

Summary
Model organisms and human studies have led to increasing empirical evidence that interactions among genes contribute broadly to genetic variation of complex traits. In the presence of gene-by-gene interactions, the dimensionality of the feature space becomes extremely high relative to the sample size. This imposes a significant methodological challenge in identifying gene-by-gene interactions. In the present paper, through a Gaussian graphical model framework, we translate the problem of identifying gene-by-gene interactions associated with a binary trait D into an inference problem on the difference of two high-dimensional precision matrices, which summarize the conditional dependence network structures of the genes. We propose a procedure for testing the differential network globally that is particularly powerful against sparse alternatives. In addition, a multiple testing procedure with false discovery rate control is developed to infer the specific structure of the differential network. Theoretical justification is provided to ensure the validity of the proposed tests and optimality results are derived under sparsity assumptions. A simulation study demonstrates that the proposed tests maintain the desired error rates under the null and have good power under the alternative. The methods are applied to a breast cancer gene expression study.
Keywords: Differential network, false discovery rate, Gaussian graphical model, gene-by-gene interaction, highdimensional precision matrix, large scale multiple testing
1. INTRODUCTION
High throughput technologies, enabling comprehensive monitoring of a biological system, have fundamentally transformed biomedical research. Studies using such technologies have led to successful molecular classifications of diseases into clinically relevant subtypes and genetic signatures predictive of disease progression and treatment response (van’t Veer et al., 2002; Gregg et al., 2008; Hu et al., 2009, e.g.). Irrespective of the technology used, analysis of high-throughput data typically considers one marker at a time and yields a list of differentially expressed genes or proteins. On the other hand, epistasis, or interactions between genes, has long been recognized as crucial to understanding the genetic architecture of disease phenotypes (Phillips, 2008; Eichler et al., 2010). Increasing empirical evidence from model organisms and human studies suggests that gene-by-gene interactions may make an important contribution to total genetic variation of complex traits (Zerba et al., 2000; Marchini et al., 2005). In this paper, we are specifically interested in gene-by-gene interactions with respect to the interactive effects of two genes on a binary disease trait D.
In the presence of gene-by-gene interactions, the dimensionality of the feature space becomes extremely high relative to the sample size. This, together with the variability of the data, imposes a significant methodological challenge in identifying gene-by-gene interactions using currently available studies, which typically have limited sample sizes and power. Recent development in interaction modeling has led to several useful methods including multi-factor dimensionality reduction (Ritchie et al., 2001; Moore, 2004), polymorphism interaction analysis (Mechanic et al., 2008), random forests (Breiman, 2001), various variations of logistic regression with interactive effects (Chatterjee et al., 2006; Chapman & Clayton, 2007; Kooperberg & Ruczinski, 2005; Kooperberg & LeBlanc, 2008) and sure independence screening (Fan & Lv, 2008). However, to overcome the high dimensionality, a majority of these methods use multistage procedures and marginal assessments of the effects of a gene pair without simultaneously accounting for the effects of other genes. Multistage procedures may have limited power in detecting genes that affect the outcome through interactions with other genes without strong main effects. The interactive effects detected through models that only consider one pair of genes at a time without conditioning on other genes may also result in false identification of interactions due to the discrepancy between conditional and unconditional effects. Furthermore, none of the existing methods provide false discovery rate control in the presence of interactions. Due to the large number of tests, the power of multiple testing procedures using the standard Bonferroni or naive false discovery rate corrections can dissipate quickly.
In this paper, through a Gaussian graphical model framework, we translate the problem of identifying gene-by-gene interactions associated with a binary trait D into the comparison of two high-dimensional precision matrices. Let G denote a p × 1 vector of genomic markers and assume that, conditional on D = d, G ~ N(μd, Σd), for d = 1, 2. Then the posterior risk given G is
where g(x) = ex/(1 + ex) and is the precision matrix for G conditional on D = d. Hence, an interaction between the gene pair (i, j) affects the disease risk if and only if δi,j= ωi,j,1 − ωi,j,2 = 0. The difference between the two precision matrices, denoted by Δ = (δi,j) = Ω1 – Ω2, is called the differential network. This type of model for a differential network has been used in Li et al. (2007) and Danaher et al. (2014). We thus propose to test for gene-by-gene interactions both by testing the global hypotheses
| (1) |
and by simultaneously testing the hypotheses
while controlling for the overall false discovery rate at a pre-specified level.
Few authors have considered testing the equality of two precision matrices in the high-dimensional setting. The global null hypothesis Δ = 0, or equivalently Ω1 = Ω2, corresponds to the hypothesis that none of the gene pairs have interactive effects on D. The equality of two precision matrices is equivalent to the equality of two covariance matrices, and the latter has been studied under various alternatives. Under the dense alternative, where Σ1 and Σ2 differ in a large number of entries, various sum-of-square type testing procedures have been proposed (Schott, 2007; Srivastava & Yanagihara, 2010; Li & Chen, 2012). Under the sparse alternative with Σ1 and Σ2 differing only in a small number of entries, Cai et al. (2013) introduced a particularly powerful test. However, in the gene-by-gene interaction setting, the goal is to identify the structure of the differential network. In such cases, it is often reasonable to assume that Δ is sparse, while Σ1 – Σ2 is not. Hence, testing procedures that can leverage information on the sparsity of Δ may improve power. Furthermore, due to the fundamental difference between conditional and unconditional dependences, the various procedures for testing the covariance matrices may not be well adapted to testing specific entries of the precision matrices.
The first goal of this paper is to develop a global test for H0 : Δ = 0 that is powerful against sparse alternatives. We then develop a multiple testing procedure for simultaneously testing the hypotheses {H0,i,j : 1 ≤ i < j ≤ p} with false discovery rate control to infer the structure of the differential network. In the high-dimensional setting, there is no sample precision matrix that one can use to approximate Ωd. We propose to infer Ωd by relating its elements to the coefficients of a set of regression models for G conditional on D = d. We then construct test statistics based on the covariances between the residuals from the fitted regression models. The testing procedures are easy to implement. A Matlab implementation is available in the Supplementary Material.
2. Global Testing of Differential Networks
2.1. Notation and Definitions
In this section we consider testing the global hypothesis (1). We begin with notation and definitions that will be used in the rest of the paper. Let Xk ε ℝp and Yk ε ℝp denote G given D = 1 and D = 2, respectively, Xk ~ N(μ1,Σ1) for k = 1,…, n1, Yk ~ N(μ1,Σ2) for k = 1,…, n2, where Σd = (σi,j,d) for d = 1, 2, and {Xk : k = 1,…, n1} and {Yk : k = 1,…, n2} are independent observations from the two populations. Let X = (X1,…, Xn1)T and Y = (Y1,…, Yn2)T denote the data matrices. Let , for d = 1,2.
