

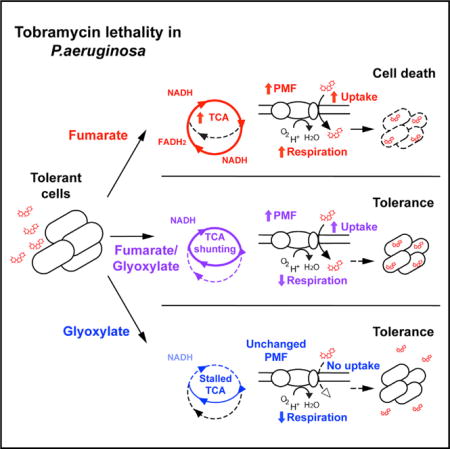
SUMMARY
Metabolically dormant bacteria present a critical challenge to effective antimicrobial therapy because these bacteria are genetically susceptible to antibiotic treatment but phenotypically tolerant. Such tolerance has been attributed to impaired drug uptake, which can be reversed by metabolic stimulation. Here, we evaluate the effects of central carbon metabolite stimulations on aminoglycoside sensitivity in the pathogen Pseudomonas aeruginosa. We identify fumarate as a tobramycin potentiator that activates cellular respiration and generates a proton motive force by stimulating the tricarboxylic acid (TCA) cycle. In contrast, we find that glyoxylate induces phenotypic tolerance by inhibiting cellular respiration with acetyl-coenzyme A diversion through the glyoxylate shunt, despite drug import. Collectively, this work demonstrates that TCA cycle activity is important for both aminoglycoside uptake and downstream lethality and identifies a potential strategy for potentiating aminoglycoside treatment of P. aeruginosa infections.
Graphical Abstract
INTRODUCTION
Bacteria can survive exposure to antibiotics by multiple mechanisms, including (1) by acquiring and expressing genetically encoded resistance elements and (2) by adopting phenotypically tolerant physiological states in response to changes in the local environment. The combination of these genetic resistance and phenotypic tolerance mechanisms underlies the overall antibiotic susceptibility profile of a bacterial population. Genetically encoded antibiotic resistance primarily acts by decreasing drug permeability, activating drug efflux, promoting drug inactivation, or directly impairing drug-target interactions (Blair et al., 2015). Although environmental factors can influence the expression of resistance elements, these activities are considered independent of the bacteria’s environment.
In contrast, phenotypic tolerance is highly sensitive to environmental conditions, which either directly interfere with antibiotics or alter bacterial physiology (McMahon et al., 2007; Poole, 2012). For example, aminoglycoside bactericidal activity requires proton motive force (PMF)-dependent transport for cell penetration (Bryan and Kwan, 1983; Taber et al., 1987). Thus, environmental conditions influencing bacterial PMF, such as anaerobic growth or nutrient starvation, can significantly affect aminoglycoside susceptibility (Bryan and Kwan, 1983). Moreover, fundamental determinants of antibiotic activity, such as cell growth and metabolism, can also stochastically change within an isogenic population, leading to a fraction of phenotypically tolerant cells called persisters (Brauner et al., 2016).
Pulmonary infections in cystic fibrosis (CF) clinically exemplify how genetic resistance and phenotypic tolerance can challenge antimicrobial therapy. CF is a genetic disease with multisystem involvement, complicated by chronic bacterial pulmonary infections that cause death in childhood if untreated (Bye et al., 1994; Folkesson et al., 2012). Modern antibiotic therapy has contributed to improving life expectancy up to 40 years (Marshall and Hazle, 2011).
Pseudomonas aeruginosa is the primary pathogen associated with CF lung infections and disease progression (Chambers et al., 2005). While early intervention may clear initial P. aeruginosa infections (Hansen et al., 2008), once chronic infection is established, antibiotic therapy can decrease the burden of P. aeruginosa but not eradicate it (Ramsey et al., 1999). Antibiotic failure in this context is thought to be due to both genetic resistance and phenotypic tolerance (Gibson et al., 2003a; Mulcahy et al., 2010). The CF airway environment is characterized by chronic inflammation, desiccated conditions, and variable nutrient availability (Boucher, 2007). P. aeruginosa adapts to these conditions by forming biofilms, decreasing central metabolism, activating the stringent response, and inducing efflux pumps, each of which can confer phenotypic tolerance (Folkesson et al., 2012; Nguyen et al., 2011; Singh et al., 2000; Son et al., 2007; Yang et al., 2011). In addition, the stringent response can promote aminoglycoside tolerance by dampening membrane potential, consequently reducing drug uptake (Nguyen et al., 2011; Verstraeten et al., 2015).
Several recent studies have sought to biochemically overcome phenotypic tolerance in model organisms (Allison et al., 2011; Barraud et al., 2013; Conlon et al., 2013; Kaushik et al., 2016; Lebeaux et al., 2014). Metabolic stimulations demonstrably potentiate aminoglycoside sensitivity in Escherichia coli and Staphylococcus aureus (Allison et al., 2011) and pH manipulations potentiate aminoglycoside sensitivity in P. aeruginosa (Kaushik et al., 2016; Lebeaux et al., 2014). Here, we sought to understand the biochemical basis for phenotypic tolerance to aminoglycoside antibiotics in P. aeruginosa. We assayed the effects of metabolic stimulations on P. aeruginosa susceptibility to tobramycin, a cornerstone CF aminoglycoside therapeutic. We found that cellular respiration critically modulates tobramycin lethality by facilitating upstream drug import and fueling downstream death processes. We describe how metabolites such as fumarate can robustly potentiate tobramycin susceptibility, while other metabolites such as glyoxylate can promote tolerance. Collectively, we reveal metabolic mechanisms by which environmental factors can phenotypically tune the susceptibility of an important clinical pathogen to antibiotic treatment.
RESULTS
Central Carbon Metabolism Perturbations Alter Tobramycin Susceptibility in P. aeruginosa
Tobramycin efficacy in P. aeruginosa is extremely sensitive to growth phase. While isogenic exponential-phase cells exhibit marked tobramycin susceptibility, non-growing stationary-phase cells exhibit tobramycin tolerance (Figure 1A), indicating a phenotypic, rather than genetic, mechanism for altered sensitivity. PMF-dependent uptake is required for aminoglycoside internalization (Bryan and Kwan, 1983; Taber et al., 1987), and stationary-phase cells are predicted to have lower metabolic activity (Kashket, 1981). We hypothesized that the differences in tobramycin sensitivity between exponential- and stationary-phase cells could in part be explained by differential uptake. To test this, we challenged exponential- and stationary-phase P. aeruginosa cells with 40 mg/L Texas red-labeled tobramycin and quantified tobramycin internalization by flow cytometry. Differences in sensitivity appeared to be significantly associated with differences in drug uptake (Figure 1B).
Figure 1. Central Carbon Metabolites Tune Tobramycin Susceptibility in P. aeruginosa.
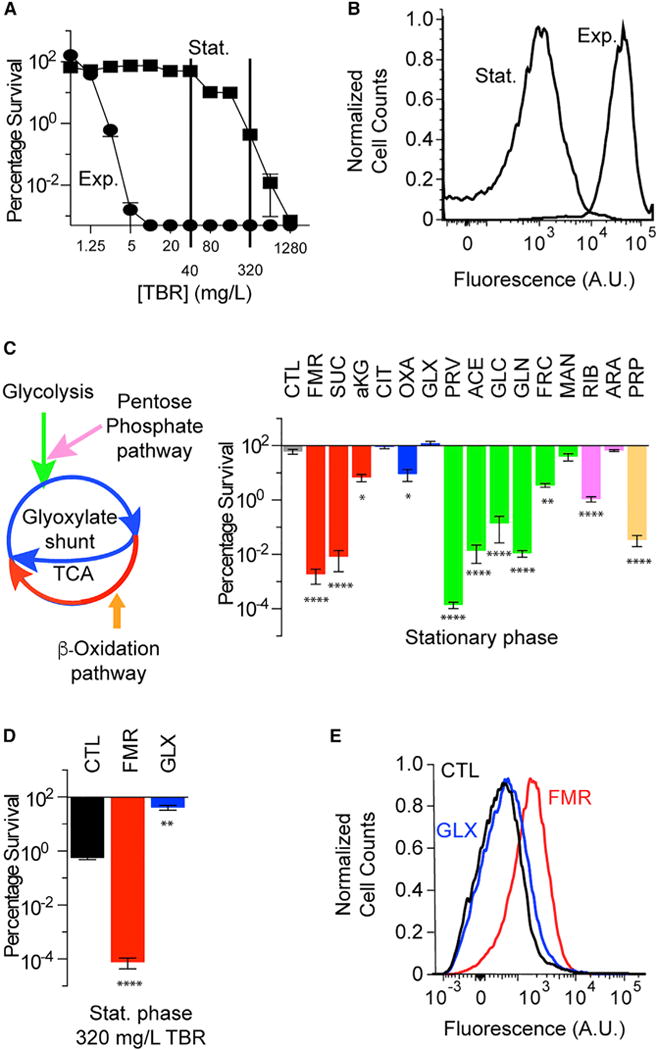
(A) Survival of exponential- (Exp.) and stationary-phase (Stat.) PAO1 cells following 4 hr tobramycin (TBR) treatment. Metabolically dormant stationary-phase cells exhibit phenotypic tolerance (n = 2).
(B) Representative flow cytometry quantification of exponential- and stationary-phase cells treated with 40 mg/L tobramycin-Texas red for 15 min. Cell counts are normalized to mode.
