

Abstract
Purpose: Implication of protein-protein interactions (PPIs) in development of many diseases such as cancer makes them attractive for therapeutic intervention and rational drug design. RON (Recepteur d’Origine Nantais) tyrosine kinase receptor has gained considerable attention as promising target in cancer therapy. The activation of RON via its ligand, macrophage stimulation protein (MSP) is the most common mechanism of activation for this receptor. The aim of the current study was to perform in silico alanine scanning mutagenesis and to calculate binding energy for prediction of hot spots in protein-protein interface between RON and MSPβ chain (MSPβ).
Methods: In this work the residues at the interface of RON-MSPβ complex were mutated to alanine and then molecular dynamics simulation was used to calculate binding free energy.
Results: The results revealed that Gln193, Arg220, Glu287, Pro288, Glu289, and His424 residues from RON and Arg521, His528, Ser565, Glu658, and Arg683 from MSPβ may play important roles in protein-protein interaction between RON and MSP.
Conclusion: Identification of these RON hot spots is important in designing anti-RON drugs when the aim is to disrupt RON-MSP interaction. In the same way, the acquired information regarding the critical amino acids of MSPβ can be used in the process of rational drug design for developing MSP antagonizing agents, the development of novel MSP mimicking peptides where inhibition of RON activation is required, and the design of experimental site directed mutagenesis studies.
Keywords: Alanine screening, Cancer, Drug design, Molecular dynamic simulation, MSP, RON
Introduction
Protein-protein interactions (PPIs) are involved in many biological processes as key regulatory steps1 and when aberrantly regulated are implicated in the development of many diseases such as cancer.2-4 These versatile roles make PPIs attractive for therapeutic intervention and rational drug design.5-7 Different classes of the approved therapeutic agents or those in development stages have been shown to interfere with PPIs in order to overcome the corresponding diseases.8-11
In PPI, the amino acids at the interaction interface have great importance in terms of starting point for the initiation of the biological and cellular functions.12 Usually several residues are exposed at the interface between the interacting proteins, but they do not contribute equally to the binding energy. The important key residues when mutated into alanine residue weaken the binding strength (increase of free energy of binding at least 2.0 kcal/mol) are called “hot spots”.13,14 Identification of the hot spots is critical in designing therapeutic agents which exert their effects by influencing PPIs. One of the experimental methods commonly used for identification of these hot spots is site-directed mutagenesis followed by comparative functional assays of the mutated and the wild type proteins; however, these experiments are time-consuming and expensive.15 Structural elucidation of the partner proteins within a complex by means of biophysical methods such as X-ray crystallography and NMR is also possible but again costly and demanding.16 In the era of modern drug discovery and development, the use of in silico methods shortens the rational drug design process in terms of both time and cost.17-21 In this regard, identifying hot spots is not an exception.22,23 Computational alanine scanning mutagenesis is a virtual method which has been extensively used for the characterization and prediction of hot spots in protein-protein, protein-DNA and protein-small molecule complexes.22,24-29 Charged, polar, or bulky amino acids are virtually mutated to a neutral, small and non-polar amino acid such as alanine and then binding free energy is calculated for both wild type and mutant forms in order to estimate the contribution of the mutated residues to the binding energy.30,31 One of the most routinely used approaches for computational estimation of binding free energy is based on accessible surface area models of implicit solvation method where molecular mechanics data are treated by Generalized Born surface area (MM-GBSA) algorithm.32-40
Tyrosine kinase receptors (TKRs) involved in well characterized protein-protein interactions are among potential candidate targets for anticancer drug development.41-46 TKRs are cell surface receptors for different polypeptide ligands and have pivotal roles in regulation of many cellular functions and physiological events47,48 and when aberrantly expressed and activated play key functions in development and progression of different types of cancers.49-54 Ligand-mediated receptor dimerization is the main mechanism of activation triggered by ligand binding to the extracellular domain of its specific receptor.55-57 This protein-protein interaction causes receptor dimerization followed by authophosphorylation of tyrosine residues located within the intracellular tyrosine kinase domain (catalytic tyrosines) followed by phosphorylation of tyrosine residues located within the C tail (docking tyrosines) that become the docking site for adaptor/effector proteins responsible of transducing the downstream signaling pathways resulting in cellular proliferation, differentiation, metabolism, survival, migration, and cell cycle control.58 In principle, all PPIs mediated by TKRs (including the downstream PPIs) could be targeted for cancer therapy59,60 but generally therapeutic PPI inhibitors interfere with the binding of endogenous ligands to the receptor.61-67 Therefore, it is obvious that uncovering the details of PPIs between TKRs and their ligands can provide useful information applicable to design of new anticancer agents.
RON (Recepteur d'Origine Nantais) is a member of TKRs superfamily, its role in tumorigenesis has been established in different cancer types and numerous studies have suggested RON as a promising target for anticancer drug development.68,69 RON also known as MSTR1 (Macrophage Stimulating Receptor1) belongs to MET proto-oncogene family,70 and is usually expressed at low levels in normal tissues while it is highly expressed in cancer cells.71 Structurally, RON is a disulfide linked heterodimer protein made of two chains, an extracellular α-chain and a β-chain which consists extracellular, transmembrane, and intracellular regions. The extracellular domain comprises three distinct domains including Sema, Plexin-Semaphorin-Integrin (PSI), and three Immunoglobulin-Plexin-Transcription factor (IPT1-IPT3) domains.68 The natural ligand of RON is MSP (Macrophage Stimulating Protein),72 a member of plasminogen-related kringle protein family73 which is a heterodimeric protein made of an α-chain composed of four kringle domains and a β-chain containing a serine protease-like domain.74 The α- and β-chains of MSP show low and high affinities to RON Sema domain, respectively.75 Several monoclonal antibodies against RON extracellular domain have been developed (in preclinical phases) to specifically inhibit the protein-protein interactions between RON and MSP.69 Identifying the key residues working as hot spots responsible for receptor-ligand (RON-MSP) interaction is of great importance for drug design and development. The aim of the current study is to identify hot spots involved in RON-MSPβ interaction using in silico alanine scanning mutagenesis by MM-GBSA method. The results can be used in anticancer drug designing where inhibition of RON is needed.
Materials and Methods
Structure preparation and in silico alanine mutagenesis
Experimental coordinates of RON complexed with MSPb (PDB ID: 4QT8) determined at 3.0 Å resolution by X-ray crystallography76 was retrieved from the Protein Data Bank at the Research Collaboratory for Structural Bioinformatics (http://www.rcsb.org/pdb/ home/home.do).77 Preparation of structures along with mutation of the residues were carried out using Swiss-Pdb Viewer (DeepView) version 4.01.78 Only one of the complexes in the reported crystal structure was used (chains B and D) for further analysis. The residues at the RON-MSPβ interface were inferred based on crystal structure reported by Chao and collaborators76 in both ligand and receptor were virtually mutated to alanine as listed in Table 1.