For subscripts, we use the convention that i stands for the ith entry of a vector and (i,j) for the entry in the ith row and jth column of a matrix, k represents the kth sample and d indexes the binary trait. Let βi,1 = (β1,i,1,…,βp−1,i,1)T denote the regression coefficients of Xk,i regressed on the rest of the entries of Xk and let βi,2 = (β1,i,2,…,βp−1,i,2)T denote the regression coefficients of Yk,i regressed on the rest of the entries of Yk.
For any vector μd with dimension p × 1, let μ−i,d denote the (p – 1) × 1 vector by removing the ith entry from μd. For a symmetric matrix A, let λmax(A) and λmin(A) denote the largest and smallest eigenvalues of A. For any p × q matrix A, Ai,−j denotes the ith row of A with its jth entry removed and A−i,j denotes the jth column of A with its ith entry removed. The matrix A−i,−j denotes a (p – 1) × (q – 1) matrix obtained by removing the ith row and jth column of A. For an n × p data matrix U = (U1,…, Un)T, let with dimension with dimension 1 × (p − 1), U(i) = (U1,i,…, Un,i)T with dimension with dimension n × 1, where , and with dimension n × (p − 1). For tuning parameters λ, let represent the ith tuning parameter for binary trait d, which depends on the sample size nd.
For a vector β = (β1,…,βp)T ε ℝp, define the ℓq norm by for 1 ≤ q ≤ ∞. A vector β is called k-sparse if it has at most k nonzero entries. For a matrix Ω = (ωi,j)p×p, the matrix 1-norm is the maximum absolute column sum, , the matrix elementwise infinity norm is defined to be ||Ω||∞ = max1≤i,j≤p |ωi,j| and the elementwise ℓ1 norm is . For a matrix Ω, we say Ω is k-sparse if each row/column has at most k nonzero entries. For a set ℋ, denote by |ℋ| the cardinality of ℋ. For two sequences of real numbers {an} and {bn}, write an = O(bn) if there exists a constant C such that |an| ≤ C|bn| holds for all n, write an = o(bn) if limn→∞ an/bn = 0, and write an ≍ bn if there are positive constants c and C such that c ≤ an/bn ≤ C for all n.
2 2. Testing Procedure
It is well known (e.g., Anderson, 2003, Section 2.5), that in the Gaussian setting the precision matrix can be described in terms of regression models. Specifically, we may write
| (2) |
| (3) |
where , are independent of Xk,−i and Yk,−i respectively, and . The regression coefficient vectors βi,d and the error terms εk,i,d satisfy
where cov(·,·) denotes the population covariance. Since the null hypothesis H0 : Δ = 0 is equivalent to the hypothesis
a natural approach to test H0 is to first construct estimators of ωi,j,d, and then base the test on the maximum standardized differences. We first construct estimators of ri,j,d
Let be estimators of βi,d satisfying
| (4) |
| (5) |
Estimators that satisfy (4) and (5) can be obtained easily via methods such as the lasso and Dantzig selector. See Section 2.3 for details. Define the residuals by
A natural estimator of ri,j,d is the sample covariance between the residuals,
| (6) |
However, when tends to be biased due to the correlation induced by the estimated parameters and it is desirable to construct a bias-corrected estimator. Lemma 2 shows that
where is the empirical covariance between {εk,i,d: k = 1,… ,nd} and {εk,j,d : k = 1,…,nd}. For 1 ≤ i ≤ j ≤ p, βi,j,d = − ωi,j,d/ωj,j,d and βj−1,i,d = −ωi,j,d/ωi,i,d Thus, we propose a bias-corrected estimator of ri,j,d as
| (7) |
The bias of is of order max{ri,j,d(log p/nd)1/2,(ndlog p)−1/2}.
For i = j, note that ri,i,d = 1/ui,i,d. We show in Lemma 2 that
which implies that is a nearly unbiased estimator of ri,i,d. A natural estimator of ωi,j,d can then be defined by
| (8) |
We test H0 : Δ = 0 based on the estimators
The estimators Ti,j,1 − Ti,j,2 in are heteroscedastic and possibly have a wide range of variability. We first standardize Ti,j,1 − Ti,j,2 before combining information from all entries in . Let and . It will be shown in Lemma 2 that, uniformly in 1 ≤ i ≤ j ≤ p,
Let . Note that
where . We then estimate θi,j,d by
Define the standardized statistics
| (9) |
Finally, we propose the following test statistic for testing the global null hypothesis H0,
| (10) |
The asymptotic properties of Mn will be studied in detail in Section 3. Intuitively, {Wi,j} are approximately standard normal variables under the null H0 and they are only weakly dependent under suitable conditions. Thus Mn is the maximum of the squares of p(p + 1)/2 such random variables, so its value should be close to 2 log{p(p + 1)/2} ≈ 4 log p under H0. We show in Section 3 that, under certain regularity conditions, Mn − 4 log p − log log p converges to a type I extreme value distribution under H0 : Δ = 0.
Based on the limiting null distribution of Mn, which will be developed in Section 3.1, we define the test ψα by
| (11) |
where qα is the 1 − α quantile of the type I extreme value distribution with the cumulative distribution function exp{(8π)−1/2e−t/2}, i.e.,
| (12) |
The hypothesis H0 is rejected whenever ψα = 1.
2.3. Data-driven estimation of regression coefficients
The testing procedure requires the estimation of regression coefficients βi,d, for i = 1,…,p and d = 1, 2. Various estimators have been studied in the literature, including the lasso and Dantizg selector. Here, we use the lasso by solving the optimization problem,
| (13) |
| (14) |
where and , d = 1,2. Then by Proposition 4.2 of Liu (2013), under Condition (C1) given in Section 3 and a mild condition on the sparsity of βi,d (i = 1,…, p, d = 1,2), the convergence rates in (4) and (5) can be guaranteed by using any κd > 2. The result is formally stated in Corollary 1. In practice, κd = 2 works well for global testing of H0 : Δ = 0, and for the multiple testing procedure with false discovery rate control, a data-driven algorithm is proposed in Section 5 to select κd adaptively.
2.4. Discussion
The global test ψα given in (11) is based on estimators of ωi,j,1 − ωi,j,2 Here we estimate ωi,j,d by first constructing estimators for ri,j,d = ωi,j,d/(ωi,i,dωj,j,d), and then estimating ri,j,d through bias correction of the residuals defined in (7).