(C) Survival of stationary-phase cells following 4 hr treatment with 40 mg/L tobramycin and supplemented with various central carbon metabolites at a total carbon concentration of 60 mM. Supplemented metabolites are as follows: no carbon source (CTL), fumarate (FMR), succinate (SUC), α-ketoglutarate (aKG), citrate (CIT), oxaloacetate (OXA), glyoxylate (GLX), pyruvate (PRV), acetate (ACE), glucose (GLC), gluconate (GLN), fructose (FRC), mannitol (MAN), ribose (RIB), arabinose (ARA), or propionate (PRP). Cells are normalized to the pre-treatment CFU/mL.
(D) Survival of stationary-phase cells following 4 hr treatment with 320 mg/L tobramycin and supplementation with 15 mM fumarate or 30 mM glyoxylate or no supplement (control).
(E) Representative flow cytometry quantification of stationary-phase cells following 15 min treatment with 40 mg/L tobramycin-Texas red and supplementation with 15 mM fumarate or 30 mM glyoxylate or no supplement (control). Cell counts are normalized to mode. Statistical analysis of geometric means is depicted in Figure S1D.
Values in all bar graphs depict the mean ± SEM with significance reported as FDR-corrected p values in comparison with untreated control (CTL): *p ≤0.01, **p ≤ 0.001, ****p ≤ 0.0001 (n = 3).
Because growth phase-dependent PMF significantly affects tobramycin sensitivity, we hypothesized that metabolic stimulations could sensitize tolerant stationary-phase P. aeruginosa cells. We therefore screened carbon source metabolites from various central metabolism pathways in minimal media against tobramycin and quantified differences in survival (Figure 1C). We challenged stationary-phase cells with 40 mg/L tobramycin (sufficient for killing exponential-phase cells) supplemented with each carbon source normalized to deliver 60 mM total carbon. Interestingly, components of the lower part of the tricarboxylic acid (TCA) cycle (fumarate, succinate) and lower glycolysis (pyruvate, acetate) most strongly sensitized stationary-phase cells to tobramycin, while upper TCA cycle metabolites (citrate, glyoxylate) appeared to have little effect. This behavior was tobramycin specific (Figure S1A).
We further explored the differences between upper and lower TCA cycle stimulation by analyzing the effects of fumarate and glyoxylate supplementation on the sensitivity of stationary-phase cells to 320 mg/L tobramycin (sufficient for killing stationary-phase cells). While fumarate significantly potentiated the killing of stationary-phase cells by tobramycin, glyoxylate protected against tobramycin lethality (Figure 1D). We also observed glyoxylate-mediated protection in exponential-phase cells (Figure S1B). We found fumarate-induced potentiation to be dose and time dependent (Figures S1A and S1C), suggesting the activation of a downstream physiological process. Because tobramycin sensitivity appeared to be explained by drug transport, we quantified uptake in stationary-phase cells treated with 40 mg/L Texas red-labeled tobramycin, with and without supplementation by either fumarate or glyoxylate. While fumarate-treated cells exhibited increased internalization, glyoxylate-treated cells did not differ from the untreated control (Figure 1E). These results were specific to tobramycin, as fumarate did not elicit any significant increase in the internalization of un-conjugated Texas red (Figure S1D).
Tobramycin Potentiation Requires Enhanced Respiratory Activity
Aminoglycoside internalization requires a PMF generated by the electron transport chain (ETC) (Figure 2A) (Taber et al., 1987). In aerobic environments, oxygen functions as the terminal electron acceptor for respiration in the ETC; oxygen consumption therefore serves as a useful analog for ETC activity. Because fumarate and glyoxylate induced marked differences in tobramycin uptake, we hypothesized that they would also exert significant differences in respiratory activity. Using the Seahorse XFe extracellular flux analyzer, we quantified changes in the oxygen consumption rate (OCR) of stationary-phase cells in response to fumarate or glyoxylate treatment. Fumarate rapidly and robustly enhanced ETC activity, while glyoxylate completely suppressed ETC activity (Figure 2B). This suggests that fumarate may sensitize stationary-phase P. aeruginosa by stimulating cellular respiration, generating a PMF, and increasing tobramycin import, while glyoxylate may do the opposite. To test this, we measured changes in fumarate- and glyoxylate-induced membrane potential with DiBAC4(3), a fluorescent molecule that accumulates in depolarized cells (Rezaeinejad and Ivanov, 2011). Fumarate re-polarized the membrane potential, while glyoxylate exhibited little effect (Figures 2C and S2A).
Figure 2. Tobramycin Potentiation Requires Increased Respiration through the Electron Transport Chain.
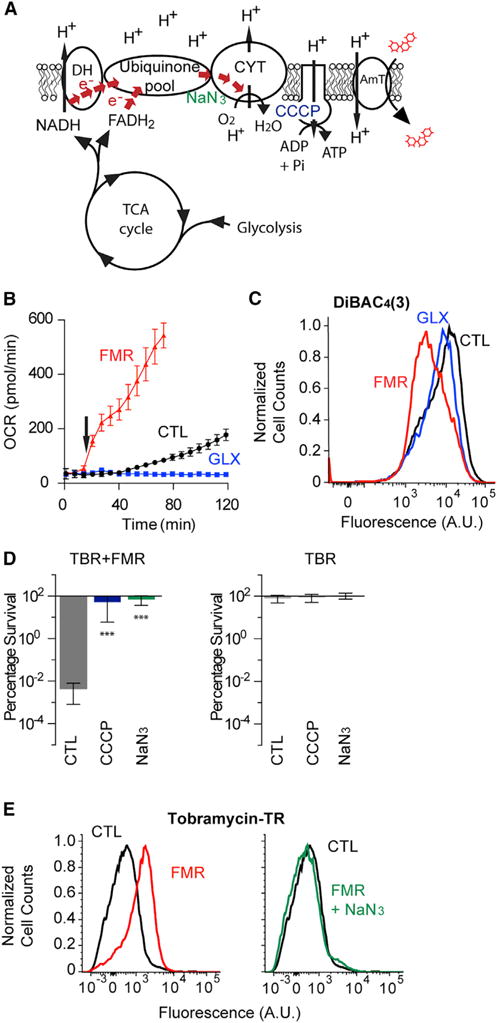
(A) Schematic of the aerobic respiratory apparatus, including glycolysis, TCA cycle, and electron transport chain (ETC) with dehydrogenases (DH), quinone pool, and cytochrome oxidases (CYT). In addition, ETC inhibitors carbonyl cyanide 3-chlorophenylhydrazone (CCCP, blue) and sodium azide (NaN3, green) are depicted near their targets. Proton motive force (PMF) generated by the ETC drives aminoglycoside uptake through its putative transporter (AmT).
(B) Respiratory activity as quantified by oxygen consumption rate (OCR) in response to treatment of stationary-phase PAO1 cells with 15 mM fumarate (FMR), 30 mM glyoxylate (GLX), or no supplementation. Carbon metabolites were added at 12 min (arrow). Data shown reflect the mean ± SEM (n ≥ 6).
(C) Representative flow cytometry quantification of PMF-dependent membrane potential by DiBAC4(3) labeling of stationary-phase cells 25 min after incubation either with no metabolite or with 15 mM fumarate or 30 mM glyoxylate. Membrane depolarization expels DiBAC4(3). Cell counts are normalized to mode. Statistical analysis of geometric means is depicted in Figure S2A.
(D) Effects of ETC inhibition by 50 μM CCCP or 0.1% NaN3 on fumarate-mediated tobramycin (TBR) resensitization. Survival of stationary-phase cells following 4 hr treatment of 40 mg/L tobramycin with (left) and without (right) 15 mM fumarate supplementation. Values depict the mean ± SEM with significance reported as FDR-corrected p values in comparison with untreated control (CTL): ***p ≤ 0.001 (n = 3).
(E) Representative flow cytometry quantification of the effects of ETC inhibition by 0.1% NaN3 on fumarate-mediated tobramycin uptake in stationary-phase cells following 15 min treatment with 40 mg/L tobramycin-Texas red and either 15 mM fumarate (FMR) or no supplementation (CTL) (left). NaN3 treatment inhibits fumarate-induced increases in tobramycin uptake (right). Cell counts are normalized to mode.
We hypothesized that respiratory enhancement was necessary for sensitization. To test this, we applied two ETC inhibitors (NaN3, a cytochrome c oxidase inhibitor, and 3-chlorophenylhy-drazone [CCCP], a proton ionophore uncoupler) to fumarate-treated cells and quantified tobramycin sensitivity. We found that NaN3 suppressed fumarate-induced enhancements to respiration (Figure S2B), membrane potential (Figure S2C), and tobramycin potentiation (Figure 2D) and confirmed that NaN3 disrupts tobramycin uptake (Figure 2E). CCCP similarly suppressed tobramycin potentiation, but also elicited the secretion of a yellow fluorescent pigment that directly confounded OCR measurements. Collectively, these data demonstrate that respiratory activity critically manages both tobramycin uptake and downstream lethality.
