

Abstract
Objectives
To characterize the epidemiology of mixed connective tissue disease (MCTD) from 1983 to 2014.
Methods
An inception cohort of patients with incident MCTD in 1985-2014 in Olmsted County, Minnesota was identified based on comprehensive individual medical record review. Diagnosis of MCTD required fulfillment of at least one of the four widely-accepted diagnostic criteria without fulfillment of classification criteria for other connective tissue diseases. Data were collected on demographic characteristics, clinical presentation, laboratory investigations and mortality.
Results
A total of 50 incident cases of MCTD were identified (mean age 48.1 years and 84% were female). The annual incidence of MCTD was 1.9 per 100,000 population. Raynaud's phenomenon was the most common initial symptoms (50%) followed by arthralgia (30%) and swollen hands (16%). The diagnosis was frequently delayed with the median time from first symptom to fulfillment of criteria of 3.6 years. At fulfillment of criteria, arthralgia was the most prevalent manifestation (86%) followed by Raynaud's phenomenon (80%), swollen hands (64%), leukopenia/lymphopenia (44%) and heartburn (38%). Evolution to other connective tissue occurred infrequently with 10-year rate of evolution of 8.5% and 6.3% for SLE and SSc, respectively. The overall mortality was not different form the general population with standardized mortality ratio: 1.1 (95% CI, 0.4-2.6).
Conclusions
This study was the first population-based study of MCTD to provide a complete picture of epidemiology and clinical characteristics of MCTD. MCTD occurred in about 2 persons per 100,000 per year. Evolution to other connective diseases occurred infrequently and the mortality was not affected.
Keywords: Epidemiology, Mixed connective tissue disease, Raynaud's phenomenon
Introduction
Mixed connective tissue disease (MCTD) is an autoimmune disorder characterized by the features of several connective tissue diseases, including systemic lupus erythematosus, systemic sclerosis, polymyositis and rheumatoid arthritis, in association with the presence of high titer of autoantibody to U1 ribonucleoprotein (RNP). It was first described as a distinct entity by Sharp et al in 1972 (1, 2).
The epidemiology of MCTD, especially the annual incidence and mortality, is not well-described as only two epidemiological studies from northern Europe have been reported. The first of these was published in 1996 (3). This study used the resource of the Finnish National Insurance database to identify incident cases of MCTD. The age- and sex-adjusted incidence of MCTD was 0.8 per 100,000 person-years. A more recent study from Norway published in 2011 suggested a lower incidence (0.2 per 100,000 person-years) (4).
The natural history and outcome of patients with MCTD are also not well-characterized as previous studies yielded inconsistent results. One study reported that over half of patients with MCTD evolved to either systemic lupus erythematosus (SLE) or systemic sclerosis (SSc) (5) while a subsequent study found such evolution in only 13% of their cohort (6).
The aim of this study was to characterize the epidemiology of MCTD, with emphasis on annual incidence and mortality, from 1985 to 2014, through the resources of the Rochester Epidemiology Project (REP). We also aimed to describe the clinical characteristics, natural history and outcome of patients with MCTD.
Methods
Data source and study population
Through the resources of the Rochester Epidemiology Project (REP), the population of Olmsted County, Minnesota, is well suited for investigation of the epidemiology of MCTD because comprehensive medical records and laboratory investigations for all residents seeking medical care for several decades are available. REP enumerates the population by linking medical records across different health care providers to create a list of unique subjects. Residency criteria were then applied to all unique subjects to describe the residency status of the subjects over time. This method ensures a complete description of a dynamic cohort of persons over time. The diagnostic codes, surgical procedure codes, laboratory values and demographic information from visits with all health care providers for the County population, including the Mayo Clinic, the Olmsted Medical Center and their affiliated hospitals, local nursing homes, and the few private practitioners, are available electronically. Only a small proportion of the population (approximately 4%) does not allow their medical records to be used in research (7). The potential of this record linkage system for use in population-based studies has previously been described (7, 8).
Approval for this study was obtained from the Mayo Clinic and Olmsted Medical Center institutional review boards and the need for informed consent was waived.
