

Abstract
Melanocyte-stimulating hormone (MSH)-induced activation of the cAMP-response element (CRE) via the CRE-binding protein in hypothalamic cells promotes expression of TRH and thereby restricts food intake and increases energy expenditure. Glucose also induces central anorexigenic effects by acting on hypothalamic neurons, but the underlying mechanisms are not completely understood. It has been proposed that glucose activates the CRE-binding protein-regulated transcriptional coactivator 2 (CRTC-2) in hypothalamic neurons by inhibition of AMP-activated protein kinases (AMPKs), but whether glucose directly affects hypothalamic CRE activity has not yet been shown. Hence, we dissected effects of glucose on basal and MSH-induced CRE activation in terms of kinetics, affinity, and desensitization in murine, hypothalamic mHypoA-2/10-CRE cells that stably express a CRE-dependent reporter gene construct. Physiologically relevant increases in extracellular glucose enhanced basal or MSH-induced CRE-dependent gene transcription, whereas prolonged elevated glucose concentrations reduced the sensitivity of mHypoA-2/10-CRE cells towards glucose. Glucose also induced CRCT-2 translocation into the nucleus and the AMPK activator metformin decreased basal and glucose-induced CRE activity, suggesting a role for AMPK/CRTC-2 in glucose-induced CRE activation. Accordingly, small interfering RNA-induced down-regulation of CRTC-2 expression decreased glucose-induced CRE-dependent reporter activation. Of note, glucose also induced expression of TRH, suggesting that glucose might affect the hypothalamic-pituitary-thyroid axis via the regulation of hypothalamic CRE activity. These findings significantly advance our knowledge about the impact of glucose on hypothalamic signaling and suggest that TRH release might account for the central anorexigenic effects of glucose and could represent a new molecular link between hyperglycaemia and thyroid dysfunction.
The hypothalamic-pituitary-thyroid (HPT) axis is of prime importance for the central regulation of appetite and energy metabolism (1, 2). The hypothalamic nucleus arcuatus receives peripheral satiety signals such as leptin or insulin and reacts by releasing melanocyte-stimulating hormone (MSH). MSH activates cAMP-response element (CRE)-binding protein (CREB) in neurons of the paraventricular nucleus (PVN) and thereby controls the expression of TRH, resulting in TSH secretion from the pituitary gland. Thus, dysfunction of hypothalamic MSH signaling in humans or mice causes a severe obesity-diabetes syndrome (3–11). Regulation of transcriptional activity is a key control mechanism within the HPT axis and CREB-mediated CRE activation has been shown to play a major role in hypothalamic gene transcription (12–14). Because of its impact on energy metabolism, dysregulation of the HPT axis has frequently been implicated in severe metabolic disorders including type 2 diabetes mellitus (T2DM) (15–18).
Glucose is the key player in diabetes and obesity and, similar to MSH, induces central anorexigenic effects by acting on hypothalamic neurons, but the underlying mechanisms are not completely understood. Besides being a pivotal energy carrier, glucose-derived ATP is an important regulator of intracellular signaling in hypothalamic neurons. In analogy to pancreatic β-cells, ATP inhibits ATP-sensitive K+ (KATP) channels and thus depolarizes hypothalamic neurons (19–24). Furthermore, ATP dampens AMP-activated protein kinase (AMPK) activity in hypothalamic neurons, thereby relieving CREB-regulated transcriptional coactivator 2 (CRTC-2), an important transcriptional coactivator of CREB, from tonic phosphorylation and inhibition by AMPK (25). Elevating extracellular glucose concentrations from 0.5mM to 15.0mM was found to enhance CRTC-2 activity due to AMPK inhibition and expression of the insulin receptor substrate-2 in hypothalamic cells (25), thus establishing the first link between hypothalamic glucose sensing and CREB/CRTC-2-dependent gene regulation. Beyond this possible association of glucose and hypothalamic insulin signaling (25), there is also some evidence suggesting that glucose affects leptin-induced signal transduction in hypothalamic cells (26). Thus, at present, it appears that in hypothalamic cells glucose promotes gene transcription via CRTC-2 and affects key players of the HPT axis, such as leptin and insulin. However, direct effects of physiological relevant fluctuations in brain glucose levels on basal or MSH-induced CRE activity or hypothalamic TRH expression have not been shown yet.
In the present study, we investigated mHypoA-2/10-CRE cells that express a wide range of hypothalamic markers and resemble PVN neurons with regard to melanocortin sensitivity and TRH production and in addition, stable express a CRE-dependent reporter construct (27–33). We dissected effects of glucose on basal and MSH-induced CRE activation in terms of kinetics, affinity, and desensitization. Physiologically relevant increases in extracellular glucose enhanced basal or MSH-induced CRE-dependent gene transcription and hypothalamic TRH expression, whereas prolonged elevated glucose concentrations reduced the sensitivity of mHypoA-2/10-CRE cells towards glucose. These findings significantly advance our knowledge of the impact of glucose on hypothalamic signaling and might reveal a new association between hyperglycaemia and hyperthyroidism.
Materials and Methods
Materials
Cell culture reagents were obtained from Invitrogen and TurboFect from Fermentas. The anti-pro-TRH (M-166) antiserum was from Santa Cruz Biotechnology. The p-AMPK (2535s) and p-CREB (9198s) antiserum were purchased from Cell Signaling, the CRTC-2 (sc-46272) or CREB (sc-240) antiserum and fructose-1,6-bisphosphate (F-1,6-BP) was from Santa Cruz Biotechnology. The peroxidase-conjugated antimouse or antirabbit antibody, both raised in goat, the histone-3 antiserum and D-glucose, 2-deoxy-D-glucose (2-DG), and tolbutamide from Sigma-Aldrich. 2-[1,2-3H(N)]-2-DG was from PerkinElmer. The firefly luciferase substrate, the signal transducers and activator of transcription (STAT), and nuclear factor of activated t cells (NFAT) reporter gene construct pGL4.47 were purchased from Promega. MSH and bradykinin were purchased from Biotrend. Dorsomorphin and metformin were purchased from Tocris. The CRE-dependent reporter, containing 6 5′-TGACCTCAC-3′ sites, has been reported previously (34).
Cell culture and transfections
The adult mouse hypothalamic cell lines mHypoA-2/10 (Clu176) were purchased from Cedarlane and cultured in DMEM (10% fetal bovine serum, 2mM L-glutamine, penicillin/streptomycin, 1mM pyruvate, and 25mM glucose) at 37°C and 5% CO2. To obtain mHypoA-2/10 cells stably expressing the CRE-dependent reporter, cells were cotransfected with the CRE reporter construct and an empty pcDNA4 vector containing the resistance gene for zeocin (27). Transfected cells were then selected with 600-μg/mL zeocin for 4 weeks and positively tested cell pools were used for further studies. To analyze effects of glucose on CRE activity DMEM without FBS, pyruvate and glucose was used. The indicated glucose concentrations were achieved by removing DMEM containing 25mM glucose, washing cells once without glucose and adding fresh DMEM containing the indicated D-glucose or 2-DG before the experiments. In order to down-regulate CRCT-2 expression, a cocktail of 3 specific siRNAs (catalog number sc-45833) and a random small interference RNA were purchased from Santa Cruz Biotechnology. To increase transfection efficacy, siRNAs were introduced into mHypoA-2/10-CRE cells via electroporation using the Neon transfection system from Invitrogen according to the manufacturer's protocol. Briefly, for 1 pulse 500 000 cells together with 10nM or 40nM the corresponding siRNA were challenged with 1450 V for 30 milliseconds and then place on a 48-well cavity for ELISA experiments or 96-well plates for reporter assays.
Whole-cell ELISA
Electroporated cells were seeded in 48-well plates (∼15 000 per well) 2 days before the experiment. After 20 hours of serum starvation, total histone-3, CRTC-2, and CREB expression was analyzed at a glucose concentration of 0.1mM. After fixation for 15 minutes with 4% paraformaldehyde, cells were permeabilized with ice-cold methanol/acetone (50:50) for 5 minutes. Next, cells were washed with PBS and then incubated with 1.0% of BSA in PBS for 15 minutes at RT to block unspecific binding sites. After an additional washing step cells were incubated for 30 minutes at RT with the anti-CREB antiserum (1:2000), the anti-CRTC-2 serum (1:2000), or the antihistone serum (1:5000) in PBS/BSA. First, antibodies were removed, cells washed and incubated with the corresponding horseradish peroxidase-conjugated secondary antibody (1:4000) in PBS/BSA for 30 minutes at RT. After several washing steps, 200 μL of the 1-Step-Ultra-TMB-ELISA substrate (Thermo Scientific) was added to the cells and incubated for 15 minutes at RT. The reaction was stopped by adding 50 mL of 1M H2SO4, 200 μL transferred to 96-well plates with clear bottom, and OD450 measured in a FLUOstar Omega plate reader.
Calcium
Calcium transients in mHypoA-2/10-CRE cells were measured with a FLUOstar Omega plate reader (BMG Labtech) as described previously (35) using the fluorescent calcium indicator fura 2.
Glucose uptake
Cells were seeded on 24-well plates (∼20 000 per well) 48 hours before the experiment. After various glucose treatments, cells were cultured for 30 minutes without glucose and then glucose uptake measured by adding 0,5 μCi 2-[1,2-3H(N)]-2-DG in the presence of 100μM glucose. Afterwards, cells were washed with 1 ml PBS, lysed with 0.5% sodium dodecyl sulfate and 0.1N NaOH and total radioactivity was measured.
Western blotting
Cells were seeded on 6-well plates (∼100 000 per well), cultured for 1 day, serum starved for 20 hours the next day, stimulated for the indicated period of time with glucose, and then lysed in Laemmli buffer. Lysates were subjected to SDS-PAGE, and protein was transferred to nitrocellulose by Western blotting. For detection of TRH expression, blots were separated in 2 parts, by a horizontal cut at 25 kDa. The upper part was used to analyze TRH expression (30 kDa) with a TRH-specific antiserum (1:2000) and the lower part to detect total histone-3 (17 kDa) proteins (1:10 000) as a loading control. To detect AMPK phosphorylation, blots were separated by a horizontal cut at 50 kDa. The upper part was used to analyze AMPK (68 kDa) phosphorylation with phospho-specific AMPK (Thr172; 1:5000 antisera and the lower part to detect total ERK-2 (42 kDa) proteins (1:10 000) as a loading control. For detection of CREB phosphorylation, blots were separated by a horizontal cut at 25 kDa. The upper part was used to analyze CREB (43 kDa) phosphorylation with phospho-specific CREB antisera (Ser133; 1:5000) and the lower part to detect total histone-3 (17 kDa) proteins (1:10 000) as a loading control. Immuno-reactivity was quantified by densitometry using ImageJ, ratios between TRH and histone-3 or p-AMPK and total-ERK-2 signals were calculated, and ligand-induced TRH expression or AMPK phosphorylation normalized to unstimulated cells.