Respiratory Enhancement Is Driven by Increased Expression of ETC and Central Metabolism Genes
For fumarate to increase PMF, respiratory activity in the ETC must be elevated at the enzyme and/or substrate level. We hypothesized that fumarate and glyoxylate may exert their opposing effects on the ETC activity by changing the expression of ETC genes. To test this, we performed microarray experiments on fumarate- and glyoxylate-treated cells and examined differences in expression of oxidative phosphorylation and terminal oxidase genes. We found that in comparison to the untreated control, fumarate significantly altered the expression of ETC genes, while glyoxylate appeared to have little effect (Figures 3A and 3B). Fumarate also significantly altered the expression of upstream central metabolism genes in both the TCA cycle and glycolysis, while glyoxylate did not (Figures 3C and 3D). Fumarate notably increased expression of succinate dehydrogenase, malate dehydrogenase, citrate synthetase, and succinyl-coenzyme A (CoA) synthetase in the TCA cycle, while glyoxylate only increased expression of succinyl-CoA synthetase and α-ketoglutarate dehydrogenase (Figure S3). Notably, fumarate also starkly increased the expression of ribosomal genes, signaling a potential increase in translational machinery (Figure 3E). Collectively, these results suggest that fumarate treatment promotes a transcriptional metabolic program in P. aeruginosa stationary-phase cells.
Figure 3. Fumarate Transcriptionally Activates Central Metabolism.
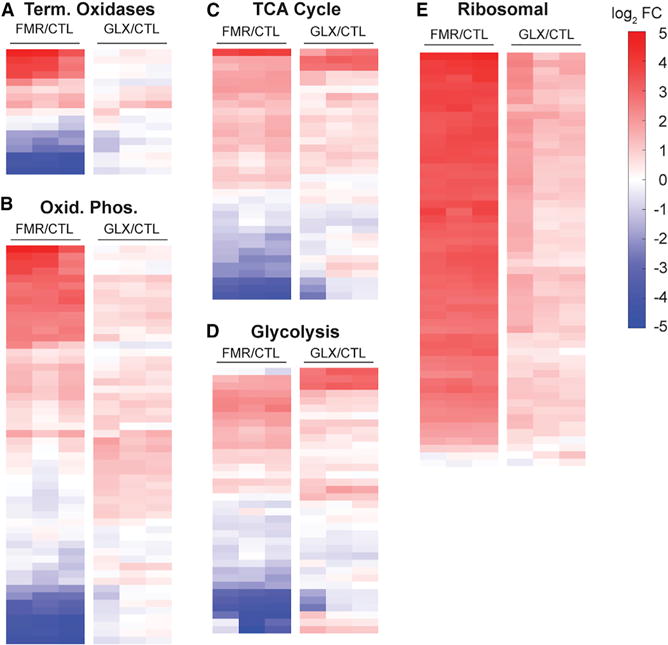
Microarray expression profiles from stationary-phase PAO1 cells treated with 15 mM fumarate (FMR) or 30 mM glyoxylate (GLX) for 1 hr. Values reported as log2 fold changes over untreated control (CTL) in the expression of (A) terminal oxidase, (B) oxidative phosphorylation, (C) TCA cycle, (D) glycolysis, and (E) ribosomal genes.
TCA Cycle Activity Drives Tobramycin Lethality
While the increased respiratory activity by fumarate may be explained by the increased expression of ETC and central metabolism genes, the suppressed respiratory activity by glyoxylate could not be explained transcriptionally. Glyoxylate has previously been documented to inhibit pyruvate dehydrogenase (Bisswanger, 1981) and isocitrate dehydrogenase (Nimmo, 1986) in E. coli and to irreversibly inhibit the α-ketoglutarate dehydrogenase of mammalian cells (Adinolfi et al., 1969); glyoxylate may therefore inhibit TCA cycle enzymatic activity in P. aeruginosa. Alternatively, but not exclusively, glyoxylate may divert carbon away from the TCA cycle, either by shunting carbon from succinate dehydrogenase (Caspi et al., 2012) or by decreasing the production of reduced electron carriers by being transformed into oxalate (Quayle et al., 1961). We therefore hypothesized that glyoxylate-mediated suppression of respiratory activity was due to an upstream inhibition of central metabolism that would reduce substrates available for generating a PMF in the ETC.
To test this hypothesis, we performed metabolite profiling on fumarate- and glyoxylate-treated cells using liquid chromatography-mass spectrometry (LC-MS). While fumarate supplementation did not significantly alter the abundance of TCA cycle metabolites over the control, glyoxylate significantly increased malate, fumarate, citrate, and α-ketoglutarate (Figure 4A). Acetyl-CoA was significantly decreased by glyoxylate treatment (below the limit of detection in four of five replicates), indicating depletion of this metabolite. The increases in citrate and α-ketoglutarate indicate that glyoxylate directly inhibits α-ketoglutarate dehydrogenase, resulting in the stalled accumulation of α-ketoglutarate; this cannot be fully resolved by reverse isocitrate dehydrogenase activity due to the reduced availability of free NADH, as supported by the respiratory collapse (Figure 2B) and insignificant changes in membrane potential (Figure 2C). The accumulation in malate suggests that glyoxylate may be driving acetyl-CoA depletion by fueling malate synthase. The net result is a bypass of the lower TCA cycle. Additionally, we detected significant increases in glycolate and oxalate (Figure 4B), confirming that these tobramycin-tolerant stationary-phase cells are actively metabolizing glyoxylate. Collectively, these data indicate that glyoxylate-mediated suppression of cellular respiration and tobramycin sensitivity occurs through a direct, enzymatic blockage of the TCA cycle. These results are also consistent with our observation that supplementation with upper TCA cycle metabolites alone is insufficient for tobramycin sensitization; intact TCA cycle activity is required (Figure 1C).
Figure 4. TCA Cycle Activity Underlies Phenotypic Sensitivity to Tobramycin Treatment.
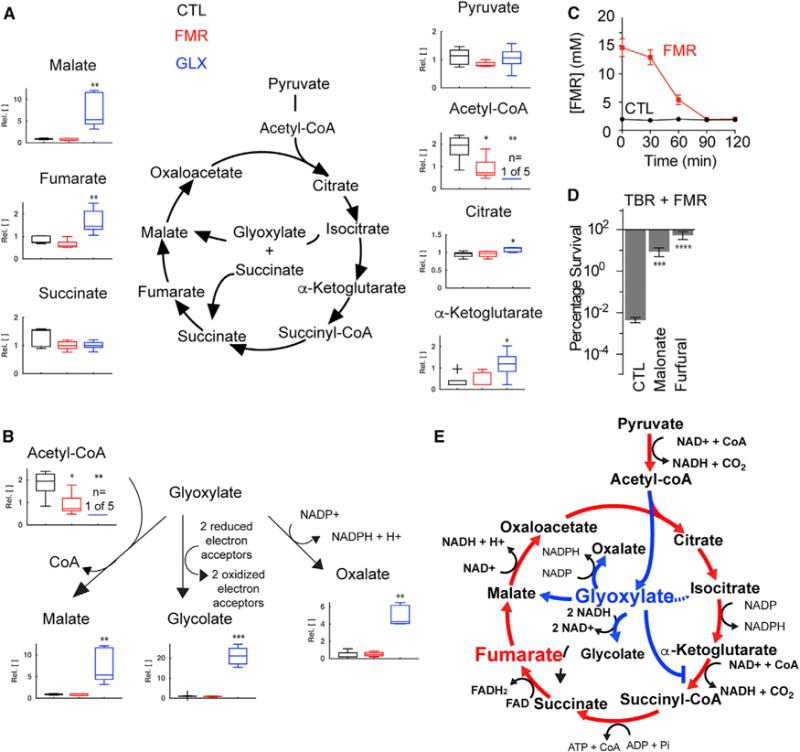
(A) Abundance of TCA cycle metabolites quantified by metabolite profiling of stationary-phase PAO1 cells with 15 mM fumarate (FMR), 30 mM glyoxylate (GLX), or no treatment (CTL). Values depict relative concentration measurements with significance reported as p values in comparison with untreated control (CTL) with FDR-computed q values ≤0.1: *p ≤ 0.05, **p ≤ 0.01 (n = 5).
(B) Abundance of downstream metabolic products of glyoxylate, quantified by metabolite profiling. Values depict relative concentration measurements with significance reported as p values in comparison with untreated control (CTL) with FDR-computed q values ≤0.1: *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001 (n = 5).
(C) Extracellular fumarate consumption following supplementation of stationary-phase cells with 15 mM fumarate.
(D) Effects of TCA cycle inhibition by 10 mM malonate or 10 mM furfural on fumarate-mediated tobramycin resensitization. Survival of stationary-phase cells following 4 hr treatment of 40 mg/L tobramycin with 15 mM fumarate supplementation. Values depict the mean ± SEM with significance reported as FDR-corrected p values in comparison with CTL: ***p ≤ 0.001, ****p ≤ 0.0001 (n = 3).
(E) Summary schematic depicting TCA cycle utilization following treatment with fumarate (red) or glyoxylate (blue).
While extracellular fumarate is rapidly consumed (Figure 4C), metabolite profiling did not reveal increases in intracellular fumarate (Figure 4A). These data indicate that fumarate stimulates increased TCA cycle activity because the relative proportions of TCA cycle metabolites are constrained by the biochemical stoichiometry of TCA cycle reactions. Because cellular fumarate was consumed and respiration (Figure 2B), PMF (Figure 2C), tobramycin uptake (Figure 1E), and lethality (Figures 1C and 1D) were all increased, we hypothesized that total TCA cycle activity must also be increased. This is supported by the increased expression of TCA cycle genes (Figure 3C), which would help process the increased substrate provided by fumarate supplementation. A corollary of this is that enhanced TCA cycle activity would require enhanced acetyl-CoA conversion into citrate. Our metabolite profiling data support this hypothesis, as acetyl-CoA significantly decreased in fumarate-treated cells. To more directly test these hypotheses, we treated fumarate-stimulated cells with two biochemical TCA cycle inhibitors: malonate, an inhibitor of succinate dehydrogenase (Pardee and Potter, 1949), and furfural, an upstream inhibitor of pyruvate dehydrogenase (Modig et al., 2002). Inhibition of either enzyme significantly inhibited fumarate sensitization (Figure 4D).