Liu (2013) considered multiple testing of entries of a single precision matrix Ω = (ωi,j). In the one-sample case, ωi,j = 0 is equivalent to ri,j= ωi,j/(ωi,iωj,j) = 0 under the null and ri,j is easier to estimate. The procedure in Liu (2013) is based on the estimation of ri,j instead of ωi,j. However, in Section 4 we will also consider multiple testing between two groups, and ωi,j,1= ωi,j,2 is not equivalent to ri,j,1= ri,j,2. Thus, it is necessary to construct testing procedures based directly on estimators of ωi,j,1 − ωi,j,2.
Testing the global hypothesis H0 : Ω1 = Ω2 is equivalent to testing H0 : Σ1 = Σ2, which has been well studied (Schott, 2007; Srivastava & Yanagihara, 2010; Li & Chen, 2012; Cai et al., 2013). In particular, Cai et al. (2013) constructed a global test for H0 : Σ1 = Σ2 that is powerful against the alternative where Σ1 − Σ2 is sparse. However, in many applications, the goal is to learn the structure of the differential network, and we are interested in both testing the global hypothesis H0 : Ω1 = Ω2 and multiple testing of the entrywise hypotheses H0,i,j : ωi,j,1 = ωi,j,2. In such cases, it is often reasonable to assume that Δ = Ω1 − Ω2 is sparse, but Σ1 − Σ2 is not. Hence, testing procedures for H0 : Σ1 = Σ2 cannot leverage information on the sparsity of Δ and more importantly do not naturally lead to a multiple testing procedure for simultaneously testing the entrywise hypotheses H0,i,j : ωi,j,1 = ωi,j,2.
3. Theoretical Results for the Global Test
3 1. Asymptotic Null Distribution of Mn
In this section, we analyze the properties of the new test for testing the global null hypothesis H0 : Δ = 0, including the null distribution of the test statistic Mn, the asymptotic size and power. We are particularly interested in the power of the new test under the alternative with Δ sparse. We further show that the power is minimax rate optimal.
Under assumptions (C1) and (C2), Theorem 1 indicates that under H0, Mn − 4 log p + log log p converges weakly to a Gumbel random variable with distribution function exp{−(8π)−1/2e−t/2}.
-
(C1)
Assume that log p = o(n1/5), n1 ≍ n2, and for some constant , for d = 1,2. There exists some τ > 0 such that | Aτ| = o(p1/16) where Aτ = {(i,j) : |ωi,j,d| ≥ (log p)−2−τ, 1 ≤ i<j ≤ p, for d = 1 or 2}.
-
(C2)
Let Dd be the diagonal of Ωd and let , for d = 1,2. Assume that max1≤i≤j≤p |ηi,j,d| ≤ ηd ≤ 1 for some constant 0 < ηd < 1.
Condition (C1) on the eigenvalues is a common assumption in the high-dimensional setting and implies that most of the variables are not highly correlated with each other. Condition (C2) is also mild. For example, if max1≤i≤j≤p |ηi,j,d| = 1, then Ωd is singular. The following theorem states the asymptotic null distribution for Mn.
Theorem 1
Suppose that (C1), (C2), (4) and (5) hold. Then under H0, for any t ε ℝ,
| (15) |
where Mn is defined in equation (10). Under H0, the convergence in (15) is uniform for all {Xk : k = 1,…, n1} and {Yk : k = 1,…, n2} satisfying (C1), (C2), (4) and (5).
Equations (4) and (5) are mild conditions on the estimator of βi,d in order to obtain the limiting distribution in Theorem 1. As discussed in Section 2 3, these conditions can be guaranteed by the lasso estimator for example.
Corollary 1
Suppose that (C1) and (C2) hold and max1≤i≤p |βi,d|0 = o{n1/2 / (log p)3/2}. Then under H0, for any κd > 2 in (13) and (14), and for any t ε ℝ,
| (16) |
where Mn is defined in (10).
3 2. Power Analysis
We now turn to an analysis of the power of the test ψα given in (11). We shall define the following class of precision matrices:
| (17) |
The next theorem shows that the null parameter set in which Ω1 = Ω2 is asymptotically distinguishable from (4) by the test ψα. That is, H0 is rejected by the test ψα with overwhelming probability if .
Theorem 2
Let the test ψα be given as in (11). Suppose that (C1), (4) and (5) hold. Then
The following result shows that this lower bound is rate-optimal. Let be the set of all α-level tests, i.e., pr(Tα = 1) ≤ α under H0 for all .
Theorem 3
Suppose that log p = o(n). Let α, β > 0 and α + β < 1. Then there exists a constant c0 > 0 such that for all sufficiently large n and p,
Theorem 3 shows that, if c0 is sufficiently small, then any α level test is unable to reject the null hypothesis correctly uniformly over with probability tending to one. So the order (logp)1/2 in the lower bound of max1≤i≤j≤p{|ωi,j,1 − ωi,j,2/(θi,j,1 + θi,j,2)1/2} in (17) cannot be improved.
4. Multiple Testing with False Discovery Rate Control
If the global null hypothesis is rejected, it is often of interest to investigate the structure of the differential network Δ. A natural approach is to carry out simultaneous testing on the elements of Δ. In this section, we introduce a multiple testing procedure with false discovery rate control for testing (p2 − p) /2 hypotheses
| (18) |
The standardized differences of Ti,j,1 and Ti,j,2 are defined by the test statistics as in (9). Let t be the threshold level such that H0,i,j is rejected if |Wi,j |≥ t. Let ℋ0 = {(i, j) : δi,j = 0,1 ≤ i < j ≤ p} be the set of true nulls. Denote by the total number of false positives, and by R(t) = Σ1≤i<j≤p I(|Wi,j|≥ t) the total number of rejections. The false discovery proportion and false discovery rate are defined as
An ideal choice of t would reject as many true positives as possible while controlling the false discovery rate and false discovery proportion at the pre-specified level α. That is, we select
Since ℋ0 is unknown, we can estimate by as in Liu (2013), where ϕ(t) is the standard normal cumulative distribution function. Note that can be estimated by (p2 − p)/2 due to the sparsity of Δ. This leads to the following multiple testing procedure.
Calculate the test statistics Wi,j.
- For given 0 ≥ α ≥ 1, calculate
If does not exists, set . For 1 ≤ i < j ≤ p, reject H0,i,i,. if and only if .
The following theorem shows that, under regularity conditions, the above procedure controls the false discovery proportion and false discovery rate at the pre-specified level α asymptotically.
