

Abstract
ST2, a member of the interleukin (IL) 1 receptor family, and its ligand IL-33 play critical roles in immune regulation and inflammatory responses. This study explores the roles of endogenous IL-33/ST2 signaling in ischemic brain injury and elucidates the underlying mechanisms of action. The expression of IL-33 rapidly increased in oligodendrocytes and astrocytes after 60 min transient middle cerebral artery occlusion (tMCAO). ST2 receptor deficiency exacerbated brain infarction 3 d after tMCAO as well as distal permanent MCAO. ST2 deficiency also aggravated neurological deficits up to 7 d after tMCAO. Conversely, intracerebroventricular infusions of IL-33 after tMCAO attenuated brain infarction. Flow cytometry analyses demonstrated high levels of ST2 expression on microglia, and this expression was dramatically enhanced after tMCAO. The absence of ST2 enhanced the expression of M1 polarization markers on microglia/macrophages, and impaired the expression of M2 polarization markers after tMCAO. In vitro studies on various types of cultures and coculture systems confirmed that IL-33/ST2 signaling potentiated expression of IL-10 and other M2 genes in primary microglia. The activation of ST2 on microglia led to a protective phenotype that enhanced neuronal survival against oxygen glucose deprivation. Further in vitro studies revealed that IL-33-activated microglia released IL-10, and that this was critical for their neuroprotective effects. Similarly, intracerebroventricular infusions of IL-33 into IL-10 knock-out mice failed to provide neuroprotection against tMCAO in vivo. These results shed new light on the IL-33/ST2 axis as an immune regulatory mechanism that serves as a natural brake on the progression of ischemic brain injury.
SIGNIFICANCE STATEMENT This is the first study to identify the function of interleukin (IL) 33/ST2 signaling in poststroke microglial responses and neuroprotection against ischemia. Using two models of ischemic stroke, we demonstrate here that ST2 deficiency shifted microglia/macrophages toward a M1-like phenotype, thereby expanding brain infarcts and exacerbating long-term behavioral deficits after stroke. Using stroke models and various in vitro culture and coculture systems, we further characterized a previously undefined mechanism whereby IL-33/ST2 engagement stimulates the production of IL-10 from microglia, which, in turn, enhances neuronal survival upon ischemic challenge. These results shed light on endogenous IL-33/ST2 signaling as a potential immune regulatory mechanism that serves to promote beneficial microglial responses and mitigate ischemic brain injury after stroke.
Keywords: IL-10, IL-33, microglia, ST2
Introduction
Ischemic strokes occur when a thrombus or an embolus in the cerebral arteries blocks blood flow to brain parenchyma. Following loss of oxygen and nutrient delivery, rapid cell loss unfolds within the ischemic core, which triggers a wave of immune responses (Iadecola and Anrather, 2011). Recent research has highlighted a central role for microglia in the immune response to ischemic stroke. Microglia represent the first line of defense against ischemic brain injury and are recruited to the site of injury soon after stroke onset. Interestingly, these mobilized microglia do not constitute a uniform population of cells. Instead, they polarize into a spectrum of phenotypes that play distinct roles at different stages after brain ischemia. Of all the assumed phenotypes, the classically activated M1 and alternatively activated M2 phenotypes are the most widely studied and are thought to represent two extreme activation states of microglia. Although the M1–M2 dichotomy is somewhat oversimplified, it remains a useful framework to understand and manipulate the functional status of microglia and macrophages. In our previous study, we reported that the M1-inducer lipopolysaccharide (LPS) caused microglia to exacerbate ischemic neurotoxicity, whereas the M2-inducer interleukin (IL) 4 caused microglia to prevent neuronal death (Hu et al., 2012). Therefore, regulating microglial phenotype is a new immunomodulatory strategy to reduce acute brain injuries, such as ischemia. However, the underlying mechanisms remain poorly explored.
ST2 is a member of the IL-1 receptor family. It has two isoforms: the soluble ST2 (sST2) and the membrane-bound ST2 (tST2). tST2 forms heterodimers with IL-1 receptor accessory protein and serves as a receptor for IL-33, which is a nuclear “alarmin” (i.e., immune activator) released after cell damage (Garlanda et al., 2013). ST2 is mainly expressed on various immune cells. The expression of ST2 on T lymphocytes has been shown to drive the production of type-2 cytokines and is responsible for protective type-2 inflammatory responses during infections and tissue repair (Garlanda et al., 2013). ST2 depletion results in enhanced Th1/Th17 responses and antitumor immunity (Jovanovic et al., 2011). In addition, IL-33–ST2 interactions are known to be important for the proliferation and functions of regulatory T cells (Arpaia et al., 2015; Lu et al., 2016). ST2 is also expressed in macrophages and upregulated in response to LPS or IL-4/IL-13 stimulation (Oshikawa et al., 2002; Espinassous et al., 2009). In the placenta and in a model of bleomycin-induced lung injury and fibrosis, macrophages are the major source of IL-33. Macrophage-derived IL-33 appears to induce M2 macrophage polarization in a paracrine and autocrine manner (Fock et al., 2013; Li et al., 2014). Administration of recombinant IL-33 also induces polarization of macrophages toward an alternatively activated M2 phenotype in adipose and lung tissue (Kurowska-Stolarska et al., 2008; Miller et al., 2010). Recent studies show that CNS damage can trigger the release of IL-33 from injured brain cells—mainly oligodendrocytes (OLs) and astrocytes (Gadani et al., 2015). Together, these observations prompt us to explore the function of endogenous IL-33/ST2 signaling in ischemic stroke and investigate whether this system is increased with injury and functions as an alarmin to activate microglia.
Using two models of ischemic stroke, we demonstrated that ST2 receptor deficiency exacerbated brain infarction and long-term behavioral deficits after stroke. Further in vivo and in vitro studies revealed the importance of the IL-33/ST2 axis in poststroke microglial responses and subsequent protection of ischemic neurons. Mechanistic studies showed that IL-33/ST2 engagement stimulates the production of IL-10 from microglia, which in turn enhances neuronal survival in experimental stroke. These findings support the view that IL-33/ST2 signal transduction activates beneficial immune responses, serving to temper a downward spiral of ischemic brain injury.
Materials and Methods
Animals.
All animal experiments were approved by the University of Pittsburgh Institutional Animal Care and Use Committee and performed in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. Wild-type (WT) and IL-10 knock-out (KO) C57BL/6 mice (8–12 weeks old) were obtained from the Jackson Laboratory. ST2 KO (Townsend et al., 2000) C57BL/6 mice were bred for experimental use at the University of Pittsburgh. ST2 KO mice were backbred to the C57BL/6 background for eight generations before use, to minimize the potential influence of genetic heterogeneity on the stroke model. WT littermates were used as controls. All mice were housed in the specific pathogen-free facility with a 12 h light/dark cycle at the University of Pittsburgh. Mice were randomly assigned to experimental groups using a lottery drawing box. All efforts were made to minimize animal suffering.
Murine models of transient or permanent focal ischemia.
Transient focal ischemia was induced by intraluminal occlusion of the left middle cerebral artery (tMCAO) for 1 h with silicone-coated sutures (Doccol) as previously described (Li et al., 2013). Physiological parameters were maintained within normal ranges. Regional cerebral blood flow (rCBF) was monitored in all stroke animals using laser Doppler flowmetry. Animals that died or failed to show ≥70% rCBF reduction of the preischemia levels were excluded from further analyses. Sham-operated animals underwent the same anesthesia and exposure of the arteries without MCAO induction. The tMCAO or sham surgeries were performed by an investigator blinded to genotype and treatment.
Distal focal ischemia was produced by permanent distal MCA occlusion (dMCAO) plus ipsilateral common carotid artery (CCA) occlusion, as previously reported (Suenaga et al., 2015). In brief, the left CCA was exposed and occluded by ligation. After opening a burr hole between the left eye and ear and performing a craniotomy, the distal part of the MCA was exposed. The dMCAO was completed at the immediate lateral part of the rhinal fissure.
A total of 235 mice (16 sham-operated and 219 ischemic mice) were used in this study, including 21 mice that were excluded from further assessments, either because of death after ischemia or failure of ischemia induction. The mortality during tMCAO surgery was 6.9% in WT male mice (4 of 58), 8.3% in ST2 KO male mice (4 of 48), 5.26% in WT female mice (1 of 19), and 6.25% in ST2 KO female mice (1 of 16). The mortality during dMCAO surgery was 0% in WT male mice (0 of 11) and 0% in ST2 KO male mice (0 of 9). The mortality during tMCAO in mice receiving intracerebroventricular infusions was 12.5% in WT male mice (3 of 24) and 7.7% in IL-10 KO male mice (1 of 13). The mortality during tMCAO surgery was 12.5% in bone-marrow chimera mice (2 of 16).