Because glyoxylate also appeared to biochemically inhibit TCA cycle activity, we hypothesized that glyoxylate would also suppress fumarate-stimulated respiration and sensitization. We measured the OCR of cells treated with both fumarate and various concentrations of glyoxylate and found progressive suppression of the fumarate-induced respiration (Figure 5A). Moreover, glyoxylate inhibited fumarate-mediated sensitization in a dose-dependent manner (Figure 5B), which could not be rescued by further fumarate supplementation (Figure S4). We also found that glyoxylate addition as late as 30 min after fumarate treatment still suppressed tobramycin sensitization (Figure 5C). Because fumarate appeared to transcriptionally enhance TCA cycle activity, we performed microarray experiments on cells treated with both fumarate and glyoxylate and found that glyoxylate did not inhibit the fumarate-induced changes in gene expression (Figure S5). These corroborate our metabolite profiling data and support our hypothesis that glyoxylate biochemically inhibits TCA cycle activity. Surprisingly, glyoxylate addition to fumarate-treated cells neither suppressed membrane re-polarization (Figure 5D) nor reduced tobramycin uptake (Figure 5E). These results indicate that drug uptake and downstream lethality are two independent steps in aminoglycoside killing and that both TCA cycle activity and accompanying cellular respiration are required for the antibiotic-induced death process.
Figure 5. Glyoxylate Exerts Dominant-Negative Suppression over Fumarate-Mediated Resensitization.
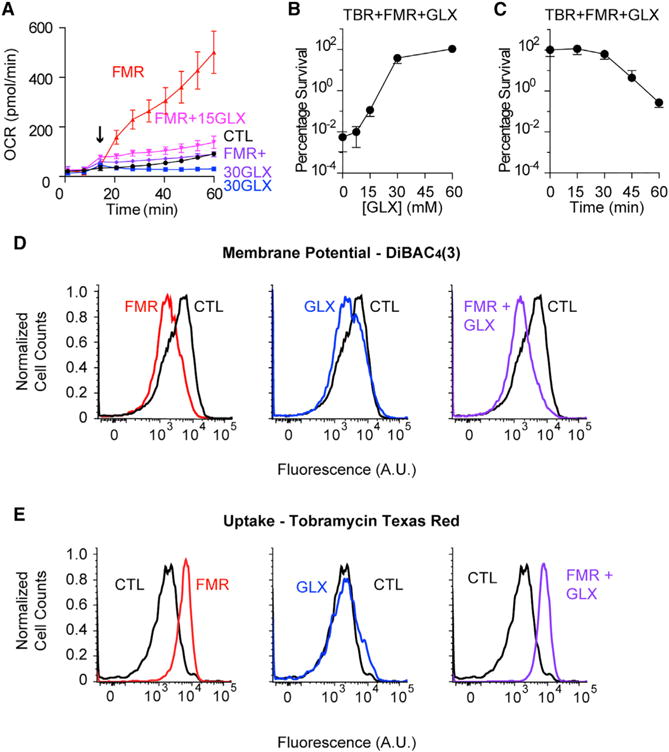
(A) Respiratory activity as quantified by oxygen consumption rate (OCR) of glyoxylate-mediated suppression of fumarate-induced respiration. Stationary-phase PAO1 cells were treated with either 15 mM fumarate (FMR) or no supplementation (CTL) with the addition of either 15 or 30 mM glyoxylate (GLX). Carbon metabolites were added at 12 min (arrow). Data shown reflect the mean ± SEM (n ≥ 6).
(B) Survival of stationary-phase cells following 4 hr treatment with 40 mg/L tobramycin (TBR), 15 mM fumarate, and supplementation with 0, 7.5, 15, 30, or 60 mM glyoxylate. Data shown reflect the mean ± SEM (n = 3).
(C) Survival of stationary-phase cells following 4 hr treatment with 40 mg/L tobramycin, 15 mM fumarate, and delayed supplementation with 30 mM glyoxylate at indicated time points following fumarate addition. Data shown reflect the mean ± SEM (n = 3).
(D) Representative flow cytometry quantification of PMF-dependent membrane potential by DiBAC4(3) labeling of stationary-phase cells 25 min after incubation with no metabolite (CTL), 15 mM fumarate (FMR), 30 mM glyoxylate (GLX), or the combination of 15 mM fumarate with 30 mM glyoxylate (FMR + GLX). Cell counts are normalized to mode.
(E) Representative flow cytometry quantification of glyoxylate suppression of fumarate-mediated tobramycin uptake in stationary-phase cells following 45 min treatment with 40 mg/L to-bramycin-Texas red and 15 mM fumarate, 30 mM glyoxylate, or their combination. Cell counts are normalized to mode.
Collectively, our data indicate that both TCA cycle and respiratory activity are required for tobramycin lethality. We propose a model where fumarate can promote TCA cycle activity, creating high levels of reduced electron carriers that fuel the ETC (Figure 6); this increases cellular respiration, which enhances PMF-dependent uptake and downstream lethality. In contrast, glyoxylate biochemically inhibits TCA cycle activity and suppresses cellular respiration (Figures 4E and 6). In cells exposed to both carbon sources, fumarate supplies enough carbon to produce NADH sufficient for promoting tobramycin uptake, but glyoxylate inhibits fumarate-induced sensitization by depleting acetyl-CoA and inhibiting α-ketoglutarate dehydro-genase, which consequently redirects carbon flow through the glyoxylate shunt and impairs downstream processes responsible for cell death.
Figure 6. TCA Cycle Activity Underlies Fumarate- and Glyoxylate-Stimulated Changes in Tobramycin Susceptibility.
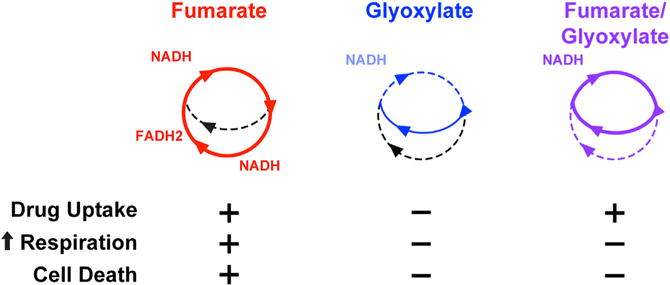
Schematic depicting TCA cycle utilization in cells treated with fumarate (red), glyoxylate (blue), or their combination (purple). The table below summarizes phenotypic data from this study measuring drug uptake, elevated respiration, and cell death (+, present; −, absent). Drug uptake is necessary but not sufficient to kill P. aeruginosa.
TCA Cycle Perturbations Control Tobramycin Susceptibility in Clinically Relevant Models
In CF patientsunder chronic antibiotic management, P.aeruginosa frequently manifests as persister cells (Mulcahy et al., 2010) or in biofilms (Singh et al., 2000). Because TCA cycle manipulations by fumarate and glyoxylate capably altered tobramycin sensitivity in stationary-phase cells, we sought to determine if such perturbations would also affect persister cells or biofilm-associated cells. We isolated P. aeruginosa persister cells by treating stationary-phase cells with 5 mg/Lciprofloxacin and then exposing the surviving fraction to 40 mg/L tobramycin in combination with 15 mM fumarate, 30 mM glyoxylate, or a no-carbon-source control (Figure 7A). We found that, like stationary-phase cells, persister cells exhibited significant sensitization by fumarate, while glyoxylate exerted a non-significant increase in survival. To determine if these effects were specific to fumarate and glyoxylate, we counter-screened the various carbon sources against tobramycin in persister cells and found their potentiation profiles (Figure S6) to be like those of stationary-phase cells (Figure 1C).
Figure 7. Fumarate and Glyoxylate Manipulate Tobramycin Sensitivity in Clinically Relevant Models of P. aeruginosa Infection.
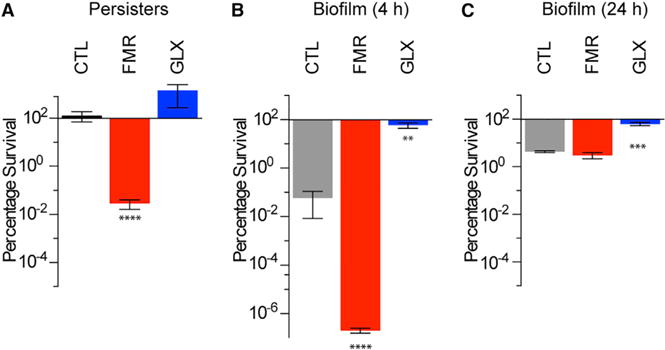
(A) Survival of ciprofloxacin persister cells following 4 hr treatment with 40 mg/L tobramycin and supplementation with no carbon metabolite (CTL), 15 mM fumarate (FMR), or 30 mM glyoxylate (GLX). (B and C) Survival of young (4 hr) (B) and mature (24 hr) (C) colony-forming biofilms treated for 24 hr on LB agar with 8 mg/L tobramycin and supplementation with no carbon metabolite (CTL), 15 mM fumarate (FMR), or 30 mM glyoxylate (GLX).
Values in all bar graphs depict the mean ± SEM with significance reported as FDR-corrected p values in comparison with untreated control (CTL): **p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.0001 (n = 3).