Table 1. List of residues mutated to alanine on RON and MSP .
| RON | MSP |
| Glu190 | Arg521 |
| Gln193 | Cys527 |
| Ser195 | His528 |
| Arg220 | Ser565 |
| Glu287 | Arg639 |
| Pro288 | Glu644 |
| Glu289 | Glu658 |
| His424 | Arg683 |
| Glu190/ Ser195* | Arg639/ Glu644* |
(* Double mutation)
Ligand-receptor binding free energy calculations using MM-GBSA method
Energy minimization and binding free energy calculation were performed using the Assisted Model Building with Energy Refinement (AMBER) suite of programs (version 14)79 operating on a Linux-based (Centus 6.8) GPU work station consisting of four Nvidia K40 M (each has 12 GB RAM and 2880 cuda cores), 2X Intel Xeon E5-2697 v2, 2.7 GHz (total of 48 cores), total RAM = 128 GB.
The energy minimization of RON-MSPb complex was carried out using AMBER-ff99 force field.80 Briefly, the usable coordinate files for AMBER (i.e. *.prmtop and *.inpcrd) were generated using leap module. Then, a correct number of counter ions (Na+ or Cl-) was added for neutralizing the total charge of the system followed by solvation of the system using TIP3P water molecules in a rectangular box with the buffering distances set to 12 Å in all directions. After that, the solvated system was submitted to an initial energy minimization process by applying Sander module (500 steps of steepest descent followed by 500 steps of conjugate gradient) followed by a 50 ps heating step where the temperature was gradually increased from 0 to 300 °K. After 50 ps of density equilibration, 500 ps of constant pressure equilibration at 300 °K with a time step of 2 fs was performed. Only bond lengths involving hydrogen atoms were constrained using the SHAKE algorithm. Final molecular dynamic simulation was individually performed for a range of 1 to 10 ns by applying the Particle Mesh Ewald (PME) method to calculate long-range electrostatic interactions. All calculations were done under periodic boundary condition where no constraint was applied to either the protein or the ligand molecules. The trajectory of the dynamic simulation was achieved by writing out the coordinates every 10 ps. After molecular dynamic simulation on receptor-ligand complex, snapshots were taken from the molecular dynamic trajectory with an interval of 10 ps. The dielectric constant values were set to 1.0 and 80 for the interior of solute and the surrounding solvent, respectively. Binding free energy was calculated for ligand–receptor complex using MM-GBSA.80 The interaction energies for the snapshots were calculated while excluding water molecules and counter ions and presented as the average value in the RON-MSPb system.
Results and Discussion
Binding free energies for the complexes of RON tyrosine kinase receptor and its ligand (i.e. MSP) as well as the mutants of either receptor or ligand (Table 1) were calculated by applying MM-GBSA method on molecular dynamic simulation data collected at different time. For this purpose, firstly the binding free energy was calculated for the RON-MSPb wild type complex, then the residues involved in RON and MSPb interaction were mutated to alanine followed by molecular dynamic simulation and re-calculation of the binding free energy for different time intervals ranging from 1 to 10 ns to estimate the contribution and the effect of individual residues in RON and MSPb binding. The binding free energies (ΔGbind) for wild type and mutant forms were calculated as follows:
ΔGbind = Gwater (complex) - Gwater (receptor) - Gwater (ligand)
where Gwater (complex), Gwater (receptor), and Gwater (ligand) denote the free energies of the complex, receptor, and ligand, respectively. The free energy (ΔG) for each term is calculated using following equation:
Gmolecule = Egas + ΔGsolvation- TS
ΔGsolvation = ΔGGB + ΔGnon-polar
Egas = Eint+ Evdw + Eelec
Eint = Ebond+ Eangle+ Etors
where G is the calculated average free energy, Egas is the standard force-field energy, including internal energy (Eint) in the gas phase as well as non-covalent van der Waals (Evdw) and electrostatic (Eelec) energies. Ebond, Eangle, and Etors demonstrate the contributions to the internal energy caused by the strain from the deviation of the bonds, angle, and torsion angle from their equilibrium values. ΔGsolvation is the solvation-free energy calculated with a numerical solution of the Poisson–Boltzmann equation and an estimate of the non-polar free energy using a surface area term.81,82
Figure 1 shows the results of binding free energy calculations for the complex of wild type RON and MSPb and their mutant forms using MM-GBSA method applied to molecular dynamic simulations ranging from 1 to 10 ns. These results have been also illustrated in Table 2. Results for ΔΔG binding (ΔGBinding-wild type - ΔGBinding-mutant) for RON and MSPb are also available in Table 3. The details of all calculations for mutants and wild types of receptor and ligand are available in appendices 1 and 2.
Figure 1.
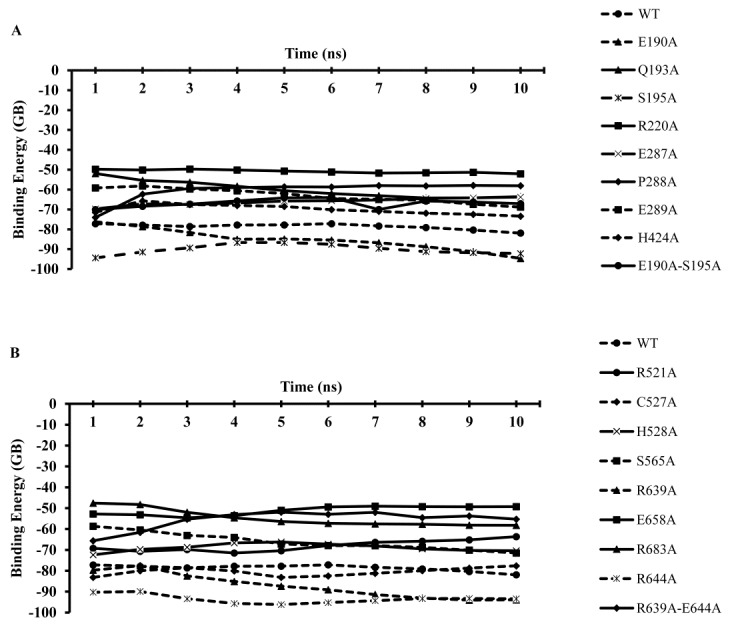
The plot of binding free energies (ΔG) for the complexes of RON-MSPb during different MD simulation time lengths (1–10 ns) using MM-GBSA calculation methods implemented in AMBER.