Cortical CBF measurements.
Regional CBF was monitored using the laser speckle technique, as previously described (Li et al., 2013). Briefly, images were acquired through the laser speckle contrast imager (PeriCam PSI System, Perimed). WT and ST2 KO male mice were subjected to repeated measurements of CBF before and during MCAO, as well as 10 min after reperfusion. CBF changes were expressed as a percentage of pre-MCAO baseline.
Intracerebroventricular IL-33 administration.
WT and IL-10 KO mice subjected to 60 min MCAO were randomly assigned to vehicle or IL-33 groups. Mice were anesthetized and stabilized in stereotaxic frames. IL-33 (Enzo Life Science; 0.1 μg/animal) or vehicle (PBS) was injected intracerebroventricularly (2 μl total volume) 30 min after MCAO. The coordinates for the infusions were 0.8 mm lateral to bregma, 0.2 mm posterior to bregma, and 2.5 mm ventral to the skull.
Irradiation and bone-marrow transplantation.
To construct bone-marrow chimeric mice, 8-week-old recipient male C57BL/6 WT and ST2 KO mice were anesthetized and exposed to lethal gamma irradiation (10 gray) with their heads shielded, followed 6 h later by intravenous transplantation of bone-marrow cells obtained from 6-week-old WT C57BL/6 donors (107 cells per recipient; Tang et al., 2012). Chimera mice were allowed to recover and subjected to brain ischemia 6 weeks after irradiation.
Measurements of infarct volume.
Measurements of infarct volumes were performed as described previously (Li et al., 2013). For 2,3,5-triphenyltetrazolium chloride (TTC) staining, brains were removed and sliced into seven 1-mm-thick coronal sections. Sections were immersed in prewarmed 2% TTC (Sigma-Aldrich) in saline for 10 min and then fixed in 4% paraformaldehyde. The neuron-specific marker microtubule associated protein 2 (MAP2) was visualized with anti-MAP2 (Santa Cruz Biotechnology) immunofluorescence staining. Infarct volumes were measured by a blinded observer using National Institutes of Health ImageJ software on TTC-stained or MAP2-stained sections. Infarct volumes were corrected for brain edema by reporting the volume of the contralateral hemisphere minus the noninfarcted volume of the ipsilateral hemisphere.
Real-time PCR.
Total RNA was extracted from cerebral tissues (ischemic hemisphere) using the RNeasy Lipid Tissue Mini Kit (Qiagen) or from cells using the RNeasy Mini Kit (Qiagen). Total RNA was reverse transcribed into cDNA using the Superscript III First-Strand Synthesis Supermix (Invitrogen) according to the manufacturer's protocols. PCR was conducted on the Opticon 2 Real-Time PCR Detection System (Bio-Rad) using corresponding primers (Table 1) and SYBR gene PCR Master Mix (Invitrogen). All data were normalized to GAPDH mRNA levels as an internal control. The expression levels of the mRNAs were then reported as percentages versus control or sham.
Table 1.
Primers for RT-PCR
| Forward | Reverse | |
|---|---|---|
| Mice | ||
| IL-33 | ||
| CD16 | AAGGCCAAACACAGCATACC | GTCTTCCTTGAGCACCTGGATC |
| CD206 | CAAGGAAGGTTGGCATTTGT | CCTTTCAGTCCTTTGCAAGC |
| IL-10 | CCAAGCCTTATCGGAAATGA | TTTTCACAGGGGAGAAATCG |
| TGF-b | TGCGCTTGCAGAGATTAAAA | CGTCAAAAGACAGCCACTCA |
| iNOSa | CAAGCACCTTGGAAGAGGAG | AAGGCCAAACACAGCATACC |
| GAPDH | AAGATGGTGAAGGTCGGTG | GTTGATGGCAACAATGTCCAC |
| Rat | ||
| CD16 | AAGGCCAAACACAGCATACC | GTCTTCCTTGAGCACCTGGATC |
| CD206 | GGTTCCGGTTTGTGGAGCAG | TCCGTTTGCATTGCCCAGTA |
| IL-10 | CATGGCCCAGAAATCAAGGA | AGCGTCGCAGCTGTATCCA |
| TGF-b | GCCTGAGTGGCTG TCTTTTGA | GAAGCGAAAGCCCTGTATTCC |
| iNOSa | CCTGGTGCAAGGGATCTTGG | GAGGGCTTGCCTGAGTGAGC |
| GAPDH | TGCTGGTGCTGAGTATGTCGTG | CGGAGATGATGACCCTTTTGG |
aiNOS, Inducible nitric oxide synthase.
Immunofluorescence staining and quantification.
Immunofluorescence staining was performed on 25 μm free-floating brain sections or 4% formaldehyde-fixed cultures, as described previously (Li et al., 2013). Primary antibodies included the following: mouse anti-IL-33 (Abcam), mouse anti-CC1 (Calbiochem), goat anti-GFAP (Abcam), rabbit anti-NeuN (Millipore), rabbit anti-NG2 (Millipore), rabbit anti-Iba1 (Wako), rabbit anti-MAP2 (Santa Cruz Biotechnology), rabbit anti-ST2 (Thermo Fisher Scientific), goat anti-Iba1 (Abcam), goat anti-CD206 (R&D Systems), rat anti-CD16 (BD Bioscience), rabbit anti-IL-10 (Abcam). F-actin staining was performed using Alexa Fluor 594-conjugated phalloidin (Invitrogen). TUNEL staining (fluorescein) was performed with the In Situ Cell Death Detection Kit (Roche). Cells were then counterstained with 40,6-diamidino-2-phenylindole for nuclear labeling. Immunostaining was analyzed with confocal microscopy (Olympus). All images were processed with ImageJ by a blinded observer for the unbiased counting of automatically recognized cells. Means of positively stained cell counts were calculated from three microscopic fields in the cortex and three in the striatum of each section, and three sections were analyzed for each brain. Data are expressed as mean numbers of cells per square millimeter.
Assessment of neurological deficits.
Neurological deficits were assessed by a blinded investigator using a five-point-scale neurological score (0, no observable deficit; 1, torso flexion to right; 2, spontaneous circling to right; 3, leaning/falling to right; 4, no spontaneous movement; 5, death).
Behavioral tests.
All behavioral tests were performed by an investigator blinded to experimental groups. The Rotarod (IITC Life Science) test was performed to determine sensorimotor coordination. Mice were placed on an accelerated rotating rod with the speed increasing from 4 to 120 rpm within 5 min. Mice were tested three times daily with an intermission of 5 min. Latency to fall off the rotating rod was recorded. Data were expressed as mean values from three trials. The adhesive removal test was performed to assess tactile responses and sensorimotor asymmetries. Two 2 × 3 mm adhesive tapes were applied to both forepaws. Tactile responses were measured by recording the time to initially contact the ipsilateral and contralateral paws, as well as the time to remove the adhesive tape from each side, with a maximum observation period of 120 s. The cylinder test was used to assess forepaw use and rotation asymmetry, as described previously (Li et al., 2013). The mouse was placed in a transparent cylinder 9 cm in diameter and 15 cm in height, and videotaped during the test. Videotapes were analyzed, and forepaw contacts (left/right/both) against the cylinder wall after rearing and during lateral exploration were recorded. A total of 20 such movements were recorded during a maximal period of 10 min. Nonimpaired forepaw (left) preference was expressed as the relative proportion of left forepaw contacts and calculated as follows: (left − right)/(right + left + both) × 100.
Flow cytometry.
Single-cell suspensions were prepared from sham or MCAO brains using the Neural Tissue Dissociation Kit (Miltenyi Biotec), according to the manufacturer's instructions. Cells were separated on a 30–70% Percoll gradient (GE Healthcare BioSciences). The mononuclear cells at the interface were stained with fluorophore-labeled anti-mouse glial glutamate–aspartate transporter (GLAST; Miltenyi Biotec), CD11b (eBioscience), CD45 (eBioscience), ST2 (MD Bioproducts), and the appropriate isotype controls (eBioscience). Cells were then permeabilized with the intracellular staining kit (eBioscience), according to the manufacturer's protocol, and stained with Olig-2 (Millipore). Flow cytometric analysis was performed using a FACS flow cytometer (BD Biosciences).
Primary cortical neuron–glia cultures and treatments.
