

Cell–extracellular matrix (ECM) and cell–cell adhesion are interdependent during dorsal closure in the fly. Cell–ECM adhesion is required for normal myosin dynamics and organization of both cell–cell adhesions and actin networks during dorsal closure. Loss of cell–cell adhesion affects cell–ECM adhesion and tissue biomechanics.
Abstract
Tissue morphogenesis relies on the coordinated action of actin networks, cell–cell adhesions, and cell–extracellular matrix (ECM) adhesions. Such coordination can be achieved through cross-talk between cell–cell and cell–ECM adhesions. Drosophila dorsal closure (DC), a morphogenetic process in which an extraembryonic tissue called the amnioserosa contracts and ingresses to close a discontinuity in the dorsal epidermis of the embryo, requires both cell–cell and cell–ECM adhesions. However, whether the functions of these two types of adhesions are coordinated during DC is not known. Here we analyzed possible interdependence between cell–cell and cell–ECM adhesions during DC and its effect on the actomyosin network. We find that loss of cell–ECM adhesion results in aberrant distributions of cadherin-mediated adhesions and actin networks in the amnioserosa and subsequent disruption of myosin recruitment and dynamics. Moreover, loss of cell–cell adhesion caused up-regulation of cell–ECM adhesion, leading to reduced cell deformation and force transmission across amnioserosa cells. Our results show how interdependence between cell–cell and cell–ECM adhesions is important in regulating cell behaviors, force generation, and force transmission critical for tissue morphogenesis.
INTRODUCTION
Throughout development, cells respond to biomechanical cues and exert forces on their neighbors and surrounding environment. In particular, tissue morphogenesis is the product of changes in the biomechanical and morphological properties of cells that are driven by interactions between the actin cytoskeleton, cell–cell adhesions, and cell–extracellular matrix (ECM) adhesions. Precise regulation of the strength and duration of cellular adhesions is therefore a critical component of tissue morphogenesis (Lecuit and Yap, 2015). A growing body of evidence supports the idea that coordination of the interactions between the actin cytoskeleton, cell–cell adhesions, and cell–ECM adhesions is a key regulatory strategy during tissue morphogenesis. Specifically, cooperation or cross-regulation between cell–cell and cell–ECM adhesion has been implicated in multiple tissues and developmental processes (Weber et al., 2011; McMillen and Holley, 2015). Of importance, cross-talk between cell–cell and cell–ECM adhesions orchestrates cell-generated mechanical forces that drive key morphogenetic processes such as cell shape changes and migration (Mui et al., 2016).
Cell–cell adhesion is typically mediated by homophilic interactions between extracellular domains of cadherin receptors (Pokutta and Weis, 2007), whereas cell–ECM adhesion is typically mediated by the binding of the extracellular domains of heterodimeric integrin receptors to ECM ligands (Hynes, 2002). Both cadherins and integrins assemble large intracellular multiprotein complexes via their cytoplasmic domains that carry out diverse functions, such as coupling to the cytoskeleton via adapter proteins and regulating cell behavior through modulation of signaling networks. Because both cadherins and integrins act as anchor points that link the actin cytoskeleton to the cell membrane, they are subject to myosin-generated forces exerted on the actin cytoskeleton and can in turn transduce forces generated in the surrounding environment to the cell interior. There is robust evidence from studies in cell culture for mechanotransduction at cell–ECM adhesions and, more recently, at cell–cell adhesions (Leckband and de Rooij, 2014; Sun et al., 2016). For example, the best-characterized type of integrin-based adhesive structures, focal adhesions, are substantially remodeled in response to internal mechanical cues, such as changes in actomyosin contractility, as well to external mechanical cues, such as substrate stiffness (Geiger et al., 2009). Similarly, cadherin-mediated adhesions are remodeled in response to changes in tension and confer changes on actin network architecture in response to the application of force (Leckband and de Rooij, 2014).