Theorem 4
Let
Suppose for some ρ > 0 and some δ > 0, . Suppose that for any ν > 0, where is given in Condition (C1). Assume that for some c > 0, and (4) and (5) hold. Let q = (p2 − p)/2. Then under (C1) with p ≤ cnr for some c > 0 and r > 0, we have
in probability, as (n, p) → ∞.
The condition in Theorem 4 is mild, since there are (p2 − p)/2 hypotheses in total and this condition only requires a few entries with the standardized difference having magnitude exceeding {(log p)1/2+ρ/n}1/2 for some constant ρ > 0. The technical condition for any ν > 0 is to ensure that most of the regression residuals are not highly correlated with each other under the null hypotheses H0,i,j : δi,j = 0.
The basic idea for the proof of Theorem 4 is similar to that in Liu (2013). However, the setting here is more complicated as ωi,j,1 and ωi,j,2 are not necessarily zero under H0,i,j : δi,j = 0. So the coordinates of the regression residuals in (2) and (3) can be correlated with each other. Thus slightly stronger conditions are needed and the proof is more involved.
5. Simulation Study
The proposed testing procedures are easy to implement, and the Matlab code is available in the Supplementary Material. We carry out a simulation study to investigate the numerical performance, including the size and power, of the global test Ψα and the false discovery rate controlled multiple testing procedure.
We first introduce the matrix models used in the simulations. Let D = (Di,j) be a diagonal matrix with Di,i = Unif(0.5, 2.5) for i = 1,…,p. The following four models under the null, , are used to study the size of the tests.
Model 1: where , , and otherwise. Ω(1) = D1/2Ω*(1)D1/2.
Model 2: where for i = 10(k − 1) + 1 and 10(k − 1) + 2 ≤ j ≤ 10(k − 1) + 10, 1 ≤ k ≤ p/10. otherwise. Ω(2) = D1/2(Ω *(2) + δI)/(1 + δ)D1/2 with δ = |λmin(Ω*(2))| + 0.05.
Model 3: where , for i < j and . Ω(3) = D1/2(Ω*(3)+ δI)/(1 + δ)D1/2 with δ = |λmin(Ω*(3))| + 0.05.
Model 4: where , for 2(k − 1) + 1 ≤ i ≠ j ≤ 2k, where k = 1,…, [p/2] and otherwise. Ω(4) = d1/2{(Σ*(4) + δI)/(1 + δ)}−1 D1/2 with δ = |λmin(Σ*(4))| + 0.05.
For global testing of H0 : Δ = 0, the sample sizes are taken to be n1 = n2 = 100, while the dimension p varies over the values 50, 100, 200 and 400. For each model, data are generated from multivariate normal distributions with mean zero and covariance matrices and The nominal significance level for all the tests is set at α1 − 0.05.
To evaluate the power of the proposed tests, let U = (ui,j) be a matrix with eight random nonzero entries. The locations of four nonzero entries are selected randomly from the upper triangle of U, each with a magnitude generated randomly and uniformly from the set [−2ω(log p/n)1/2, −ω(log p/n)1/2] ∪ [ω(log p/n)1/2,2 ω(log p/n)1/2], where . The other four nonzero entries in the lower triangle are determined by symmetry. We use the following four pairs of precision matrices , to show the power of the test, where and , with δ = |min{λmin(Ω(m) + U), λmin(Ω(m))}| + 0.05. The actual sizes and powers in percentage for the four models, reported in Table 1, are estimated from 1000 replications.
Table 1.
Empirical sizes and powers (%) for global testing with α1 = 0.05, n1 = n2 = 100, and 1000 replications.
| p | Model 1 | Model 2 | Model 3 | Model 4 | |
|---|---|---|---|---|---|
| Size | |||||
|
| |||||
| 50 | 3.8 | 3.9 | 5.4 | 4.4 | |
| 100 | 3.6 | 4.4 | 4.1 | 3.8 | |
| 200 | 3.4 | 3.6 | 3.7 | 3.9 | |
| 400 | 3.5 | 3.7 | 3.6 | 3.5 | |
|
| |||||
| Power | |||||
|
| |||||
| 50 | 100 | 98.7 | 95.6 | 81.6 | |
| 100 | 99.7 | 96.6 | 95.1 | 77.8 | |
| 200 | 93.1 | 88.2 | 93.6 | 72.1 | |
| 400 | 86.3 | 73.1 | 77.7 | 70.7 | |
Table 1 shows that the sizes of the global test are close to the nominal level in all cases. This reflects the fact that the null distribution of the test statistic Mn is well approximated by its asymptotic distribution. The empirical sizes are slightly below the nominal level in some models, due to the correlation among the variables. Similar phenomena have also been observed in Cai et al. (2013) and are theoretically justified by their Proposition 1. Table 1 shows that the proposed test is powerful in all settings, although the two precision matrices differ only in eight entries with the magnitude of the difference of the order (log p/n)1/2.
In addition, we consider nearer alternatives by generating the nonzero entries randomly and uniformly from the set [−ω(2 log p/n)1/2, ω(2 log p/n)1/2]. The power results are summarized in Table 2. Under the nearer alternatives, the magnitude of the standardized difference of Ω1 − Ω2 is smaller and as a result the power is lower.
Table 2.
Empirical power (%) for global testing under nearer alternatives.
| p | Model 1 | Model 2 | Model 3 | Model 4 |
|---|---|---|---|---|
| Power under nearer alternative | ||||
|
| ||||
| 50 | 90.3 | 71.6 | 58.9 | 20.6 |
| 100 | 89.4 | 70.3 | 60.8 | 22.8 |
| 200 | 81.9 | 55.2 | 54.2 | 21.7 |
| 400 | 73.5 | 54.7 | 57.7 | 17.5 |
More extensive simulation results are presented in the Supplementary Material. The proposed test significantly outperforms both that of Cai et al. (2013), which is powerful when Σ1 − Σ2 is sparse under the alternative, and that of Li & Chen (2012), which is powerful when Σ1 − Σ2 is dense under the alternative.
For simultaneous testing of the individual entries of the differential network Δ with false discovery rate control, we select in (13) and (14) adaptively with the principle of making and as close as possible. The algorithm is as follows.
For any given i ∈{1,…,p}, let and for s = 1,…, 40. For each s, calculate and d = 1,2. Based on the estimated regression coefficients, construct the corresponding standardized difference for each s.
- Choose
The tuning parameters are chosen to be and .
