic alcohol substrate was also employed successfully to furnish 18 in high yield. We observed that the piperidine ARC can effectively discriminate between two electronically differentiated olefins present in the same substrate. Specifically, the more electron-rich trisubstituted olefin in the terpene linalool could be aminated selectively in the presence of the terminal allylic alcohol (19). Similarly, geraniol derivatives bearing two sterically similar trisubstituted alkenes could be selectively aminated at the more electron-rich olefin distal to the allylic ester moiety (20). Cyclooctadiene was also a successful substrate for amination, providing 21 in 86% yield. Notably, no [3.3.0] bicyclic products resulting from trans-annular radical addition were observed, suggesting that this C–C bond forming process is slow relative to reductive HAT from the thiol. We also found that the bicyclic olefin norbornene could be aminated efficiently, though with relatively poor diastereoselectivity (22) - an outcome likely related to an early transition state for C–N bond formation. Lastly, we found that electron-rich styrene derivatives could be aminated to furnish anti-Markovnikov addition products 23 and 24 in good yield. Within this collection of olefins, we found that the most nucleophilic members could be used in slight excess (1.5 equivalents) to obtain high yields of amination product. However, for less electron-rich alkenes, which are known to react more slowly with ARCs (24), 3 to 5 equivalents were required to obtain optimal yields. The higher concentration in these examples enables C–N bond formation to remain kinetically competitive with unproductive charge recombination between the ARC and the reduced Ir(II) complex.
Figure 3.
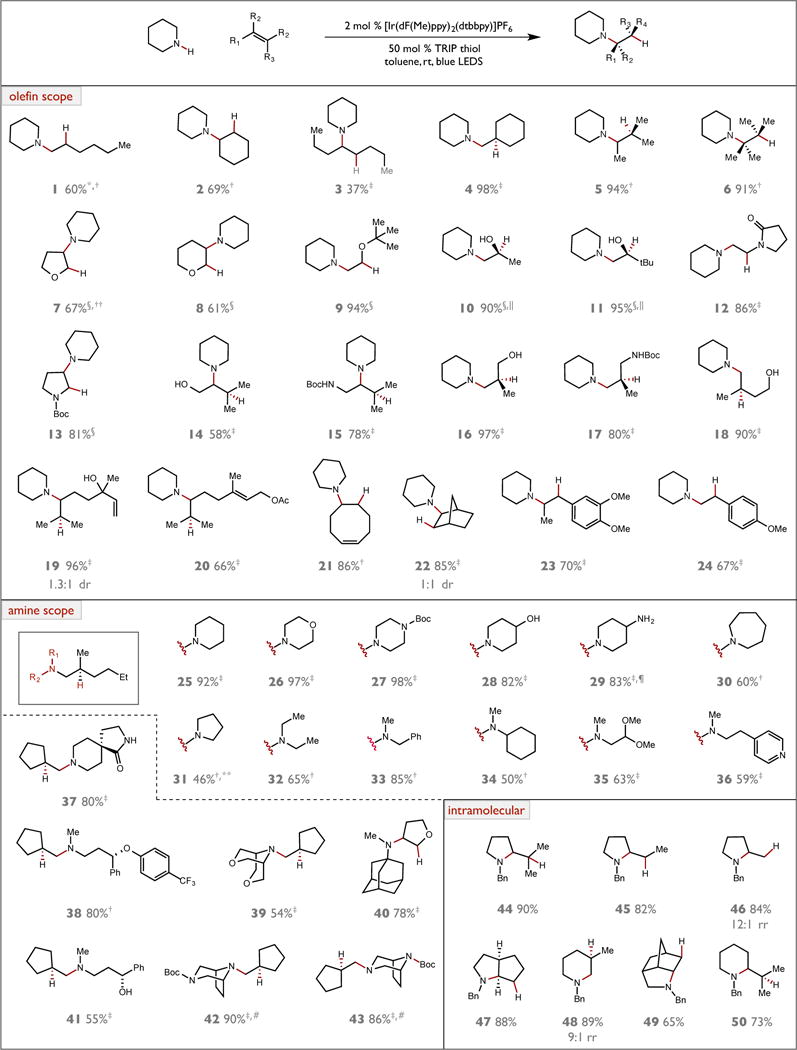
Reaction scope. Reactions run on 0.5 mmol scale unless otherwise noted. Yields are for isolated material following purification unless otherwise noted and are the average of two experiments. All chiral products in this study are formed as racemates. Irradiation from two 34 W Kessel LED lamps. rt, room temperature. *Reaction run at 45 °C. †5.0 equivalents of olefin. ‡3.0 equivalents of olefin. §1.5 equivalents of olefin. ||Substrate is a trimethylsilyl enol ether. ¶Isolated yield following acylative workup with Boc2O to facilitate purification. #Reactions run on 0.2 mmol scale. **2-methylnon-1-ene as the olefin partner. ††NMR yield.