The interplay between cell adhesion complexes and cytoskeletal networks is critical for the precise regulation of changes to cell and tissue architecture during morphogenesis. A diverse array of cellular behaviors are used to achieve changes in tissue morphology, including cell intercalation, apical constriction, and collective migration (Roper, 2015). In epithelia, constriction of the apical domain of the cell is a commonly used cellular program that drives morphogenetic processes such as tissue bending, reduction in tissue surface area, and cell ingression. Pulsatile contraction of actomyosin networks are believed to be one of the main drivers of developmentally programmed incremental apical constriction, a process like ratcheting (Martin et al., 2009; Blanchard et al., 2010). During Drosophila dorsal closure (DC), constriction of apical cell area drives shrinkage of an extraembryonic tissue called the amnioserosa (AS; Solon et al., 2009; Blanchard et al., 2010). Constriction of the AS is additionally driven by cell extrusion (Toyama et al., 2008) and gradual cell volume loss (Saias et al., 2015). The AS initially occupies an eye-shaped hole in the dorsal epidermis of the embryo. As the AS constricts, a supracellular actin cable forms in the leading edge of the lateral epidermis, which may provide an additional tensile force to shrink the dorsal hole (Hutson et al., 2003; Solon et al., 2009). DC is divided into three stages based on cellular behaviors and the rate of tissue contraction (Gorfinkiel et al., 2009). During the early phase, AS cells oscillate in the apical domain but do not constrict, and the tissue area remains constant. At the onset of closure, the slow phase begins; the actin cable forms in the leading edge, and pulsatile apical constriction of AS cells leads to gradual decrease of tissue area. Finally, DC enters the fast phase, which is characterized by more rapid constriction of AS cell area and ends with zippering of the epithelium (Jacinto et al., 2000; Gorfinkiel et al., 2009).
Although the relative contributions of the different force-generating processes driving DC have been debated, it has become increasingly clear that the AS plays the central role, whereas the actin cable in the leading edge is not required for closure per se (Wells et al., 2014; Ducuing and Vincent, 2016; Pasakarnis et al., 2016). For contraction of the AS to occur, forces must be generated within and transmitted across the apical plane of the tissue. Given the importance of cell–cell and cell–ECM adhesions in regulating cellular force generation, it is unsurprising that both cadherins and integrins are required for DC (Narasimha and Brown, 2004; Gorfinkiel and Arias, 2007). However, the exact mechanical roles of cell–cell and cell–ECM adhesions in regulating cell behaviors during DC have not yet been extensively characterized. We recently showed that cell–ECM adhesion localizes to focal adhesion–like structures (FALS) on the basal surface of AS cells and that these FALS provide resistance to both cell deformation and force transmission in the apical plane of the tissue (Goodwin et al., 2016). Consequently genetically altering the amount of cell–ECM adhesion alters tissue biomechanical properties and cell behaviors in the AS, leading to defective DC (Goodwin et al., 2016).
The roles of cadherin-mediated adhesion, integrin-mediated adhesion, and myosin contractility have all been explored in the context of DC, but the relationships between them have not been fully elucidated. Here we uncover functional interactions and phenotypic overlaps between modulation of cell–cell and cell–ECM adhesion during DC. First, we sought to understand the consequences of reduced cell–ECM adhesion for myosin dynamics in AS cells. Using live imaging and quantitative analysis, we find that myosin dynamics are altered in contracting AS cells of embryos lacking integrin. Second, further investigations revealed that localization and stability of cadherin along AS cell membranes are perturbed when cell–ECM adhesion is compromised and that cortical and medial actin is aberrantly distributed. Third, given the effects of reducing cell–ECM adhesion on cadherin-mediated adhesions, we examined the opposite scenario and report that loss of cell–cell adhesion results in increased recruitment of cell–ECM adhesions and defective DC. Cell deformation and force transmission are both inhibited when cell–cell adhesion is compromised, suggesting a role for cross-regulation of cadherin- and integrin-based adhesions in regulating cellular behaviors that are required for tissue morphogenesis.
RESULTS
Loss of cell–ECM adhesion results in abnormal myosin dynamics, cadherin localization, and actin organization
DC is driven by pulsatile constriction of AS cells, which leads to a decrease in overall tissue area. Constriction of AS cells is mediated by the assembly and contraction of actomyosin networks. These pulsatile contractions are coupled to the cell membrane by cadherin-based adhesions, which allow for force transmission between adhering cells. Previous work from our group showed that loss of integrin-based cell–ECM adhesion resulted in increased cell deformation and force transmission in the apical plane of AS cells (Goodwin et al., 2016). To explore further the mechanisms underlying DC phenotypes of flies lacking integrin-based cell-ECM adhesion, we examined in detail whether reduced cell–ECM adhesion could also affect force generation in AS cells. Specifically, we analyzed myosin dynamics in fly embryos homozygous for a null mutation in the gene myospheroid (henceforth referred to as mys −/− embryos) encoding the Drosophila βPS-integrin subunit. To measure myosin dynamics, we tracked the movement of myosin in z-projected images spanning the apical and basal domains of AS cells using fluorescently tagged regulatory myosin light chain encoded by the gene spaghetti squash (sqh-mCherry reporter). To measure changes in cell morphology at the apical side of the cell, we used the E-cadherin–green fluorescent protein (GFP) reporter. From early to slow phases of closure, AS cell area decreased and mean myosin intensity increased in heterozygous mys +/– controls, consistent with stabilization of actomyosin networks and with previous reports (Figure 1, a–d; Blanchard et al., 2010). As actomyosin networks stabilize from early to slow phases, we expect to see reduced speed of myosin flows. Indeed, measurement of the movement of fluorescently tagged myosin using particle image velocimetry (PIV), a technique used to visualize and measure flow, showed lower speed in the slow phase of closure compared with the early phase (Figure 1, a and e).