Reporter gene assays
mHypoA-2/10-CRE cells were seeded in white 96-well plates with clear bottom (∼5000 per well) for the experiments shown in figures 1, 2, 4, or 5 below and in 24-well plates (∼20 000 per well) for the experiments shown in figures 3 or 4 below. For the experiments shown in figure 1 below, mHypoA-2/10 cells were seeded in 12-well plates (∼20 000 per well) 24 hours before the experiment and transfected with various luciferase reporter genes constructs using the Turbofect reagent (R0531) from Thermo Scientific according to the manufacturers' protocol the next day. After stimulation, cells were lysed (25mM Tris-HCl [pH 7.4], 4mM EGTA, 8mM MgCl2, 1mM dithiothreitol, and 1% Triton X-100) and luciferase activity measured in white bottom 96-well plates after automatically injecting luciferase substrate. Resulting total light emission was detected every s for 10 seconds after injection in a FLUOstar Omega plate reader. Maximal light emission during this period was recorded and is indicated relative to unstimulated cells (0mM or 0.1mM glucose).
cAMP accumulation
To determine agonist-induced cAMP accumulation, 200 000 cells were seeded in 12-well dishes 24 hours before the experiment and labeled in serum-free DMEM containing 2 μCi/mL of [3H]adenine for 4 hours. Cells were stimulated for 30 minutes at 37°C in DMEM containing 1μM 3-isobutyl-1-methylxanthine and MSH. The reaction was terminated by removing the medium and adding ice-cold 5% trichloroacetic acid. [3H]ATP and [3H]cAMP were then purified by sequential chromatography (dowex-resin/aluminum oxide columns), and the accumulation of [3H]cAMP was expressed as the ratio of [3H]cAMP to ([3H]cAMP + [3H]ATP).
Immunostaining
Approximately 4000 mHypoA-2/10-CRE cells per well were seeded on 0.1% poly-L-lysine-coated black 96-well plates with clear bottom. Twenty-four hours later, cells were cultured with distinct glucose concentrations and, after additional 24 hours, fixed with 4.0% paraformaldehyde for 10 minutes, permeabilized with 0.5% Triton X-100 in PBS for 10 minutes, and unspecific binding sites blocked with 2.0% BSA in PBS with 0.02% Tween 20 (PBST) for 45 minutes. A monoclonal anti-CRTC-2 antiserum (clone number 628430 from R&D Systems) was incubated for 60 minutes at 37°C in PBST with a final concentration of 100 μg/mL and detected by an Alexa Fluor 488-conjugated goat antiserum raised against mouse IgG also for 60 minutes at 37°C in PBST with a final concentration of 4 μg/mL. Nuclei were stained using 4′,6-diamidino-2-phenylindole (0.1 μg/mL) for 30 minutes at 37°C. After intense washing cells were conserved with a drop of Ibidi mounting medium and analyzed using an Olympus IX 71 microscope. Alexa Fluor 488 immunostaining in nuclei and background areas was quantified by densitometry using ImageJ.
Quantitative RT-PCR (qRT-PCR)
Total RNA was isolated from mHypoA-2/10-CRE cells using the TriFast reagent (Invitrogen) according to the manufacturer's instructions. First strand synthesis was carried out with oligo(dT)18 primer using 2 μg of total RNA and the RevertAid H Minus First Strand cDNA Synthesis kit (Fermentas). qRT-PCR was done using the LightCycler 480 SybrGreen I Master Mix (Roche), intron-spanning primer pairs at a final concentration of 1μM each and 0.08 μL of the first strand synthesis reaction in a LightCycler 480 (Roche) using the following conditions: initial denaturation for 15 minutes at 94°C, 55 cycles of 94°C for 10 seconds, 55°C for 10 seconds, and 72°C for 10 seconds. Crossing points (Cps) were determined by the software supplied with the LightCycler 480. Sequences of primer pairs were 5′-ccaaccgcgagaagatga-3′ and 5′-ccagaggcgtacagggatag-3′ for β-actin, 5′-tgcagagtctccaccttgc-3′ and 5′-ggggataccagttagcacga-3′ for TRH. Data were analyzed by 2−ΔΔCp calculations: Cp values measured for TRH under stimulated conditions (either increased glucose concentrations or MSH) were subtracted from those measured for actin under the same conditions (ΔCp stimulated). Cp values measured for TRH under basal conditions were subtracted from those measured for actin under the same conditions (ΔCp basal). Afterwards, ΔCp-basal values were subtracted from ΔCp-stimulated values.
Data analysis
Data were analyzed using Prism4.0 (GraphPad Software, Inc). Statistical significance of differences was assessed by 1- or 2-sample Student's t test. Asterisks (***, P < .001; **, P < .01; *, P < .05) in the case of the 2-sample and hash signs (###, P < .001; ##, P < .01; #, P < .05) in the case of the 1-sample test were used to indicate significant differences.
Results
Glucose exerts specific effects on CRE-dependent reporter gene activity in murine hypothalamic mHypoA-2/10 cells
In vivo brain glucose concentrations vary between 0.2mM and 2.5mM under physiological conditions (19, 36–40). mHypoA-2/10 cells are cultured under high-glucose conditions (25.0mM), clearly exceeding physiological extracellular brain glucose concentrations. To investigate the effects of extracellular glucose on gene expression in hypothalamic cells, we transfected NFAT, STAT, or CREB-dependent reporter gene constructs into mHypoA-2/10 cells and cultured these cells with reduced glucose concentrations for 24 hours. As shown in Figure 1A, complete removal of glucose decreased reporter activity independently from the promoter region tested, indicating that a minimal glucose concentration is required to maintain salient cell functions such as transcription and translation. In contrast, no differences in STAT- or NFAT-dependent reporter activity were observed when glucose concentrations were varied between 0.1mM and 25.0mM, indicating that the activity of both promoters is glucose insensitive. However, the activity of the CRE reporter construct increased incrementally from 0.1mM to 25.0mM glucose, providing first evidence that glucose specifically affects CRE-dependent gene expression in mHypoA-2/10 cells. Next, we analyzed how the different reporter gene constructs would react to complete glucose withdrawal over time. As opposed to NFAT or STAT reporters, we observed decreased CRE reporter activity after 4 and 6 hours (Figure 1B), confirming the selective impact of glucose on CRE-dependent promoter activity and further illustrating that glucose withdrawal for up to 6 hours does not compromise protein biosynthesis in general. In order to gain deeper insight into glucose-induced regulation of CRE-dependent reporter activity, we used a previously established mHypoA-2/10 cell pool that stably expresses the CRE reporter construct (mHypoA-2/10-CRE cells). After growing mHypoA-2/10-CRE cells in 0.1mM instead of 25.0mM glucose for 24 hours, an approximately 50% reduction in basal CRE activity was observed that could not be recovered by addition of 1.0mM pyruvate (Figure 1C). These data suggest that cytosolic, but not mitochondrial, glucose metabolism affects CRE-dependent reporter activity. Thus, we next tested effects of the cytosolic glucose metabolite F-1,6-BP on CRE activity. As shown in Figure 1D, F-1,6-BP significantly enhanced the CRE reporter, albeit with reduced efficacy compared with glucose most probably due to less efficient uptake into cells. However, because glucose and F-1,6-BP, but not pyruvate, affects the CRE-dependent reporter, we assume that cytosolic products of glucose metabolism are required to activate CRE-dependent gene expression.
Figure 1. Glucose exerts specific effects on CRE-dependent reporter gene activity in mHypoA-2/10 cells.
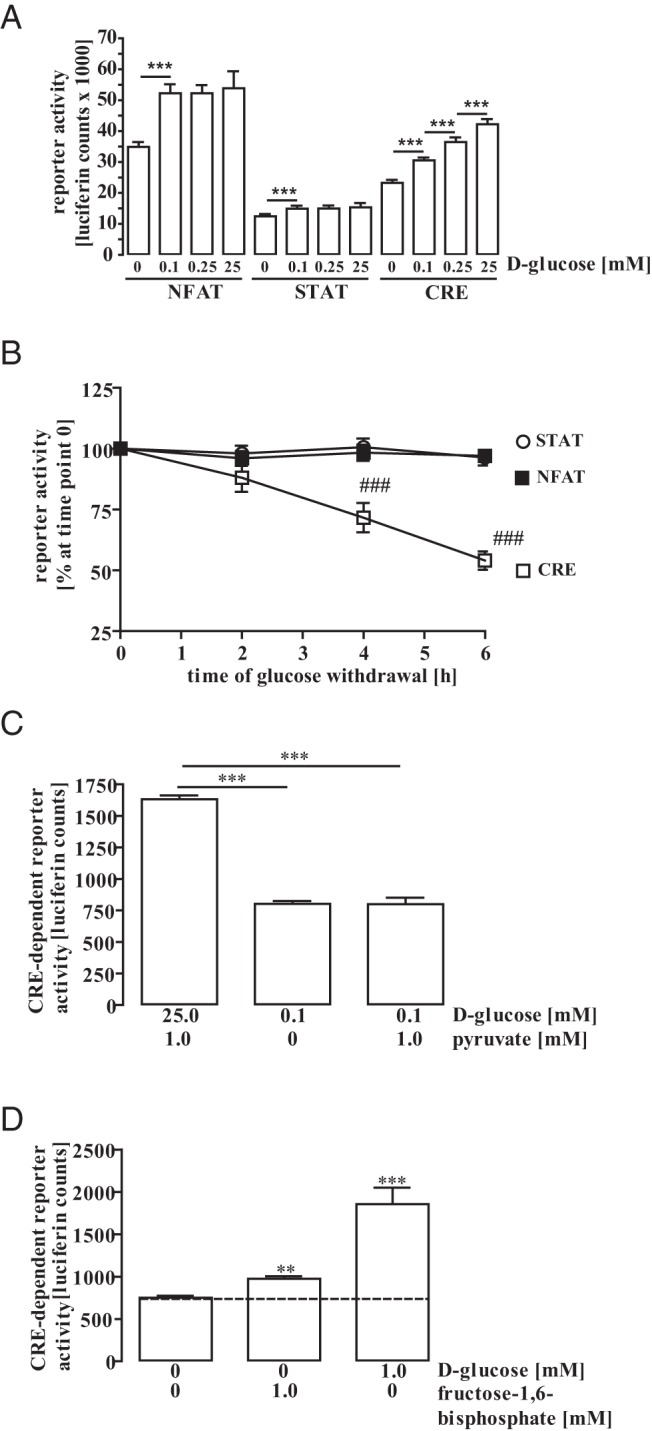
A and B, Reporter activity was measured after transient expression of a CRE-, NFAT-, or STAT-dependent reporter construct. A, Cells were cultured with the indicated glucose concentration for 24 hours. Data of 5 independent experiments performed in quadruplicates are presented as the mean ± SEM. Asterisks indicate a significant difference to the next lower glucose concentration. B, Cells were cultured without glucose for the indicated period of time. Data of 3 independent experiments performed in quadruplicates were compiled by setting the corresponding reporter activity measured at time point 0 to 100% and expressed as the mean ± SEM. Hash signs indicate a significant difference to 100%. C, mHypoA-2/10-CRE cells stably expressing the CRE-dependent reporter were cultured with the indicated glucose or pyruvate concentration for 24 hours. Data of 15 independent experiments performed in quadruplicates are presented as the mean ± SEM. Asterisks indicate a significant difference to the values of 25mM glucose. D, mHypoA-2/10-CRE cells were cultured with 1.0mM glucose or fructose-1,6-bisphophate for 24 hours. Data of 5 independent experiments performed in quadruplicates are presented as the mean ± SEM. Asterisks indicate a significant difference to the values of 25mM glucose.