We next tested the effects of fumarate and glyoxylate on P. aeruginosa biofilms. We treated both young (4 hr) and mature (24 hr) biofilms grown on Luria Broth (LB) agar (Walters et al., 2003) for 24 hr with 8 mg/L tobramycin supplemented with 15 mM fumarate, 30 mM glyoxylate, or a no-carbon-source control. While fumarate strongly potentiated tobramycin sensitivity in young biofilms (Figure 7B), it had no effect on mature biofilms (Figure 7C). In contrast, glyoxylate significantly protected both young and mature biofilms against tobramycin treatment. The lack of fumarate-mediated sensitization in mature biofilms is likely explained by spatially restricted diffusion of oxygen, tobramycin, and fumarate, as we did find striking fumarate-mediated potentiation of mature biofilms grown on M9 agar, which are smaller and spatially less complex than those grown on LB agar (Figure S7). Collectively, these results suggest that TCA cycle perturbations may be efficacious for eradicating P. aeruginosa in clinical settings.
DISCUSSION
Here, we investigated the metabolic basis for tobramycin susceptibility in the clinically important pathogen P. aeruginosa. We found that TCA cycle activity determines tobramycin sensitivity in conjunction with cellular respiration, which is required for both drug uptake and downstream lethality. This supports prior studies, which noted that drug uptake requires an active ETC (Bryan and Kwan, 1983; Taber et al., 1987) and that amino-glycoside lethality is associated with increased metabolism (Allison et al., 2011; Kohanski et al., 2008). We propose a model in which some metabolites, such as fumarate, potentiate tobramycin lethality by directly fueling the TCA cycle to induce cellular respiration, while other metabolites such as glyoxylate may protect against tobramycin treatment by shunting flux out of the TCA cycle through carbon redirection and acetyl-CoA depletion (Figures 4E and 6). This is supported by the suppression of fumarate-induced potentiation by the TCA cycle inhibitors malonate and furfural (Figure 4D).
Fumarate as a Potential Antibiotic Adjuvant
Antibiotic development is daunting (Cooper and Shlaes, 2011) and has led to interest in enhancing existing antibiotics with adjuvant molecules (Allison et al., 2011; Barraud et al., 2013; Brynildsen et al., 2013; Hentzer et al., 2003; Lebeaux et al., 2014; Morones-Ramirez et al., 2013). In addition to bolstering antibiotic efficacy, an effective adjuvant must be safe for the patient and not stimulate virulence. Fumarate appears to be a potentially appealing tobramycin adjuvant for treating Pseudomonas pulmonary infections as it is already approved by the US Food and Drug Administration for treating asthma (e.g., in inhalers with β-adrenergic agonists such as formoterol), removing some regulatory hurdles in the drug development pipeline. Our work suggests that the potentiating effect is likely to be specific to amino-glycosides, as fumarate did not potentiate either the β-lactam ceftazidime or the quinolone ciprofloxacin in our experiments (Figure S1A). P. aeruginosa infections associated with CF are challenging due to persistence (Mulcahy et al., 2010), and inhaled tobramycin is a cornerstone of antimicrobial therapy (McKeage, 2013). Co-administration of fumarate with tobramycin could potentially increase the success rate of eradication and significantly improve pulmonary function (Gibson et al., 2003b; Ramsey et al., 1999).
Prior studies have pursued other strategies to potentiate aminoglycosides for treating P. aeruginosa. In particular, mannitol (Barraud et al., 2013) and arginine (Lebeaux et al., 2014) have been shown to resensitize P. aeruginosa biofilms to aminoglycosides; however, in the case of mannitol, most of the effect seemed to be due to osmotic effects, and arginine required very high aminoglycoside concentrations. In contrast, fumarate appears to potentiate tobramycin in the physiologically relevant concentration ranges achievable by intravenous (Vandenbussche and Homnick, 2012) or inhaled administration (Ruddy et al., 2013). Finally, although the present work primarily focuses on fumarate, we anticipate that other lower TCA cycle and lower glycolysis carbon sources could also have therapeutic value as potential adjuvants.
Glyoxylate as an Inducer of Antibiotic Tolerance
Our data also reveal glyoxylate as a biochemical inducer of tolerance to aminoglycoside in both metabolically active cells and quiescent cells, through its suppression of the TCA cycle activity and cellular respiration. While phenotypic tolerance is generally thought to be transcriptionally mediated by induction of bacterial stress responses (Maisonneuve et al., 2011; Nguyen et al., 2011; Verstraeten et al., 2015), our present study indicates that tolerance can also be conferred on a purely post-translational level. Work in Pseudomonas fluorescens has revealed that oxidative stress alters carbon flux in the TCA cycle from isocitrate dehydrogenase to isocitrate lyase to produce glyoxylate, which is transformed into oxalate with concomitant production of NADPH as a reduced electron carrier (Singh et al., 2009). Consequently, P. fluorescens adapts its TCA cycle to minimize production of reduced electron carriers feeding the ETC, which would further expose the bacterium to reactive oxygen species (Mailloux et al., 2011; Singh et al., 2009). Similarly, the coincident conversion of malate to pyruvate could produce NADPH to additionally combat the oxidative stress resulting from metabolic activation (Mailloux et al., 2011). Such scenarios would tip the relative balance of electron carriers toward NADPH in protecting cells against oxidative stress. These observations in P. fluorescens support our overall model whereby metabolic activity downstream of drug uptake is critical for cell fate.
Our metabolite profiling of glyoxylate-treated cells highlights the role of glyoxylate-mediated shunting of the lower TCA cycle, bypassing the production of two reducing equivalents (NADH and FADH2) and accounting for the observed decrease in cellular respiration (Figures 2B and 5A). This would also explain the dominant inhibition of glyoxylate over fumarate. Fumarate, when shunted by glyoxylate, may produce substrates for NADH production by malate dehydrogenase that enable sufficient PMF buildup and drug uptake. However, decreased enzymatic activity in the lower part of the TCA cycle causes a drop in NADH and FADH2 production rate, decreasing downstream respiration. Consequently, energy production is insufficient to sustain processes leading to death (Figure 6). Although genetic deletion of the glyoxylate shunt was recently reported to protect against aminoglycoside lethality (Ahn et al., 2016), those strains exhibited a transcriptional switch toward anaerobic respiration and coincident decrease in oxygen consumption. Those findings complement and support our principal findings here that both TCA cycle activity and downstream respiration are important for aminoglycoside lethality and support our prior observations that antibiotic efficacy is associated with cellular respiration (Lobritz et al., 2015). Interestingly, some clinical isolates from urinary tract infections and the CF lung exhibit upregulation of the glyoxylate shunt (Berger et al., 2014; Hoboth et al., 2009; Son et al., 2007), suggesting that the glyoxylate shunt may be a clinically important in vivo tolerance mechanism for managing antibiotic stress; the glyoxylate shunt may warrant further investigation as a therapeutic target.
Bacterial Metabolism and Antibiotic Efficacy
Here, we demonstrate how central carbon metabolites may differentially potentiate or protect against tobramycin lethality in P. aeruginosa by manipulating drug uptake and downstream death processes through their actions on the TCA cycle and cellular respiration. We report that tobramycin may be imported without eliciting cell death (Figure 5), which supports previous work from our lab demonstrating that membrane damage due to the accumulation of mistranslated proteins induced by aminoglycosides may lead to a metabolic response detrimental to the cell (Kohanski et al., 2008). In this model, an increase in translational activity, as supported by our microarray data, would act as a catalyst, further increasing the metabolic response. It is plausible that glyoxylate, by shunting of the lower part of the TCA cycle and suppressing respiration, may additionally undermine this metabolic response. This could contribute to glyoxylate-mediated protection following delayed supplementation to fumarate treatment (Figure 5C), when the PMF responsible for uptake has already been established. In summary, our work here supports the growing body of evidence that altered metabolism downstream of the primary antibiotic-target interaction critically influences the overall susceptibility phenotype (Dwyer et al., 2015).
EXPERIMENTAL PROCEDURES
Reagents
Antibiotics, most carbon sources, CCCP, NaN3, malonate, furfural, DMSO, K2CO3, and N,N-dimethylformamide were purchased from Sigma-Aldrich. Arabinose and gluconic acid potassium were purchased from Acros Organics. D-Glucose, disodium citrate, mannitol, LB (Miller formulation), and PBS were purchased from Fisher Scientific. M9 minimal medium (parts A and B, containing 7 g/L K2HPO4, 3 g/L KH2HPO4, 1 g/L (NH4)2SO4, 0.1 g/L MgSO4, 0.588 g/L sodium citrate) was purchased from MP Biomedicals. DiBAC4(3) and Texas red sulfonyl chloride were purchased from Life Technologies.
Strains and Culture Conditions
For all experiments, the P. aeruginosa strain used in this study was PAO1 Ochsner (S.M.M. lab). Unless specified otherwise, all cells were cultured at 37°C, 60% humidity, with 300 rpm shaking in their respective media. Overnight PAO1 cultures were grown by inoculating cells from frozen stock into fresh LB. Stationary-phase cells were prepared by growing cells in LB to OD600 ~0.25–0.3 and then re-inoculating these cultures at 1:1,000 dilution into 250 mL flasks and incubating for 16 hr, at which point maximal carrier capacity is reached (stationary phase OD600 ~1.8, density ~5 × 109 colony-forming units [CFU]/mL). Exponential-phase cells were obtained by diluting overnight cells 1:50,000 in fresh LB and growing until OD600 ~0.07–0.1, at which point carbon sources and antibiotics were added. For stationary-phase cells, carbon sources and antibiotics were added to the overnight medium. Persister cells were prepared by treating 25 mL stationary-phase cells for 4 hr with 5 mg/L ciprofloxacin in the spent medium. In all cases, cells grown in LB were washed in PBS and then resuspended in the indicated culture media for each experiment.