Study design
A cohort containing adult Olmsted County, Minnesota residents age 18 years and older diagnosed with MCTD between January 1, 1985 and December 31, 2014 was identified. Potential cases were initially identified from the laboratory database of positive autoantibody to RNP. Tests were performed using enzyme-linked immunosorbent assay (ELISA) with the value of more than or equal to one unit considered positive and more than or equal to two units considered high positive. Medical records of these potential cases were individually reviewed. Diagnosis of MCTD required fulfillment of at least one set of the four widely-accepted diagnostic criteria (Sharp, Alarcon-Segovia, Kasukawa or Kahn criteria) (9, 12) without fulfillment of classification criteria for systemic lupus erythematosus (SLE), systemic sclerosis (SSc), idiopathic inflammatory myositis (IIM) or rheumatoid arthritis (RA) (13-17). Cases with a diagnosis of MCTD prior to residency in Olmsted County or prior to the study period were not included.
A standardized data extraction form was utilized to record the following information: date of fulfillment of criteria, date of birth, sex, self-reported ethnicity, date of last follow-up, vital status at last follow-up (dead or alive), cause of death and serology including antinuclear antibody (ANA), anti-ribonucleoprotein (RNP), anti SS-A, anti SS-B, anti-Sm, anti-Jo-1, anti-Scl-70, anti-centromere, anti-ribonucleic acid (RNA) polymerase III, anti-double-stranded deoxyribonucleic acid (DNA), rheumatoid factor (RF), anti-cyclic citrullinated peptide (CCP) and complement level(s) at fulfillment of criteria. The presence of the following manifestations at fulfillment of criteria was documented: Raynaud's phenomena, arthritis/arthralgia, swollen hands, sclerodactyly, dysphagia, heartburn, myositis/myalgia, interstitial lung disease, pulmonary hypertension, pleuritis, pericarditis, facial erythema, lymphadenopathy, leukopenia, lymphopenia, thrombocytopenia, renal disease and neurological involvement. The presence of the aforementioned manifestations any time during follow-up, date of first appearance of those manifestations, and evolution to SLE, SSc, polymyositis (PM)/dermatomyositis (DM) or RA defined by fulfillment of the 2012 SLICC classification criteria for SLE (13), the 2013 ACR/EULAR classification criteria for SSc (14), Bohan and Peter criteria for PM /DM (15, 16) and 2010 ACR/EULAR classification criteria for RA (17) were collected. Arthritis was diagnosed by joint pain and joint swelling on examination; Arthralgia was diagnosed by joint pain without joint swelling on examination. Interstitial lung disease was diagnosed based on the presence of abnormal pulmonary interstitial infiltration, without any evidence of pulmonary infection. Pulmonary hypertension was diagnosed based on the presence of mean pulmonary artery pressure of ≥25 mmHg at rest by right heart catheterization or pulmonary artery systolic pressure of > 50 mmHg by transthoracic echocardiogram. Pleuritis was diagnosed based on the presence of pleuritic chest pain in the lateral chest wall in the absence of better alternative diagnosis; Pericarditis was diagnosed based on the presence of at least 2 of the following: 1) Typical pleuritic chest pain 2) Pericardial friction rub 3) Suggestive changes on the electrocardiogram 4) New or worsening pericardial effusion. Myositis was diagnosed based the presence of at least one of the following: 1) Muscle biopsy evidence of myositis 2) Myositis pattern on electromyogram 3) Muscle pain or weakness in conjunction with elevated muscle enzyme.
Statistical analysis
Descriptive statistics (means, percentages, etc.) were used to summarize the data. Age- and sex-specific incidence rates were calculated by using the number of incident cases as the numerator and adult population estimates based on decennial census counts as the denominator, with linear interpolation used to estimate population size for intercensal years. Overall incidence rates were age- and/or sex-adjusted to the 2010 white population of the United States. The annual incidence rates were graphically illustrated using a 3-year, centered, moving average to reduce the random fluctuations over time. In order to compute 95% confidence intervals (95% CI) for incidence rates, it was assumed that the number of incident cases followed a Poisson distribution. Poisson regression models were used to examine trends over time in the incidence of MCTD. Smoothing splines were used to represent the calendar year effect to allow for the possibility of non-linear trends.
Multi-state methods were used to estimate the percentage of patients who evolved from MCTD to SLE or SSc with adjustment for competing risk of death during follow-up. These methods are similar to Kaplan-Meier methods, but they allow for more than one possible outcome. Kaplan-Meier methods were also used to estimate the cumulative incidence of each symptom including those patients who had the symptom at the time of fulfillment of diagnostic criteria for MCTD.