Physiologically relevant changes in extracellular glucose concentrations enhance basal CRE-dependent gene expression in mHypoA-2/10-CRE cells
So far, we had established that glucose withdrawal from 25.0mM to 0.1mM for 24 hours selectively reduced CRE-dependent promoter activity. Next, we sought to explore how mHypoA-2/10-CRE cells that are adapted to low-glucose concentrations, would react when stimulated with increasing physiologically relevant glucose concentrations. To this end, we weaned cells off in 0.1mM glucose for 18 hours and then stimulated these cells with increasing glucose concentrations (0mM–2.0mM) for an additional 6-hour period (Figure 2, A and C). An incremental increase in extracellular glucose concentration dependently enhanced CRE-dependent reporter activity in low-glucose-adapted cells with an EC50 value of 0.4 ± 0.1mM. In parallel, we administered the nonmetabolizable glucose derivative 2-DG that had no impact on CRE reporter activity, providing further evidence that the effects of glucose depend on metabolism. In a second approach, we tested how glucose might affect basal CRE-dependent activity if extracellular glucose was adapted to physiologically relevant levels over a longer time period. Thus, we cultured cells with glucose concentrations between 0.1mM and 2.0mM for 24 hours without a previous weaning period (Figure 2, B and C). We also observed dose-dependent changes in CRE-dependent activity, albeit with a slightly increased EC50 value of 1.6 ± 0.8mM compared with “stimulation” conditions. Similarly, 2-DG did not mimic the effects of glucose on the CRE-dependent reporter under “basal” conditions.
Figure 2. Physiological-relevant changes in glucose concentrations enhance basal CRE-dependent reporter activity in mHypoA-2/10-CRE cells.
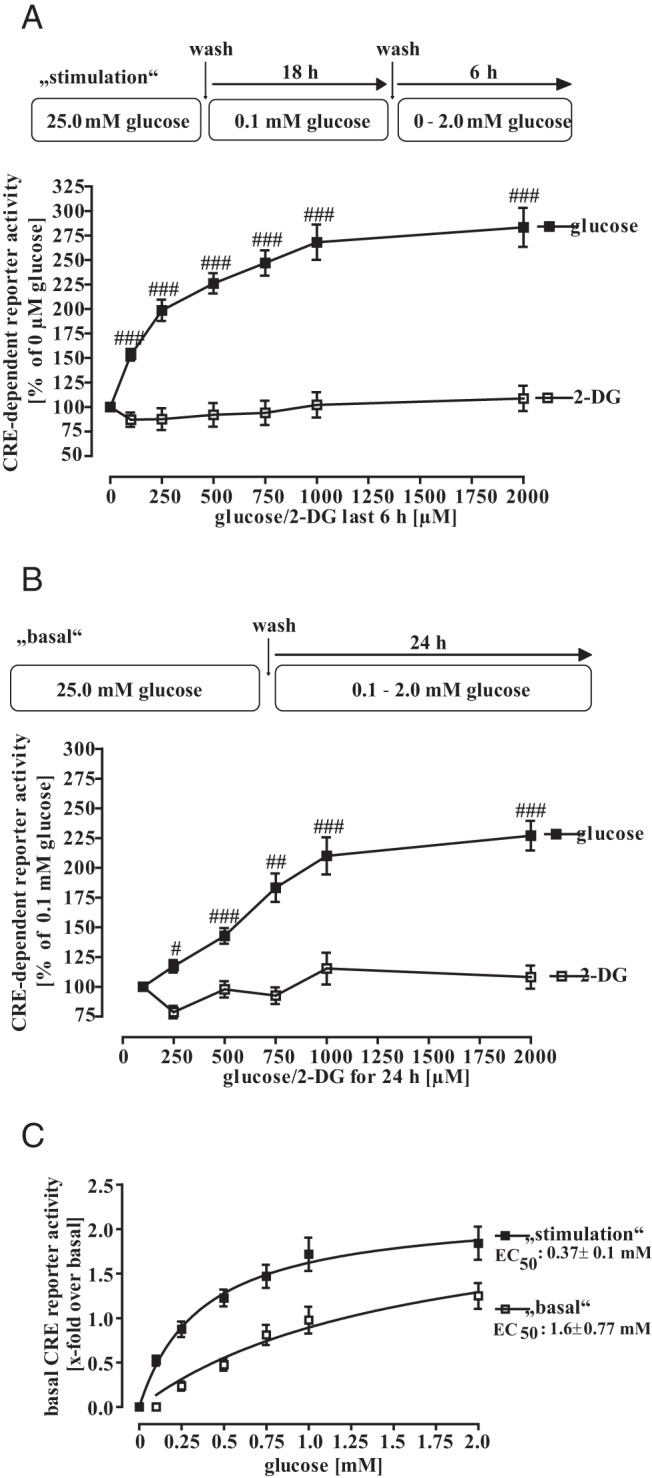
A, Cells were cultured for 18 hours with 0.1mM glucose and afterwards with 0mM, 0.1mM, 0.25mM, 0.5mM, 1.0mM, or 2.0mM glucose for additional 6 hours. Data of 10 independent experiments performed in quadruplicates were compiled by setting CRE activity of no glucose condition to 100% and expressed as the mean ± SEM. Hash signs indicate a significant difference to 100%. B, Cells were cultured with 0.1mM, 0.25mM, 0.5mM, 1.0mM, or 2.0mM glucose for 24 hours. Data of 10 independent experiments performed in quadruplicates were compiled by setting CRE activity of 0.1mM glucose to 100% and expressed as the mean ± SEM. Hash signs indicate a significant difference to 100%. C, Data obtained in A and B are presented as x-fold over basal and fitted to a 1-site saturation curve to calculate corresponding EC50 values.
Prolonged elevated glucose levels reduce the sensitivity of hypothalamic cells towards glucose
Based on data shown in Figure 2, it appeared that low-glucose-adapted mHypoA-2/10-CRE cells displayed higher CRE-dependent reporter activity when stimulated with 1.0mM glucose for 6 hours compared with cells that had been cultured in 1.0mM or even 25.0mM glucose for 24 hours (Figure 1C), suggesting that sustained elevated glucose concentrations desensitize glucose-induced CRE signaling. To rigorously test this hypothesis, we performed the basal and stimulation experiment with 1.0mM glucose in parallel (Figure 3A). Under stimulation conditions, 1.0mM glucose promoted significantly more CRE-dependent reporter activity compared with the basal experiment and even more compared with cells permanently cultured in 25.0mM glucose. In order to analyze whether reduced sensitivity of mHypoA-2/10 cells towards glucose is a dynamic process, we adapted cells to low-glucose conditions (0.1mM) for 12 hours, elevated glucose concentrations afterwards for additional 12 hours (0.5mM to 5.0mM), and then measured CRE-dependent reporter activity induced by 1.0mM glucose in parallel with time-dependent 3H-2-DG uptake (Figure 3B). Preincubation of glucose-starved cells with physiologically relevant brain glucose concentrations resulted in a stepwise decrease of glucose-induced CRE activation with a maximal desensitization of approximately 63% at 5.0mM. Interestingly, concentrations higher than 1.0mM also reduced glucose uptake with a maximal reduction of approximately 24%.
Figure 3. Prolonged elevated glucose levels reduced sensitivity of hypothalamic cells towards glucose.

A, mHypoA-2/10-CRE cells were incubated with the indicated glucose concentration for 24 hours. Data of 5 independent experiments performed in quadruplicates were compiled by calculating the x-fold over the values obtained for 0.1mM glucose (24 h) and expressed as the mean ± SEM. Asterisks indicate a significant difference to the values obtained for 0.1 (18 h) and 1.0mM (6 h) glucose. B, mHypoA-2/10-CRE cells were incubated with 0.1mM glucose for 12 hours. Afterwards, cells were incubated with 0.1mM, 0.5mM, 1.0mM, 2.5mM, or 5.0mM glucose for additional 12 hours. Next, cells were cultured for 30 minutes without glucose, stimulated or not with 1.0mM glucose for 6 hours, and CRE-dependent reporter activity measured (left panel) or glucose uptake monitored by measuring 3H-2-DG uptake for 1 hour (right panel). Left panel, Data of 4 independent experiments performed in quadruplicates were compiled by calculating the x-fold over basal of 1.0mM glucose for each condition and expressed as the mean ± SEM. Asterisks indicate a significant difference to the values obtained for 0.1mM glucose preincubation. Right panel, Data of 4 independent experiments performed in quadruplicates were compiled by calculating glucose uptake as percentage per hour for each condition and expressed as the mean ± SEM. Asterisks indicate a significant difference to the values obtained for 0.1mM glucose preincubation. Lower panel, Data of both experiments was normalized by setting values obtained with 0.1mM glucose as 100%. Hash signs indicate a significant difference to 100%.