Pairwise comparisons among these four models are considered. The sample sizes are n1 = n2 = 100, while the dimension p = 50, 100, and 200. The false discovery rate level is set at α2 = 0.1, and the empirical false discovery rate and the power of false discovery rate control in percentage, summarized in Table 3, are estimated from 100 replications. We examine the power based on the average powers for 100 replications as follows
where Wi,j,l denotes standardized difference for the lth replication and denotes the nonzero locations. For all six cases, the false discovery rates are close to α across all dimensions. For empirical power, the procedure is powerful when the dimension p is low, and retains high power for the comparisons between Model 1 and Models 2 and 4. However, for the comparison between Model 2 and Model 3, the power is low when dimension is high and this is because all of | ωi,j,1 − ωi,j,2|/(θi,j,1n1 + θi,j,2n2)1/2 is smaller than 0.25 when p = 200 and D = I. Similarly, most nonzero entries of the standardized difference for Model 2 and 4 are smaller than 0.24. Thus it is difficult to detect nonzero locations. Furthermore, under the same scenario, ωi,j/(θi,jn)1/2 is always smaller than 0.16 for Model 3, and thus the detection becomes harder when we compare Model 3 with other models. Thus, the power results are not good when Model 3 is included in the comparison.
Table 3.
Empirical false discovery rate and power (%) with α2 = 0.1, n1 = n2 = 100, and 100 replications.
| p | Models 1, 2 | Models 1, 3 | Models 2, 3 | Models 1, 4 | Models 2, 4 | Models 3, 4 |
|---|---|---|---|---|---|---|
| Empirical False Discovery Rate | ||||||
|
| ||||||
| 50 | 10.5 | 11.0 | 12.6 | 12.2 | 11.5 | 10.2 |
| 100 | 9.5 | 10.0 | 12.1 | 11.8 | 11.4 | 9.5 |
| 200 | 9.7 | 10.4 | 11.2 | 11.7 | 11.6 | 10.3 |
|
| ||||||
| Power | ||||||
|
| ||||||
| 50 | 67.9 | 65.6 | 35.7 | 55.0 | 30.2 | 26.1 |
| 100 | 64.2 | 38.3 | 19.3 | 51.4 | 25.1 | 18.2 |
| 200 | 61.1 | 20.6 | 17.1 | 46.1 | 21.7 | 11.3 |
6. Real Data Analysis
The high throughput technology and massively parallel measurement of mRNA expression catalyzed a new area of genomic biomarkers. A number of prominent genomic markers have been identified to assist in predicting breast cancer patient survival in clinical practice, and increasingly, pharmacogenomic endpoints are being incorporated into the design of clinical trials (Olopade et al., 2008). Molecular pathways of pathogenesis for breast cancer have also been increasingly discovered and curated (Nathanson et al., 2001). However, the role of gene-by-gene interactions, within and across pathways, in breast cancer survival remain unclear. Here, we apply our procedures to identify gene-by-gene interactions important for breast cancer survival.
For illustration, we consider 32 pathways from the molecular signature database that are related to breast cancer survival. Examples include the MAPK/ERK, WNT, TGF-β, P13k-AKT-mTOR and ATRBRCA pathways. Existing literature has indicated that a defect in the MAPK pathway may lead to uncontrolled growth, which is a step necessary for the development of all cancers (Santen et al., 2002; Downward, 2003). Mutations or deregulated expression of genes in the Wnt pathway can induce cancer (Klaus & Birchmeier, 2008). The TGF-β signaling pathway is critical to a plethora of cellular processes including cell proliferation, apoptosis and differentiation (Shi & Massagué, 2003). An increase in the TGF-β2 expression is associated with response to tamoxifen for breast cancer patients (Buck & Knabbe, 2006). The ATRBRCA pathway describes the role of BRCA1, BRCA2 and ATR in cancer susceptibility (Venkitaraman, 2002). BRCA1 and BRCA2 are the best-known genes linked to breast cancer risk. Hence, these pathways may play critical roles in breast cancer progression. To examine the interactions between genes in these pathways, we applied our procedure to a recent breast cancer gene expression study of 295 patients with primary breast carcinomas from the Netherlands Cancer Institute (van de Vijver et al., 2002). Out of the 32 pathways, there are a total of p = 754 genes with available data in this study. The two populations we consider are the short term survivors, defined as those 78 patients who died within 5 years; and the long term survivors, defined as those 69 patients who survived more than 10 years. We are particularly interested in identifying gene pairs with interactive effects on the binary cancer survival trait using the proposed procedures. In this setting, the sparsity assumption about βi,k’s is reasonable as it is generally believed that transcriptional regulation of a single gene is generally defined by a small set of regulatory elements (Segal et al., 2003; Dobra et al., 2004).
Based on our proposed procedures, we identified nine pairs of gene-by-gene interactions as significant at a false discovery rate level of 0.1. An interaction here does not simply indicate a co-expression between a pair of genes, but instead represents a difference between the co-expression patterns among the long terms survivors and among the short term survivors. As shown in Figure 1, the majority of the genes involved in these interactions belong to five major pathways, the MAPK, WNT, TGF-β, Apoptosis, and ATRBRCA pathways, although many of these genes belong to multiple pathways. One pair of the identified interactions represent gene-by-gene interactions within pathways and the remaining eight pairs represent cross-talk between these pathways, some of which are previously documented. A total of five interactions are between the MAPK signaling pathway and the WNT and TGF-β, Apoptosis, ATRBRCA and MTA3 pathways. These cross-talks are not surprising since MAPK modulates a wide range of processes including gene expression, mitosis, proliferation, metabolism and apoptosis (Wada & Penninger, 2004). Several recent studies suggest extensive crosstalk between WNT and MAPK signaling pathways in cancer. For example, hyper-activation of MAPK signaling results in down-regulation of the WNT signal transduction pathway in melanoma, suggesting a negative crosstalk between the two pathways; while in colorectal cancer, stimulating the WNT pathway leads to activation of the MAPK pathway through Ras stabilization, representing a positive crosstalk (Guardavaccaro & Clevers, 2012). The observed interactive effect between the WNT and MAPK pathways suggests that the cross-talk between these two pathways may play an important role in breast cancer survival. The interaction between the tumor suppressor gene BRCA2 and the MAPK pathway has been documented in experiments with prostate cancer cells with upregulation of BRCA2 linked to an increase in MAPK activity (Moro et al., 2007). In the WNT pathway, the WNT1 gene promotes cell survival in various cell types and it has been experimentally shown that blocking WNT1 signaling can induce apoptotic cell death (You et al., 2004). Thus the interaction between WNT1 gene and the PRKACB gene in the Apoptosis pathway may also be crucial for breast cancer.