Mortality rates were estimated using the Kaplan-Meier method and compared to expected mortality for persons of the same age, sex and calendar year estimated using Minnesota population life tables. The standardized mortality rate (SMR) was estimated as the ratio of the observed and expected number of deaths. Ninety-five percent confidence intervals for the SMR were calculated assuming that the expected rates are fixed and the observed rates followed a Poisson distribution.
Results
Between January 1, 1985 and December 31, 2014, 264 Olmsted county residents had a positive RNP test. Most of the residents with a positive test (214/264) did not have MCTD as they had other rheumatologic disorders/connective tissue diseases such as SLE, RA, SSc and Sjogren's syndrome or were tested for RNP without a clear rheumatologic symptom. The details of the diagnosis of those patients are provided in Supplementary Table 1. It should be noted that 2 out of 214 excluded cases had a clinical diagnosis of MCTD but the patients did not fulfill the diagnostic criteria for MCTD and, thus, were not included in this cohort. 50 of them fulfilled at least one of the diagnostic criteria sets for MCTD (72% Kasukawa, 72% Alarcon-Segovia, 54% Kahn and 28% Sharp's criteria). 11 of them (22%) fulfilled one set of criteria; 23 (46%) fulfilled 2 sets of criteria; 8 (16%) fulfilled 3 sets of criteria and 8 (16%) fulfilled 4 sets of criteria. The mean age of our cohort at fulfillment of the criteria was 48.1 years; 84% were female. The majority of the patients who fulfilled at least one of the diagnostic criteria sets also had clinical diagnosis of MCTD (42/50) while 7 of them had clinical diagnosis of undifferentiated connective tissue disease (UCTD). One patient had clinical diagnosis of possible SLE but did not fulfill the SLICC classification criteria for SLE. In contrast, the patient had Raynaud's phenomena, esophageal dysmotility and polyarthritis fulfilling the Kasukawa criteria for MCTD. All of the patients were evaluated by rheumatologists. Table 1 describes the demographic characteristics of the study population.
Table 1. Characteristics of Olmsted County, Minnesota residents with incident mixed connective tissue disease in 1985-2014.
| Female (N=42) | Male (N=8) | Total (N=50) | |
|---|---|---|---|
| Age at fulfillment of criteria, years (SD) | 47.9 (16.2) | 49.2 (13.8) | 48.1(15.7) |
| Race, n (%) | |||
| White | 38 (90%) | 6 (75%) | 44 (88%) |
| African-American | 0 (0%) | 1 (13%) | 1 (2%) |
| Asian | 4 (10%) | 0 (0%) | 4 (8%) |
| Other | 0 (0%) | 1 (13%) | 1 (2%) |
| Median length of follow-up, years, median (interquartile range) | 10.0 (4.9, 14.3) | 1.6 (1.1, 4.2) | 8.3 (3.4, 14.1) |
| Time from first symptoms to fulfillment of criteria, years, median (interquartile range) | 3.6 (1.0, 10.3) | 3.8 (1.8, 5.2) | 3.6 (1.0, 10.0) |
Abbreviations: SD=standard deviation
Overall, the annual incidence of MCTD among adults aged 18 years and older in 1985-2014 was 1.9 per 100,000 population (3.1 per 100,000 population among females and 0.7 per 100,000 population among males; Table 2). The female to male ratio was 5:1. Figure 1 demonstrates the annual incidence of MCTD per calendar year. The incidence rate appeared to be higher in year 2000 to 2002. However, there were no significant calendar year trends in the incidence rate (p=0.20).
Table 2. Incidence of mixed connective tissue disease in Olmsted County, Minnesota residents in 1985-2014.
| Female | Male | Total | ||||
|---|---|---|---|---|---|---|
| Age Group | N | Rate (per 100,000) | N | Rate (per 100,000) | N | Rate (per 100,000) |
| 18-29 | 8 | 2.5 | 1 | 0.3 | 9 | 1.4 |
| 30-39 | 6 | 2.0 | 1 | 0.3 | 7 | 1.2 |
| 40-49 | 9 | 3.3 | 2 | 0.8 | 11 | 2.1 |
| 50-59 | 9 | 4.2 | 2 | 1.0 | 11 | 2.6 |
| 60-69 | 6 | 4.3 | 2 | 1.6 | 8 | 3.0 |
| 70-79 | 2 | 2.0 | 0 | 0.0 | 2 | 1.1 |
| 80-110 | 2 | 2.4 | 0 | 0.0 | 2 | 1.6 |
| Total | 42 | 2.8* (1.4 – 4.2) | 8 | 0.9* (0.0, 1.7) | 50 | 1.9** (1.0 – 2.7) |
Age-adjusted to US white 2010 population
Age- and sex-adjusted to US white 2010 population
Figure 1.