Potential role of AMPK in glucose-induced CRE-dependent reporter activation
So far, we discovered that glucose and F-1,6-BP, but not 2-DG or pyruvate, enhance CRE-dependent reporter activity, suggesting that cytosolic products of glucose metabolism are involved. Metabolized glucose decreases the cytosolic AMP to ATP ratio and thus inhibits AMPK activity. In line with this notion, we observed decreased AMPK phosphorylation under both glucose stimulation and basal conditions with EC50 values of 0.4 ± 0.07mM and 1.0 ± 0.2mM, respectively (Figure 4A). To further evaluate a possible role of AMPK for glucose-induced CRE-dependent reporter activation, we applied the AMPK inhibitor dorsomorphin or the activator metformin and monitored CRE-dependent reporter activity (Figure 4B). Dorsomorphin enhanced and metformin inhibited CRE-dependent gene transcription in mHypoA-2/10-CRE cells, underscoring the role of AMPK as a negative regulator of CRE activity in hypothalamic cells. Furthermore, although metformin inhibited glucose-induced CRE-dependent reporter activation, blocking KATP channels by the sulfonylurea tolbutamide or neuronal NO synthase activity by NS-2028 had no effect (Figure 4C), strongly suggesting a role for AMPK in glucose-induced CRE activation. Tonic phosphorylation of hypothalamic CRTC-2 by AMPK has been reported to retain CRTC-2 in the cytosol (25). In line with these data, CRTC-2 was almost exclusively located in the cytosol of mHypoA-2/10-CRE cells cultured with 0.1mM glucose but translocated to the nucleus when cells were cultured with 1.0mM glucose (Figure 5A), suggesting that CRTC-2 is a target of glucose in hypothalamic cells. To further assess the role of CRCT-2 in glucose-induced CRE-dependent reporter activation, we aimed at down-regulating CRTC-2 protein expression by specific siRNAs. A cocktail of 3 distinct siRNAs against CRTC-2 dose dependently decreased CRCT-2, but not CREB expression, when compared with cells receiving control siRNA (Figure 5B, upper panel). Of note, decreased CRTC-2 expression inhibited CRE-dependent reporter activation induced by glucose and to a lesser extent by MSH activating CRE via CREB (Figure 5B, lower panel). Hence, we provide first experimental data to support the concept that enhancing effects of glucose on CRE-dependent gene expression require activation of the CRTC-2 transcription factor, most probably via inhibition of AMPK.
Figure 4. Role for AMPK in glucose-induced CRE-dependent reporter activation.
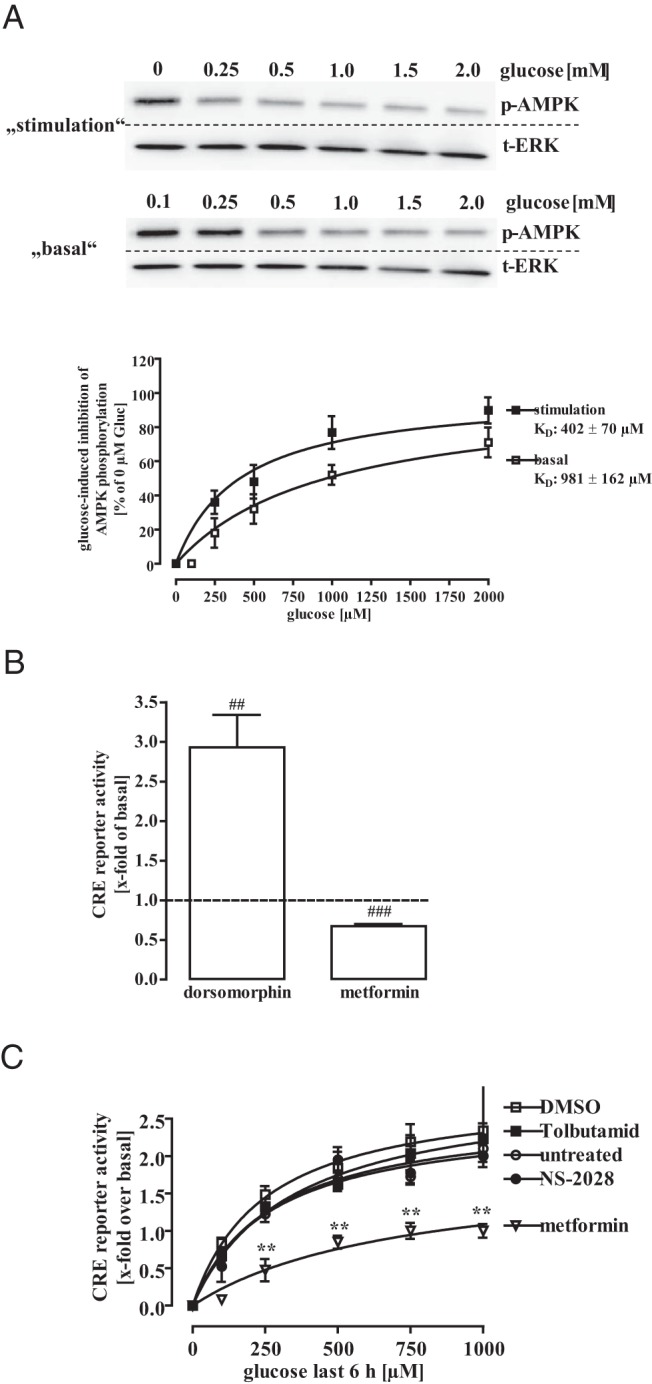
A, Lysates of mHypoA-2/10-CRE cells, either cultured for 18 hours with 0.1mM glucose and then stimulated with 0mM, 0.25mM, 0.5mM, 1.0mM, 1.5mM, or 2.0mM glucose for additional 6 hours or with 0.1mM, 0.25mM, 0.5mM, 1.0mM, 1.5mM, or 2.0mM glucose for 24 hours were subjected to Western blot analysis using either a phospho-specific antiserum against p-AMPK or against the total ERK-2 protein to control for the total protein amount. One representative blot is shown. Data of 5 independent experiments were quantified by densitometry, ratios between p-AMPK and t-ERK-2 signals calculated, glucose-induced AMPK dephosphorylation normalized to not stimulated cells (no glucose), and data fitted to a 1-site saturation curve to calculate corresponding EC50 values. B, mHypoA-2/10-CRE cells were cultured under high-glucose conditions (25.0mM) and either stimulated with 10μM dorsomorphin or 100μM metformin. Data of 5 independent experiments performed in quadruplicates were compiled by calculation the x-fold of basal expressed as the mean ± SEM. Hash signs indicate a significant difference to 1.0. C, mHypoA-2/10-CRE cells were cultured for 18 hours with 0.1mM glucose and then with 0mM, 0.25mM, 0.5mM, 1.0mM, or 2.0mM glucose for additional 6 hours. Before glucose stimulation, cells were treated with 500μM tolbutamide, 10μM NS-2028, 100μM metformin, or the carrier control (0.1% DMSO). Data of 4 independent experiments performed in quadruplicates were compiled and expressed as the mean ± SEM. Asterisks indicate a significant difference between the control and metformin-treated cells.
Figure 5. Role for CRTC-2 in glucose-induced CRE-dependent reporter activation.
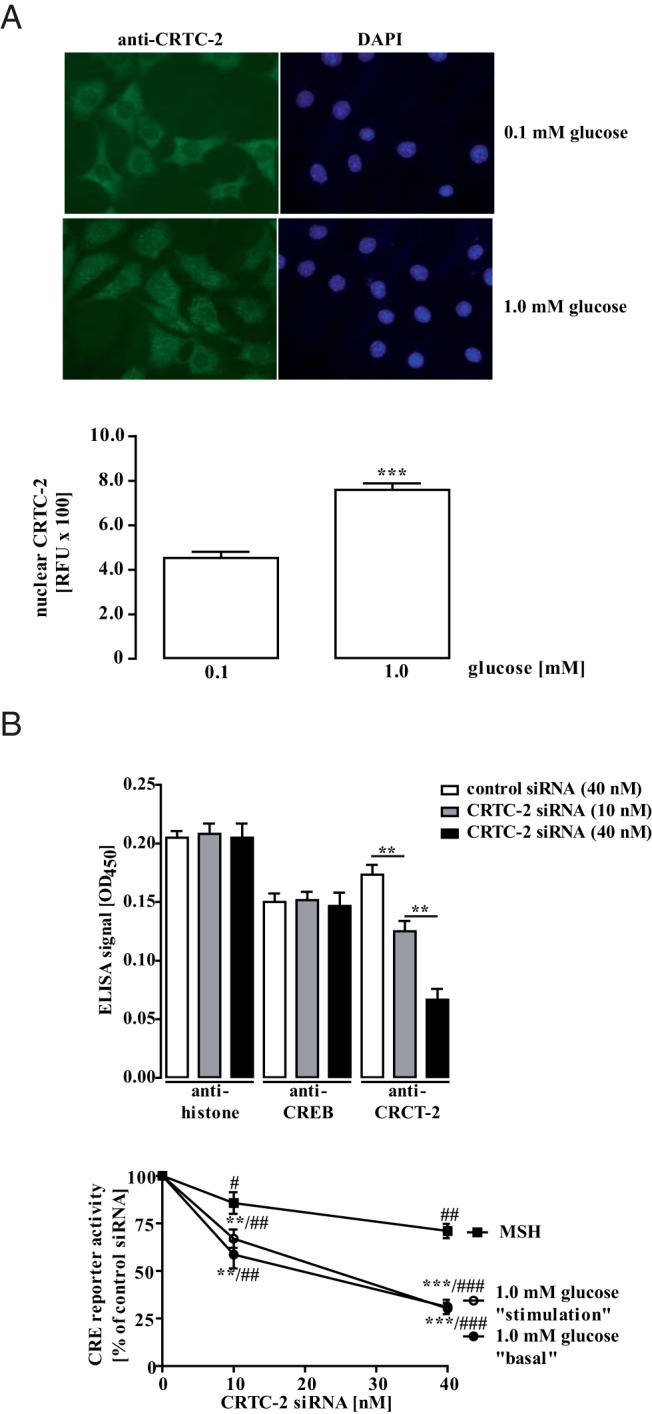
A, Immunostaining of mHypoA-2/10-CRE cells cultured with 0.1mM or 1.0mM glucose for 24 hours was performed using anti-CRCT-2/ Alexa Fluor 488-conjugated antimouse antibodies or DAPI. Immunostaining of cells cultured in 0.1mM (78 cells) or 1.0mM (82 cells) glucose was quantified and the corresponding background values subtracted. Random fluorescence units (RFUs) are expressed as the mean ± SEM, and asterisks indicate a significant difference between both glucose concentrations. B, mHypoA-2/10-CRE cells were transfected with 10nM or 40nM a cocktail of 3 distinct siRNAs against the murine CRCT-2 protein or with 40nM a random control siRNA. Cells were seeded on 48-well plates to monitor CRTC-2, CREB, or histone expression by whole cell ELISA (upper panel) and on 96-well plates to determine CRE-dependent reporter activation (lower panel). Upper panel, Cells were cultured for 24 hours with 0.1mM glucose. One representative experiment performed in quadruplicates is shown as the ± SEM. Background values of nonpermeabilized cells were subtracted. Lower panel, Cells were cultured either for 24 with 0.1mM or 1.0mM glucose (basal), for 18 hours with 0.1mM glucose and then 6 hours with no glucose or 1.0mM glucose (stimulation) or for 24 hours with 1.0mM and treated or not with 1μM MSH for the last 6 hours. Data of 5 independent experiments performed in quadruplicates were normalized by setting CRE-dependent reporter activation after expression of the control siRNA to 100% and are expressed as the mean ± SEM. Hash signs indicate a significant difference to 100%. Asterisks indicate a significant difference between MSH and glucose-treated cells.