We then evaluated the scope of amines that could be employed in this protocol. Using 2-methylhex-1-ene as a model olefin, we observed that numerous cyclic secondary amines were successfully accommodated, including piperidine, morpholine, N-Boc piperazine, and unprotected 4-hydroxypiperidine (25–28). Unprotected 4-amino piperidine was alkylated with complete selectivity for the secondary nitrogen center to furnish 29 in 83% yield. Azepane and pyrrolidine were also viable substrates, though with somewhat diminished yields relative to their 6-membered ring analog (30, 31). Excitingly, a range of acyclic secondary amines could also be used as aminating partners. Alkylation of diethylamine provided tertiary amine 32 in 65% yield. The use of N-methyl benzylamine was also successful (33). Branching adjacent to the nitrogen center was also tolerated, as demonstrated by the use of N-methyl cyclohexylamine (34). Secondary amines bearing acetals and electron-deficient aromatics could also be alkylated in good yield (35, 36). More structurally complex and sterically demanding amines, including the antidepressant drug fluoxetine, [3.2.1] bicyclic amines and diamines, spirocyclic amines, and N-methyl adamantylamine, were also readily accommodated (37–43). Lastly, we found that intramolecular variants of this transformation were successful, as a variety of N-benzyl protected substrates cyclized under the standard reaction conditions to afford a range of 5- and 6-membered heterocyclic products (44–50). With respect to limitations, this method has thus far not proven successful with aromatic amines, α-amino acid derivatives, or tetramethylpiperidine. Efforts to address these limitations are currently ongoing.
A number of experimental observations provide support for the proposed mechanism. Stern-Volmer analysis in dioxane revealed that piperidine (Ep/2 = 0.56 V vs Fc/Fc+ in MeCN) (32) efficiently quenches (fig. S2, Ksv = 200 M−1) the excited state of catalyst C (*E1/2 = 0.59 V vs Fc/Fc+ in MeCN) (33), consistent with the proposed electron transfer event. In contrast, we observed that numerous representative olefin classes (1-hexene, cyclohexene, 2-methylhex-1-ene, tetramethylethylene, and dihydrofuran) do not decrease the luminescence intensity of *C, suggesting that olefin oxidation mechanisms are not operative in these reactions. As such, this protocol is mechanistically orthogonal to the seminal photocatalytic anti-Markovnikov hydroamination methods reported by Nicewicz (34), which proceed through alkene radical cation intermediates (35). The tertiary amine products (Ep/2 = 0.43 V vs Fc/Fc+ in MeCN for NEt3) can also be oxidized (fig. S8, Ksv = 180 M−1 for 25) by the excited state of catalyst C, but the generally high yields observed in these reactions suggest that these processes are reversible and do not lead to meaningful amounts of product decomposition. We postulate that this outcome may result from the protective action of the thiol cocatalyst, which could reduce any α-amino radical intermediates resulting from tertiary amine oxidation and deprotonation before they can engage in further deleterious side reactions. As these reactions function best in toluene and dioxane, we considered whether reduction of the radical generated after C–N bond formation might occur via H-atom transfer from weak solvent C–H bonds. However, 1H-NMR analysis of the reaction between piperidine and tetramethyl ethylene in d8-toluene revealed no evidence of deuterium incorporation into the tertiary amine product 6. Lastly, the formation of 6 is calculated to be +4.8 kcal/mol (CBS-QB3) endergonic relative to the amine and olefin starting materials (14). This penalty is offset by the favorable energetics of photon absorption (emission λmax = 512 nm = +55.8 kcal/mol for the excited state of C (32), providing a clear demonstration of the ability of excited state redox catalysts to enable endergonic bond constructions (36).