Fig. 1.
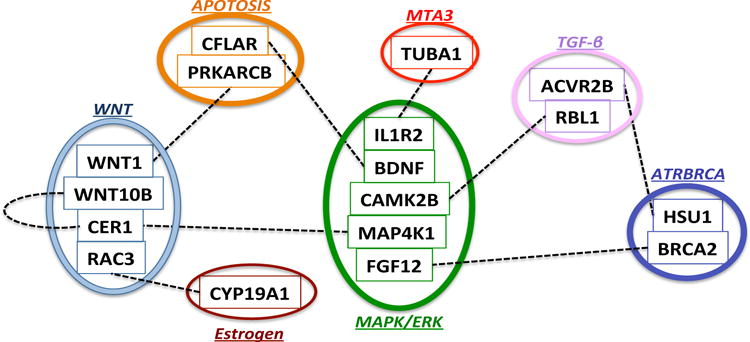
Identified gene-by-gene interactions for the breast cancer example. The dashed lines between gene-paris represent detected interactions. Genes inside each circle belong to the same pathway whose name is also shown.
A. Appendix: Proofs
A·1. Technical Lemmas
We prove the main results in this section. We begin by collecting technical lemmas proved in the supplementary material. The first lemma is the classical Bonferroni inequality.
Lemma A1 (Bonferroni inequality)
Let . For any k < [p/2], we have
where .
For d = 1, 2, let , and define for 1 ≤ i < j ≤ p and .
Lemma A2
Suppose that Conditions (C1), (4) and (5) hold. Then
and
for 1 ≤ i < j ≤ p, where is the empirical covariance between {εk,i,d : k = 1, …, nd} and {εk,j,d : k = 1, …, nd}. Consequently, uniformly in 1 ≤ i < j ≤ p,
and uniformly in 1 ≤ i ≤ p,
where is defined in (7), and .
Lemma A3
Let Xk ~ N(μ1, Σ1) for k = 1, …, n1 and Yk ~ N(μ2, Σ2) for k = 1, …, n2. Define
Then, for some constant C > 0, satisfies the large deviation bound
uniformly for 0 ≤ x ≤ (8 log p)1/2 and any subset .
The following lemma is needed for false discovery rate control in Theorem 4.
Lemma A4
Let Vi,j = (Ui,j,2−Ui,j,1){var(εk,i,1εk,j,1)/n1 + var(εk,i,2εk,j,2)/n2}−1/2. Under the same conditions as in Theorem 4, we have for any ε > 0 that,
where tp = (4 log p − log2 p – log3 p)1/2 and vp = 1/{log p(log4 p)2}1/2.
A·2. Proof of Theorem 1
Without loss of generality, throughout this section, we assume that ωi,i,d = 1 for d = 1, 2 and i = 1,…, p. Let A = {(i, j) : 1 ≤ i ≤ j ≤ p}. (C1) implies |Aτ|=o(p1/16). To prove Theorem 1, we first show that the terms in Aτ are negligible. Then we use Lemma 1, together with the Gaussian approximation technique, to show that , where Wi,j is defined in equation (9).
For d = 1, 2, let Vi,j = (Ui,j,2 − Ui,j,1)/{var(εk,i,1εk,j,1)/n1 + var(εk,i,d εk,j,d)/n2}1/2, where with . The proof of Lemma 2 yields
| (A1) |
and , where n = max{n1, n2}. Note that
| (A2) |
and Also note that for (i, j) ∈ A\Aτ, we have |ωi,j,d| = o{(log p)−1}. Then by Lemma 2, it is easy to see that, under conditions (C1), (4) and (5), we have, for (i, j) ∈ A\Aτ, . For (i,j) ∈ Aτ as a result of Lemma 2, we have Wi,j = Vi,j + bi,j + op 1og p–1/2), where , , and . Note that
where . Thus, we have
where the last equality is a direct result of Lemma 3. Thus it suffices to prove that
We arrange the indices {(i, j) : (i, j) ∈ A\Aτ} in any ordering and set them as {(im, jm) : m = 1, …, q} with q =Card(A\Aτ). Let n1/n2 ≤ K with K ≥ 1, , for d = 1, 2 and define for 1 ≤ k ≤ n2, for n2 + 1 ≤ k ≤ n1 + n2, and , where , and τn = 32K1 log(p + n). Note that , and that
Hence, . By the fact that , it suffices to prove that for any t ∈ ℝ, as n, p → ∞,
| (A3) |
By Lemma 1, for any integer l with 0 < l < q/2,
| (A4) |
where yp = 4 log p − log log p + t and . Let for m = 1, …, q and , for 1 ≤ k ≤ n1 + n2. Define for any vector a ∈ Rd. Then we have
Then it follows from Theorem 1 in Zaïtsev (1987) that
| (A5) |
where c1 > 0 and c2 > 0 are constants, εn → 0 which will be specified later and is a normal random vector with E(Nd) = 0 and . Recall that d is a fixed integer which does not depend on n, p. Because , we can let εn → 0 sufficiently slowly that, for any large M > 0
| (A6) |
Combining (A4), (A5) and (A6) we have
| (A7) |
Similarly, using Theorem 1 in Zaïtsev (1987) again, we can get
| (A8) |
We recall the following lemma, which is shown in the supplementary material of Cai et al. (2013).
Lemma A5
For any fixed integer d ≥ 1 and real number t ∈ ℝ,
| (A9) |
It then follows from Lemma 5, (A7) and (A8) that
for any positive integer l. By letting l → ∞, we obtain (A3) and Theorem 1 is proved.
A·3. Proof of Theorem 2
Let . It follows from the proof of Theorem 1 that , as n, p → ∞. By (A1), (A2) and the inequalities , and , we have pr(Mn ≥ qα + 4 log p − log log p) → 1 as n, p → ∞.
A·4. Proof of Theorem 3
To prove the lower bound result, we first construct the worst case scenario to test between Ω1 and Ω2, and then apply the arguments as shown in Baraud (2002).