Primary neuron–glia (N/G) cultures were prepared from the brains of mixed-sex embryonic day 17 ± 0.5 Sprague Dawley rats or WT and ST2 KO mice, as previously described (Hu et al., 2008). Seven-day-old cultures were subjected to 3 h oxygen–glucose deprivation (OGD) or control treatment. OGD or control N/G cultures were treated with 12.5, 25, 50, or 100 ng/ml IL-33 (Enzo Life Science) or PBS immediately after OGD for 24 h. In some experiments, OGD or control N/G cultures were treated with PBS or 50 ng/ml IL-33 in the presence of IL-10 neutralizing antibodies (R&D Systems), TGF-β neutralizing antibodies (R&D Systems), IL-10 rat isotype IgG (R&D Systems), or TGF-β rabbit isotype IgG (R&D Systems) immediately after OGD for 24 h. Cultures were fixed with 4% formaldehyde for further analysis.
Primary cortical neuronal cultures and treatments.
Primary cortical neuronal cultures were prepared from 17-d-old mixed-sex Sprague Dawley rat embryos or WT and ST2 KO mice embryos, as previously described (Shi et al., 2016). OGD (1.5 h) and control neurons were treated with 12.5, 25, 50, or 100 ng/ml IL-33 or PBS for 24 h. Neuronal survival was assessed by the MTT assay.
OGD and conditioned media collection.
To model ischemia in vitro, mixed N/G cultures or neuron-enriched cultures were exposed to transient OGD for 3 or 1.5 h, respectively (Shi et al., 2016). Control cultures were incubated for the same period of time at 37°C in humidified 95% air and 5% CO2. Conditioned media (CM) were collected from OGD neurons (OGD-CM) or healthy control neurons (CON-CM) 24 h later and concentrated using centrifugal filters (Millipore).
Primary microglia-enriched cultures and treatments.
Primary microglia-enriched cultures were prepared from whole brains of 1-d-old mixed-sex Sprague Dawley rat pups or WT and ST2 KO mice pups, as described previously (Hu et al., 2008). Microglia were treated with 50 ng/ml IL-33 or PBS for 24 h. Cells were collected for real-time reverse-transcription PCR (RT-PCR) or ELISA or fixed for immunofluorescence staining. In some experiments, microglia were treated with OGD-CM in the presence or absence of 50 ng/ml IL-33 and CON-CM for 24 h. Cells were then collected for RT-PCR.
Neuron–microglia cocultures.
Primary microglia (1 × 105/well) were seeded in culture inserts and treated with OGD-CM or CON-CM in the presence or absence of IL-33 for 24 h. These pretreated microglia were then added to 11-d-old neuron-enriched cultures that had been subjected to 1.5 h OGD or sham conditions in the presence of 50 ng/ml IL-33, with or without IL-10 antibodies (R&D Systems) or IL-10 rat isotype IgG (R&D Systems). Neuronal survival was analyzed 24 h after coculture using TUNEL staining.
Quantitative MAP2 ELISA.
N/G cultures were fixed in 96-well plates with 4% paraformaldehyde. MAP2 ELISA was performed, as previously described (Hu et al., 2012).
IL-10 ELISA.
Cell-culture supernatant and cell lysates were harvested as described above. Protein concentrations were measured using IL-10 commercial ELISA quantification kits (R&D Systems), according to the manufacturer's instructions.
Statistical analysis.
All data were reported as mean ± SEM. The two-tailed Student's t test was used for the comparison of two groups. Differences in means across multiple groups were analyzed using one-way ANOVA. Differences in means across multiple groups with multiple measurements over time were analyzed using repeated-measures ANOVA. When the ANOVA revealed significant differences, the post hoc Bonferroni's test was used for pairwise comparisons between means. When parametric assumptions were not met, the nonparametric two-tailed Mann–Whitney U was applied. A P value of ≤0.05 was considered statistically significant.
Results
Ischemic injury induces an early elevation of IL-33 in the brain
IL-33 mRNA levels were measured in sham and stroke mice at various time points after the onset of ischemia. A dramatic but transient increase in IL-33 mRNA was detected 1 d after stroke (Fig. 1A). Recent studies suggest that spinal cord injury rapidly triggers the release of IL-33 predominantly from injured OLs and astrocytes (Gadani et al., 2015). Similarly, in our stroke model, we observed a robust increase in IL-33 protein expression in CC-1+ OLs (Fig. 1C,D) and in GFAP+ astrocytes (Fig. 1E,F) 1 d after stroke. In contrast, IL-33 expression was not observed in NeuN+ neurons, NG2+ oligodendrocyte precursor cells, or Iba1+ microglia after stroke (Fig. 1G).
Figure 1.
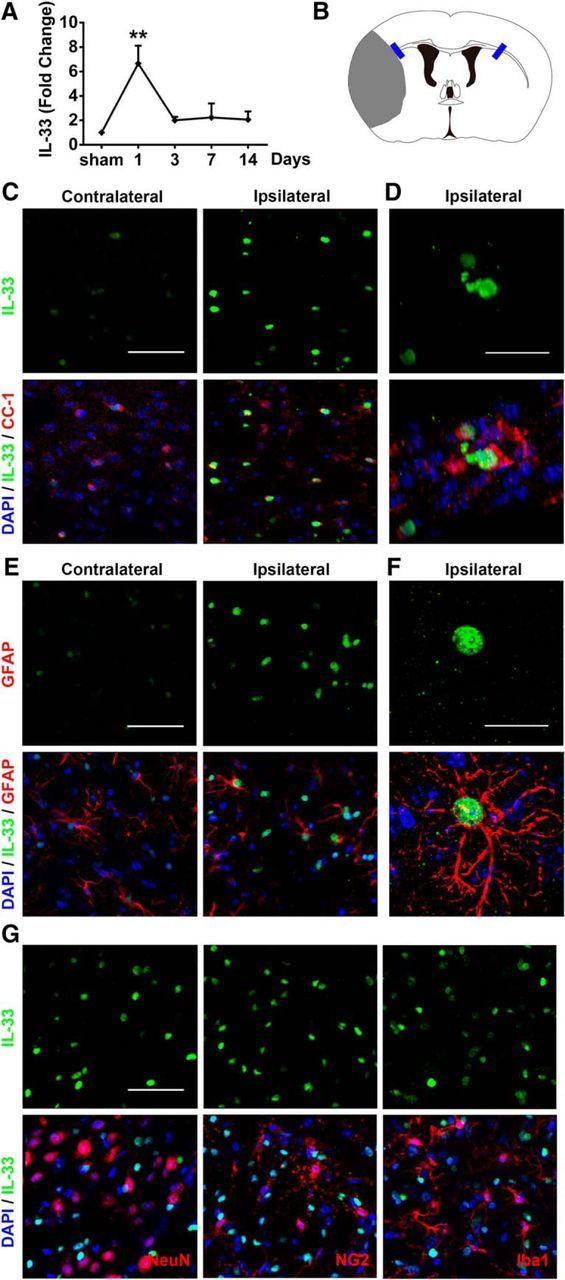
IL-33 expression is elevated in mature oligodendrocytes and astrocytes after ischemic brain injury. A, Real-time reverse-transcription PCR (RT-PCR) of IL-33 was performed using total RNA extracted from the ischemic brain at 1, 3, 7, and 14 d after 60 min tMCAO or sham operation. n = 3 per group, **p < 0.01 versus sham (1-way ANOVA followed by the Bonferroni's post hoc test). B, Blue rectangles illustrate the anatomical location of images in C–G in the ipsilateral peri-infarct regions and contralateral corresponding areas. C, Representative images of IL-33 (green) and CC-1 (red) labeling in WT mice at 1 d after ischemic injury. Scale bar, 50 μm. D, Colocalization of IL-33 in CC-1+ cells was observed in the ischemic brain. Scale bar, 20 μm. E, Representative images of IL-33 (green) and GFAP (red) labeling in WT mice at 1 d after ischemic injury. Scale bar, 50 μm. F, Colocalization of IL-33 in GFAP+ cells. Scale bar, 20 μm. G, No IL-33 staining was observed in NeuN+, NG2+, or Iba-1+ cells 1 d after ischemia. Scale bar, 50 μm. Nuclei were stained blue with 4′,6-diamidino-2-phenylindole (DAPI). All images are representative of three independent experiments.
ST2 is critical in restricting acute brain injury in mouse models of stroke
As mentioned above, IL-33 binds to the ST2 receptor. Thus, to evaluate the role of endogenous IL-33/ST2 in acute ischemic injury, we subjected male ST2-deficient mice and their male WT littermates to 60 min tMCAO (Fig. 2A). rCBF was monitored before and during ischemia, as well as 10 min after reperfusion. There were no significant differences in rCBF between the two mouse lines, suggesting that both groups were exposed to the same degree of ischemia (Fig. 2B,C). Physiological parameters were also monitored and maintained at similar levels in ST2 KO and WT mice. Infarct volumes were quantified at 3 d after tMCAO using TTC staining. As shown in Figure 2D–F, lack of ST2 expression enlarged the brain infarct sizes. NeuN and TUNEL double staining confirmed a significant increase in neuronal death (NeuN+TUNEL+) 3 d after tMCAO in ST2-deficient mice (Fig. 2G–I).