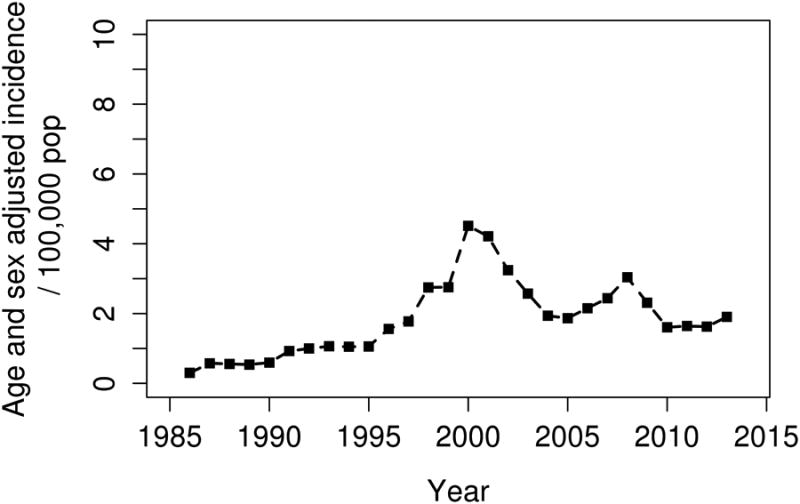
Incidence of mixed connective tissue disease among Olmsted County, Minnesota residents in 1985-2014 by calendar year.
Raynaud's phenomenon was the most common initial symptom (50%) followed by arthralgia (30%) and swollen hands (16%). However, at fulfillment of criteria, arthralgia was the most prevalent manifestation (86%) followed by Raynaud's phenomenon (80%), swollen hands (64%), leukopenia/lymphopenia (44%) and heartburn (38%). The median time from first symptom to fulfillment of criteria was 3.6 years (interquartile range: 1-10 years).
All patients had positive ANA and RNP as a positive RNP was required for disease identification in this cohort. A high titer for RNP was present in 121 of 264 cases (46%). The overall proportion of patients with positive RNP who had MCTD was 50/264 (19%) and the proportion of patients with high RNP who had MCTD was 35/121 (29%). Rheumatoid factor was positive in 24% of tested patients. Other serologies including SSA, SSB, anti-Sm and anti Jo-1 were positive in a small percentage of patients. Table 3 demonstrates the serological findings at fulfillment of criteria.
Table 3. Frequency of serology at fulfillment of criteria for mixed connective disease among Olmsted County, Minnesota residents in 1985-2014.
| Numbers positive/numbers tested (%) | |
|---|---|
| ANA | 50/50 (100) |
| Anti-RNP | 50/50 (100) |
| Anti SS-A | 7/49 (14) |
| Anti SS-B | 0/49 (0) |
| Anti Sm | 9/50 (18) |
| Anti Scl-70 | 2/40 (5) |
| Anti-centromere | 0/13 (0) |
| Anti-ds-DNA | 1/35 (3) |
| Anti Jo-1 | 1/41 (2) |
| Anti RNA polymerase III | 0/2 (0) |
| Hypocomplementemia | 1/32 (3) |
| Rheumatoid factor | 8/34 (24) |
| Anti CCP | 0/10 (0) |
Abbreviations: ANA indicates antinuclear antibody; RNP, ribonucleoprotein; ds-DNA, double-stranded deoxyribonucleic acid; RNA, ribonucleic acid; CCP, cyclic citrullinated peptide
The scleroderma-like symptoms became more common during follow-up. The cumulative incidence of sclerodactyly almost doubled compared with its presence at fulfillment of criteria (27% versus 14%). The cumulative incidence of esophageal symptoms also increased, though to a lesser extent (48% versus 38% for heartburn and 33% versus 26% for dysphagia). Three more cases of myositis (6%) and six more cases of serositis (12%) were also identified during follow-up. Table 4 demonstrates the prevalence of each manifestation at fulfillment of criteria and the cumulative incidence during follow up (including those with the manifestation present at fulfillment of criteria).