Let ℳ denote the set of all subsets of {1,…, p} with cardinality pr, for r < 1/2. Let be a random subset of {1,…, p}, which is uniformly distributed on ℳ. We construct a class of Ω1, , such that ωi,j = 0 for i ≠ j and , for i, j = 1,…, p and ρ = c(log p/n)1/2, where c > 0 will be specified later. Let Ω2 = I and Ω1 be uniformly distributed on . Let μρ be the distribution of Ω1 − I. Note that μρ is a probability measure on , where is the class of matrices with pr nonzero entries. Let dpr1({Xn, Yn}) and dpr2({Xn, Yn}) be the functions with precision matrices Ω1 and Ω2 respectively, likelihood then we have
where is the expectation on Ω1. By the arguments in Baraud (2002), it suffices to show that . It is easy to check that
where and . Thus, we have
Set Ωm + Ωm′ − 2I = (ai,j). It is easy to show that ai,j = 0 for i ≠ j, aj,j = 0 if j ε (m ∪ m′)c, aj,j = 2(1/(1 + ρ)−1) if j ε m ∩ m′ and aj,j = 1/(1 + ρ) −1 if j ε m \ m′ \ m. Let t = | m ∩ m′|. Then
for r < 1/2. Thus, by letting c be sufficiently small, we have
A·5. Proof of Theorem 4
We first show that , as defined in Section 4, is obtained in the range (0, 2(log p)1/2). Then we illustrate that R0(t), defined in Section 4, is close to 2 {1 − Φ(t)}|ℋ0| by first showing the terms in Aτ are negligible. We then focus on the set ℋ0 \ Aτ and prove the result based on Lemma 4.
Under the condition of Theorem 4, we have Σ1≤i<j≤p I{|Wi,j| ≥ 2(log p)1/2} ≥ [1/{(8π)1/2 α} + δ](log2 p)1/2, with probability going to one. Hence we have with probability going to one,
Let tp = (4 log p − log2 p − log3 p)1/2. Because , we have according to the definition of in the false discovery rate control algorithm in Section 4. Note that, for , we have
Thus to prove Theorem 4, it suffices to prove that in probability, for 0 ≤ t ≤ {4 log p + o(log p)}1/2, where G(t) = 2{1 − Φ(t)}. Now we consider two cases.
- If t = {4 log p + o(log p)}1/2, the proof of Theorem 1 yields that . Thus, it suffices to prove that probability. For (i, j) ∊ ℋ0 \ Aτ, we have from the proof of Theorem 1 that max1≤i<j≤p | Wi,j − Vi,j | = op {(log p)−1/2}. Thus, it suffices to show that
in probability, where εi,j(t) = I(|Vi,j |≥ t) − G(t).(A10) - If t ≤ (C log p)1/2 with C < 4, we have
in probability. Thus, it is again enough to show that
in probability. Define . Let 0 ≤ t0 < ⋯ < tm = tp such that tl − tl−1 = vp for l = 1,…, m − 1 and tm − tm−1 ≤ vp. Thus we have m·~ tp/vp. For any t such that tl−1 ≤ t ≤ tl, we have(A11)
Thus it suffices to prove in probability. Note that
Thus by (A5) with d = 1 and Lemma 4, Theorem 4 is proved.
Footnotes
Supplementary Material
Supplementary material available at Biometrika online includes more extensive simulation esults comparing the numerical performance of the proposed global test with that of other tests, the proofs of Lemmas 2, 3 and 4, and the Matlab code for numerical implementation.
Contributor Information
Yin Xia, Department of Statistics & Operations Research, University of North Carolina at Chapel Hill, Chapel Hill, North Carolina 27514, USA.
Tianxi Cai, Department of Biostatistics, Harvard School of Public Health, Harvard University, Boston, Massachusetts 02115, USA.
T. Tony Cai, Department of Statistics, The Wharton School, University of Pennsylvania, Philadelphia, Pennsylvania 19104, USA.
References
- Anderson TW. An Introduction To Multivariate Statistical Analysis. 3rd New York: Wiley-Intersceince; 2003. [Google Scholar]
- Baraud Y. Non-asymptotic minimax rates of testing in signal detection. Bernoulli. 2002;8:577–606. [Google Scholar]
- Breiman L. Random forests. Mach Learn. 2001;45:5–32. [Google Scholar]
- Buck MB, Knabbe C. TGF-Beta signaling in breast cancer. Ann N Y Acad Sci. 2006;1089:119–126. doi: 10.1196/annals.1386.024. [DOI] [PubMed] [Google Scholar]
- Cai T, Liu W, Xia Y. Two-sample covariance matrix testing and support recovery in high-dimensional and sparse settings. J Am Statist Assoc. 2013;108:265–277. [Google Scholar]
- Chapman J, Clayton D. Detecting association using epistatic information. Genet Epidemiol. 2007;31:894–909. doi: 10.1002/gepi.20250. [DOI] [PubMed] [Google Scholar]
- Chatterjee N, Kalaylioglu Z, Moslehi R, Peters U, Wacholder S. Powerful multilocus tests of genetic association in the presence of gene-gene and gene-environment interactions. Am J Hum Genet. 2006;79:1002–1016. doi: 10.1086/509704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danaher P, Wang P, Witten DM. The joint graphical lasso for inverse covariance estimation across multiple classes. J R Statist Soc B. 2014;76:373–397. doi: 10.1111/rssb.12033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobra A, Hans C, Jones B, Nevins JR, Yao G, West M. Sparse graphical models for exploring gene expression data. J Multivariate Anal. 2004;90:196–212. [Google Scholar]
- Downward J. Targeting RAS signalling pathways in cancer therapy. Nat Rev Cancer. 2003;3:11–22. doi: 10.1038/nrc969. [DOI] [PubMed] [Google Scholar]
- Eichler EE, Flint J, Gibson G, Kong A, Leal SM, Moore JH, Nadeau JH. Missing heritability and strategies for finding the underlying causes of complex disease. Nat Rev Genet. 2010;11:446–450. doi: 10.1038/nrg2809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan J, Lv J. Sure independence screening for ultra-high dimensional feature space (with discussion) J R Statist Soc B. 2008;70:849–911. doi: 10.1111/j.1467-9868.2008.00674.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregg JP, Lit L, Baron CA, Hertz-Picciotto I, Walker W, Davis RA, Croen LA, Ozonoff S, Hansen R, Pessah IN, et al. Gene expression changes in children with autism. Genomics. 2008;91:22–29. doi: 10.1016/j.ygeno.2007.09.003. [DOI] [PubMed] [Google Scholar]
- Guardavaccaro D, Clevers H. Wnt/β-Catenin and MAPK Signaling: Allies and enemies in different battlefields. Sci Signal. 2012;5 doi: 10.1126/scisignal.2002921. pe15. [DOI] [PubMed] [Google Scholar]