Figure 2.

ST2 deficiency exacerbates ischemic brain injury after MCAO. A, Timeline of experimental design. B, rCBF was monitored by laser speckle imaging before and during 60 min tMCAO, as well as 10 min after reperfusion. C, Quantification of rCBF. n = 4 per group. D, Representative TTC-stained coronal sections illustrate larger brain infarct sizes in ST2 KO male mice than in WT male mice at 3 d after tMCAO. E, Infarct volumes at 3 d after tMCAO in WT and ST2 KO male mice (n = 9 per group). F, Infarct areas of consecutive coronal sections spaced 1 mm apart throughout the MCA territory at 3 d after tMCAO (2-way ANOVA followed by the Bonferroni's post hoc test). G, Blue squares illustrate the anatomical location of images in H in the ipsilateral peri-infarct regions and contralateral corresponding areas. H, Representative images of NeuN and TUNEL at 3 d after tMCAO in WT (top) and ST2 KO (bottom) male mice. I, The quantification of NeuN and TUNEL dual-labeled cells demonstrates significantly increased neuronal death (NeuN+TUNEL+) at 3 d after tMCAO in ST2-deficient mice. J, Representative TTC-stained coronal sections illustrate larger brain infarct sizes in ST2 KO male mice than in WT male mice at 3 d after dMCAO. K, Infarct volumes in WT and ST2 KO male mice at 3 d after dMCAO (n = 9–11 per group). L, TTC staining and infarct volumes in WT and ST2 KO female mouse brains at 3 d after tMCAO (n = 6–9 per group). M, TTC staining and infarct volumes in WT and ST2 KO OVX female mouse brains at 3 d after tMCAO (n = 8–9 per group). N, TTC staining and infarct volumes in ST2 KO mice reconstituted with WT bone marrow (ST2KO/WT) and in WT mice reconstituted with WT bone marrow (WT/WT) at 3 d after tMCAO (n = 7 per group). Data are mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001 versus WT or WT/WT (2-tailed Student's t test).
To confirm the reproducibility of our findings, we evaluated whether the effects of ST2 on ischemic brain injury can be generalized to other stroke models. To this end, male ST2 KO and WT mice were subjected to permanent dMCAO. Once again, TTC staining revealed the expected deterioration of brain infarcts in ST2 KO mice at 3 d after dMCAO (Fig. 2J,K).
Next, we investigated the effects of ST2 in female stroke mice. Female animals are thought to be protected against ischemic brain injury by the neurosupportive effects of estrogen (Koellhoffer and McCullough, 2013). Therefore, vaginal smears were performed to choose females in proestrus or estrus phases, when estrogen levels are high. Similar to the findings in males, female ST2-deficient mice exhibited enlarged brain infarct volumes 3 d after 60 min tMCAO (Fig. 2L), suggesting that ST2 is also protective in female mice. We also performed experiments using ovariectomized (OVX) female mice to examine ST2-mediated neuroprotection without the effects of estrogen (Gibson et al., 2011; Cai et al., 2014). Eight-week-old female WT and ST2 KO mice were subjected to OVX, and then to tMCAO 3 weeks after OVX. As shown in Figure 2M, infarct volumes in ST2-deficient OVX female mice were larger than in WT OVX female mice. These results confirm that ST2 is protective in mice of both genders, and that it mitigates ischemic injury in the female brain under conditions of low or high estrogen levels.
To confirm that ST2 expression specifically on CNS cells is critical for the mitigation of brain infarction early after stroke, we used bone-marrow chimeras. Two groups were studied: (1) WT marrow transplantation into WT recipients, designated as WT/WT, and (2) WT marrow transplantation to ST2 KO recipients, designated as ST2KO/WT. The success of hematopoietic cell reconstitution was confirmed by white blood cell counts (data not shown). ST2KO/WT mice, whose CNS cells were ST2KO and peripheral immune cells were ST2 positive, exhibited enlarged brain infarcts compared with WT/WT mice (Fig. 2N). These findings confirm the importance of ST2 expression on CNS cells in mitigating ischemic injury in the acute phase of stroke. It is noted that the infarct volume in ST2KO/WT mice was smaller than that in the ST2 KO mice (Fig. 2D–F), suggesting that ST2 expression on peripheral immune cells may also influence stroke outcomes.
Together, the data gathered thus far suggested that there is a robust increase in IL-33 shortly after stroke and that the IL-33 receptor ST2 mitigates the severity of the ischemic lesion in mice of both genders and in multiple stroke models.
ST2 deficiency aggravates neurological deficits and tissue loss 7 d after stroke
Neurological functions after stroke were assessed in WT and ST2 KO mice. ST2 KO mice exhibited inferior neurological function 2 and 3 d after tMCAO (Fig. 3A). Several sensorimotor tests, including the Rotarod (Fig. 3B), adhesive removal (Fig. 3C,D), and cylinder tests (Fig. 3E) consistently revealed worse neurological performance in ST2 KO mice up to 7 d after stroke. The sham WT and sham ST2 KO mice exhibited no significant differences in any behavioral tests, suggesting equivalent baselines in both genotypes. Thus, data from these two groups were combined.
Figure 3.

ST2 deficiency exacerbates neurobehavioral deficits and brain damage up to 7 d after tMCAO. A, ST2 loss worsened neurological deficits over 3 d after tMCAO (n = 13–14 per group, 2-tailed Mann–Whitney U test). B–E, Sensorimotor function was evaluated up to 7 d after tMCAO in ST2 KO and WT mice. B, Rotarod test. Data are expressed as the duration on the Rotarod. n = 9–12 per group (repeated-measures ANOVA). C, D, Adhesive removal test. Data are expressed as the latency to contact (C) and remove (D) the tape from the impaired forepaw. n = 10–12 per group (repeated-measures ANOVA). E, Cylinder test. The number of left, right, or bilateral forepaw contacts were counted, and performance asymmetry was expressed as follows: (left − right) / (left + right + both) × 100% paw use in 10 min. n = 8–12 per group (repeated-measures ANOVA). F, Representative MAP2-stained coronal sections illustrate larger infarct sizes in ST2 KO mouse brains, compared with WT mouse brains at 7 d after tMCAO. Dotted lines encompass infarct zone. G, Infarct volumes in WT and ST2 KO mouse brains at 7 d after tMCAO (n = 8–12 per group, 2-tailed Student's t test). H, 7 d survival rate. ST2 KO mice exhibited lower 7 d survival rates after tMCAO, compared with WT mice (n = 15 per group). Data are mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001 versus WT.
Consistent with poor behavioral performance, ST2 deficiency exacerbated MAP2+ brain tissue loss 7 d after stroke (Fig. 3F,G). Notably, mortality rates were also elevated in ST2 KO mice after tMCAO. Only 53.3% of ST2 KO mice survived to 7 d after tMCAO, compared with significantly higher survival rates (93.3%) in WT littermates (Fig. 3H). These findings confirm the critical nature of the ST2 receptor, both for survival after stroke and for mitigating subsequent tissue loss.
ST2 is highly expressed on microglia under physiological and pathological conditions
To determine the cellular targets of IL-33/ST2 signaling in the brain, we used flow cytometry to explore the expression of ST2 on different CNS cells in the normal and the ischemic brain at 3 d after stroke induction. A combination of cell-specific markers was used to identify Glast+ astrocytes, Glast−CD11b+CD45intermediate microglia, Glast−CD11b+CD45high infiltrating myeloid cells (mainly macrophages and neutrophils), Glast−CD11b−CD45−Oligo2+ OLs and other Glast−CD11b−CD45−Oligo2− CNS cells (Fig. 4A). In normal brains, ST2 was mainly expressed in Glast−CD11b+CD45intermediate microglia and Glast+ astrocytes (Fig. 4B–D). After stroke, the expression of ST2 was dramatically increased in these two populations. Ischemic stroke also robustly elevated the number of ST2+CD11b+CD45high macrophages/neutrophils in the ischemic brain (data not shown). Immunohistochemical staining confirmed the expression of ST2 on Iba1+ microglia/macrophages 3 d after stroke (Fig. 4G–I). There was no expression of ST2 in OLs or other CNS cells before or after stroke (Fig. 4B,E,F). Together, the data gathered thus far suggest that stroke increases the expression of the ST2 receptor on microglia after stroke, which may facilitate the microglial response to a dramatic and rapid increase in IL-33 expression after ischemic injury.