FIGURE 1:
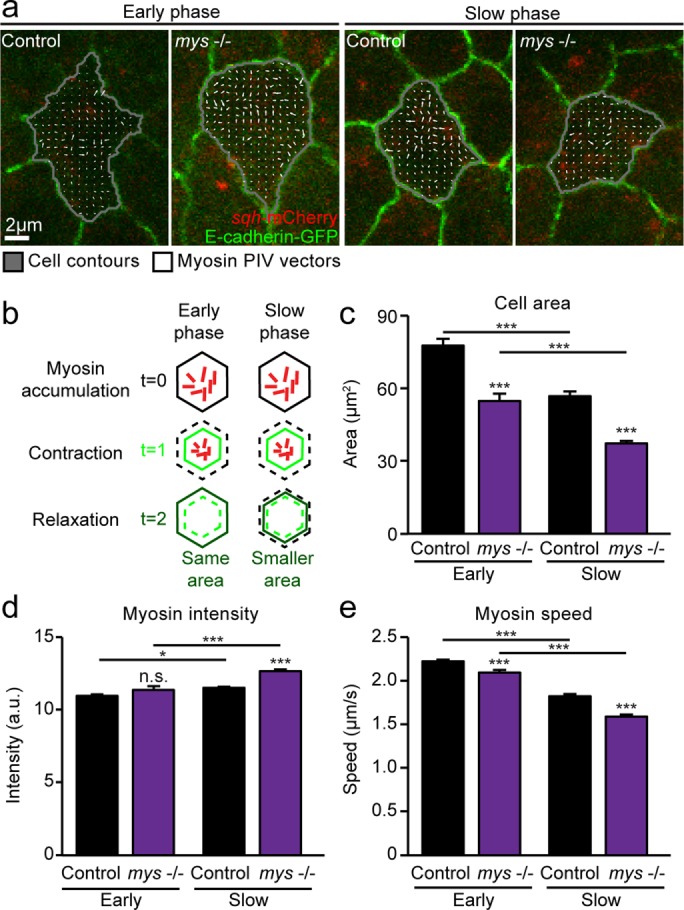
Myosin dynamics in AS cells are perturbed in the absence of cell–ECM adhesion. (a) Images from z-projected time-lapse movies of control and mys −/− embryos expressing E-cadherin–GFP and sqh-mCherry during the early and slow phases of DC, with overlaid cell contours and PIV vectors representing myosin movement over 20 s. Vectors are uniformly scaled across samples. (b) Schematic illustrating cell area oscillations at early and slow phases of closure. Myosin accumulation drives cell contraction, and, as pulses dissipate, the cell relaxes either to the same area (early phase) or to a smaller area (slow phase). (c–e) Mean apical cell area (c), mean myosin intensity (d), and medial myosin speed measured by PIV (e) in control and mys −/− embryos at early and slow phases of closure (two to four embryos, 26–78 cells). Error bars indicate SEM. *p < 0.05, ***p < 0.0001.
We performed similar analysis in mys −/− embryos expressing sqh-mCherry and E-cadherin–GFP. This analysis showed that AS cells in mys −/− embryos had smaller apical areas at both early and slow phases of DC, possibly as a result of abnormal apical constriction and/or changes to cell shape due to reduced adhesion to the ECM (Figure 1, a–c). Moreover, at slow phases, mean myosin intensity was significantly greater than with controls (Figure 1d). Apical constriction and increased myosin intensity are consistent with lower myosin flow speed. Indeed, in mys −/− mutants, we found that mean myosin speed measured by PIV was reduced compared with controls at both early and slow phases (Figure 1, a and e). Taken together, the smaller apical areas, increased mean myosin intensity, and reduced flows suggest that mys −/− cells experience more apical constriction than controls. Overall myosin accumulation and dynamics are altered in the absence of cell–ECM adhesion, leading to smaller apical area and potentially contributing to the previously observed changes to cell behavior and tissue biomechanics in the AS of mys mutants.