Physiologically relevant increases in extracellular glucose concentrations enhance MSH-induced CRE activation
MSH is a key player within the HPT axis and the only known physiological enhancer of hypothalamic CRE activity so far. Because data shown in Figure 5B propose that glucose and MSH induce CRE-dependent reporter activation via distinct cellular mechanisms, we next assessed putative effects of glucose on MSH-induced CRE activation. To this end we first constructed concentration-response curves in mHypoA-2/10-CRE cells cultured in 25.0mM or 0.1mM glucose for 24 hours (Figure 6A). Reducing extracellular glucose concentrations had no effect on the potency of MSH to activate CRE but significantly reduced its efficacy. Thus, we determined concentration-response relationships of MSH-induced CRE activation under stimulation conditions, applying glucose concentrations between 0.1mM and 1.0mM (Figure 6B). Increasing extracellular glucose gradually improved the efficacy of MSH-induced CRE activation without affecting potency. Under basal conditions, 0.25mM glucose was sufficient to yield the maximal MSH effect on the CRE promoter (Figure 6C).
Figure 6. Physiological-relevant changes in glucose concentrations enhance MSH-induced CRE-dependent reporter activation.
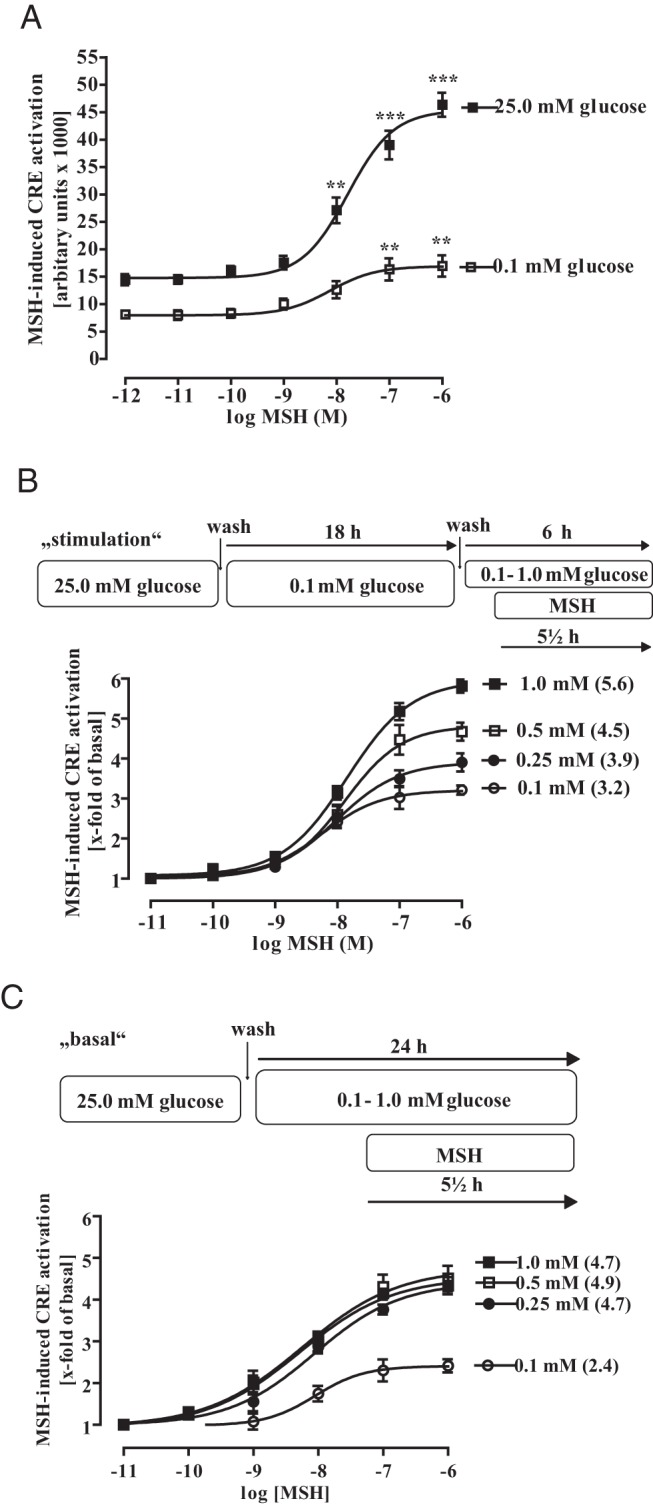
A, mHypoA-2/12-CRE cells were either cultured with 25.0mM or 0.1mM glucose for 18 and then stimulated with various MSH concentrations for additional 6 hours without changing the glucose level. Data of 5 independent experiments performed in quadruplicates were compiled, expressed as the mean ± SEM, and fitted to dose-response curves with fixed slopes. Asterisks indicate a significant difference to the basal values obtained with no MSH. B, Cells were cultured for 18 hours with 0.1mM glucose and then with 0.1mM, 0.25mM, 0.5mM, or 1.0mM glucose (as indicated) for additional 6 hours to obtain basal CRE activity. To measure MSH-induced CRE-dependent reporter activation distinct pools of cells were also stimulated with the indicated MSH concentration for the last 5.5 hours without changing the glucose level. Data of 5 independent experiments performed in quadruplicates were compiled, by calculating the corresponding x-fold of basal value, expressed as the mean ± SEM, and fitted to dose-response curves with fixed slopes. Numbers in parenthesis indicate the x-fold over basal for 1.0μM MSH. C, Cells were cultured with 0.1mM, 0.25mM, 0.5mM, or 1.0mM glucose (as indicated) for 24 hours to obtain basal CRE activity. To measure MSH-induced CRE-dependent reporter activation, distinct pools of cells were stimulated or not with the indicated MSH concentrations for the last 5.5 hours without changing the glucose concentration. Data of 5 independent experiments performed in quadruplicates were compiled, by calculating the corresponding x-fold of basal value, expressed as the mean ± SEM, and fitted to dose-response curves with fixed slopes. Numbers in parenthesis indicate the x-fold over basal for 1.0μM MSH.
MSH acts via G protein-coupled receptors dependent on the nucleoside triphosphate GTP whose metabolism is closely linked to glucose and ATP. Therefore, we wondered whether glucose deprivation would affect G protein-dependent signaling in general and measured Gq-dependent calcium signaling induced by bradykinin (Figure 7A) and Gs-dependent cAMP accumulation by MSH (Figure 7B). Both signaling pathways were unaltered when extracellular glucose concentrations were reduced to 0.1mM for 24 hours, indicating that the stimulatory effects of glucose on MSH-induced CRE activation are not caused by impaired G protein signaling. To garner further evidence that the potentiation of MSH-induced CRE activation by glucose is not due to direct effects on MSH signaling, we measured MSH-induced phosphorylation of CREB. As shown in Figure 7C, MSH-induced CREB phosphorylation was undistinguishable, when cells were cultured either with 0.1mM or 25.0mM glucose for 24 hours.
Figure 7. Effects of glucose on MSH-induced CRE-dependent reporter activation are not due to direct effects on MSH signaling.
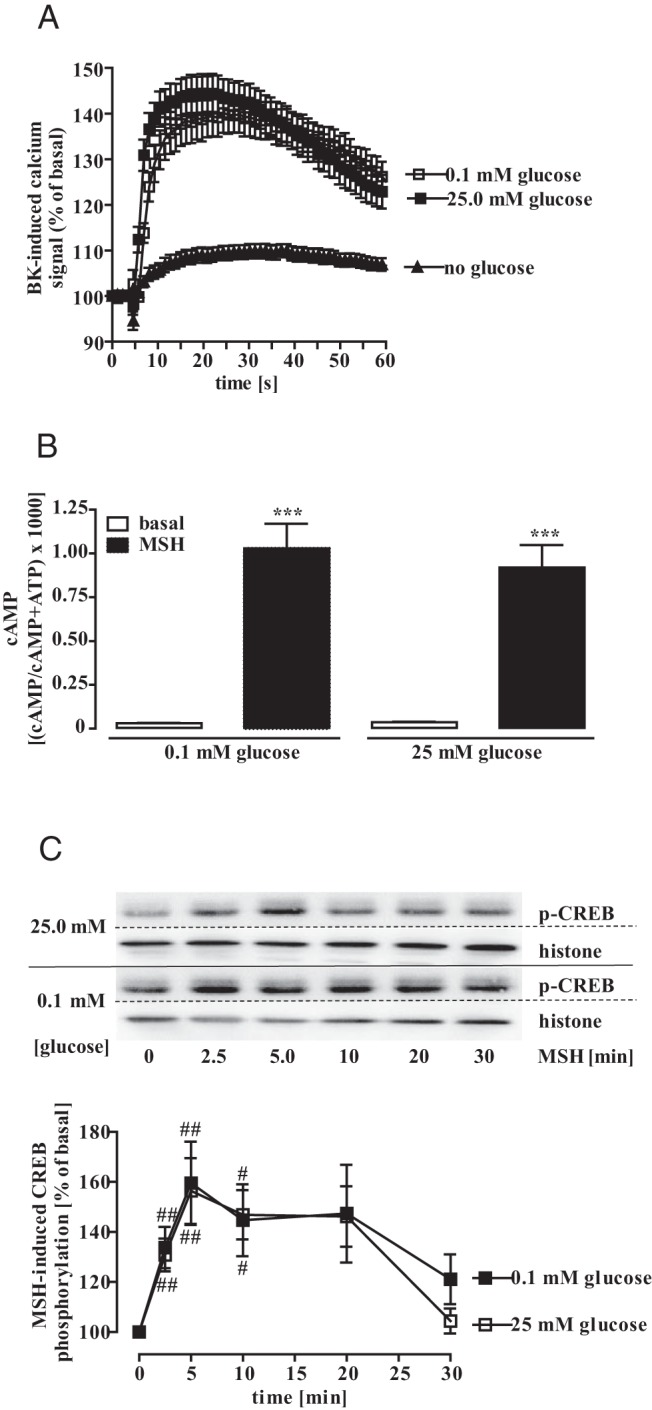
A, Bradykinin-induced calcium signaling (1.0μM) was analyzed in fura 2-loaded mHypoA-2/12-CRE cells cultured with 25.0mM, 0.1mM, or no glucose for 24 hours. Data of 3 independent experiments performed in quadruplicates were compiled by setting the first value measured to 100% and expressed as the mean ± SEM. B, MSH-induced cAMP accumulation was measured in mHypoA-2/12-CRE cells cultured with 25.0mM or 0.1mM glucose for 24 hours. Data of 4 independent experiments performed in quadruplicates were compiled and expressed as the mean ± SEM. Asterisks indicate a significant difference to basal. C, Lysates of mHypoA-2/10-CRE cells, cultured for 24 hours with 0.1mM or 25mM glucose and then stimulated with 1.0μM MSH for the indicated period of time were subjected to Western blot analysis using either a phospho-specific antiserum against p-CREB or against the total histone-3 protein to control for the total protein amount. One representative blot is shown. Data of 5 independent experiments were quantified by densitometry, ratios between p-AMPK and histone signals calculated, MSH-induced CREB phosphorylation normalized to not stimulated cells, and expressed as the mean ± SEM. Hash signs indicate a significant difference to 100%.