Table 2. Effects of alanine substitution on RON (A) and MSP (B) to contribution of binding energy (∆GBind) for RON-MSP complex calculated using MM-GBSA method in a 1 to 10 ns molecular dynamic simulation.
| 1ns | 2ns | 3ns | 4ns | 5ns | 6ns | 7ns | 8ns | 9ns | 10ns | 1ns | ||
| A) | WT | -77.17 | -77.85 | -78.58 | -77.81 | -77.78 | -77.17 | -78.33 | -79.10 | -80.36 | -81.91 | |
| E 190 A | -76.19 | -78.62 | -81.58 | -85.02 | -84.85 | -85.41 | -86.74 | -88.76 | -91.26 | -94.65 | ||
| Q 193 A | -51.91 | -55.39 | -56.27 | -58.29 | -60.57 | -62.00 | -63.08 | -64.22 | -64.15 | -63.56 | ||
| S 195 A | -94.36 | -91.40 | -89.31 | -86.62 | -86.64 | -87.50 | -89.57 | -91.20 | -91.76 | -92.19 | ||
| R 220 A | -49.73 | -50.15 | -49.67 | -50.17 | -50.67 | -51.18 | -51.70 | -51.52 | -51.33 | -52.06 | ||
| E 287 A | -69.70 | -67.19 | -67.55 | -66.75 | -65.81 | -65.60 | -65.24 | -64.51 | -64.20 | -63.79 | ||
| P 288 A | -74.01 | -62.30 | -59.40 | -59.00 | -58.62 | -58.67 | -58.01 | -58.14 | -57.92 | -58.12 | ||
| E 289 A | -59.17 | -58.18 | -59.67 | -60.61 | -61.96 | -64.52 | -64.92 | -65.35 | -67.40 | -68.80 | ||
| H 424 A | -71.45 | -65.70 | -67.52 | -67.99 | -68.51 | -70.10 | -70.94 | -71.88 | -72.46 | -73.37 | ||
| E 190 A/S 195 A | -70.09 | -68.40 | -67.30 | -65.73 | -64.15 | -64.02 | -69.99 | -65.81 | -66.26 | -67.10 | ||
| B) | WT | -77.17 | -77.85 | -78.58 | -77.81 | -77.78 | -77.17 | -78.33 | -79.10 | -80.36 | -81.91 | |
| R 521 A | -69.23 | -70.67 | -69.80 | -71.45 | -70.41 | -67.89 | -66.31 | -65.83 | -65.14 | -63.65 | ||
| C 527 A | -83.18 | -79.98 | -78.55 | -80.10 | -83.16 | -82.49 | -81.23 | -79.92 | -78.73 | -77.63 | ||
| H 528 A | -72.37 | -69.73 | -68.66 | -66.65 | -66.17 | -67.20 | -68.09 | -69.49 | -70.27 | -70.38 | ||
| S 565 A | -58.67 | -60.44 | -63.05 | -64.01 | -67.13 | -67.90 | -67.93 | -68.80 | -70.12 | -71.47 | ||
| R 639 A | -79.74 | -77.52 | -82.47 | -85.12 | -87.36 | -89.09 | -91.37 | -93.07 | -94.06 | -93.91 | ||
| E 644 A | -90.27 | -89.90 | -93.40 | -95.66 | -96.18 | -95.22 | -94.30 | -93.31 | -93.29 | -93.42 | ||
| E 658 A | -52.77 | -53.15 | -54.63 | -53.59 | -51.00 | -49.41 | -49.05 | -49.24 | -49.31 | -49.28 | ||
| R 683 A | -47.54 | -48.21 | -51.97 | -54.61 | -56.40 | -57.25 | -57.53 | -57.68 | -58.10 | -58.24 | ||
| R 639 A/E 644 A | -65.58 | -61.56 | -55.29 | -53.06 | -51.88 | -52.91 | -51.99 | -54.55 | -53.77 | -55.28 |
Table 3. The binding energy differences (DDGbinding= DGwildtype -DGmutant) for wild type and mutant forms of RON-MSP complex. The mutations are performed on RON (A) and MSP (B) using in silico alanine substitution.
| 1ns | 2ns | 3ns | 4ns | 5ns | 6ns | 7ns | 8ns | 9ns | 10ns | ||
| A) | E 190 A | -0.98 | 0.78 | 3.01 | 7.21 | 7.07 | 8.24 | 8.41 | 9.66 | 10.90 | 12.74 |
| Q 193 A | -25.26 | -22.45 | -22.30 | -19.52 | -17.22 | -15.17 | -15.25 | -14.88 | -16.21 | -18.35 | |
| S 195 A | 17.19 | 13.55 | 10.73 | 8.81 | 8.86 | 10.33 | 11.25 | 12.11 | 11.41 | 10.28 | |
| R 220 A | -27.44 | -27.70 | -28.90 | -27.64 | -27.12 | -25.99 | -26.63 | -27.58 | -29.03 | -29.85 | |
| E 287 A | -7.47 | -10.65 | -11.03 | -11.06 | -11.97 | -11.57 | -13.09 | -14.59 | -16.16 | -18.12 | |
| P 288 A | -3.16 | -15.54 | -19.18 | -18.81 | -19.17 | -18.50 | -20.32 | -20.96 | -22.44 | -23.79 | |
| E 289 A | -18.00 | -19.67 | -18.90 | -17.19 | -15.82 | -12.64 | -13.41 | -13.75 | -12.96 | -13.12 | |
| H 424 A | -5.72 | -12.15 | -11.05 | -9.82 | -9.27 | -7.06 | -7.38 | -7.22 | -7.90 | -8.54 | |
| E 190 A/ S 195 A | -7.08 | -9.44 | -11.28 | -12.08 | -13.63 | -13.15 | -8.33 | -13.28 | -14.10 | -14.81 | |
| B) | R 521 A | -7.94 | -7.17 | -8.77 | -6.36 | -7.37 | -9.28 | -12.01 | -13.26 | -15.21 | -18.26 |
| C 527 A | 6.01 | 2.14 | -0.03 | 2.29 | 5.37 | 5.32 | 2.90 | 0.82 | -1.62 | -4.28 | |
| H 528 A | -4.80 | -8.12 | -9.91 | -11.16 | -11.61 | -9.97 | -10.24 | -9.60 | -10.09 | -11.53 | |
| S 565 A | -18.50 | -17.40 | -15.53 | -13.80 | -10.66 | -9.27 | -10.40 | -10.30 | -10.24 | -10.44 | |
| R 639 A | 2.57 | -0.33 | 3.89 | 7.31 | 9.58 | 11.93 | 13.04 | 13.97 | 13.70 | 12.00 | |
| E 644 A | 13.10 | 12.06 | 14.82 | 17.85 | 18.40 | 18.06 | 15.98 | 14.21 | 12.94 | 11.51 | |
| E 658 A | -24.40 | -24.70 | -23.95 | -24.21 | -26.78 | -27.76 | -29.28 | -29.86 | -31.04 | -32.63 | |
| R 683 A | -29.63 | -29.64 | -26.61 | -23.19 | -21.38 | -19.92 | -20.79 | -21.42 | -22.26 | -23.68 | |
| R 639 A-E 644 A | -11.59 | -16.29 | -23.29 | -24.75 | -25.91 | -24.26 | -26.33 | -24.55 | -26.59 | -26.63 |
Cancer is one of the most important causes of death in the world83 and several strategies including pharmacotherapy protocols are employed to control this devastating condition.84 Due to the importance of protein-protein interactions in cancer initiation and development, many efforts have been dedicated to target cancer cells by inhibition of those PPIs involved in cancer progression.5,85,86 RON a tyrosine kinase receptor has gained considerable attention as promising target in cancer therapy.68 Most of the therapeutic agents developed so far against RON interfere with RON and MSP binding highlighting the importance of PPIs.69 Therefore, the identification of hot spots involved in the interface of RON-MSP complex is of great importance in rational drug design.