Exponential-Phase, Stationary-Phase, Persister Cell Survival
Exponential-phase and stationary-phase cell survival against antibiotics was quantified by introducing tobramycin titrated at different concentrations into LB cultures and incubating for 4 hr. Cells were then washed in PBS and CFU/mL enumerated by 1:10 serial dilution in PBS and plating onto LB agar with 72 hr incubation. For experiments with ceftazidime and ciprofloxacin, stationary-phase cells were treated with 120 mg/L ceftazidime or 5 mg/L ciprofloxacin. Percentage survival was calculated by normalizing to the pretreatment CFU/mL. Experiments were performed in triplicate. Statistical significance between treatment groups was determined by pairwise t tests between each condition and its corresponding control, with multiple-comparison false-discovery rate (FDR) corrections.
Carbon Source Screening
Carbon source screening in stationary-phase and persister cells was performed as previously described (Allison et al., 2011). Stationary-phase and persister cells were washed in PBS and then resuspended in M9 simultaneously containing 40 mg/L tobramycin with either no carbon supplementation or the addition of the carbon source normalized to achieve a total carbon concentration of 60 mM as described previously (Allison et al., 2011), yielding 10 mM glucose, 10 mM mannitol, 10 mM fructose, 20 mM propionate, 30 mM acetate, 15 mM fumarate, 15 mM succinate, 15 mM α-ketoglutarate, 12 mM oxaloacetate, 20 mM pyruvate, 10 mM gluconate, 12 mM arabinose, and 12 mM ribose. After 4 hr incubation, 100 μL was sampled for CFU enumeration. Experiments were performed in triplicate. Statistical significance between treatment groups was determined by pairwise t tests between each condition and its corresponding control, with multiple-comparison FDR corrections.
Proton Motive Force
In experiments using respiration inhibitors, samples were pre-treated for 5 min with 50 μM CCCP or 0.1% NaN3, before adding fumarate, glyoxylate, and/or tobramycin. DiBAC4(3) quantification was performed as previously described (Andres et al., 2005). DiBAC4(3) was resuspended in DMSO to form a 1 mM stock and then diluted to a final concentration of 1 μM. Cells grown to stationary phase as previously described were treated in M9 minimal medium for 25 min with various concentrations of fumarate and/or glyoxylate and CCCP as described above. DiBAC4(3) was added for an additional 10 min and cells were analyzed on a BD Cytoflex S flow cytometer for fluorescein isothiocyanate (FITC)-A fluorescence. Experiments were performed in triplicate.
Tobramycin-Texas Red Uptake
Tobramycin was conjugated to Texas red by adapting a previously described protocol for gentamicin (Sandoval et al., 1998). One milligram of Texas red sulfonyl chloride was resuspended in 50 μL of anhydrous N,N-dimethylformamide on ice. The solution was added slowly to 2.3 mL of 100 mM K2CO3 at pH 8.5, with or without 10 mg/mL tobramycin, on ice. To quantify tobramycin-Texas red uptake, stationary-phase cells were induced by adding carbon sources and incubating for 30 min. Concentrated tobramycin-Texas red was then added to achieve a working concentration of 40 mg/L and samples were incubated for 15 or 45 min as indicated. Cells were prepared for flow cytometry by washing 100 μL of each sample in 1 mL PBS and then resuspending in 1 mL 2% paraformaldehyde. Two hundred microliters of the paraformaldehyde-fixed samples was then diluted into 800 μL of PBS in flow tubes. Samples were analyzed on a BD FACS Aria II flow cytometer for mCherry fluorescence or a Cytoflex 3000 for ECD fluorescence with 30,000 events. Histograms are representative of triplicate experiments.
Bacterial Respiration
Bacterial respiration, estimated by the OCR, was quantified as previously described (Dwyer et al., 2014) using the Seahorse XFe96 extracellular flux analyzer (Agilent Technologies). Briefly, cells were grown to stationary phase, washed in PBS, and resuspended in M9. Cells were diluted 1:100 and then adhered to poly-D-lysine-coated XFe96 microplates and OCR was measured. Basal OCR was measured for 12 min prior to fumarate, glyoxylate, and/or tobramycin injection to verify uniform cell adhesion. OCR measurements were acquired at 6 min intervals. For PMF blockade experiments, CCCP and NaN3 were added 12 min after fumarate.
Extracellular Fumarate Quantification
Stationary-phase cells were resuspended in M9 with or without 15 mM fumarate. For each time point, samples were collected and centrifuged at high speed, and the supernatant was passed through a 0.2 μm cellulose acetate filter. The filtered supernatants were then quantified using a fumarate assay kit (Sigma-Aldrich). Experiments were performed in triplicate.
Biofilms
Colony biofilms were grown as previously described (Walters et al., 2003). Briefly, ~5 × 105 CFU/mL from an overnight culture was inoculated onto an autoclave-sterilized 0.22 μm polycarbonate filter (GE Osmotics) and placed on 1.5% agar LB or M9 plates and incubated for 24 hr at 37°C. Membranes were then transferred to plates containing M9 ± tobramycin, ± fumarate or glyoxylate using sterilized forceps and incubated for another 24 hr at 37°C. Viable cells were enumerated by resuspending the biofilm biomass in PBS with vigorous vortexing and serial dilution for CFU enumeration. Experiments were performed in triplicate and statistical significance between treatment groups was determined by sets of pairwise t tests between every condition and the control, with multiple-comparison FDR corrections.
Microarray Analysis
Stationary-phase cells were washed in PBS and resuspended in M9 with no carbon supplementation, 15 mM fumarate, or 30 mM glyoxylate, or their combination. Samples for RNA extraction were collected following 1 hr incubation. Total RNA was obtained using RNAprotect (QIAGEN), the RNeasy Protect Bacteria Mini Kit (QIAGEN), and Turbo DNA-free (Life Technologies) DNase treatment, according to the respective manufacturer’s instructions. cDNA preparation and hybridization to Affymetrix GeneChip P. aeruginosa microarrays were performed as previously described (Dwyer et al., 2007). CEL files for the resulting expression profiles were background adjusted and normalized using robust multiarray averaging (Bolstad et al., 2003). Statistical significance was computed using Welch’s t test. Triplicate measurements from treated PAO1 samples were compared with the triplicate untreated samples. For each set of comparisons, p values were corrected for FDR (Storey and Tibshirani, 2003). Genes with FDR-corrected p values ≤0.05 were deemed statistically significant. Microarray data collected in this study are available for download on the Gene Expression Omnibus (GEO: GSE90620).
Metabolite Profiling
Stationary-phase cells were washed in PBS and resuspended in M9 with no carbon supplementation, 15 mM fumarate, or 30 mM glyoxylate. After 1 hr incubation, cells were pelleted (5 min at 1,400 × g, 4°C), washed with cold PBS, and snap frozen in liquid nitrogen. Quintuplicate samples were collected and sent for analysis by Metabolon (Shakoury-Elizeh et al., 2010). Relative concentrations for each metabolite were normalized by Bradford protein concentration and scaled to have a median equal to 1. Metabolites detected in fewer than 50% of the total samples (i.e., undetected in ≥8/15 samples) were excluded from analysis. The remaining missing data points were imputed using the lowest detected value across all samples for that metabolite. To quantify significant changes in metabolite abundance between conditions, Welch’s two-sample t test was performed on log2-transformed relative concentration measurements; the MATLAB mafdr function was used to correct for multiple hypothesis testing using the Storey method (Storey and Tibshirani, 2003). Metabolites with a p value ≤0.05 and q value ≤0.1 were deemed statistically significant. All data processing and analyses were done in MATLAB R2014b (MathWorks).
Supplementary Material
SIGNIFICANCE.
Phenotypic tolerance to antibiotic treatment contributes to the growing deficit in effective antimicrobial therapies for chronic infections. Here, we have taken a systems approach, integrating chemical screening, microarray profiling, metabolite profiling, and biochemical measurements to investigate the metabolic mechanisms underlying aminoglycoside susceptibility in Pseudomonas aeruginosa, an important cystic fibrosis pathogen. By screening various central carbon metabolites against the aminoglycoside tobramycin, we discover that lower tricarboxylic acid (TCA) cycle metabolites such as fumarate can potentiate tobramycin lethality in metabolically dormant and phenotypically tolerant stationary-phase cells. This occurs through the potentiation of TCA cycle activity, which increases downstream cellular respiration and proton motive force. Induction of these metabolic processes enhances drug uptake and downstream lethality. In contrast, upper TCA cycle metabolites such as glyoxylate elicit a dominant-negative and protective effect against tobramycin lethality by diverting carbon flux away from the TCA cycle, collapsing cellular respiration, and thereby inhibiting both uptake and lethality. Using biochemical inhibitors of the TCA cycle and electron transport chain, we discover that drug uptake and downstream killing are two separate processes in aminoglycoside lethality and that both TCA cycle activity and cellular respiration are required for downstream killing, even in the presence of imported drug. Collectively, these data identify lower TCA cycle stimulation as a potential strategy for potentiating aminoglycoside therapeutics.
Highlights.