Glucose-induced TRH expression in hypothalamic cells
After having established that glucose enhances CRE-dependent reporter activity in mHypoA-2/10-CRE cells, we next investigated the effect of glucose on the expression of the CRE-controlled gene TRH. To this end, we monitored TRH protein expression by Western blot analysis under basal and glucose stimulation conditions (Figure 8). Glucose induced TRH expression under both conditions in a concentration-dependent manner with an EC50 value of 699 ± 342μM and 1203 ± 877μM, indicating that changes in glucose levels that affect CRE-dependent reporter activity also affect TRH protein expression. In order to investigate whether these effects occur on the transcriptional or translational level, we additionally analyzed the effects of glucose on TRH mRNA levels. As shown in Figure 9A, glucose significantly increased TRH mRNA levels under basal and stimulation conditions, clearly demonstrating that glucose affects TRH expression on the transcriptional level. Glucose also enhanced effects of MSH on TRH mRNA levels (Figure 9B), highlighting the important role of glucose for TRH expression and providing first evidence that glucose may engage the HPT axis by fostering hypothalamic TRH expression.
Figure 8. Glucose-induced TRH protein expression in hypothalamic cells.
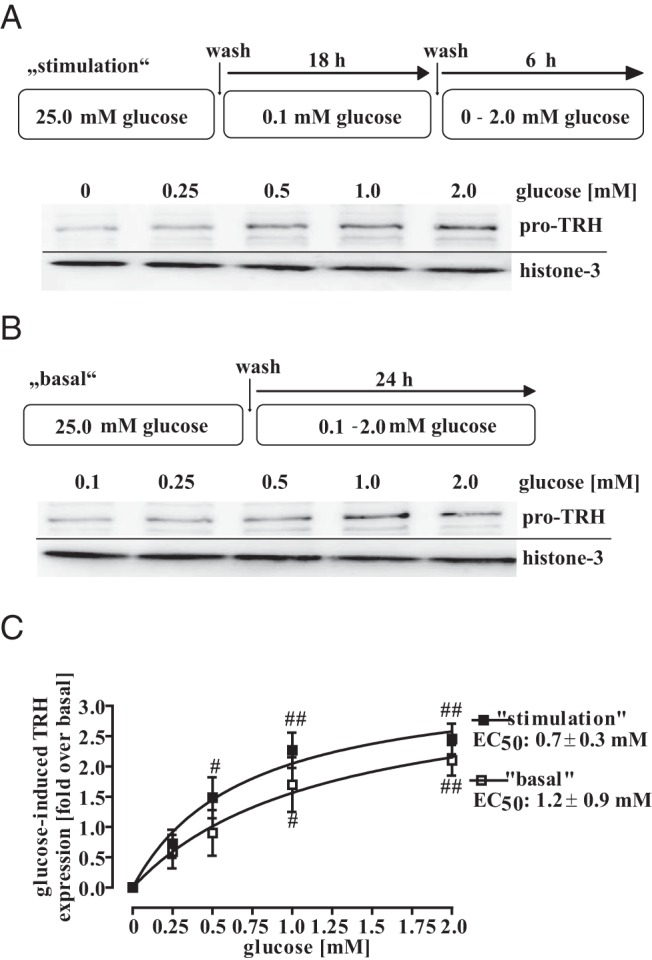
A, Cells were cultured for 18 hours with 0.1mM glucose and afterwards with 0mM, 0.25mM, 0.5mM, 1.0mM, or 2.0mM glucose for additional 6 hours. B, Cells were cultured with 0mM, 0.25mM, 0.5mM, 1.0mM, or 2.0mM glucose for 24 hours. Lysates were subjected to Western blot analysis using either a specific antiserum against pro-TRH or against the total histone-3 protein to control for the total protein amount. One representative blot is shown. C, Data of 5 independent experiments were quantified by densitometry, ratios between TRH and histone signals calculated, glucose-induced TRH expression normalized to not stimulated cells (no glucose), and data fitted to a 1-site saturation curve to calculate the EC50 value. Hash signs indicate a significant difference to 0 fold over basal.
Figure 9. Glucose-induced TRH mRNA expression in hypothalamic cells.

A, Cells were either cultured for 18 hours with 0.1mM glucose and afterwards with 0.0mM or 2.0mM glucose for 50 minutes (stimulation) or for 24 hours with 0.1mM or 2.0mM glucose (basal). Total mRNA was isolated and TRH mRNA specifically detected by qRT-PCR. Cp values measured for TRH after increasing glucose concentrations to 2.0mM were subtracted from those measured for actin (ΔCp stimulated). Cp values measured for TRH under basal conditions (0.0mM glucose for stimulation and 0.1mM glucose for basal) were subtracted from those measured for actin under the same conditions (ΔCp basal). Afterwards, ΔCp-basal values were subtracted from ΔCp-stimulated values and presented as 2−ΔΔCp. B, Cells were first cultured for 18 hours with 0.1mM glucose, then for 30 minutes with 0.0mM or 2.0mM glucose (stimulation) and afterwards treated or not with 1μM MSH for 20 minutes or for 24 hours with 0.1mM or 2.0mM glucose (basal) and then treated or not with 1μM MSH for 20 minutes. Total mRNA was isolated and TRH mRNA specifically detected by qRT-PCR. Cp values measured for TRH after MSH stimulation were subtracted from those measured for actin under the same conditions (ΔCp stimulated). Cp values measured for TRH under basal conditions (no MSH) were subtracted from those measured for actin under the same conditions (ΔCp basal). Afterwards, ΔCp-basal values were subtracted from ΔCp-stimulated values and presented as 2−ΔΔCp. Data of 4 independent experiments performed in triplicates were compiled and expressed as the mean ± SEM. Hash signs indicate a significant difference to 0; asterisks between basal and MSH-stimulated cells.
Discussion
Glucose has been reported to act on hypothalamic nuclei such as the nucleus arcuatus, the PVN and the ventromedial or lateral hypothalamus to affect the central regulation of food intake (19–23, 41–45). Interstitial hypothalamic glucose concentrations vary depending on food intake or insulin levels suggesting that naturally occurring fluctuations in hypothalamic glucose concentrations may directly induce anorexigenic signaling or modulate the impact of food intake-regulating hormones like leptin and insulin (19, 36, 37, 39, 40). However, little is known about the cellular events underlying the glucose-induced regulation of food intake.
Effects of glucose on central appetite control have frequently been attributed to the same mechanisms mediating glucose effects in pancreatic β-cells. Extracellular glucose is transported into the cell, metabolized to ATP that inhibits KATP channels and thereby depolarizes the plasma membrane leading to reduced insulin secretion (19–24). Besides this well-established effect of glucose on KATP channels, glucose-derived ATP also activates the CREB coactivator CRTC-2 in hypothalamic neurons (25). So far, however, it has remained elusive whether physiologically relevant fluctuations of extracellular glucose concentrations would activate CREB via CRTC-2 and whether glucose-induced CRTC-2/CREB activation would subsequently lead to CRE-controlled gene expression. Here, we conclusively show that physiologically relevant variations of extracellular brain glucose concentrations 1) fine-tune basal and MSH-induced CRE activity, 2) induce CRTC-2 translocation, 3) reduce glucose sensitivity of hypothalamic cells because of desensitization and diminished uptake, and 4) control hypothalamic TRH expression.
Our study shows that glucose selectively induces CRE-dependent reporter activity in hypothalamic cells; because incremental changes in the extracellular glucose concentration selectively enhance CRE but not NFAT- or STAT-dependent reporter activity. Thus, the effects of glucose on reporter activity cannot simply be explained by altered protein biosynthesis, but appear to reflect an intrinsic property of the CRE-dependent reporter. In vivo, brain glucose concentrations vary between 0.2mM and 2.5mM under physiological and between 0.1mM and 5.0mM under pathophysiological conditions (19, 36–40). It has been shown that interstitial hypothalamic glucose concentrations of freely feed rats were 1.42mM and of food-deprived (12 h) animals 0.84mM (36). Thus, EC50 values for glucose-induced CRE activation obtained in mHypoA-2/10-CRE cells (0.4mM and 1.6mM) indicate that physiological changes of extracellular glucose concentration may affect hypothalamic CRE activity. The same study showed that refeeding of food-deprived animals, led to a profound increase (5- to 10-fold) in hypothalamic glucose levels 2 hours after feeding lasting for an additional 2 hours. Thus, the kinetics of glucose-induced CRE activation in mHypoA-2/10-CRE cells mirrors that of food-induced changes in hypothalamic glucose levels observed in vivo.
Intracerebroventricular administration of glucose, but not of 2-DG, depressed food intake in mice (45). Concordant with these in vivo data, we show that glucose and its cytosolic metabolite F-1,6-BP, but not 2-DG, regulates CRE-dependent reporter activity in mHypoA-2/10-CRE cells. Hence, the effects of glucose on the CRE-dependent reporter depend on its metabolism, most probably due to the increase of cytosolic ATP levels. This observation highlights AMPK as a potential molecular target mediating glucose-induced CRE-dependent reporter activation. In accord with this assumption, we demonstrated that glucose induces AMPK dephosphorylation at Thr172, pertinent to its catalytic activity, with EC50 values (0.4mM and 1.0mM) similar to glucose-promoted CRE activation (46). Furthermore, we discovered that the AMPK inhibitor dorsomorphin enhances and the activator metformin decreases CRE-dependent reporter activity, illustrating an inverse correlation between AMPK and CRE activity in hypothalamic cells and suggesting that inhibition of AMPK activity is responsible for glucose-induced CRE activation. This notion is supported by our findings that 1) metformin inhibited glucose-induced CRE-dependent reporter activation, 2) glucose induces translocation of CRCT-2 to the nucleus, and 3) down-regulation of the CRTC-2 protein diminishes glucose-induced CRE-dependent reporter activation. However, at this stage, additional mechanisms underlying glucose-induced CRTC-2 activation cannot be ruled out. Alternative glucose targets could be KATP channels, neuronal NO synthase or glucose-sensitive TAS1 receptors, which were recently found in hypothalamic neurons (47). A KATP channel blocker (tolbutamide) or a NO synthase inhibitor (NS-2028) did not affect basal or glucose-induced CRE activity, excluding KATP channels and NO synthase as potential targets for glucose-induced CRE activation. At present, we cannot rigorously rule out a role for TAS1 receptors, but glucose did not interfere with known TAS1-induced second messengers such as calcium or cAMP in mHypoA-2/10 cells (data not shown).