In the current study, the residues reported to be involved in RON-MSPb interactions (Figure 2) were virtually mutated to alanine one at the time to determine the contribution of each residue using MM-GBSA approach. The binding free energy difference between the mutant and the wild type complexes was obtained as follows:
Figure 2.
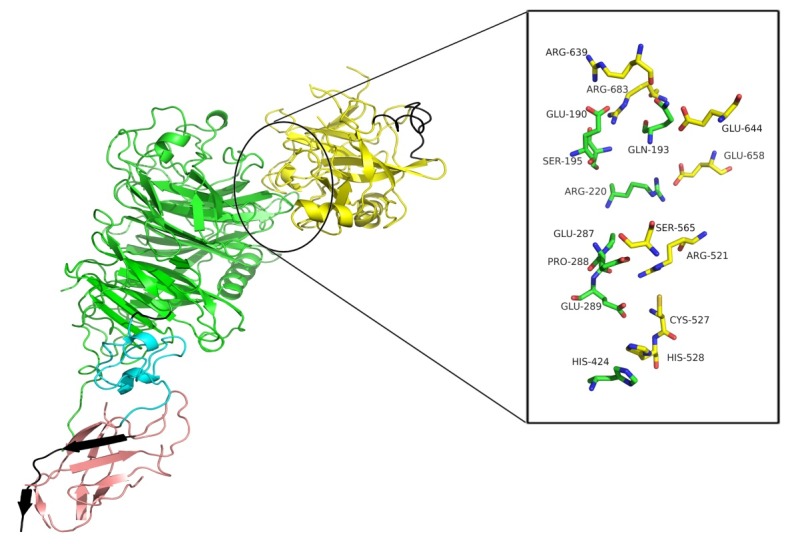
Cartoon and stick representation of RON-MSPb complex generated in PyMol (version 1.5.0.3).
ΔΔGBinding = ΔGBinding-wild type - ΔGBinding-mutant
In this expression, negative ΔΔGbinding value implies that the substitution of the corresponding amino acid with alanine is an unfavorable substitution whereas a positive value indicates a favorable substitution in terms of binding free energy compared to the wild type complex.87
The results of molecular dynamic simulation of RON indicated that all receptor (except for Glu190 and Ser195) and ligand (except for Arg639 and Glu644) mutants have low affinity compared to the wild type as deduced from the negative ΔΔG values shown in Figure 2 and Table 3.
One of the crucial residues at the interface of RON-MSPb complex is RON Gln193; its side chain NH2 group makes two ionic interactions with carboxylate group of MSP Glu644and carbonyl group of Arg639. In addition, Arg639of MSPis involved in another interaction with RON Glu1 which will be discussed later.76 The MM-GBSA based binding energy calculations on the wild type and Q193A mutant showed that this amino acid is important in the binding (also confirmed by Chao and coworkers)76 while the calculations did not support the importance of its partners MSP, i.e. Arg639 and Glu644. To shade more light on this issue, an in silico R639A/E644A double mutation was introduced on MSP and then the binding energy calculated. Surprisingly, results showed that the double mutation caused unfavorable effect on binding energy for RON-MSPβ complex formation highlighting the importance of simultaneous interaction established between both Arg639 and Glu644with Gln193.
The RON Arg220 is another key residue involved in charge-charge interaction with Glu658of MSP.76 The ΔΔG values calculated for R220A mutant during 1 to 10 ns molecular dynamic simulation range from ~ -25 to -30 Kcal/mol, which are the highest negative values obtained for all RON mutants. This observation implies the great importance of this residue as a hot spot in the interaction between RON and MSPb. Interestingly, the ΔΔG values for E658A mutant has also high negative value (Table 2 and 3). This is in agreement with experimental observation reported previously.76
According to the study of Chao et al, MSP Arg521 simultaneously interacts with three residues of RON namely Glu287, Pro288and Glu289.76 Additionally, RON Glu287 forms a hydrogen bond interaction with the hydroxyl group of MSP Ser656 whereas Glu289 of RON establishes an ionic interaction with MSP His528 as well as interaction with the backbone NH group of MSP Cys527. Moreover, MSP His528, located in proximity of RON Glu289 is engaged in aromatic interaction with His424. The results of computational alanine scanning reported here revealed that Glu287, Pro288, Glu289, and His424 of RON located at the interface of RON-MSPb complex are crucial residues for its binding to MSPb.76 According to ΔΔG values, Pro288, Glu287, and Glu289 of RON are the next most important amino acids after Arg220 (see Table 3).It seems that the importance of these residues is related to their interactions with more than one residues on MSP (except for Pro288). In the case of Pro288, it interacts only with MSP Arg521 which in turn is highly important due to its participation in multiple interactions with RON Glu287, and Glu289.76 Based on binding ΔΔG values, His424 seems to be less important in comparison to other RON residues at the interface. However, this residue can also be considered as a hot spot on RON (Table 2 and 3). Additionally, MSP His528 and Ser565 are suggested to be important residues for RON binding despite the fact that their ΔΔG values are not as significant as those mentioned above (Table 2 and 3). The ΔΔG binding calculated for MSP Cys527 using different molecular dynamic simulation intervals includes both positive (1 to 8 ns) and negative (9 to 10 ns) values, making it difficult to extrapolate its importance in the binding. It seems that interaction via Cys527switches on and off during molecular dynamic simulation. However, the ΔΔG values toward end of simulation reach -4 kcal/mol which indicates positive contribution of this residue in RON-MSP binding.