Central carbon perturbations alter aminoglycoside susceptibility in P. aeruginosa
TCA cycle activation of respiration and PMF overcomes aminoglycoside tolerance
Shunting of the TCA cycle biochemically protects against aminoglycoside lethality
Aminoglycoside lethality requires both drug uptake and downstream TCA cycle activity
Acknowledgments
We wish to thank Caleb Bashor for insightful comments. This work was supported by a generous gift from Anita and Josh Bekenstein and by the Societe Academique Vaudoise, the Freiwillige Akademische Gesellschaft Basel, Defense Threat Reduction Agency grant HDTRA1-15-1-0051, NIH K99GM118907, the Broad Institute of MIT and Harvard, and the Wyss Institute of Biologically Inspired Engineering, Harvard University. S.M., S.M.M., and J.J.C. are co-inventors on a patent related to the potential adjuvant therapies here discussed. S.M.M. is alsoa medical director at Vertex Pharmaceuticals. S.M.M. reports receiving fees for serving on advisory boards from Vertex Pharmaceuticals; consulting fees from EnBiotix, MCIC Vermont, Kala Pharmaceuticals, Pfizer, and Cowen Group; and grant support from Gilead Sciences, Novartis, Rempex Pharmaceuticals, Kala Pharmaceuticals, Vertex Pharmaceuticals, and Savara Pharmaceuticals. J.J.C. is scientific co-founder and SAB chair of EnBiotix, which is an antibiotics startup company that has licensed the patent.
Footnotes
AUTHOR CONTRIBUTIONS
Conceptualization, S.M., J.H.Y., A.G., S.M.M., and J.J.C.; Methodology, S.M., J.H.Y., A.G., C.B.M.P., S.M.M., and J.J.C; Investigation, S.M., J.H.Y., C.B.M.P, J.P., P.B., and S.H.K.; Writing – Original Draft, S.M. and A.G.; Writing – Review & Editing, S.M., A.G., J.H.Y., M.A.L., C.B.M.P., and J.J.C; Funding Acquisition, S.M. and J.J.C.; Resources, J.J.C. and S.M.M.; Supervision, J.J.C. and S.M.M. C.B.M.P. and J.H.Y. contributed equally to this work.
SUPPLEMENTAL INFORMATION
Supplemental Information includes seven figures and can be found with this article online at http://dx.doi.org/10.1016/j.chembiol.2016.12.015.
References
- Adinolfi A, Moratti R, Olezza S, Ruffo A. Control of the citric acid cycle by glyoxylate. The mechanism of inhibition of oxoglutarate dehydrogenase, isocitrate dehydrogenase and aconitate hydratase. Biochem J. 1969;114:513–518. doi: 10.1042/bj1140513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn S, Jung J, Jang IA, Madsen EL, Park W. Role of glyoxylate shunt in oxidative stress response. J Biol Chem. 2016;291:11928–11938. doi: 10.1074/jbc.M115.708149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allison KR, Brynildsen MP, Collins JJ. Metabolite-enabled eradication of bacterial persisters by aminoglycosides. Nature. 2011;473:216–220. doi: 10.1038/nature10069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andres MT, Viejo-Diaz M, Perez F, Fierro JF. Antibiotic tolerance induced by lactoferrin in clinical Pseudomonas aeruginosa isolates from cystic fibrosis patients. Antimicrob Agents Chemother. 2005;49:1613–1616. doi: 10.1128/AAC.49.4.1613-1616.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barraud N, Buson A, Jarolimek W, Rice SA. Mannitol enhances antibiotic sensitivity of persister bacteria in Pseudomonas aeruginosa biofilms. PLoS One. 2013;8:e84220. doi: 10.1371/journal.pone.0084220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger A, Dohnt K, Tielen P, Jahn D, Becker J, Wittmann C. Robustness and plasticity of metabolic pathway flux among uropathogenic isolates of Pseudomonas aeruginosa. PLoS One. 2014;9:e88368. doi: 10.1371/journal.pone.0088368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bisswanger H. Substrate specificity of the pyruvate dehydrogenase complex from Escherichia coli. J Biol Chem. 1981;256:815–822. [PubMed] [Google Scholar]
- Blair JM, Webber MA, Baylay AJ, Ogbolu DO, Piddock LJ. Molecular mechanisms of antibiotic resistance. Nat Rev Microbiol. 2015;13:42–51. doi: 10.1038/nrmicro3380. [DOI] [PubMed] [Google Scholar]
- Bolstad BM, Irizarry RA, Astrand M, Speed TP. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics. 2003;19:185–193. doi: 10.1093/bioinformatics/19.2.185. [DOI] [PubMed] [Google Scholar]
- Boucher RC. Airway surface dehydration in cystic fibrosis: pathogenesis and therapy. Annu Rev Med. 2007;58:157–170. doi: 10.1146/annurev.med.58.071905.105316. [DOI] [PubMed] [Google Scholar]
- Brauner A, Fridman O, Gefen O, Balaban NQ. Distinguishing between resistance, tolerance and persistence to antibiotic treatment. Nat Rev Microbiol. 2016;14:320–330. doi: 10.1038/nrmicro.2016.34. [DOI] [PubMed] [Google Scholar]
- Bryan LE, Kwan S. Roles of ribosomal binding, membrane potential, and electron transport in bacterial uptake of streptomycin and gentamicin. Antimicrob Agents Chemother. 1983;23:835–845. doi: 10.1128/aac.23.6.835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brynildsen MP, Winkler JA, Spina CS, MacDonald IC, Collins JJ. Potentiating antibacterial activity by predictably enhancing endogenous microbial ROS production. Nat Biotechnol. 2013;31:160–165. doi: 10.1038/nbt.2458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bye MR, Ewig JM, Quittell LM. Cystic fibrosis. Lung. 1994;172:251–270. doi: 10.1007/BF00164308. [DOI] [PubMed] [Google Scholar]
- Caspi R, Altman T, Dreher K, Fulcher CA, Subhraveti P, Keseler IM, Kothari A, Krummenacker M, Latendresse M, Mueller LA, et al. The MetaCyc database of metabolic pathways and enzymes and the BioCyc collection of pathway/genome databases. Nucleic Acids Res. 2012;40:D742–D753. doi: 10.1093/nar/gkr1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambers D, Scott F, Bangur R, Davies R, Lim A, Walters S, Smith G, Pitt T, Stableforth D, Honeybourne D. Factors associated with infection by Pseudomonas aeruginosa in adult cystic fibrosis. Eur Respir J. 2005;26:651–656. doi: 10.1183/09031936.05.00126704. [DOI] [PubMed] [Google Scholar]
- Conlon BP, Nakayasu ES, Fleck LE, LaFleur MD, Isabella VM, Coleman K, Leonard SN, Smith RD, Adkins JN, Lewis K. Activated ClpP kills persisters and eradicates a chronic biofilm infection. Nature. 2013;503:365–370. doi: 10.1038/nature12790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper MA, Shlaes D. Fix the antibiotics pipeline. Nature. 2011;472:32. doi: 10.1038/472032a. [DOI] [PubMed] [Google Scholar]
- Dwyer DJ, Kohanski MA, Hayete B, Collins JJ. Gyrase inhibitors induce an oxidative damage cellular death pathway in Escherichia coli. Mol Syst Biol. 2007;3:91. doi: 10.1038/msb4100135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dwyer DJ, Belenky PA, Yang JH, MacDonald IC, Martell JD, Takahashi N, Chan CT, Lobritz MA, Braff D, Schwarz EG, et al. Antibiotics induce redox-related physiological alterations as part of their lethality. Proc Natl Acad Sci USA. 2014;111:E2100–2109. doi: 10.1073/pnas.1401876111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dwyer DJ, Collins JJ, Walker GC. Unraveling the physiological complexities of antibiotic lethality. Annu Rev Pharmacol Toxicol. 2015;55:313–332. doi: 10.1146/annurev-pharmtox-010814-124712. [DOI] [PubMed] [Google Scholar]
- Folkesson A, Jelsbak L, Yang L, Johansen HK, Ciofu O, Hoiby N, Molin S. Adaptation of Pseudomonas aeruginosa to the cystic fibrosis airway: an evolutionary perspective. Nature reviews. Microbiology. 2012;10:841–851. doi: 10.1038/nrmicro2907. [DOI] [PubMed] [Google Scholar]
- Gibson RL, Burns JL, Ramsey BW. Pathophysiology and management of pulmonary infections in cystic fibrosis. Am J Respir Crit Care Med. 2003a;168:918–951. doi: 10.1164/rccm.200304-505SO. [DOI] [PubMed] [Google Scholar]
- Gibson RL, Emerson J, McNamara S, Burns JL, Rosenfeld M, Yunker A, Hamblett N, Accurso F, Dovey M, Hiatt P, et al. Significant microbiological effect of inhaled tobramycin in young children with cystic fibrosis. Am J Respir Crit Care Med. 2003b;167:841–849. doi: 10.1164/rccm.200208-855OC. [DOI] [PubMed] [Google Scholar]
- Hansen CR, Pressler T, Hoiby N. Early aggressive eradication therapy for intermittent Pseudomonas aeruginosa airway colonization in cystic fibrosis patients: 15 years experience. J Cyst Fibros. 2008;7:523–530. doi: 10.1016/j.jcf.2008.06.009. [DOI] [PubMed] [Google Scholar]
- Hentzer M, Wu H, Andersen JB, Riedel K, Rasmussen TB, Bagge N, Kumar N, Schembri MA, Song Z, Kristoffersen P, et al. Attenuation of Pseudomonas aeruginosa virulence by quorum sensing inhibitors. EMBO J. 2003;22:3803–3815. doi: 10.1093/emboj/cdg366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoboth C, Hoffmann R, Eichner A, Henke C, Schmoldt S, Imhof A, Heesemann J, Hogardt M. Dynamics of adaptive microevolution of hypermutable Pseudomonas aeruginosa during chronic pulmonary infection in patients with cystic fibrosis. J Infect Dis. 2009;200:118–130. doi: 10.1086/599360. [DOI] [PubMed] [Google Scholar]