In conclusion, we have developed a general photo-driven protocol for the intermolecular anti-Markovnikov hydroamination of unactivated internal olefins with dialkyl amines that proceeds via aminium radical cation intermediates. We anticipate that this method will help address a long standing synthetic challenge in hydroamination chemistry and simplify the design and construction of complex tertiary alkyl amine products.
Supplementary Material
One sentence summary.
A general catalytic method for the intermolecular anti-Markovnikov hydroamination of unactivated olefins with dialkyl amines has been developed.
Acknowledgments
Support for this work was provided by the NIH (R01 GM120530) and Bristol-Myers Squibb. We thank David C. Miller (Princeton) for calculations. We also thank William (Rick) Ewing (BMS), Lou Lombardo (BMS), Martin Eastgate (BMS), and Robert Borzilleri (BMS) for helpful discussions. RRK is the recipient of a Sloan Foundation Research Fellowship, Amgen Young Investigator Award, and Eli Lilly Grantee Award.
Footnotes
References and Notes
- 1.Coulson DR. Tetrahedron Lett. 1971;12:429–430. [Google Scholar]
- 2.Müller TE, Beller M. Chem Rev. 1998;98:675–704. doi: 10.1021/cr960433d. [DOI] [PubMed] [Google Scholar]
- 3.Huang L, Arndt M, Gooßen K, Heydt H, Gooßen LJ. Chem Rev. 2015;115:2596–2697. doi: 10.1021/cr300389u. [DOI] [PubMed] [Google Scholar]
- 4.Hong S, Marks T. Acc Chem Res. 2004;37:673–686. doi: 10.1021/ar040051r. [DOI] [PubMed] [Google Scholar]
- 5.Utsunomiya M, Kuwano R, Kawatsura M, Hartwig JF. J Am Chem Soc. 2003;125:5608–5609. doi: 10.1021/ja0293608. [DOI] [PubMed] [Google Scholar]
- 6.Bender CF, Widenhoefer RA. J Am Chem Soc. 2005;127:1070–1071. doi: 10.1021/ja043278q. [DOI] [PubMed] [Google Scholar]
- 7.Waser J, Gaspar B, Nambu H, Carreira EM. J Am Chem Soc. 2006;128:11693–11712. doi: 10.1021/ja062355+. [DOI] [PubMed] [Google Scholar]
- 8.LaLonde RL, Sherry BD, Kang EJ, Toste FD. J Am Chem Soc. 2007;129:2452–2453. doi: 10.1021/ja068819l. [DOI] [PubMed] [Google Scholar]
- 9.Crimmin MR, et al. J Am Chem Soc. 2009;131:9670–9685. doi: 10.1021/ja9003377. [DOI] [PubMed] [Google Scholar]
- 10.Reznichenko AL, Nguyen HN, Hultzsch KC. Angew Chem Int Edit. 2010;49:8984–8987. doi: 10.1002/anie.201004570. [DOI] [PubMed] [Google Scholar]
- 11.Ickes AR, Ensign SC, Gupta AK, Hull KL. J Am Chem Soc. 2014;136:11256–11259. doi: 10.1021/ja505794u. [DOI] [PubMed] [Google Scholar]
- 12.Sevov CS, Zhou JS, Hartwig JF. J Am Chem Soc. 2014;136:3200–3207. doi: 10.1021/ja412116d. [DOI] [PubMed] [Google Scholar]
- 13.Johns AM, Sakai N, Ridder A, Hartwig JF. J Am Chem Soc. 2006;128:9306–9307. doi: 10.1021/ja062773e. [DOI] [PubMed] [Google Scholar]
- 14.For details, see supplementary material.