E190A and S195A mutations can be considered exception as the binding affinities toward ligand were improved after mutation to alanine. The crystallography studies on RON-MSPb complex showed that RON Glu190 is involved in two salt bridges via its carboxylate group with guanidinium group of Arg639 and Arg683,76 however, our results do not attribute positive contribution for this residue as inferred from its positive ΔΔG values in MM-GBSA calculations upon mutation to alanine (See Table 3). Such disagreement between the reported experimental results and our in silico estimates may be due to the fact that Glu190 interacts with two different MSP residues (i.e. Arg639 and Arg683), which are already interacting with other RON residues.76 Therefore, lack of their interactions with Glu190may not contribute favorably in the overall binding energy.
RON Ser195 is shown to be involved in a charge-charge interaction with MSP Arg683,76 however, our results did not identify this amino acid as an important residue in RON-MSPb complex (Table 3). Again the disagreement between our in silico estimates and the crystallographic data may be due to the formation of another interaction by Arg683 with RON via Glu190 which renders the interaction between Ser195 and Arg683 less important.76 The only previous experimental site directed mutagenesis studies on the residues at the interface of RON-MSP complex was carried out for Arg683 and results obtained are in agreement with the ones discussed below.75 This amino acid is an important residue in the interaction of RON-MSPβ complex based on in silico calculation despite the results obtained for its partners on MSP (i.e Glu190 and Ser195). In order to gain more information regarding these residues an in silico double mutation (E190A/S195A) study was performed. This double mutation lead to a positive ΔΔG value indicative of their harmonic interplay in the interaction with Arg683.
Conclusion
In modern drug design and discovery process, computational approaches have streamlined a promising perspective by supplying useful and supportive information. In this context, identification of hot spots in biomolecules’ interactions through the estimating the binding affinity of molecules towards targets of interest can provide valuable information where protein-protein interactions are important initiators in cancer pathogenesis. Virtual alanine scanning mutagenesis is one of the tools that are commonly employed for this purpose. Therefore, in the current investigation, amino acids reported to be at the interface of RON-MSPb complex were evaluated using the MM-GBSA method and some of them were assigned as hot spots in the interaction. Taken together, in silico alanine scanning mutagenesis results revealed that Gln193, Arg220, Glu287, Pro288, Glu289 and His424 residues from RON and Arg521, His528, Ser565, Glu658, and Arg683 form MSPβ may play important roles in protein-protein interaction between RON and MSP. Identification of these RON hot spots is important in designing anti-RON drugs when the aim is disruption of RON-MSP interaction. In the same way, the acquired information regarding the critical amino acids of MSPβ can be used in the process of rational drug design for developing MSP antagonizing agents, the development of novel MSP mimicking peptides where inhibition of RON activation is required, and the design of experimental site directed mutagenesis studies.
Acknowledgments
This work is a part of Ph.D thesis of Omid Zarei at Tabriz university of Medical Sciences.
Ethical Issues
Not applicable.
Conflict of Interest
The authors declare no conflict of interests.
References
- 1.Ngounou Wetie AG, Sokolowska I, Woods AG, Roy U, Loo JA, Darie CC. Investigation of stable and transient protein-protein interactions: Past, present, and future. Proteomics. 2013;13(3-4):538–57. doi: 10.1002/pmic.201200328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ryan DP, Matthews JM. Protein-protein interactions in human disease. Curr Opin Struct Biol. 2005;15(4):441–6. doi: 10.1016/j.sbi.2005.06.001. [DOI] [PubMed] [Google Scholar]
- 3.Gonzalez MW, Kann MG. Chapter 4: Protein interactions and disease. PLoS Comput Biol. 2012;8(12):e1002819. doi: 10.1371/journal.pcbi.1002819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lage K. Protein-protein interactions and genetic diseases: The interactome. Biochim Biophys Acta. 2014;1842(10):1971–80. doi: 10.1016/j.bbadis.2014.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ivanov AA, Khuri FR, Fu H. Targeting protein-protein interactions as an anticancer strategy. Trends Pharmacol Sci. 2013;34(7):393–400. doi: 10.1016/j.tips.2013.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Skwarczynska M, Ottmann C. Protein-protein interactions as drug targets. Future Med Chem. 2015;7(16):2195–219. doi: 10.4155/fmc.15.138. [DOI] [PubMed] [Google Scholar]
- 7.Ottmann C. New compound classes: Protein-protein interactions. Handb Exp Pharmacol. 2016;232:125–38. doi: 10.1007/164_2015_30. [DOI] [PubMed] [Google Scholar]
- 8.Whitby LR, Boger DL. Comprehensive peptidomimetic libraries targeting protein-protein interactions. Acc Chem Res. 2012;45(10):1698–709. doi: 10.1021/ar300025n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang J, Rabbitts TH. Intracellular antibody capture: A molecular biology approach to inhibitors of protein-protein interactions. Biochim Biophys Acta. 2014;1844(11):1970–6. doi: 10.1016/j.bbapap.2014.05.009. [DOI] [PubMed] [Google Scholar]
- 10.Helmer D, Schmitz K. Peptides and peptide analogs to inhibit protein-protein interactions. Adv Exp Med Biol. 2016;917:147–83. doi: 10.1007/978-3-319-32805-8_8. [DOI] [PubMed] [Google Scholar]
- 11.Ivan T, Enkvist E, Viira B, Manoharan GB, Raidaru G, Pflug A. et al. Bifunctional ligands for inhibition of tight-binding protein-protein interactions. Bioconjug Chem. 2016;27(8):1900–10. doi: 10.1021/acs.bioconjchem.6b00293. [DOI] [PubMed] [Google Scholar]