Table 4. Frequency of manifestations of mixed connective tissue disease at fulfillment of criteria and during follow-up among Olmsted County, Minnesota residents in 1985-2014.
| At fulfillment of criteria (%) | Cumulative incidence at 10 years ± SE (%) | |
|---|---|---|
| Raynaud's phenomena | 40 (80) | 83.3 ± 5.6 |
| Arthritis/arthralgia | 43 (86) | 86.0 ± 4.9 |
| Dysphagia | 13 (26) | 32.6 ± 6.7 |
| Heartburn | 19 (38) | 47.6 ± 8.0 |
| Sclerodactyly | 7 (14) | 26.8 ± 7.1 |
| Interstitial lung disease | 9 (18) | 27.8 ± 6.8 |
| Pulmonary hypertension | 1 (2) | 6.9 ± 3.9 |
| Pleuritis/pericarditis | 3 (6) | 14.9 ± 5.2 |
| Swollen hands | 32 (64) | 64.0 ± 6.8 |
| Lymphadenopathy | 2 (4) | 6.1 ± 3.4 |
| Myositis | 12 (24) | 30.6 ± 6.6 |
| Leukopenia/lymphopenia | 22 (44) | 62.5 ± 7.2 |
| Thrombocytopenia | 0 (0) | 5.8 ± 4,2 |
| Renal disease | 0 (0) | 6.0 ± 4.1 |
| CNS involvement | 0 (0) | 0 |
| PNS involvement | 0 (0) | 4.1 ± 2.9 |
| Facial erythema | 2 (4) | 4.0 ± 2.8 |
Major internal organ involvement including interstitial lung disease (ILD), pulmonary hypertension and pleuritis/pericarditis was relatively uncommon at fulfillment of criteria (prevalence of 18%, 2% and 6%, respectively), but these manifestations became more prevalent during follow-up. The most common subtype of ILD was nonspecific interstitial pneumonia (12 cases) followed by organizing pneumonia (2 cases). No case of central nervous system (CNS) involvement was observed in this cohort, while two cases of glomerulonephritis were observed during follow-up. However, those two cases evolved to SLE before developing renal involvement.
Evolution to another connective tissue disease occurred in 5 patients (3 to SLE and 2 to SSc), which corresponded to 10-year rate (+ standard error) of evolution of 8.5% + 4.7% and 6.3% + 4.6% for SLE and SSc, respectively. We did not conduct an evaluation of potential predictors of evolution as the number of patients whose disease evolved into a separate CTD was too low to analyze.
During a median length of follow of 8.3 years, 5 patients died (2 patients died of heart failure, 1 patient died of pulmonary disease, 1 patient died of cancer and no information on the cause of death in 1 patient). The overall mortality of patients with MCTD was not different from the general population (standardized mortality ratio: 1.1; 95% CI, 0.4-2.6).
Discussion
The incidence of MCTD observed in this study was higher compared with the previous studies that reported the incidence of 0.2 to 0.8 per 100,000 person-years (3, 4) .This difference might reflect the true difference in incidence rates between different populations as a result of different genetic/environmental risk factors or might reflect the different methods utilized to identify the cases. The study by Kaipiainen-Seppanen et al. (3) used an administrative database to identify their cases which had inherent limitations of coding inaccuracy and incompleteness, particularly the diseases that most physicians are not familiar with like MCTD. The study by Gunnarsson et al. (4) identified their cohort from the department of rheumatology of all public hospitals in Norway from 2005 to 2008 and retrospectively identified the year of diagnosis. The approach utilized in this study had several limitations as it could miss patients who were not seen by rheumatologists, and those who died or were lost to follow-up before inclusion period or who evolved to SLE, SSc, DM/PM or RA before inclusion period.
Patients with MCTD in this cohort were predominately female, although the female to male ratio of 5:1 was lower than that reported in previous referral-based cohort studies which had female-to-male ratios of approximately 10 to 1 (6, 18). It is possible that our population-based design might capture a more complete spectrum of the disease, in contrast to referral-based cohorts that tend to capture the more severe cases and, hence, may have a differing female-to-male ratio.