- 15.Zhu S, Niljianskul N, Buchwald SL. J Am Chem Soc. 2013;135:15746–15749. doi: 10.1021/ja4092819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Miki Y, Hirano K, Satoh T, Miura M. Angew Chem Int Edit. 2013;52:10830–10834. doi: 10.1002/anie.201304365. [DOI] [PubMed] [Google Scholar]
- 17.Gui J, et al. Science. 2015;348:886–891. doi: 10.1126/science.aab0245. [DOI] [PubMed] [Google Scholar]
- 18.Stella L. Angew Chem Int Ed. 1983;22:337–350. [Google Scholar]
- 19.Newcomb M, Deeb TM, Marquardt DJ. Tetrahedron. 1990;46:2317–2328. [Google Scholar]
- 20.Horner JH, Martinez FN, Musa OM, Newcomb M, Shahin HE. J Am Chem Soc. 1995;117:11124–11133. [Google Scholar]
- 21.Musacchio AJ, Nguyen LQ, Beard GH, Knowles RR. J Am Chem Soc. 2014;136:12217–12220. doi: 10.1021/ja5056774. [DOI] [PubMed] [Google Scholar]
- 22.Maity S, Zheng N. Angew Chem Int Edit. 2012;51:9562–9566. doi: 10.1002/anie.201205137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Prier CK, Rankic DA, MacMillan DWC. Chem Rev. 2013;113:5322–5363. doi: 10.1021/cr300503r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wagner BD, Ruel G, Lusztyk J. J Am Chem Soc. 1996;118:13–19. [Google Scholar]
- 25.Singh K, Staig S, Weaver JD. J Am Chem Soc. 2014;136:5275–5278. doi: 10.1021/ja5019749. [DOI] [PubMed] [Google Scholar]
- 26.Margrey KA, Nicewicz DA. Acc Chem Res. 2016;49:1997–2006. doi: 10.1021/acs.accounts.6b00304. [DOI] [PubMed] [Google Scholar]
- 27.Guin J, Mück-Lichtenfeld C, Grimme S, Studer A. J Am Chem Soc. 2007;129:4498–4503. doi: 10.1021/ja0692581. [DOI] [PubMed] [Google Scholar]
- 28.Miller DC, Choi GJ, Orbe HS, Knowles RR. J Am Chem Soc. 2015;137:13492–13495. doi: 10.1021/jacs.5b09671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yayla HG, Wang H, Tarantino KT, Orbe HS, Knowles RR. J Am Chem Soc. 2016;138:10794–10797. doi: 10.1021/jacs.6b06517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Roberts BP, Steel AJ. J Chem Soc Perkin Trans. 1994;2:2155–2162. [Google Scholar]
- 31.Goldsmith JI, Hudson WR, Lowry MS, Anderson TH, Bernhard S. J Am Chem Soc. 2005;127:7502–7510. doi: 10.1021/ja0427101. [DOI] [PubMed] [Google Scholar]
- 32.Roth HG, Romero NA, Nicewicz DA. Synlett. 2016;27:714–723. [Google Scholar]
- 33.Ladouceur S, Fortin D, Zysman-Colman E. Inorg Chem. 2011;50:11514–11526. doi: 10.1021/ic2014013. [DOI] [PubMed] [Google Scholar]
- 34.Nguyen TM, Nicewicz DA. J Am Chem Soc. 2013;135:9588–9591. doi: 10.1021/ja4031616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.N-Boc dihydropyrrole was observed to quench the excited state of E, suggesting that olefin radical cation chemistry may be operative in the reactions of that substrate.
- 36.For calculated hydroamination thermochemistry of other representative olefin classes, see supplementary material
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.