- Hu VW, Sarachana T, Kim KS, Nguyen A, Kulkarni S, Steinberg ME, Luu T, Lai Y, Lee NH. Gene expression profiling differentiates autism case–controls and phenotypic variants of autism spectrum disorders: evidence for circadian rhythm dysfunction in severe autism. Autism Res. 2009;2:78–97. doi: 10.1002/aur.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klaus A, Birchmeier W. Wnt signalling and its impact on development and cancer. Nat Rev Cancer. 2008;8:387–398. doi: 10.1038/nrc2389. [DOI] [PubMed] [Google Scholar]
- Kooperberg C, Leblanc M. Increasing the power of identifying gene × gene interactions in genomewide association studies. Genet Epidemiol. 2008;32:255–263. doi: 10.1002/gepi.20300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kooperberg C, Ruczinski I. Identifying interacting SNPs using Monte Carlo logic regression. Genet Epidemiol. 2005;28:157–170. doi: 10.1002/gepi.20042. [DOI] [PubMed] [Google Scholar]
- Li J, Chen SX. Two sample tests for high-dimensional covariance matrices. Ann Statist. 2012;40:908–940. [Google Scholar]
- Li KC, Palotie A, Yuan S, Bronnikov D, Chen D, Wei X, Choi OW, Saarela J, Peltonen L. Finding disease candidate genes by liquid association. Genome Biol. 2007;8:R205. doi: 10.1186/gb-2007-8-10-r205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W. Gaussian graphical model estimation with false discovery rate control. Ann Statist. 2013;41:2948–2978. [Google Scholar]
- Marchini J, Donnelly P, Cardon L. Genome-wide strategies for detecting multiple loci that influence complex diseases. Nat Genet. 2005;37:413–417. doi: 10.1038/ng1537. [DOI] [PubMed] [Google Scholar]
- Mechanic L, Luke B, Goodman J, Chanock S, Harris C. Polymorphism Interaction Analysis (PIA): a method for investigating complex gene-gene interactions. BMC Bioinformatics. 2008;9:146. doi: 10.1186/1471-2105-9-146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore J. Computational analysis of gene-gene interactions using multifactor dimensionality reduction. Expert Rev Mol Diagn. 2004;4:795–803. doi: 10.1586/14737159.4.6.795. [DOI] [PubMed] [Google Scholar]
- Moro L, Arbini AA, Marra E, Greco M. Constitutive activation of MAPK/ERK inhibits prostate cancer cell proliferation through upregulation of BRCA2. Int J Oncol. 2007;30:217–224. doi: 10.3892/ijo.30.1.217. [DOI] [PubMed] [Google Scholar]
- Nathanson K, Wooster R, Weber B. Breast cancer genetics: what we know and what we need. Nat Med. 2001;7:552–556. doi: 10.1038/87876. [DOI] [PubMed] [Google Scholar]
- Olopade O, Grushko T, Nanda R, Huo D. Advances in Breast Cancer: Pathways to Personalized Medicine. Clin Cancer Res. 2008;14:7988. doi: 10.1158/1078-0432.CCR-08-1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips PC. Epistasisthe essential role of gene interactions in the structure and evolution of genetic systems. Nat Rev Genet. 2008;9:855–867. doi: 10.1038/nrg2452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie M, Hahn L, Roodi N, Bailey L, Dupont W, Parl F, Moore J. Multifactor-dimensionality reduction reveals high-order interactions among estrogen-metabolism genes in sporadic breast cancer. Am J Hum Genet. 2001;69:138–147. doi: 10.1086/321276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santen RJ, Song RX, Mcpherson R, Kumar R, Adam L, Jeng MH, Yue W. The role of mitogen-activated protein (MAP) kinase in breast cancer. J Steroid Biochem Mol Biol. 2002;80:239–256. doi: 10.1016/s0960-0760(01)00189-3. [DOI] [PubMed] [Google Scholar]
- Schott JR. A test for the equality of covariance matrices when the dimension is large relative to the sample sizes. Comput Stat Data An. 2007;51:6535–6542. [Google Scholar]
- Segal E, Shapira M, Regev A, Pe’er D, Botstein D, Koller D, Friedman N. Module networks: identifying regulatory modules and their condition-specific regulators from gene expression data. Nat Genet. 2003;34:166–176. doi: 10.1038/ng1165. [DOI] [PubMed] [Google Scholar]
- Shi Y, Massagué J. Mechanisms of TGF-β signaling from cell membrane to the nucleus. Cell. 2003;113:685–700. doi: 10.1016/s0092-8674(03)00432-x. [DOI] [PubMed] [Google Scholar]
- Srivastava MS, Yanagihara H. Testing the equality of several covariance matrices with fewer observations than the dimension. J Multivariate Anal. 2010;101:1319–1329. [Google Scholar]
- van de Vijver M, He Y, Van’t Veer L, et al. A gene-expression signature as a predictor of survival in breast cancer. N Engl J Med. 2002;347:1999–2009. doi: 10.1056/NEJMoa021967. [DOI] [PubMed] [Google Scholar]
- van’t Veer LJ, Dai H, Van De Vijver MJ, He YD, Hart AA, Mao M, Peterse HL, Van Der Kooy K, Marton MJ, Witteveen AT, et al. Gene expression profiling predicts clinical outcome of breast cancer. Nature. 2002;415:530–536. doi: 10.1038/415530a. [DOI] [PubMed] [Google Scholar]
- Venkitaraman AR. Cancer susceptibility and the functions of BRCA1 and BRCA2. Cell. 2002;108:171–182. doi: 10.1016/s0092-8674(02)00615-3. [DOI] [PubMed] [Google Scholar]
- Wada T, Penninger JM. Mitogen-activated protein kinases in apoptosis regulation. Oncogene. 2004;23:2838–2849. doi: 10.1038/sj.onc.1207556. [DOI] [PubMed] [Google Scholar]
- You L, He B, Uematsu K, Xu Z, Mazieres J, Lee A, Mccormick F, Jablons DM. Inhibition of wnt-1 signaling induces apoptosis in β-catenin-deficient mesothelioma cells. Cancer Res. 2004;64:3474–3478. doi: 10.1158/0008-5472.CAN-04-0115. [DOI] [PubMed] [Google Scholar]
- Zaïtsev AY. On the gaussian approximation of convolutions under multidimensional analogues of sn bernstein’s inequality conditions. Probab Theory Rel. 1987;74:535–566. [Google Scholar]
- Zerba K, Ferrell R, Sing C. Complex adaptive systems and human health: the influence of common genotypes of the apolipoprotein E (ApoE) gene polymorphism and age on the relational order within a field of lipid metabolism traits. Hum Genet. 2000;107:466–475. doi: 10.1007/s004390000394. [DOI] [PubMed] [Google Scholar]