- 12.Yan C, Wu F, Jernigan RL, Dobbs D, Honavar V. Characterization of protein-protein interfaces. Protein J. 2008;27(1):59–70. doi: 10.1007/s10930-007-9108-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Keskin O, Ma B, Nussinov R. Hot regions in protein--protein interactions: The organization and contribution of structurally conserved hot spot residues. J Mol Biol. 2005;345(5):1281–94. doi: 10.1016/j.jmb.2004.10.077. [DOI] [PubMed] [Google Scholar]
- 14.Moreira IS, Fernandes PA, Ramos MJ. Hot spots--a review of the protein-protein interface determinant amino-acid residues. Proteins. 2007;68(4):803–12. doi: 10.1002/prot.21396. [DOI] [PubMed] [Google Scholar]
- 15.Bradshaw RT, Patel BH, Tate EW, Leatherbarrow RJ, Gould IR. Comparing experimental and computational alanine scanning techniques for probing a prototypical protein-protein interaction. Protein Eng Des Sel. 2011;24(1-2):197–207. doi: 10.1093/protein/gzq047. [DOI] [PubMed] [Google Scholar]
- 16.Shi Y. A Glimpse of Structural Biology through X-Ray Crystallography. Cell. 2014;159(5):995–1014. doi: 10.1016/j.cell.2014.10.051. [DOI] [PubMed] [Google Scholar]
- 17.Sliwoski G, Kothiwale S, Meiler J, Lowe EW Jr. Computational methods in drug discovery. Pharmacol Rev. 2014;66(1):334–95. doi: 10.1124/pr.112.007336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang X, Chen H, Yang F, Gong J, Li S, Pei J. et al. Idrug: A web-accessible and interactive drug discovery and design platform. J Cheminform. 2014;6:28. doi: 10.1186/1758-2946-6-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Macalino SJ, Gosu V, Hong S, Choi S. Role of computer-aided drug design in modern drug discovery. Arch Pharm Res. 2015;38(9):1686–701. doi: 10.1007/s12272-015-0640-5. [DOI] [PubMed] [Google Scholar]
- 20.Wang T, Wu MB, Lin JP, Yang LR. Quantitative structure-activity relationship: Promising advances in drug discovery platforms. Expert Opin Drug Discov. 2015;10(12):1283–300. doi: 10.1517/17460441.2015.1083006. [DOI] [PubMed] [Google Scholar]
- 21.de Ruyck J, Brysbaert G, Blossey R, Lensink MF. Molecular docking as a popular tool in drug design, an in silico travel. Adv Appl Bioinform Chem. 2016;9:1–11. doi: 10.2147/aabc.s105289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Moal IH, Jimenez-Garcia B, Fernandez-Recio J. Ccharppi web server: Computational characterization of protein-protein interactions from structure. Bioinformatics. 2015;31(1):123–5. doi: 10.1093/bioinformatics/btu594. [DOI] [PubMed] [Google Scholar]
- 23.Sukhwal A, Sowdhamini R. PPCheck: A webserver for the quantitative analysis of protein-protein interfaces and prediction of residue hotspots. Bioinform Biol Insights. 2015;9:141–51. doi: 10.4137/bbi.s25928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Perez MA, Sousa SF, Oliveira EF, Fernandes PA, Ramos MJ. Detection of farnesyltransferase interface hot spots through computational alanine scanning mutagenesis. J Phys Chem B. 2011;115(51):15339–54. doi: 10.1021/jp205481y. [DOI] [PubMed] [Google Scholar]
- 25.De Rienzo F, Barbosa AJ, Perez MA, Fernandes PA, Ramos MJ, Menziani MC. The extracellular subunit interface of the 5-HT3 receptors: A computational alanine scanning mutagenesis study. J Biomol Struct Dyn. 2012;30(3):280–98. doi: 10.1080/07391102.2012.680029. [DOI] [PubMed] [Google Scholar]
- 26.Ramos RM, Moreira IS. Computational alanine scanning mutagenesis-an improved methodological approach for protein-DNA complexes. J Chem Theory Comput. 2013;9(9):4243–56. doi: 10.1021/ct400387r. [DOI] [PubMed] [Google Scholar]
- 27.Gesto DS, Cerqueira NM, Ramos MJ, Fernandes PA. Discovery of new druggable sites in the anti-cholesterol target hmg-coa reductase by computational alanine scanning mutagenesis. J Mol Model. 2014;20(4):2178. doi: 10.1007/s00894-014-2178-8. [DOI] [PubMed] [Google Scholar]
- 28.Duan L, Liu X, Zhang JZ. Interaction entropy: A new paradigm for highly efficient and reliable computation of protein-ligand binding free energy. J Am Chem Soc. 2016;138(17):5722–8. doi: 10.1021/jacs.6b02682. [DOI] [PubMed] [Google Scholar]
- 29.Yang Z, Wu F, Yuan X, Zhang L, Zhang S. Novel binding patterns between ganoderic acids and neuraminidase: Insights from docking, molecular dynamics and MM/PBSA studies. J Mol Graph Model. 2016;65:27–34. doi: 10.1016/j.jmgm.2016.02.006. [DOI] [PubMed] [Google Scholar]
- 30.Moreira IS, Fernandes PA, Ramos MJ. Computational alanine scanning mutagenesis--an improved methodological approach. J Comput Chem. 2007;28(3):644–54. doi: 10.1002/jcc.20566. [DOI] [PubMed] [Google Scholar]
- 31.Martins SA, Perez MA, Moreira IS, Sousa SF, Ramos MJ, Fernandes PA. Computational alanine scanning mutagenesis: MM-PBSA vs TI. J Chem Theory Comput. 2013;9(3):1311–9. doi: 10.1021/ct4000372. [DOI] [PubMed] [Google Scholar]
- 32.Zoete V, Irving MB, Michielin O. MM-GBSA binding free energy decomposition and T cell receptor engineering. J Mol Recognit. 2010;23(2):142–52. doi: 10.1002/jmr.1005. [DOI] [PubMed] [Google Scholar]
- 33.Hou T, Wang J, Li Y, Wang W. Assessing the performance of the mm/pbsa and mm/gbsa methods1The accuracy of binding free energy calculations based on molecular dynamics simulations. J Chem Inf Model. 2011;51(1):69–82. doi: 10.1021/ci100275a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mulakala C, Viswanadhan VN. Could MM-GBSA be accurate enough for calculation of absolute protein/ligand binding free energies? J Mol Graph Model. 2013;46:41–51. doi: 10.1016/j.jmgm.2013.09.005. [DOI] [PubMed] [Google Scholar]
- 35.Rathore RS, Sumakanth M, Reddy MS, Reddanna P, Rao AA, Erion MD. et al. Advances in binding free energies calculations: QM/MM-based free energy perturbation method for drug design. Curr Pharm Des. 2013;19(26):4674–86. doi: 10.2174/1381612811319260002. [DOI] [PubMed] [Google Scholar]
- 36.Wang DD, Zhou W, Yan H, Wong M, Lee V. Personalized prediction of egfr mutation-induced drug resistance in lung cancer. Sci Rep. 2013;3:2855. doi: 10.1038/srep02855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Reddy MR, Reddy CR, Rathore RS, Erion MD, Aparoy P, Reddy RN. et al. Free energy calculations to estimate ligand-binding affinities in structure-based drug design. Curr Pharm Des. 2014;20(20):3323–37. doi: 10.2174/13816128113199990604. [DOI] [PubMed] [Google Scholar]