Figure 4.

ST2 expression on microglia/macrophages is elevated after tMCAO. A, Representative gating strategy for Glast+ astrocytes, Glast−CD11b+CD45intermediate microglia, Glast−CD11b+CD45high macrophages/neutrophils, Glast−CD11b−CD45−Oligo2+ oligodendrocyte populations, and other Glast−CD11b−CD45−Oligo2− CNS cells in the brain 3 d after tMCAO. B, Histograms showing ST2 expression on microglia, astrocytes, oligodendrocytes, and other CNS cells at 3 d after tMCAO or sham operation. The green and red lines represent staining in sham and ischemic brains, respectively, at 3 d after surgery. The gray line represents isotype control staining. C–F, Quantification of mean fluorescence intensity (MFI) of ST2 on microglia (C), astrocytes (D), oligodendrocytes (E), and other CNS cells (F) in sham brain and ischemic brain at 3 d after surgery. Data are mean ± SEM, n = 5–7 per group, **p < 0.01, ***p < 0.001 versus sham (2-tailed Student's t test). G, Blue square illustrates the anatomical location of images in H and I in ipsilateral peri-infarct regions at 3 d after tMCAO. H, Representative images of ST2 (green) and Iba1 (red) labeling in the ischemic brain. Scales bars: top, 30 μm; bottom, 5 μm. I, Colocalization of ST2 and Iba1 was observed in the ischemic brain at 3 d after tMCAO. Scale bar, 4 μm.
ST2 signaling is protective in mixed N/G cultures but not in neuron-enriched cultures
We used in vitro cultures to extend our in vivo observations and further explore the cell-specific mechanisms of ST2-afforded protection. First, we prepared mixed N/G cultures. Rat N/G cultures were subjected to 3 h OGD and then treated with different concentrations of IL-33 or vehicle (PBS). Neuronal survival was measured 24 h later using MAP2 ELISA. There was a dramatic loss of neuronal MAP2 levels after OGD, which was partially rescued by IL-33 after treatment at concentrations ranging from 25 to 100 ng/ml (Fig. 5A). The maximal efficacy of IL-33 was achieved at 50 ng/ml, which was used for all subsequent in vitro experiments. NeuN immunostaining and Hoechst labeling were in agreement with the MAP2 ELISA results. Dramatic neuronal death, as demonstrated by prominent nuclear fragmentation in NeuN+ neurons, was observed 24 h after OGD. However, treatment with the optimal dose of IL-33 reduced the numbers of dying neurons after OGD (Fig. 5B). In the next experiments, N/G cultures prepared from WT or ST2 KO pups were subjected to 3 h OGD. As expected, there was greater neuronal MAP2 loss in KO N/G cultures (Fig. 5C).
Figure 5.

Activation of IL-33/ST2 signaling protects against OGD in mixed N/G cultures but not in neuron-enriched cultures. A, B, Rat mixed N/G cultures with or without 3 h OGD were treated with a range of concentrations of IL-33 or PBS (control) for 24 h. A, Neuronal survival was quantified using MAP2 ELISA. n = 8 per group. Data represent three independent experiments. B, Representative images of Hoechst and NeuN double staining in N/G cultures treated with 50 ng/ml IL-33 or PBS (control) for 24 h. Scale bar, 30 μm. C, ELISA quantification of MAP2 expression at 24 h after OGD (3 h) treatments in N/G cultures prepared from WT and ST2 KO mice. Data represent three independent experiments. D, MTT assay in rat neuron-enriched cultures subjected to 1.5 h OGD or sham conditions followed by treatment with PBS or a range of concentrations of IL-33 for another 24 h. Data represent three independent experiments. E, F, MTT and LDH assays were performed at 24 h after OGD (1.5 h) in neuron-enriched cultures prepared from WT and ST2 KO mice. G, Quantification of RT-PCR for M2 markers IL-10, TGF-β, CD206, and M1 marker inducible nitric oxide synthase (iNOS) in microglial cultures treated with 50 ng/ml IL-33 or PBS (control) for 24 h. n = 8–9 per group. Data are mean ± SEM. *p < 0.05, **p < 0.01, ****p < 0.0001 (2-tailed Student's t test or 1-way ANOVA followed by the Bonferroni's post hoc).
Next, we subjected neuron-enriched cultures to 1.5 h OGD, which resulted in similar neuronal loss (∼40–45%), as observed in mixed N/G cultures subjected to 3 h OGD. Post-treatment with various concentrations of IL-33 offered no protection to OGD neurons compared with vehicle treatment, as demonstrated by the MTT assay (Fig. 5D). Furthermore, WT and ST2 KO neuron-enriched cultures were equally sensitive to 1.5 h OGD challenge and exhibited no difference in either the MTT or LDH assay (Fig. 5E,F). Together, these results confirm that ST2/IL-33 signaling is protective in mixed N/G cultures but not in neuron-enriched cultures, suggesting that non-neuronal cells might be the primary target of the IL-33 alarmin.
ST2 signaling is essential for the M2 phenotype in microglia/macrophages after ischemia
As microglia express high levels of ST2 under normal conditions and after stroke (Fig. 4), we assessed the direct effects of IL-33 on microglia. IL-33 treatment of microglia-enriched cultures for 24 h significantly induced the expression of M2 markers by RT-PCR, particularly the expression of IL-10, and inhibited the expression of M1 markers (Fig. 5G).
We further explored the effect of IL-33/ST2 on microglia/macrophage phenotypic responses in stroke mice. As shown in Figure 6A–E, ST2 deficiency impaired the mRNA expression of M2-like markers (IL-10, TGF-β, and CD206) and increased the expression of M1-like markers (inducible nitric oxide synthase and CD16) at 3 d after tMCAO. Immunostaining confirmed the reduced expression of M2 marker CD206 (Fig. 6F–H) and increased expression of M1 marker CD16 (Fig. 6I–K) in the ischemic cortex and striatum of ST2-deficient mice compared with WT littermates at 3 and 7 d after tMCAO.
Figure 6.

ST2 deficiency shifts microglial polarization toward M1 at 3 and 7 d after tMCAO. A–E, mRNA expression of M2 markers (A, IL-10; B, TGF-β; C, CD206) and M1 markers [inducible nitric oxide synthase (D, iNOS); CD16 (E)] were quantified by RT-PCR in the WT and ST2 KO brain at 3 d after tMCAO or sham operation. Data are expressed as percentage of sham. n = 4 per group. *p < 0.05 versus WT group (2-tailed Student's t test). F, Representative images of double immunofluorescent staining for microglia/macrophage marker Iba1 with M2 marker CD206 in the cortex and striatum of sham, WT tMCAO, and ST2 KO tMCAO mice at 3 d after surgery. Blue squares in the schematic diagram illustrate the anatomical location of images in F and I in ipsilateral peri-infarct regions. Scale bar, 50 μm. G, H, The number of CD206+Iba1+ microglia/macrophage was quantified in ipsilesional cortex (left) and striatum (right) 3 and 7 d after tMCAO. n = 3–4 per group. I, Representative images of double immunofluorescent staining for microglia/macrophage marker Iba1 with M1 marker CD16 in the cortex and striatum of sham, WT MCAO, and ST2 KO tMCAO mice at 3 d after surgery. Scale bar, 50 μm. J, K, The number of CD16+Iba1+ microglia/macrophages was quantified in ipsilesional cortex (left) and striatum (right) 3 and 7 d after tMCAO. n = 4–5 per group. Data are mean ± SEM. *p < 0.05, **p < 0.01 versus WT group (2-tailed Student's t test).
ST2 signaling in microglia is essential for their neuroprotective effects in vitro
Finally, we investigated whether activation of IL-33/ST2 signaling in microglia protects ischemic neurons in vitro (Fig. 7A). Our previous study showed that ischemic neurons primed microglia toward the M1 phenotype (Hu et al., 2012). In the present study, we subjected neuron-enriched cultures to 1.5 h OGD, then collected CM from OGD neurons (OGD-CM) or from healthy control neurons (CON-CM), and applied the media to primary microglia cells. As expected, OGD-CM shifted the microglial phenotype toward M1 (Fig. 7B,C). However, the application of IL-33 shifted microglia toward the M2 phenotype upon stimulation with OGD-CM.
Figure 7.