- Kashket ER. Effects of aerobiosis and nitrogen source on the proton motive force in growing Escherichia coli and Klebsiella pneumoniae cells. J Bacteriol. 1981;146:377–384. doi: 10.1128/jb.146.1.377-384.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaushik SK, Stolhandske K, Shindell O, Smyth HD, Gordon VD. Tobramycin and bicarbonate synergise to kill planktonic Pseudomonas aeruginosa, but antigonise to promote biofilm survival. npj Biofilms Microbiomes. 2016;2:16006. doi: 10.1038/npjbiofilms.2016.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohanski MA, Dwyer DJ, Wierzbowski J, Cottarel G, Collins JJ. Mistranslation of membrane proteins and two-component system activation trigger antibiotic-mediated cell death. Cell. 2008;135:679–690. doi: 10.1016/j.cell.2008.09.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebeaux D, Chauhan A, Letoffe S, Fischer F, de Reuse H, Beloin C, Ghigo JM. pH-mediated potentiation of aminoglycosides kills bacterial persisters and eradicates in vivo biofilms. J Infect Dis. 2014;210:1357–1366. doi: 10.1093/infdis/jiu286. [DOI] [PubMed] [Google Scholar]
- Lobritz MA, Belenky P, Porter CB, Gutierrez A, Yang JH, Schwarz EG, Dwyer DJ, Khalil AS, Collins JJ. Antibiotic efficacy is linked to bacterial cellular respiration. Proc Natl Acad Sci USA. 2015;112:8173–8180. doi: 10.1073/pnas.1509743112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mailloux RJ, Lemire J, Appanna VD. Metabolic networks to combat oxidative stress in Pseudomonas fluorescens. Antonie Van Leeuwenhoek. 2011;99:433–442. doi: 10.1007/s10482-010-9538-x. [DOI] [PubMed] [Google Scholar]
- Maisonneuve E, Shakespeare LJ, Jorgensen MG, Gerdes K. Bacterial persistence by RNA endonucleases. Proc Natl Acad Sci USA. 2011;108:13206–13211. doi: 10.1073/pnas.1100186108. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Marshall BC, Hazle L. Cystic Fibrosis Foundation Patient Registry: 2011 Annual Data Report. Cystic Fibrosis Foundation; 2011. [Google Scholar]
- McKeage K. Tobramycin inhalation powder: a review of its use in the treatment of chronic Pseudomonas aeruginosa infection in patients with cystic fibrosis. Drugs. 2013;73:1815–1827. doi: 10.1007/s40265-013-0141-0. [DOI] [PubMed] [Google Scholar]
- McMahon MA, Xu J, Moore JE, Blair IS, McDowell DA. Environmental stress and antibiotic resistance in food-related pathogens. Appl Environ Microbiol. 2007;73:211–217. doi: 10.1128/AEM.00578-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Modig T, Liden G, Taherzadeh MJ. Inhibition effects of furfural on alcohol dehydrogenase, aldehyde dehydrogenase and pyruvate dehydrogenase. Biochem J. 2002;363:769–776. doi: 10.1042/0264-6021:3630769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morones-Ramirez JR, Winkler JA, Spina CS, Collins JJ. Silver enhances antibiotic activity against gram-negative bacteria. Sci Transl Med. 2013;5:190ra181. doi: 10.1126/scitranslmed.3006276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulcahy LR, Burns JL, Lory S, Lewis K. Emergence of Pseudomonas aeruginosa strains producing high levels of persister cells in patients with cystic fibrosis. J Bacteriol. 2010;192:6191–6199. doi: 10.1128/JB.01651-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen D, Joshi-Datar A, Lepine F, Bauerle E, Olakanmi O, Beer K, McKay G, Siehnel R, Schafhauser J, Wang Y, et al. Active starvation responses mediate antibiotic tolerance in biofilms and nutrient-limited bacteria. Science. 2011;334:982–986. doi: 10.1126/science.1211037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nimmo HG. Kinetic mechanism of Escherichia coli isocitrate dehydro-genase and its inhibition by glyoxylate and oxaloacetate. Biochem J. 1986;234:317–323. doi: 10.1042/bj2340317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardee AB, Potter VR. Malonate inhibition of oxidations in the Krebs tricarboxylic acid cycle. J Biol Chem. 1949;178:241–250. [PubMed] [Google Scholar]
- Poole K. Bacterial stress responses as determinants of antimicrobial resistance. J Antimicrob Chemother. 2012;67:2069–2089. doi: 10.1093/jac/dks196. [DOI] [PubMed] [Google Scholar]
- Quayle JR, Keech DB, Taylor GA. Carbon assimilation by Pseudomonas oxalaticus (OXI). 4 Metabolism of oxalate in cell-free extracts of the organism grown on oxalate. Biochem J. 1961;78:225–236. doi: 10.1042/bj0780225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramsey BW, Pepe MS, Quan JM, Otto KL, Montgomery AB, Williams-Warren J, Vasiljev KM, Borowitz D, Bowman CM, Marshall BC, et al. Intermittent administration of inhaled tobramycin in patients with cystic fibrosis. Cystic Fibrosis Inhaled Tobramycin Study Group. N Engl J Med. 1999;340:23–30. doi: 10.1056/NEJM199901073400104. [DOI] [PubMed] [Google Scholar]
- Rezaeinejad S, Ivanov V. Heterogeneity of Escherichia coli population by respiratory activity and membrane potential of cells during growth and long-term starvation. Microbiol Res. 2011;166:129–135. doi: 10.1016/j.micres.2010.01.007. [DOI] [PubMed] [Google Scholar]
- Ruddy J, Emerson J, Moss R, Genatossio A, McNamara S, Burns JL, Anderson G, Rosenfeld M. Sputum tobramycin concentrations in cystic fibrosis patients with repeated administration of inhaled tobramycin. J Aerosol Med Pulm Drug Deliv. 2013;26:69–75. doi: 10.1089/jamp.2011.0942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandoval R, Leiser J, Molitoris BA. Aminoglycoside antibiotics traffic to the Golgi complex in LLC-PK1 cells. J Am Soc Nephrol. 1998;9:167–174. doi: 10.1681/ASN.V92167. [DOI] [PubMed] [Google Scholar]
- Shakoury-Elizeh M, Protchenko O, Berger A, Cox J, Gable K, Dunn TM, Prinz WA, Bard M, Philpott CC. Metabolic response to iron deficiency in Saccharomyces cerevisiae. J Biol Chem. 2010;285:14823–14833. doi: 10.1074/jbc.M109.091710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh PK, Schaefer AL, Parsek MR, Moninger TO, Welsh MJ, Greenberg EP. Quorum-sensing signals indicate that cystic fibrosis lungs are infected with bacterial biofilms. Nature. 2000;407:762–764. doi: 10.1038/35037627. [DOI] [PubMed] [Google Scholar]
- Singh R, Lemire J, Mailloux RJ, Chenier D, Hamel R, Appanna VD. An ATP and oxalate generating variant tricarboxylic acid cycle counters aluminum toxicity in Pseudomonas fluorescens. PLoS One. 2009;4:e7344. doi: 10.1371/journal.pone.0007344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Son MS, Matthews WJ, Jr, Kang Y, Nguyen DT, Hoang TT. In vivo evidence of Pseudomonas aeruginosa nutrient acquisition and pathogenesis in the lungs of cystic fibrosis patients. Infect Immun. 2007;75:5313–5324. doi: 10.1128/IAI.01807-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storey JD, Tibshirani R. Statistical significance for genomewide studies. Proc Natl Acad Sci USA. 2003;100:9440–9445. doi: 10.1073/pnas.1530509100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taber HW, Mueller JP, Miller PF, Arrow AS. Bacterial uptake of aminoglycoside antibiotics. Microbiol Rev. 1987;51:439–457. doi: 10.1128/mr.51.4.439-457.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandenbussche HL, Homnick DN. Evaluation of serum concentrations achieved with an empiric once-daily tobramycin dosage regimen in children and adults with cystic fibrosis. J Pediatr Pharmacol Ther. 2012;17:67–77. doi: 10.5863/1551-6776-17.1.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verstraeten N, Knapen WJ, Kint CI, Liebens V, Van den Bergh B, Dewachter L, Michiels JE, Fu Q, David CC, Fierro AC, et al. Obg and membrane depolarization are part of a microbial bet-hedging strategy that leads to antibiotic tolerance. Mol Cell. 2015;59:9–21. doi: 10.1016/j.molcel.2015.05.011. [DOI] [PubMed] [Google Scholar]
- Walters MC, 3rd, Roe F, Bugnicourt A, Franklin MJ, Stewart PS. Contributions of antibiotic penetration, oxygen limitation, and low metabolic activity to tolerance of Pseudomonas aeruginosa biofilms to ciprofloxacin and tobramycin. Antimicrob Agents Chemother. 2003;47:317–323. doi: 10.1128/AAC.47.1.317-323.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Jelsbak L, Molin S. Microbial ecology and adaptation in cystic fibrosis airways. Environ Microbiol. 2011;13:1682–1689. doi: 10.1111/j.1462-2920.2011.02459.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.