- 38.Genheden S, Ryde U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin Drug Discov. 2015;10(5):449–61. doi: 10.1517/17460441.2015.1032936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Suri C, Naik PK. Combined molecular dynamics and continuum solvent approaches (MM-PBSA/GBSA) to predict noscapinoid binding to gamma-tubulin dimer. SAR QSAR Environ Res. 2015;26(6):507–19. doi: 10.1080/1062936x.2015.1070200. [DOI] [PubMed] [Google Scholar]
- 40.Chen J, Wang J, Zhang Q, Chen K, Zhu W. Probing origin of binding difference of inhibitors to MDM2 and MDMX by polarizable molecular dynamics simulation and QM/MM-GBSA calculation. Sci Rep. 2015;5:17421. doi: 10.1038/srep17421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Margulies D, Opatowsky Y, Fletcher S, Saraogi I, Tsou LK, Saha S. et al. Surface binding inhibitors of the SCF-KIT protein-protein interaction. Chembiochem. 2009;10(12):1955–8. doi: 10.1002/cbic.200900079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Banappagari S, Corti M, Pincus S, Satyanarayanajois S. Inhibition of protein-protein interaction of HER2-EGFR and HER2-HER3 by a rationally designed peptidomimetic. J Biomol Struct Dyn. 2012;30(5):594–606. doi: 10.1080/07391102.2012.687525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Banappagari S, McCall A, Fontenot K, Vicente MG, Gujar A, Satyanarayanajois S. Design, synthesis and characterization of peptidomimetic conjugate of BODIPY targeting HER2 protein extracellular domain. Eur J Med Chem. 2013;65:60–9. doi: 10.1016/j.ejmech.2013.04.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gogate PN, Ethirajan M, Kurenova EV, Magis AT, Pandey RK, Cance WG. Design, synthesis, and biological evaluation of novel FAK scaffold inhibitors targeting the FAK-VEGFR3 protein-protein interaction. Eur J Med Chem. 2014;80:154–66. doi: 10.1016/j.ejmech.2014.04.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kanthala S, Gauthier T, Satyanarayanajois S. Structure-activity relationships of peptidomimetics that inhibit PPI of HER2-HER3. Biopolymers. 2014;101(6):693–702. doi: 10.1002/bip.22441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tognolini M, Lodola A. Targeting the Eph-ephrin System with Protein-Protein Interaction (PPI) Inhibitors. Curr Drug Targets. 2015;16(10):1048–56. doi: 10.2174/1389450116666150825144457. [DOI] [PubMed] [Google Scholar]
- 47.Choura M, Rebai A. Receptor tyrosine kinases: From biology to pathology. J Recept Signal Transduct Res. 2011;31(6):387–94. doi: 10.3109/10799893.2011.625425. [DOI] [PubMed] [Google Scholar]
- 48.Vasudevan HN, Soriano P. A thousand and one receptor tyrosine kinases: Wherein the specificity? Curr Top Dev Biol. 2016;117:393–404. doi: 10.1016/bs.ctdb.2015.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rettew AN, Getty PJ, Greenfield EM. Receptor tyrosine kinases in osteosarcoma: Not just the usual suspects. Adv Exp Med Biol. 2014;804:47–66. doi: 10.1007/978-3-319-04843-7_3. [DOI] [PubMed] [Google Scholar]
- 50.Morishita A, Gong J, Masaki T. Targeting receptor tyrosine kinases in gastric cancer. World J Gastroenterol. 2014;20(16):4536–45. doi: 10.3748/wjg.v20.i16.4536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chen J, Song W, Amato K. Eph receptor tyrosine kinases in cancer stem cells. Cytokine Growth Factor Rev. 2015;26(1):1–6. doi: 10.1016/j.cytogfr.2014.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gluck AA, Aebersold DM, Zimmer Y, Medova M. Interplay between receptor tyrosine kinases and hypoxia signaling in cancer. Int J Biochem Cell Biol. 2015;62:101–14. doi: 10.1016/j.biocel.2015.02.018. [DOI] [PubMed] [Google Scholar]
- 53.Segaliny AI, Tellez-Gabriel M, Heymann MF, Heymann D. Receptor tyrosine kinases: Characterisation, mechanism of action and therapeutic interests for bone cancers. J Bone Oncol. 2015;4(1):1–12. doi: 10.1016/j.jbo.2015.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Raval SH, Singh RD, Joshi DV, Patel HB, Mody SK. Recent developments in receptor tyrosine kinases targeted anticancer therapy. Vet World. 2016;9(1):80–90. doi: 10.14202/vetworld.2016.80-90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Adrain C, Freeman M. Regulation of receptor tyrosine kinase ligand processing. Cold Spring Harb Perspect Biol 2014;6(1). [DOI] [PMC free article] [PubMed]
- 56.Maruyama IN. Mechanisms of activation of receptor tyrosine kinases: Monomers or dimers. Cells. 2014;3(2):304–30. doi: 10.3390/cells3020304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Schlessinger J. Receptor tyrosine kinases: Legacy of the first two decades. Cold Spring Harb Perspect Biol 2014;6(3). [DOI] [PMC free article] [PubMed]
- 58.Lemmon MA, Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell. 2010;141(7):1117–34. doi: 10.1016/j.cell.2010.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Roskoski R Jr. A historical overview of protein kinases and their targeted small molecule inhibitors. Pharmacol Res. 2015;100:1–23. doi: 10.1016/j.phrs.2015.07.010. [DOI] [PubMed] [Google Scholar]
- 60.Zhang X, Wang Y, Wang J, Sun F. Protein-protein interactions among signaling pathways may become new therapeutic targets in liver cancer (Review) Oncol Rep. 2016;35(2):625–38. doi: 10.3892/or.2015.4464. [DOI] [PubMed] [Google Scholar]
- 61.Matzke A, Herrlich P, Ponta H, Orian-Rousseau V. A five-amino-acid peptide blocks Met- and Ron-dependent cell migration. Cancer Res. 2005;65(14):6105–10. doi: 10.1158/0008-5472.can-05-0207. [DOI] [PubMed] [Google Scholar]
- 62.Pedersen MW, Jacobsen HJ, Koefoed K, Hey A, Pyke C, Haurum JS. et al. Sym004: A novel synergistic anti-epidermal growth factor receptor antibody mixture with superior anticancer efficacy. Cancer Res. 2010;70(2):588–97. doi: 10.1158/0008-5472.can-09-1417. [DOI] [PubMed] [Google Scholar]