Il-33/ST2 signaling in microglia is essential for their neuroprotective effects in vitro. A, In vitro experimental design for B and C. Neuron-enriched cultures were subjected to 1.5 h OGD. CM from OGD neurons (OGD-CM) or healthy control neurons (CON-CM) were collected 24 h later. The CM was then applied to primary microglial cells in the presence or absence of IL-33. RT-PCR for the expression of M1 and M2 markers was performed on microglial lysates 24 h later. B, C, mRNA expression of M2 markers (IL-10, TGF-β, CD206; B) and M1 markers [CD16, inducible nitric oxide synthase (iNOS); C] was quantified using RT-PCR. The expression levels in CON-CM-treated microglia were set as 100% (dotted line in the bar graphs). Data are expressed as a percentage of CON-CM-treated microglia. n = 5 per group. *p < 0.05 versus vehicle control group (2-tailed Student's t test). D, In vitro experimental design for E and F. Microglia in culture inserts were pretreated with OGD-CM or CON-CM in the presence or absence of IL-33 for 24 h. These pretreated microglia were then added to the neuron-enriched cultures that had been subjected to 1.5 h OGD or non-OGD conditions. TUNEL staining was used to quantify neuronal death at 24 h after coculture. E, Representative image of Hoechst and TUNEL double staining in neurons. Scale bar, 50 μm. F, Quantification of TUNEL+ neurons. n = 3 per group. Data are mean ± SEM. *p < 0.05, **p < 0.01 (1-way ANOVA followed by the Bonferroni's post hoc test).
In the next experiment, microglia seeded in culture inserts were treated with OGD-CM or CON-CM in the presence or absence of IL-33 for 24 h. These pretreated microglia were then added to the neuron-enriched cultures that had been subjected to 1.5 h OGD (Fig. 7D). TUNEL staining was used to quantify neuronal death at 24 h after coculture. As shown in Figure 7E,F, cocultures with CON-CM+IL-33-treated microglia (shifted to M2) significantly reduced neuronal death after OGD. In contrast, OGD-CM-treated M1-like microglia dramatically increased neuronal death. The addition of IL-33 to OGD-CM-treated microglia maintained the M2 phenotype (Fig. 7B,C) and significantly inhibited the toxicity of OGD-CM-treated microglia toward ischemic neurons.
Together, these in vitro studies strongly suggest that activation of the IL-33/ST2 axis in microglia promotes their polarization toward M2 and protects neighboring neurons from ischemic injury.
IL-10 is an essential protective factor released by IL-33-treated microglia
As IL-10 mRNA was dramatically upregulated in IL-33-treated microglia, we sought to explore whether IL-10 is critical for the neuroprotective effect of IL-33-treated microglia. First, we showed increased expression of IL-10 protein in primary microglia after IL-33 treatment (Fig. 8A). IL-10 protein levels were significantly increased in both the cell lysates and in the culture media of IL-33-treated microglia, as measured by ELISA (Fig. 8B). Furthermore, addition of IL-10 neutralizing antibodies, but not TGF-β neutralizing antibodies or isotype control antibodies, abolished the protective effects of IL-33 against OGD in mixed N/G cultures (Fig. 8C). None of these antibodies had any effect on baseline cell survival in cultures without OGD (data not shown). Similarly, IL-10-neutralizing antibodies abolished the protective effects of IL-33 against OGD in neuron–microglia cocultures in transwell systems (Fig. 8D,E).
Figure 8.

IL-10 is an essential protective factor released by IL-33-treated microglia. A, Representative image of 4′,6-diamidino-2-phenylindole (DAPI; blue), actin (red), and IL-10 (green) triple staining in primary microglia treated with 50 ng/ml IL-33 or PBS (control). Scare bars: top, 50 μm; bottom, 10 μm. B, IL-10 protein levels were measured in cell lysates (left) and culture media (right) of IL-33-treated (50 ng/ml) or PBS-treated (control) microglia. *p < 0.05 versus control group (2-tailed Student's t test). C, Rat N/G mixed culture was subjected to 3 h OGD, then treated with PBS (control) or IL-33 (50 ng/ml) in the presence or absence of IL-10 neutralizing antibodies, TGF-β neutralizing antibodies, or isotype controls. Quantification of MAP2 expression was performed by ELISA. Rat Iso, IL-10 neutralizing antibody isotype control; Rabbit Iso, TGF-β neutralizing antibody isotype control. n = 3 per group. ***p < 0.001 (1-way ANOVA followed by the Bonferroni's post hoc test). D, E, Microglia seeded in inserts were placed on top of non-OGD neurons or neurons subjected to 1.5 h OGD. The coculture system was treated with PBS (control) or IL-33 (50 ng/ml) in the presence or absence of IL-10-neutralizing antibodies or isotype control. TUNEL staining was performed 24 h after treatment. D, Representative images of Hoechst and TUNEL double staining in neurons. Scale bar, 50 μm. E, Quantification of TUNEL+ neurons. Data are mean ± SEM. *p < 0.05, ***p < 0.001 (1-way ANOVA followed by the Bonferroni's post hoc test).
To confirm the importance of IL-10 in the neuroprotective effects of IL-33 in vivo, we injected IL-33 (0.1 μg/animal) intracerebroventricularly 30 min after the onset of ischemia in WT and IL-10 KO animals. Consistent with a previous study (Perez-de Puig et al., 2013), we observed that IL-10 deficiency resulted in slightly, but not significantly, enlarged brain infarct volumes after tMCAO when animals were housed in a pathogen-free environment (Fig. 9). Importantly, IL-33 reduced brain infarct sizes in WT mice but not in IL-10 KO mice, confirming the importance of IL-10 in IL-33-afforded neuroprotection in vivo.
Figure 9.

IL-10 is essential for IL-33-afforded protection in the ischemic brain. WT or IL-10 KO mice were subjected to 60 min MCAO. IL-33 or PBS was injected intracerebroventricularly 30 min after the induction of ischemia. A, Representative images of MAP2-stained coronal sections show infarct volumes at 3 d after reperfusion. B, Quantification of infarct volumes in all groups shown in A. n = 6–10 per group. Data are mean ± SEM. *p < 0.05 (1-way ANOVA followed by the Bonferroni's post hoc test).
Discussion
Recent studies have shown that IL-33 is released from CNS cells rapidly after injury and contributes to the activation of immune responses in lesion areas (Gadani et al., 2015). Consistent with these observations, the present study demonstrates that IL-33 mRNA levels were transiently elevated early (1 d) after stroke and subsequently returned to baseline within 3 d. Increased IL-33 expression was observed in OLs and astrocytes 1 d after tMCAO. These observations prompted us to explore the role of the IL-33/ST2 axis in acute ischemic brain injury. We found that loss of ST2 enlarged brain infarct sizes in multiple acute models of stroke and in mice of both genders. The activation of IL-33/ST2 signaling in the ischemic brain resulted in M2 microglial polarization, which in turn afforded protection to ischemic neurons in an IL-10-dependent manner.
It has become increasingly evident that microglia are not a uniform population of cells, and that activated microglia with distinct functions and gene expression profiles differentially and profoundly influence stroke outcomes in vivo and in vitro. For example, M2-like microglia with elevated expression of anti-inflammatory and trophic factors exert protective effects in the ischemic brain. Although both M1 and M2 microglia/macrophages are present in the ischemic brain, the M1 response predominates with stroke progression (Hu et al., 2012). Aged mice exhibit further impairments in M2 polarization after stroke, and this deficiency is closely correlated with enlarged brain infarcts and worse long-term functional deficits (Suenaga et al., 2015). Therefore, elucidating the mechanism(s) underlying microglial regulation in the ischemic brain may accelerate the development of novel therapeutic targets for stroke. The current study demonstrated that ST2 deficiency shifts microglial responses toward M1-like states and exacerbates ischemic brain injury, suggesting that ST2/IL-33 signaling is an important neuroprotective mechanism that naturally maintains beneficial microglial responses after injury. In line with these interpretations, several recent studies have shown that IL-33/ST2 activation can mediate macrophage M2 polarization under pathological conditions (Fock et al., 2013; Li et al., 2014). Thus, IL-33 supplementation or agonism might be a promising new strategy to promote beneficial microglial responses after stroke.