Overall, the frequency of each MCTD manifestation was similar to previous studies (6, 18) with the exception of sclerodactyly (observed in approximately one-third in those studies but only 14% in our cohort) and pleuritis/pericarditis (34% in one study (6), 22% in the other study (18) but only 6% in the current cohort). Lungs were the most common site of major internal organ involvement in this cohort but the cumulative frequency observed in this study was significantly lower compared with studies that specifically investigated pulmonary involvement and performed chest imaging in every patient (19, 20). However, our cumulative frequency was comparable with other observational cohorts (4, 6, 18). This might suggest that subclinical ILD is common and a more pro-active screening strategy should be employed in clinical practice. We also confirmed the rarity of CNS and renal involvement, similar to the original description by Sharp et al (2, 21, 22).
ANA and anti-RNP were positive in all patients as they were the diagnostic criteria requirement used to identify the cohort. Interestingly, anti-Sm, which is generally considered to be an SLE-specific antibody, was positive in 9 (18%) patients. While negative anti-Sm was one of the major criteria for MCTD in Sharp's original report, the clinical presentations of those with positive anti-Sm in this cohort were similar to those without this autoantibody and evolution to SLE occurred in only 2 out of 9 patients. The details of clinical characteristics of these patients are provided in Supplementary Table 2.
The existence of MCTD as a distinct clinical entity has been a subject of debate. Earlier studies had suggested that the majority of patients with MCTD would evolve to other connective tissue diseases, especially SLE and SSc (5, 21). However, those two studies recruited patients based primarily on the presence of RNP in conjunction with the absence of anti-Sm without taking clinical phenotype into consideration. Therefore, it was possible that the two studies might misclassify patients with other connective tissue diseases as MCTD, leading to a higher rate of evolution. In fact, more recent studies that utilized diagnostic criteria to define their cohorts showed a different result as evolution was observed in less than one-third of patients (13% in one study (6) and 26% in the other study (18)). Our results were more consistent with the more recent studies as evolution to other connective tissue diseases was observed in only 10% of our cohort (4% to SSc and 6% to SLE), corresponding to a 10-year rate of evolution of 8.5% and 6.3% for SLE and SSc, respectively. Thus, the findings from the current study would serve as another evidence to support the existence of MCTD as distinct entity, not just a transitional stage to other connective tissue diseases.
To date, four diagnostic criteria sets for MCTD have been developed (9-12). A recent study involving 161 patients with a clinical diagnosis of MCTD demonstrated the Alarcon-Segovia and Kasukawa criteria to be the most sensitive for MCTD diagnosis (73% and 75% of patients classified as having MCTD, respectively), while the Sharp criteria were the least sensitive (42%). The results of our study were line with the previous work as we found that both Alarcon-Segovia and Kasukawa criteria were equally sensitive with the sensitivity of 72%. Sharp criteria remained the least sensitive criteria (28%). The outdated definition of pulmonary involvement in the Sharp criteria (proliferative vascular lesion on lung biopsy or diffuse capacity of less than 70%) was part of the reason for its lack of sensitivity as this definition could not classify patients with early ILD observed from pulmonary imaging.
MCTD is generally considered as a milder connective tissue disease with a relatively benign prognosis. A large prospective cohort study of 280 patients with MCTD has demonstrated favorable survival rates as the 5, 10 and 15-year survival rates after the diagnosis were 98%, 96% and 88%, respectively. Our study is the first study to report the standardized mortality ratio of patients with MCTD compared with general population. We found only 5 deaths during the 440 person-years of follow-up, corresponding to a standardized mortality ratio of 1.1, not significantly different from the general population.
The major strengths of this study are that it is a population-based study that utilized a comprehensive record-linkage system to capture nearly all the cases of MCTD in the community with verification of diagnosis by medical record review. This approach minimizes the likelihood of misclassification, a common concern in coding-based studies. As the accuracy of the incidence rate calculation relies on the comprehensiveness of identification of incident cases, this study design provides a precise result. Referral bias, a common concern in referral-based cohort, is also minimized by the population-based design which provides an accurate picture of clinical manifestation and mortality of MCTD in the general population. This study also had a long period of follow-up that allowed the investigations of the cumulative incidence of each manifestation and evolution to other connective tissue diseases. The major limitations are those inherent in the study design that was retrospective in nature as the case ascertainment depends on diagnosis being made by the health-care providers. The incidence rate may be an underestimate as cases were identified using a positive RNP test and we may have missed cases in whom RNP testing was not performed. Thus, the exact burden of undiagnosed disease remains unclear. Also, the data on clinical manifestations and laboratory investigation were not systematically collected and, therefore, all the pertinent information might not be recorded. This limitation is of particularly importance in this study as those missing data could have a significant impact on the performance of the diagnostic criteria. The results of this study might not be generalizable to other populations as the population of Olmsted County is predominately of Northern European ancestry. There are also a higher proportion of workers in healthcare industry and correspondingly higher education level and socioeconomic status in this population.