Activation of melanocortin-4-receptors (MC4Rs) by MSH induces the expression of TRH via CRE activation in PVN neurons (48, 49). This sequel of events is of prime importance for proper functioning of the HPT axis and the regulation of human metabolism, as highlighted by the finding that mutations in the MC4R gene are the most frequent monogenic cause of severe obesity (3–9). Accordingly, targeted disruption of the MC4R or the MSH gene in mice causes an obesity-diabetes syndrome characterized by hyperphagia, hyperinsulinemia and hyperglycemia (10, 11). Here, we complement the overall picture by revealing that glucose and the melanocortin system functionally interact on the level of hypothalamic cells. It appears that under stimulation conditions, mimicking a high caloric meal after a longer fasting period, the efficacy of MSH-induced signaling is closely linked to the glucose concentration. Hence we propose that anorexigenic actions of MSH are controlled by glucose in that high glucose concentrations enhance MSH-mediated depression of food intake. Enhanced anorexigenic signaling by MSH in the presence of elevated glucose concentrations appears to be reasonable in the physiological context, but in the case of dysregulation or malfunction, might also contribute to pathophysiological consequences such as obesity. However, further in vivo studies are required to analyze the physiological outcome of the functional interactions between glucose and MSH in the hypothalamus.
With regard to putative signaling events responsible for the effects of glucose on MSH, we provide data indicating that glucose does not directly affect MSH signaling on the level of G protein activation or CREB phosphorylation. However, our study supports a model in which CRE activity in hypothalamic cells is controlled by CREB and CRTC-2, thus coactivation of both transcription factors might be required for full CRE activation. In the framework of such a model, glucose coadministration may support MSH-promoted CREB/CRE activation by inducing significant nuclear CRTC-2 translocation due to its strong inhibitory effects on AMPK.
With this report we provide new insight into the actions of glucose on cellular signaling that affect hypothalamic gene expression. Assuming that our conclusions can be extended to the physiological level, some of them may be of clinical interest (1). Several studies reported that metformin reduces serum TSH levels in patients with T2DM, a phenomenon not observed for sulfonylureas, such as tolbutamide (50–56). If hypothalamic CRE activity is correlated with serum TSH levels, as shown for MSH, differential effects of metformin and tolbutamide in hypothalamic cells may contribute to the distinct actions of the 2 drug classes in T2DM patients (2). The present paper provides first experimental evidence that physiologically relevant glucose fluctuations enhance TRH expression on the mRNA and protein levels in hypothalamic cells. Interestingly, glucose concentrations above 5mM have also been reported to depress TRH release from rat hypothalamic nuclei, suggesting a negative feed-back loop (57). Further, glucose also stimulates the release of pancreatic TRH in rats, indicating that glucose affects central and peripheral TRH production/release (58). Because of the established anorexigenic actions of TRH, our data contribute to a better understanding of glucose-induced appetite control and thus may pave the way for new strategies to treat obesity. Furthermore, it has been recognized that T2DM severely disturbs the regulation of the HPT axis and that suboptimally controlled patients show increased serum TSH levels (17, 18). In line with this notion, it has been recommended that the diagnosis of thyroid function in T2DM patients be delayed until improvement of the metabolic status (15). Thus, glucose-induced hypothalamic TRH expression could likely be responsible for the association between hyperglycaemia and hyperthyroidism (3). We show that incubation of cells, which have been adapted to low-glucose concentrations, with increasing glucose concentrations exhibited a significant loss of sensitivity towards glucose. At this point we can only speculate about the molecular basis of glucose-induced desensitization. Of note, AMPK has been shown to enhance glucose uptake into skeletal muscle cells (59), thus sustained inhibition of AMPK activity due to high glucose levels could reduce glucose uptake into hypothalamic cells and thus decrease glucose-induced CRE activation. Indeed, we observed reduced glucose uptake when cells were incubated with glucose concentrations above 1.0mM, but these effects were small and concentrations below 1.0mM, also eliciting CRE desensitization, had no effect on glucose uptake. Therefore, we assume that reduced uptake is not sufficient to fully account for glucose desensitization implicating uptake-independent additional mechanisms. However, these data suggest that prolonged hyperglycaemic conditions, for instance due to uncontrolled eating behavior, weaken the central anorexigenic effects of glucose and thus may provide a new association between T2DM and obesity. Hence, it will be enlightening in future studies to see whether prolonged elevations of hypothalamic glucose concentrations impair glucose sensitivity of food intake-controlling neurons in vivo and thereby interfere with central appetite control.
Acknowledgments
This work was supported by the “FöFoLe” Programe of the Medicine Department of the Medicine Department of the Ludwig-Maximilians-Universität München Grant 28/2014.
Disclosure Summary: The authors have nothing to disclose.
Funding Statement
This work was supported by the “FöFoLe” Programe of the Medicine Department of the Medicine Department of the Ludwig-Maximilians-Universität München Grant 28/2014.
Footnotes
- AMPK
- AMP-activated protein kinase
- Cp
- crossing point
- CRE
- cAMP-response element
- CREB
- CRE-binding protein
- CRTC-2
- CREB-regulated transcriptional coactivator 2
- 2-DG
- 2-deoxy-D-glucose
- F-1,6-BP
- fructose-1,6-bisphosphate
- HPT
- hypothalamic-pituitary-thyroid
- KATP
- ATP-sensitive K+
- MC4R
- melanocortin-4-receptor
- MSH
- melanocyte-stimulating hormone
- NFAT
- nuclear factor of activated t cells
- NO
- nitrogen monoxide
- p
- probability
- PBST
- PBS with 0.02% Tween 20
- PVN
- paraventricular nucleus
- qRT-PCR
- quantitative RT-PCR
- RT
- room temperature
- STAT
- signal transducers and activator of transcription
- T2DM
- type 2 diabetes mellitus
- TAS1
- taste receptor type 1.
References
- 1. Amin A, Dhillo WS, Murphy KG. The central effects of thyroid hormones on appetite. J Thyroid Res. 2011;2011:306510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Nillni EA. Regulation of the hypothalamic thyrotropin releasing hormone (TRH) neuron by neuronal and peripheral inputs. Front Neuroendocrinol. 2010;31:134–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Biebermann H, Krude H, Elsner A, Chubanov V, Gudermann T, Grüters A. Autosomal-dominant mode of inheritance of a melanocortin-4 receptor mutation in a patient with severe early-onset obesity is due to a dominant-negative effect caused by receptor dimerization. Diabetes. 2003;52:2984–2988. [DOI] [PubMed] [Google Scholar]
- 4. Govaerts C, Srinivasan S, Shapiro A, et al. Obesity-associated mutations in the melanocortin 4 receptor provide novel insights into its function. Peptides. 2005;26:1909–1919. [DOI] [PubMed] [Google Scholar]
- 5. Hinney A, Bettecken T, Tarnow P, et al. Prevalence, spectrum, and functional characterization of melanocortin-4 receptor gene mutations in a representative population-based sample and obese adults from Germany. J Clin Endocrinol Metab. 2006;91:1761–1769. [DOI] [PubMed] [Google Scholar]
- 6. Hinney A, Hohmann S, Geller F, et al. Melanocortin-4 receptor gene: case-control study and transmission disequilibrium test confirm that functionally relevant mutations are compatible with a major gene effect for extreme obesity. J Clin Endocrinol Metab. 2003;88:4258–4267. [DOI] [PubMed] [Google Scholar]
- 7. Tao YX, Segaloff DL. Functional characterization of melanocortin-4 receptor mutations associated with childhood obesity. Endocrinology. 2003;144:4544–4551. [DOI] [PubMed] [Google Scholar]
- 8. Vaisse C, Clement K, Guy-Grand B, Froguel P. A frameshift mutation in human MC4R is associated with a dominant form of obesity. Nat Genet. 1998;20:113–114. [DOI] [PubMed] [Google Scholar]
- 9. Yeo GS, Lank EJ, Farooqi IS, Keogh J, Challis BG, O'Rahilly S. Mutations in the human melanocortin-4 receptor gene associated with severe familial obesity disrupts receptor function through multiple molecular mechanisms. Hum Mol Genet. 2003;12:561–574. [DOI] [PubMed] [Google Scholar]
- 10. Huszar D, Lynch CA, Fairchild-Huntress V, et al. Targeted disruption of the melanocortin-4 receptor results in obesity in mice. Cell. 1997;88:131–141. [DOI] [PubMed] [Google Scholar]
- 11. Balthasar N, Dalgaard LT, Lee CE, et al. Divergence of melanocortin pathways in the control of food intake and energy expenditure. Cell. 2005;123:493–505. [DOI] [PubMed] [Google Scholar]
- 12. Légrádi G, Holzer D, Kapcala LP, Lechan RM. Glucocorticoids inhibit stress-induced phosphorylation of CREB in corticotropin-releasing hormone neurons of the hypothalamic paraventricular nucleus. Neuroendocrinology. 1997;66:86–97. [DOI] [PubMed] [Google Scholar]
- 13. Sarkar S, Légrádi G, Lechan RM. Intracerebroventricular administration of α-melanocyte stimulating hormone increases phosphorylation of CREB in TRH- and CRH-producing neurons of the hypothalamic paraventricular nucleus. Brain Res. 2002;945:50–59. [DOI] [PubMed] [Google Scholar]
- 14. Díaz-Gallardo MY, Cote-Vélez A, Carreón-Rodríguez A, Charli JL, Joseph-Bravo P. Phosphorylated cyclic-AMP-response element-binding protein and thyroid hormone receptor have independent response elements in the rat thyrotropin-releasing hormone promoter: an analysis in hypothalamic cells. Neuroendocrinology. 2010;91:64–76. [DOI] [PubMed] [Google Scholar]