The neuroprotective role of IL-33 has recently been reported in animal models of CNS injuries. In models of spinal cord injury (Gadani et al., 2015; Pomeshchik et al., 2015), intraperitoneal application of IL-33 reduced tissue loss and ameliorated white matter demyelination (Pomeshchik et al., 2015). Two other stroke studies similarly demonstrated that IL-33 injections before or immediately after MCAO protected the ischemic brain (Korhonen et al., 2015; Luo et al., 2015). Most of these studies used systemic approaches to deliver IL-33 to the site of CNS injury, and attributed the IL-33-afforded neuroprotection to its effects on peripheral immune cells, such as T lymphocytes and macrophages. However, the direct effects of IL-33 on CNS cells remain largely unexplored and the underlying mechanisms of action have yet to be characterized. In our study, we identified a direct effect of IL-33 on microglia. As the first line of evidence, we discovered that ST2 was highly expressed on microglia and that microglial ST2 expression was dramatically induced after ischemic stroke. Furthermore, direct delivery of IL-33 into the ventricular compartment ameliorated acute ischemic brain damage, suggesting that the elevation of IL-33 within the brain could exert neuroprotection by directly targeting CNS cells. In vitro studies confirmed the direct effects of IL-33 on microglial polarization under ischemic conditions and subsequent protection of ischemic neurons. Thus, IL-33 secreted from injured OLs or astrocytes soon after brain injury (Gadani et al., 2015) may directly target local microglia and promote an anti-inflammatory phenotype. This self-defensive effect, together with other immunomodulatory effects of IL-33 on peripheral immune cells, preserves brain tissue and improves stroke outcomes.
Some authors have argued that the M1–M2 dichotomy is an arbitrary and oversimplified framework, as there are many other phenotypes in between these two extremes and their functions may partly overlap. Therefore, it is critical to identify the specific effectors or functions of the IL-33-stimulated microglial phenotype. In line with this goal, our mechanistic studies have identified IL-10 as a key molecule released by IL-33-stimulated microglia and a major neuroprotective factor in the ischemic brain. Among all the M2 microglial markers assessed in the present study, the expression of IL-10 was most robustly induced by IL-33. Strikingly, blocking the function of IL-10 almost completely abolished the protective effects of IL-33 in neuron–microglia cocultures. As IL-10 is a well established anti-inflammatory cytokine, administration of recombinant IL-10 or gene transfer of IL-10 has been shown to reduce acute neuronal injury after stroke (Spera et al., 1998; Grilli et al., 2000; Ooboshi et al., 2005). Many immunomodulatory cells, including regulatory T cells and regulatory B cells, have also been shown to exert neuroprotection by releasing IL-10 (Liesz et al., 2009; Ren et al., 2011). Therefore, the capacity of IL-33 to induce IL-10 in microglia may serve as an important mediator of IL-33-afforded CNS protection. Importantly, intracerebroventricular injections of IL-33 into IL-10 KO mice failed to provide any significant protection against ischemia. In agreement with these results, a recent study documented that peripheral IL-33 administration elevated the expression of IL-10 in the spleen (Korhonen et al., 2015). That study also showed that IL-4, another anti-inflammatory factor, was induced by IL-33. IL-4 is a well established promoter of Th2 responses in T lymphocytes and of M2 polarization in macrophages. Studies of IL-4 deficiency have demonstrated that IL-4 signaling is an endogenous neuroprotective mechanism both in the acute (Xiong et al., 2011, 2015) and late stages of stroke (Liu et al., 2016). It is therefore not surprising that IL-4 has been identified as an important mediator of peripheral IL-33-afforded neuroprotection after stroke by targeting peripheral T cells (Korhonen et al., 2015). However, in our study, we did not observe obvious effects of IL-33/ST2 on IL-4 expression in the ischemic brain or in IL-33-treated microglial cultures (data not shown), suggesting that microglia are not a source of IL-4 induction upon ST2/IL-33 activation. In light of a recent discovery that injured neurons may release IL-4 at acute stages of stroke (Zhao et al., 2015), it is possible that IL-4 (released from neurons) and IL-33 (released from astrocytes and OLs) exert synergistic effects to regulate microglial responses after stroke and to provide maximal natural protection to the ischemic brain.
Although the current study revealed a protective effect of tST2 on microglia in stroke, potential effects of sST2, a decoy IL-33 receptor, cannot be excluded. Recent clinical data have shown increased levels of sST2 in the plasma of human stroke patients. The elevation of sST2 was correlated with worse stroke outcomes, suggesting a detrimental role for sST2 in stroke (Korhonen et al., 2015). This is consistent with reports of a harmful role of sST2 in acute myocardial infarction (Januzzi, 2013). However, in global ST2 KO mice, in which tST2 and sST2 are both absent, overall stroke outcomes were worse. These results suggest that the protective effects of tST2 may outweigh the detrimental effects of sST2, at least in acute stages of stroke.
In addition to microglia, astrocytes also express high levels of ST2 under normal conditions and even higher levels upon ischemic challenge. Therefore, astrocytes might also be alarmin responders sensitive to IL-33/ST2 signaling. Systemic application of IL-33 has been reported to inhibit astrocyte activation after stroke (Korhonen et al., 2015). However, it is unclear whether this is a direct effect of IL-33 on astrocytes or an indirect effect through other immune cells, such as T cells. Further studies are warranted to elucidate the direct effect of IL-33 on astrocytes in the ischemic brain.
In conclusion, the current study identified IL-33/ST2 signaling as a potential immune regulatory mechanism that enhances the expression of IL-10 in M2 microglia and reduces acute ischemic brain injury after stroke. Considered together with recent studies addressing the modulatory effects of IL-33 on peripheral immune cells, the activation of ST2/IL-33 signaling might represent a potent new immunotherapy for stroke patients.
Footnotes
This work was supported by grants from the National Institutes of Health/National Institute of Neurological Disorders and Stroke (NS094573 and NS092618 to X.H.; NS045048 and NS095671 to J.C.), and the American Heart Association (13SDG14570025 to X.H.). J.C. is a recipient of the Department of Veterans Affairs Senior Research Career Scientist Award. We thank Joycelyne Johnson for excellent editorial assistance.
The authors declare no competing financial interests.
References
- Arpaia N, Green JA, Moltedo B, Arvey A, Hemmers S, Yuan S, Treuting PM, Rudensky AY (2015) A distinct function of regulatory T cells in tissue protection. Cell 162:1078–1089. 10.1016/j.cell.2015.08.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai M, Ma YL, Qin P, Li Y, Zhang LX, Nie H, Peng Z, Dong H, Dong HL, Hou WG, Xiong LZ (2014) The loss of estrogen efficacy against cerebral ischemia in aged postmenopausal female mice. Neurosci Lett 558:115–119. 10.1016/j.neulet.2013.11.007 [DOI] [PubMed] [Google Scholar]
- Espinassous Q, Garcia-de-Paco E, Garcia-Verdugo I, Synguelakis M, von Aulock S, Sallenave JM, McKenzie AN, Kanellopoulos J (2009) IL-33 enhances lipopolysaccharide-induced inflammatory cytokine production from mouse macrophages by regulating lipopolysaccharide receptor complex. J Immunol 183:1446–1455. 10.4049/jimmunol.0803067 [DOI] [PubMed] [Google Scholar]
- Fock V, Mairhofer M, Otti GR, Hiden U, Spittler A, Zeisler H, Fiala C, Knöfler M, Pollheimer J (2013) Macrophage-derived IL-33 is a critical factor for placental growth. J Immunol 191:3734–3743. 10.4049/jimmunol.1300490 [DOI] [PubMed] [Google Scholar]
- Gadani SP, Walsh JT, Smirnov I, Zheng J, Kipnis J (2015) The glia-derived alarmin IL-33 orchestrates the immune response and promotes recovery following CNS injury. Neuron 85:703–709. 10.1016/j.neuron.2015.01.013 [DOI] [PubMed] [Google Scholar]