Conclusion
MCTD is a rare condition, incident in about 2 persons per 100,000 person-years. Raynaud phenomena, arthralgia and swollen hands were the most common manifestations. Evolution to other connective diseases was infrequent. Mortality of these patients was unimpaired compared with the general population.
Supplementary Material
Significance and innovation.
-
-
The annual incidence of MCTD was 1.9 per 100,000 population.
-
-
Raynaud's phenomenon, arthralgia and swollen hands were the most common initial symptoms.
-
-
Evolution to other connective tissue diseases was infrequent.
Acknowledgments
Funding: This study was made possible using the resources of the Rochester Epidemiology Project, which is supported by the National Institute on Aging of the National Institutes of Health under Award Number R01AG034676, and CTSA Grant Number UL1 TR000135 from the National Center for Advancing Translational Sciences (NCATS), a component of the National Institutes of Health (NIH). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Conflict of interest: All authors have disclosed no conflict of interest.
Contribution: All authors have role in conducting the research and writing the manuscript. The final version of this manuscript is approved by all authors.
Ethic Approval: This study is approved by Institutional Review Board of Mayo Clinic and Olmsted Medical Center. The need for informed consent was waived.
References
- 1.Aringer M, Steiner G, Smolen JS. Does mixed connective tissue disease exist? Yes Rheum Dis Clin North Am. 2005;31:411–20. doi: 10.1016/j.rdc.2005.04.007. [DOI] [PubMed] [Google Scholar]
- 2.Sharp GC, Irvin WS, Tan EM, Gould RG, Holman HR. Mixed connective tissue disease – an apparently distinct rheumatic disease syndrome associated with a specific antibody to an extractable nuclear antigen (ENA) Am J Med. 1972;52:148–59. doi: 10.1016/0002-9343(72)90064-2. [DOI] [PubMed] [Google Scholar]
- 3.Kaipiainen-Seppanen O, Aho K. Incidence of rare systemic rheumatic and connective tissue diseases in Finland. J Intern Med. 1996;240:81–4. doi: 10.1046/j.1365-2796.1996.14843000.x. [DOI] [PubMed] [Google Scholar]
- 4.Gunnarsson R, Molberg O, Gilboe IM, Gran JT. The prevalence and incidence of mixed connective tissue disease: a national multicentre survey of Norwegian patients. Ann Rheum Dis. 2011;70:1047–51. doi: 10.1136/ard.2010.143792. [DOI] [PubMed] [Google Scholar]
- 5.Gendi NST, Welsh KI, van Venrooij WJ, Vancheeswaran R, Gilroy J, Black CK. HLA type as a predictor of mixed connective tissue disease differentiation: ten-year clinical and immunogenetic followup of 46 patients. Arthritis Rheum. 1995;38:259–66. doi: 10.1002/art.1780380216. [DOI] [PubMed] [Google Scholar]
- 6.Burdt MA, Hoffman RW, Deutscher SL, Wang GS, Johnson JC, Sharp GC. Long-term outcome in mixed connective tissue disease. Arthritis Rheum. 1999:899–909. doi: 10.1002/1529-0131(199905)42:5<899::AID-ANR8>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 7.St Sauver JL, Grossardt BR, Leibson CL, Yawn BP, Melton LJ., III Generalizability of epidemiological findings and public health decision: An Illustration from the Rochester Epidemiology Project. Mayo Clin Proc. 2012;87:151–64. doi: 10.1016/j.mayocp.2011.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.St Sauver JL, Grossardt BR, Leibson CL, Yawn BP, Melton LJ, III, Rocca WA. Use of medical records linkage system to enumerate a dynamic population over time: The Rochester Epidemiology Project. Am J Epidemiol. 2011;173:1059–68. doi: 10.1093/aje/kwq482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sharp GC. Diagnostic criteria for classification of MCTD. In: Kasukawa R, Sharp GC, editors. Mixed connective tissue disease and anti-nuclear antibodies. Amsterdam: Elsevier Science Publishers B.V. (Biomedical Division); 1987. pp. 23–30. [Google Scholar]