- 63.Gunes Z, Zucconi A, Cioce M, Meola A, Pezzanera M, Acali S. et al. Isolation of fully human antagonistic RON antibodies showing efficient block of downstream signaling and cell migration. Transl Oncol. 2011;4(1):38–46. doi: 10.1593/tlo.10211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Veggiani G, Ossolengo G, Aliprandi M, Cavallaro U, de Marco A. Single-domain antibodies that compete with the natural ligand fibroblast growth factor block the internalization of the fibroblast growth factor receptor 1. Biochem Biophys Res Commun. 2011;408(4):692–6. doi: 10.1016/j.bbrc.2011.04.090. [DOI] [PubMed] [Google Scholar]
- 65.Banappagari S, Corti M, Pincus S, Satyanarayanajois S. Inhibition of protein-protein interaction of HER2-EGFR and HER2–HER3 by a rationally designed peptidomimetic. J Biomol Struct Dyn. 2012;30(5):594–606. doi: 10.1080/07391102.2012.687525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.D'Souza JW, Reddy S, Goldsmith LE, Shchaveleva I, Marks JD, Litwin S. et al. Combining anti-ERBB3 antibodies specific for domain I and domain III enhances the anti-tumor activity over the individual monoclonal antibodies. PLoS One. 2014;9(11):e112376. doi: 10.1371/journal.pone.0112376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Vigna E, Comoglio PM. Targeting the oncogenic met receptor by antibodies and gene therapy. Oncogene. 2015;34(15):1883–9. doi: 10.1038/onc.2014.142. [DOI] [PubMed] [Google Scholar]
- 68.Yao HP, Zhou YQ, Zhang R, Wang MH. MSP-RON signalling in cancer: Pathogenesis and therapeutic potential. Nat Rev Cancer. 2013;13(7):466–81. doi: 10.1038/nrc3545. [DOI] [PubMed] [Google Scholar]
- 69.Zarei O, Benvenuti S, Ustun-Alkan F, Hamzeh-Mivehroud M, Dastmalchi S. Strategies of targeting the extracellular domain of RON tyrosine kinase receptor for cancer therapy and drug delivery. J Cancer Res Clin Oncol. 2016;142(12):2429–46. doi: 10.1007/s00432-016-2214-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ronsin C, Muscatelli F, Mattei MG, Breathnach R. A novel putative receptor protein tyrosine kinase of the met family. Oncogene. 1993;8(5):1195–202. [PubMed] [Google Scholar]
- 71.Gaudino G, Avantaggiato V, Follenzi A, Acampora D, Simeone A, Comoglio PM. The proto-oncogene RON is involved in development of epithelial, bone and neuro-endocrine tissues. Oncogene. 1995;11(12):2627–37. [PubMed] [Google Scholar]
- 72.Wang MH, Ronsin C, Gesnel MC, Coupey L, Skeel A, Leonard EJ. et al. Identification of the ron gene product as the receptor for the human macrophage stimulating protein. Science. 1994;266(5182):117–9. doi: 10.1126/science.7939629. [DOI] [PubMed] [Google Scholar]
- 73.Yoshimura T, Yuhki N, Wang MH, Skeel A, Leonard EJ. Cloning, sequencing, and expression of human macrophage stimulating protein (MSP, MST1) confirms MSP as a member of the family of kringle proteins and locates the MSP gene on chromosome 3. J Biol Chem. 1993;268(21):15461–8. [PubMed] [Google Scholar]
- 74.Donate LE, Gherardi E, Srinivasan N, Sowdhamini R, Aparicio S, Blundell TL. Molecular evolution and domain structure of plasminogen-related growth factors (HGF/SF and HGF1/MSP) Protein Sci. 1994;3(12):2378–94. doi: 10.1002/pro.5560031222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Danilkovitch A, Miller M, Leonard EJ. Interaction of macrophage-stimulating protein with its receptor. Residues critical for beta chain binding and evidence for independent alpha chain binding. J Biol Chem. 1999;274(42):29937–43. doi: 10.1074/jbc.274.42.29937. [DOI] [PubMed] [Google Scholar]
- 76.Chao KL, Gorlatova NV, Eisenstein E, Herzberg O. Structural basis for the binding specificity of human Recepteur d'Origine Nantais (RON) receptor tyrosine kinase to macrophage-stimulating protein. J Biol Chem. 2014;289(43):29948–60. doi: 10.1074/jbc.M114.594341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Berman HM, Kleywegt GJ, Nakamura H, Markley JL. The protein data bank archive as an open data resource. J Comput Aided Mol Des. 2014;28(10):1009–14. doi: 10.1007/s10822-014-9770-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kaplan W, Littlejohn TG. Swiss-PDB Viewer (Deep View) Brief Bioinform. 2001;2(2):195–7. doi: 10.1093/bib/2.2.195. [DOI] [PubMed] [Google Scholar]
- 79.Case DA, Cheatham TE 3rd, Darden T, Gohlke H, Luo R, Merz KM Jr. et al. The amber biomolecular simulation programs. J Comput Chem. 2005;26(16):1668–88. doi: 10.1002/jcc.20290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Miller BR 3rd, McGee TD Jr, Swails JM, Homeyer N, Gohlke H, Roitberg AE. Mmpbsa.Py: An efficient program for end-state free energy calculations. J Chem Theory Comput. 2012;8(9):3314–21. doi: 10.1021/ct300418h. [DOI] [PubMed] [Google Scholar]
- 81.Kollman PA, Massova I, Reyes C, Kuhn B, Huo S, Chong L. et al. Calculating structures and free energies of complex molecules: Combining molecular mechanics and continuum models. Acc Chem Res. 2000;33(12):889–97. doi: 10.1021/ar000033j. [DOI] [PubMed] [Google Scholar]
- 82.Wong S, Amaro RE, McCammon JA. MM-PBSA captures key role of intercalating water molecules at a protein-protein interface. J Chem Theory Comput. 2009;5(2):422–9. doi: 10.1021/ct8003707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65(2):87–108. doi: 10.3322/caac.21262. [DOI] [PubMed] [Google Scholar]
- 84.Miller KD, Siegel RL, Lin CC, Mariotto AB, Kramer JL, Rowland JH. et al. Cancer treatment and survivorship statistics, 2016. CA Cancer J Clin. 2016;66(4):271–89. doi: 10.3322/caac.21349. [DOI] [PubMed] [Google Scholar]
- 85.Ferreira LG, Oliva G, Andricopulo AD. Protein-protein interaction inhibitors: Advances in anticancer drug design. Expert Opin Drug Discov. 2016;11(10):957–68. doi: 10.1080/17460441.2016.1223038. [DOI] [PubMed] [Google Scholar]
- 86.Cierpicki T, Grembecka J. Targeting protein-protein interactions in hematologic malignancies: Still a challenge or a great opportunity for future therapies? Immunol Rev. 2015;263(1):279–301. doi: 10.1111/imr.12244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Teng S, Srivastava AK, Schwartz CE, Alexov E, Wang L. Structural assessment of the effects of amino acid substitutions on protein stability and protein protein interaction. Int J Comput Biol Drug Des. 2010;3(4):334–49. doi: 10.1504/ijcbdd.2010.038396. [DOI] [PMC free article] [PubMed] [Google Scholar]