- Garlanda C, Dinarello CA, Mantovani A (2013) The interleukin-1 family: back to the future. Immunity 39:1003–1018. 10.1016/j.immuni.2013.11.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson CL, Coomber B, Murphy SP (2011) Progesterone is neuroprotective following cerebral ischaemia in reproductively ageing female mice. Brain 134:2125–2133. 10.1093/brain/awr132 [DOI] [PubMed] [Google Scholar]
- Grilli M, Barbieri I, Basudev H, Brusa R, Casati C, Lozza G, Ongini E (2000) Interleukin-10 modulates neuronal threshold of vulnerability to ischaemic damage. Eur J Neurosci 12:2265–2272. 10.1046/j.1460-9568.2000.00090.x [DOI] [PubMed] [Google Scholar]
- Hu X, Zhang D, Pang H, Caudle WM, Li Y, Gao H, Liu Y, Qian L, Wilson B, Di Monte DA, Ali SF, Zhang J, Block ML, Hong JS (2008) Macrophage antigen complex-1 mediates reactive microgliosis and progressive dopaminergic neurodegeneration in the MPTP model of Parkinson's disease. J Immunol 181:7194–7204. 10.4049/jimmunol.181.10.7194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu X, Li P, Guo Y, Wang H, Leak RK, Chen S, Gao Y, Chen J (2012) Microglia/macrophage polarization dynamics reveal novel mechanism of injury expansion after focal cerebral ischemia. Stroke 43:3063–3070. 10.1161/STROKEAHA.112.659656 [DOI] [PubMed] [Google Scholar]
- Iadecola C, Anrather J (2011) The immunology of stroke: from mechanisms to translation. Nat Med 17:796–808. 10.1038/nm.2399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Januzzi JL., Jr (2013) ST2 as a cardiovascular risk biomarker: from the bench to the bedside. J Cardiovasc Transl Res 6:493–500. 10.1007/s12265-013-9459-y [DOI] [PubMed] [Google Scholar]
- Jovanovic I, Radosavljevic G, Mitrovic M, Juranic VL, McKenzie AN, Arsenijevic N, Jonjic S, Lukic ML (2011) ST2 deletion enhances innate and acquired immunity to murine mammary carcinoma. Eur J Immunol 41:1902–1912. 10.1002/eji.201141417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koellhoffer EC, McCullough LD (2013) The effects of estrogen in ischemic stroke. Transl Stroke Res 4:390–401. 10.1007/s12975-012-0230-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korhonen P, Kanninen KM, Lehtonen Š, Lemarchant S, Puttonen KA, Oksanen M, Dhungana H, Loppi S, Pollari E, Wojciechowski S, Kidin I, García-Berrocoso T, Giralt D, Montaner J, Koistinaho J, Malm T (2015) Immunomodulation by interleukin-33 is protective in stroke through modulation of inflammation. Brain Behav Immun 49:322–336. 10.1016/j.bbi.2015.06.013 [DOI] [PubMed] [Google Scholar]
- Kurowska-Stolarska M, Kewin P, Murphy G, Russo RC, Stolarski B, Garcia CC, Komai-Koma M, Pitman N, Li Y, Niedbala W, McKenzie AN, Teixeira MM, Liew FY, Xu D (2008) IL-33 induces antigen-specific IL-5+ T cells and promotes allergic-induced airway inflammation independent of IL-4. J Immunol 181:4780–4790. 10.4049/jimmunol.181.7.4780 [DOI] [PubMed] [Google Scholar]
- Li D, Guabiraba R, Besnard AG, Komai-Koma M, Jabir MS, Zhang L, Graham GJ, Kurowska-Stolarska M, Liew FY, McSharry C, Xu D (2014) IL-33 promotes ST2-dependent lung fibrosis by the induction of alternatively activated macrophages and innate lymphoid cells in mice. J Allergy Clin Immunol 134:1422–1432.e11. 10.1016/j.jaci.2014.05.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li P, Gan Y, Sun BL, Zhang F, Lu B, Gao Y, Liang W, Thomson AW, Chen J, Hu X (2013) Adoptive regulatory T-cell therapy protects against cerebral ischemia. Ann Neurol 74:458–471. 10.1002/ana.23815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liesz A, Suri-Payer E, Veltkamp C, Doerr H, Sommer C, Rivest S, Giese T, Veltkamp R (2009) Regulatory T cells are key cerebroprotective immunomodulators in acute experimental stroke. Nat Med 15:192–199. 10.1038/nm.1927 [DOI] [PubMed] [Google Scholar]
- Liu X, Liu J, Zhao S, Zhang H, Cai W, Cai M, Ji X, Leak RK, Gao Y, Chen J, Hu X (2016) Interleukin-4 is essential for microglia/macrophage M2 polarization and long-term recovery after cerebral ischemia. Stroke 47:498–504. 10.1161/STROKEAHA.115.012079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu B, Yang M, Wang Q (2016) Interleukin-33 in tumorigenesis, tumor immune evasion, and cancer immunotherapy. J Mol Med (Berl) 94:535–543. 10.1007/s00109-016-1397-0 [DOI] [PubMed] [Google Scholar]
- Luo Y, Zhou Y, Xiao W, Liang Z, Dai J, Weng X, Wu X (2015) Interleukin-33 ameliorates ischemic brain injury in experimental stroke through promoting Th2 response and suppressing Th17 response. Brain Res 1597:86–94. 10.1016/j.brainres.2014.12.005 [DOI] [PubMed] [Google Scholar]
- Miller AM, Asquith DL, Hueber AJ, Anderson LA, Holmes WM, McKenzie AN, Xu D, Sattar N, McInnes IB, Liew FY (2010) Interleukin-33 induces protective effects in adipose tissue inflammation during obesity in mice. Circ Res 107:650–658. 10.1161/CIRCRESAHA.110.218867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ooboshi H, Ibayashi S, Shichita T, Kumai Y, Takada J, Ago T, Arakawa S, Sugimori H, Kamouchi M, Kitazono T, Iida M (2005) Postischemic gene transfer of interleukin-10 protects against both focal and global brain ischemia. Circulation 111:913–919. 10.1161/01.CIR.0000155622.68580.DC [DOI] [PubMed] [Google Scholar]
- Oshikawa K, Yanagisawa K, Tominaga Si, Sugiyama Y (2002) ST2 protein induced by inflammatory stimuli can modulate acute lung inflammation. Biochem Biophys Res Commun 299:18–24. 10.1016/S0006-291X(02)02578-0 [DOI] [PubMed] [Google Scholar]
- Pérez-de Puig I, Miró F, Salas-Perdomo A, Bonfill-Teixidor E, Ferrer-Ferrer M, Márquez-Kisinousky L, Planas AM (2013) IL-10 deficiency exacerbates the brain inflammatory response to permanent ischemia without preventing resolution of the lesion. J Cereb Blood Flow Metab 33:1955–1966. 10.1038/jcbfm.2013.155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pomeshchik Y, Kidin I, Korhonen P, Savchenko E, Jaronen M, Lehtonen S, Wojciechowski S, Kanninen K, Koistinaho J, Malm T (2015) Interleukin-33 treatment reduces secondary injury and improves functional recovery after contusion spinal cord injury. Brain Behav Immun 44:68–81. 10.1016/j.bbi.2014.08.002 [DOI] [PubMed] [Google Scholar]
- Ren X, Akiyoshi K, Dziennis S, Vandenbark AA, Herson PS, Hurn PD, Offner H (2011) Regulatory B cells limit CNS inflammation and neurologic deficits in murine experimental stroke. J Neurosci 31:8556–8563. 10.1523/JNEUROSCI.1623-11.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Zhang L, Pu H, Mao L, Hu X, Jiang X, Xu N, Stetler RA, Zhang F, Liu X, Leak RK, Keep RF, Ji X, Chen J (2016) Rapid endothelial cytoskeletal reorganization enables early blood–brain barrier disruption and long-term ischaemic reperfusion brain injury. Nat Commun 7:10523. 10.1038/ncomms10523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spera PA, Ellison JA, Feuerstein GZ, Barone FC (1998) IL-10 reduces rat brain injury following focal stroke. Neurosci Lett 251:189–192. 10.1016/S0304-3940(98)00537-0 [DOI] [PubMed] [Google Scholar]
- Suenaga J, Hu X, Pu H, Shi Y, Hassan SH, Xu M, Leak RK, Stetler RA, Gao Y, Chen J (2015) White matter injury and microglia/macrophage polarization are strongly linked with age-related long-term deficits in neurological function after stroke. Exp Neurol 272:109–119. 10.1016/j.expneurol.2015.03.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang XN, Zheng Z, Yenari MA (2012) Bone marrow chimeras in the study of experimental stroke. Transl Stroke Res 3:341–347. 10.1007/s12975-012-0169-6 [DOI] [PubMed] [Google Scholar]
- Townsend MJ, Fallon PG, Matthews DJ, Jolin HE, McKenzie AN (2000) T1/ST2-deficient mice demonstrate the importance of T1/ST2 in developing primary T helper cell type 2 responses. J Exp Med 191:1069–1076. 10.1084/jem.191.6.1069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong X, Barreto GE, Xu L, Ouyang YB, Xie X, Giffard RG (2011) Increased brain injury and worsened neurological outcome in interleukin-4 knock-out mice after transient focal cerebral ischemia. Stroke 42:2026–2032. 10.1161/STROKEAHA.110.593772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong X, Xu L, Wei L, White RE, Ouyang YB, Giffard RG (2015) IL-4 is required for sex differences in vulnerability to focal ischemia in mice. Stroke 46:2271–2276. 10.1161/STROKEAHA.115.008897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X, Wang H, Sun G, Zhang J, Edwards NJ, Aronowski J (2015) Neuronal interleukin-4 as a modulator of microglial pathways and ischemic brain damage. J Neurosci 35:11281–11291. 10.1523/JNEUROSCI.1685-15.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]