- 10.Alarcón-Segovia D, Villarreal M. Classification and diagnostic criteria for mixed connective tissue disease. In: Kasukawa R, Sharp GC, editors. Mixed connective tissue disease and anti-nuclear antibodies. Amsterdam: Elsvier Science Publishers B.V. (Biomedical Division); 1987. pp. 33–40. [Google Scholar]
- 11.Kasukawa R, Tojo T, Miyawaki S. Preliminary diagnostic criteria for classification of mixed connective tissue disease. In: Kasukawa R, Sharp GC, editors. Mixed connective tissue disease and anti-nuclear antibodies. Amsterdam: Elsevier Science Publishers B.V. (Biomedical Division); 1987. pp. 41–7. [Google Scholar]
- 12.Kahn MF, Appelboom T. Syndrome de Sharp. In: Kahn MF, Peltier AP, Mayer O, Piette JC, editors. Les maladies systémiques. 3rd. Paris: Flammarion; 1991. pp. 545–56. [Google Scholar]
- 13.Petri M, Orbai AM, Alarcon GS, Gordon C, Merrill JT, Fortin PR, et al. Derivation and validation of the Systemic Lupus International Collaborating Clinics classification criteria for systemic lupus erythematosus. Arthritis Rheum. 2012;64:2677–86. doi: 10.1002/art.34473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.van den Hoogen F, Khanna D, Fransen J, Johnson SR, Baron M, Tyndall A, et al. 2013 Classification Criteria for Systemic Sclerosis: An American College of Rheumatology/European League Against Rheumatism Collaborative Initiative. Arthritis Rheum. 2013;65:2737–47. doi: 10.1002/art.38098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bohan A, Peter JB. Polymyositis and dermatomyositis-(part 1) New Engl J Med. 1975;292:344–7. doi: 10.1056/NEJM197502132920706. [DOI] [PubMed] [Google Scholar]
- 16.Bohan A, Peter JB. Polymyositis and dermatomyositis (part II) New Engl J Med. 1975;292:403–7. doi: 10.1056/NEJM197502202920807. [DOI] [PubMed] [Google Scholar]
- 17.Aletaha D, Neogi T, Silman AJ, Funovits J, Felson DT, Bingham CO, 3rd, et al. 2010 rheumatoid arthritis classification criteria: an American College of Rheumatology/European League Against Rheumatism collaborative initiative. Ann Rheum Dis. 2010;69:1580–8. doi: 10.1136/ard.2010.138461. [DOI] [PubMed] [Google Scholar]
- 18.Cappelli S, Randone SB, Martinovic D, Tamas MM, Pasalic K, Allanore Y, et al. “To be or not to be,” Ten years after: Evidence for mixed connective tissue disease as distinct entity. Semin Arthritis Rheum. 2012;41:589–98. doi: 10.1016/j.semarthrit.2011.07.010. [DOI] [PubMed] [Google Scholar]
- 19.Gunnarsson R, Aaløkken TM, Molberg Ø, Lund MB, Mynarek GK, Lexberg AS, et al. Prevalence and severity of interstitial lung disease in mixed connective tissue disease: a nationwide, cross-sectional study. Ann Rheum Dis. 2012;71:1966–72. doi: 10.1136/annrheumdis-2011-201253. [DOI] [PubMed] [Google Scholar]
- 20.Fagundes MN, Caleiro MT, Navarro-Rodriguez T, Baldi BG, Kavakama J, Salge JM, et al. Esophageal involvement and interstitial lung disease in mixed connective tissue disease. Respir Med. 2009;103:854–60. doi: 10.1016/j.rmed.2008.12.018. [DOI] [PubMed] [Google Scholar]
- 21.Nimelstein SH, Brody S, McShane D, Holman HR. Mixed connective tissue disease: A subsequent evaluation of the original 25 patients. Medicine (Baltimore) 1980;59:239–48. [PubMed] [Google Scholar]
- 22.Hajas A, Szodoray P, Nakken B, Gaal J, Zold E, Laczik R, et al. Clincial course, prognosis and causes of death in mixed connective tissue disease. J Rheumatol. 2013;40:1134–42. doi: 10.3899/jrheum.121272. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.