- 15. Celani MF, Bonati ME, Stucci N. Prevalence of abnormal thyrotropin concentrations measured by a sensitive assay in patients with type 2 diabetes mellitus. Diabetes Res. 1994;27:15–25. [PubMed] [Google Scholar]
- 16. Wilber JF, Banerji A, Prasad C, Mori M. Alterations in hypothalamic-pituitary-thyroid regulation produced by diabetes mellitus. Life Sci. 1981;28:1757–1763. [DOI] [PubMed] [Google Scholar]
- 17. Hage M, Zantout MS, Azar ST. Thyroid disorders and diabetes mellitus. J Thyroid Res. 2011;2011:439463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Nascimento-Saba CC, Breitenbach MM, Rosenthal D. Pituitary-thyroid axis in short- and long-term experimental diabetes mellitus. Braz J Med Biol Res. 1997;30:269–274. [DOI] [PubMed] [Google Scholar]
- 19. Routh VH. Glucose sensing neurons in the ventromedial hypothalamus. Sensors. 2010;10:9002–9025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Melnick IV, Price CJ, Colmers WF. Glucosensing in parvocellular neurons of the rat hypothalamic paraventricular nucleus. Eur J Neurosci. 2011;34:272–282. [DOI] [PubMed] [Google Scholar]
- 21. Burdakov D, Gerasimenko O, Verkhratsky A. Physiological changes in glucose differentially modulate the excitability of hypothalamic melanin-concentrating hormone and orexin neurons in situ. J Neurosci. 2005;25:2429–2433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Song Z, Routh VH. Differential effects of glucose and lactate on glucosensing neurons in the ventromedial hypothalamic nucleus. Diabetes. 2005;54:15–22. [DOI] [PubMed] [Google Scholar]
- 23. Wang R, Liu X, Hentges ST, et al. The regulation of glucose-excited neurons in the hypothalamic arcuate nucleus by glucose and feeding-relevant peptides. Diabetes. 2004;53:1959–1965. [DOI] [PubMed] [Google Scholar]
- 24. Lee K, Dixon AK, Richardson PJ, Pinnock RD. Glucose-receptive neurones in the rat ventromedial hypothalamus express KATP channels composed of Kir6.1 and SUR1 subunits. J Physiol. 1999;515(pt 2):439–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lerner RG, Depatie C, Rutter GA, Screaton RA, Balthasar N. A role for the CREB co-activator CRTC2 in the hypothalamic mechanisms linking glucose sensing with gene regulation. EMBO Rep. 2009;10:1175–1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Su H, Jiang L, Carter-Su C, Rui L. Glucose enhances leptin signaling through modulation of AMPK activity. PLoS One. 2012;7:e31636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Breit A, Besik V, Solinski HJ, et al. Serine-727 phosphorylation activates hypothalamic STAT-3 independently from tyrosine-705 phosphorylation. Mol Endocrinol. 2015;29:445–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Dalvi PS, Belsham DD. Glucagon-like peptide-2 directly regulates hypothalamic neurons expressing neuropeptides linked to appetite control in vivo and in vitro. Endocrinology. 2012;153:2385–2397. [DOI] [PubMed] [Google Scholar]
- 29. Dalvi PS, Erbiceanu FD, Irwin DM, Belsham DD. Direct regulation of the proglucagon gene by insulin, leptin, and cAMP in embryonic versus adult hypothalamic neurons. Mol Endocrinol. 2012;26:1339–1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Belsham DD, Fick LJ, Dalvi PS, et al. Ciliary neurotrophic factor recruitment of glucagon-like peptide-1 mediates neurogenesis, allowing immortalization of adult murine hypothalamic neurons. FASEB J. 2009;23:4256–4265. [DOI] [PubMed] [Google Scholar]
- 31. Dhillon SS, Gingerich S, Virtanen C, Belsham DD. Gene array analysis of embryonic- versus adult-derived hypothalamic NPY-expressing cell lines. Mol Cell Endocrinol. 2012;358:116–126. [DOI] [PubMed] [Google Scholar]
- 32. Dhillon SS, Belsham DD. Leptin differentially regulates NPY secretion in hypothalamic cell lines through distinct intracellular signal transduction pathways. Regul Pept. 2011;167:192–200. [DOI] [PubMed] [Google Scholar]
- 33. Dhillon SS, Belsham DD. Estrogen inhibits NPY secretion through membrane-associated estrogen receptor (ER)-α in clonal, immortalized hypothalamic neurons. Int J Obes. 2011;35:198–207. [DOI] [PubMed] [Google Scholar]
- 34. Himmler A, Stratowa C, Czernilofsky AP. Functional testing of human dopamine D1 and D5 receptors expressed in stable cAMP-responsive luciferase reporter cell lines. J Recept Res. 1993;13:79–94. [DOI] [PubMed] [Google Scholar]
- 35. Solinski HJ, Boekhoff I, Bouvier M, Gudermann T, Breit A. Sensory neuron-specific MAS-related gene-X1 receptors resist agonist-promoted endocytosis. Mol Pharmacol. 2010;78:249–259. [DOI] [PubMed] [Google Scholar]
- 36. Mayer CH, Fink H, Rex A, Voigt JP. Changes in extracellular hypothalamic glucose in relation to feeding. Eur J Neurosci. 2006;24:1695–1701. [DOI] [PubMed] [Google Scholar]
- 37. Dunn-Meynell AA, Sanders NM, Compton D, et al. Relationship among brain and blood glucose levels and spontaneous and glucoprivic feeding. J Neurosci. 2009;29:7015–7022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Diggs-Andrews KA, Zhang X, Song Z, Daphna-Iken D, Routh VH, Fisher SJ. Brain insulin action regulates hypothalamic glucose sensing and the counterregulatory response to hypoglycemia. Diabetes. 2010;59:2271–2280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Silver IA, Ereciska M. Extracellular glucose concentration in mammalian brain: continuous monitoring of changes during increased neuronal activity and upon limitation in oxygen supply in normo-, hypo-, and hyperglycemic animals. J Neurosci. 1994;14:5068–5076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. DeVries JH, Snoek FJ, Kostense PJ, Heine RJ. Improved glycaemic control in type 1 diabetes patients following participation per se in a clinical trial–mechanisms and implications. Diabetes Metab Res Rev. 2003;19:357–362. [DOI] [PubMed] [Google Scholar]
- 41. Burdakov D, Gonzalez JA. Physiological functions of glucose-inhibited neurones. Acta Physiol (Oxf). 2009;195:71–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. de Gortari P, Cisneros M, Joseph-Bravo P. Chronic ethanol or glucose consumption alter TRH content and pyroglutamyl aminopeptidase II activity in rat limbic regions. Regul Pept. 2005;127:141–150. [DOI] [PubMed] [Google Scholar]
- 43. de Gortari P, Cisneros M, Medellín MA, Joseph-Bravo P. Chronic ingestion of ethanol or glucose solutions affects hypothalamic and limbic TRH metabolism in dams and their pups. Neurochem Int. 2002;41:237–249. [DOI] [PubMed] [Google Scholar]
- 44. de Andrade IS, Zemdegs JC, de Souza AP, et al. Diet-induced obesity impairs hypothalamic glucose sensing but not glucose hypothalamic extracellular levels, as measured by microdialysis. Nutr Diabetes. 2015;5:e162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Bady I, Marty N, Dallaporta M, et al. Evidence from glut2-null mice that glucose is a critical physiological regulator of feeding. Diabetes. 2006;55:988–995. [DOI] [PubMed] [Google Scholar]
- 46. Hawley SA, Davison M, Woods A, et al. Characterization of the AMP-activated protein kinase kinase from rat liver and identification of threonine 172 as the major site at which it phosphorylates AMP-activated protein kinase. J Biol Chem. 1996;271:27879–27887. [DOI] [PubMed] [Google Scholar]
- 47. Ren X, Zhou L, Terwilliger R, Newton SS, de Araujo IE. Sweet taste signaling functions as a hypothalamic glucose sensor. Front Integr Neurosci. 2009;3:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Cyr NE, Toorie AM, Steger JS, et al. Mechanisms by which the orexigen NPY regulates anorexigenic α-MSH and TRH. Am J Physiol Endocrinol Metab. 2013;304:E640–E650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Lechan RM, Fekete C. Role of melanocortin signaling in the regulation of the hypothalamic-pituitary-thyroid (HPT) axis. Peptides. 2006;27:310–325. [DOI] [PubMed] [Google Scholar]
- 50. Cappelli C, Rotondi M, Pirola I, et al. Thyreotropin levels in diabetic patients on metformin treatment. Eur J Endocrinol. 2012;167:261–265. [DOI] [PubMed] [Google Scholar]
- 51. Cappelli C, Rotondi M, Pirola I, et al. Metformin-induced thyrotropin suppression is not associated with cardiac effects. Hormones. 2014;13:252–258. [DOI] [PubMed] [Google Scholar]
- 52. Cappelli C, Rotondi M, Pirola I, et al. TSH-lowering effect of metformin in type 2 diabetic patients: differences between euthyroid, untreated hypothyroid, and euthyroid on L-T4 therapy patients. Diabetes Care. 2009;32:1589–1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Krysiak R, Okopien B. The effect of metformin on the hypothalamic-pituitary-thyroid axis in women with polycystic ovary syndrome and subclinical hypothyroidism. J Clin Pharmacol. 2015;55:45–49. [DOI] [PubMed] [Google Scholar]
- 54. Krysiak R, Szkrobka W, Okopien B. The effect of metformin on the hypothalamic-pituitary-thyroid axis in patients with type 2 diabetes and subclinical hyperthyroidism. Exp Clin Endocrinol Diabetes. 2015;123:205–208. [DOI] [PubMed] [Google Scholar]
- 55. Fournier JP, Yin H, Yu OH, Azoulay L. Metformin and low levels of thyroid-stimulating hormone in patients with type 2 diabetes mellitus. CMAJ. 2014;186:1138–1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Karimifar M, Aminorroaya A, Amini M, et al. Effect of metformin on thyroid stimulating hormone and thyroid volume in patients with prediabetes: a randomized placebo-controlled clinical trial. J Res Med Sci. 2014;19:1019–1026. [PMC free article] [PubMed] [Google Scholar]
- 57. Lewis BM, Ismail IS, Issa B, Peters JR, Scanlon MF. Desensitisation of somatostatin, TRH and GHRH responses to glucose in the diabetic (Goto-Kakizaki) rat hypothalamus. J Endocrinol. 1996;151:13–17. [DOI] [PubMed] [Google Scholar]
- 58. Benický J, Strbák V. Glucose stimulates and insulin inhibits release of pancreatic TRH in vitro. Eur J Endocrinol. 2000;142:60–65. [DOI] [PubMed] [Google Scholar]
- 59. Musi N, Hayashi T, Fujii N, Hirshman MF, Witters LA, Goodyear LJ. AMP-activated protein kinase activity and glucose uptake in rat skeletal muscle. Am J Physiol Endocrinol Metab. 2001;280:E677–E684. [DOI] [PubMed] [Google Scholar]