In several developmental contexts, perturbation of myosin dynamics has been linked to changes in cell–cell adhesion (Levayer et al., 2011; Levayer and Lecuit, 2013; Duque and Gorfinkiel, 2016). We therefore investigated cadherin-mediated adhesion in mys −/− mutants (Figure 2). Examining the localization of E-cadherin in AS cells of mys −/− mutants using immunohistochemistry revealed an abnormal punctate distribution along the apical membrane, in stark contrast to the relatively uniform distribution seen in controls (Figure 2a). We quantified these differences by measuring E-cadherin staining intensity along the entire cell contour and identifying peaks in which fluorescence exceeded the mean intensity by at least one SD (Figure 2b). In mys −/− embryos, the number of peaks per micrometer along the cell contour (referred to as peak density) was lower than in controls, but mean peak intensity was much greater (Figure 2, c and d). In comparison, the overall mean intensity of E-cadherin along the entire contour was similar between mys −/− mutants and controls (Figure 2e). Taken together, these results suggest that in the absence of cell–ECM adhesion, the same overall levels of E-cadherin reach the membrane, but the protein distribution along cell–cell contacts is irregular. Another factor that may influence the ability of cadherins to anchor the actomyosin network is their dynamics at the plasma membrane. We therefore examined E-cadherin stability at cell–cell junctions using fluorescence recovery after photobleaching (FRAP) experiments. Fluorescence recovery in terms of overall mobile fraction of E-cadherin in AS cells was not drastically different between mys −/− and control embryos (Figure 2, f–h). However, although mobile fraction was not significantly different than with controls, the half-time of recovery was lower in mys −/− embryos. This is consistent with the notion that cadherin-based junctions may be more dynamic in mys −/− mutants.
FIGURE 2:

Cadherin localization and stability is disrupted in mys −/− mutants. (a) Sample z-projected images of control and mys −/− embryos fixed during the early phase of DC and stained for E-cadherin. (b) Quantification of E-cadherin staining intensity along the entire cell contour for sample cells in control and mys −/− embryos. (c) Mean density of E-cadherin intensity peaks along cell contour measured as number of peaks per micrometer for control and mys −/− embryos. (d, e) Mean peak intensity and mean overall E-cadherin intensity along cell contour for control and mys −/− embryos (three embryos, 8–11 cells). (f–h) Mean FRAP curves (f), half-time of fluorescence recovery (g), and mobile fraction (h) for control and mys −/− embryos (10–12 embryos). Error bars indicate SEM. *p < 0.05.
Myosin pulses and flows are intrinsically linked to medial actin networks, and the binding of cadherin to actin can change the distribution of cortical actin. Given that myosin dynamics and cadherin distribution were disrupted in the absence of integrin-mediated adhesion, we sought to determine whether actin networks were also perturbed in the AS of mys −/− embryos by quantifying differences in both cortical and medial actin in mys −/− embryos. Using an approach similar to the one described earlier for measuring E-cadherin intensity, we measured cortical F-actin intensity along cell contours (Figure 3, a and b). We found that, similar to E-cadherin distribution, F-actin intensity was irregularly distributed along cell–cell contacts in mys −/− mutants, as evidenced by lower peak density and higher peak intensity (Figure 3, c and d). Again, mean intensity along the entire cell contour was the same as controls, suggesting that the overall recruitment of F-actin to the membrane was normal, but its distribution was perturbed (Figure 3e). Using z-projected images spanning the apical and basal domains of AS cells, we found that medial actin networks throughout the cell were also affected by disruption of cell–ECM adhesion. F-actin in mys −/− mutants appeared denser and was arranged in large bundles, whereas in controls it was, in general, uniformly spread (Figure 3a). To provide further support for these qualitative observations, we quantified the density and area of bundles, defined as large, bright accumulations of immunofluorescence intensity, and found that they were both significantly increased in mys −/− AS cells (Figure 3, f and g, and Supplemental Figure S1). In addition, mean F-actin intensity across the entire cell was increased compared with controls (Figure 3 h). Overall these results show that actin networks are irregularly organized in the absence of cell–ECM adhesion.
FIGURE 3:

Cortical and medial F-actin organization is perturbed in mys −/− mutants. (a) Sample z-projected images of control and mys −/− embryos fixed during the early phase of DC and stained for F-actin (using rhodamine–phalloidin) and E-cadherin. (b) Quantification of F-actin staining intensity along the entire cell contour for sample cells in control and mys −/− embryos. (c) Mean density of cortical F-actin intensity peaks along cell contour measured as number of peaks per micrometer for control and mys −/− embryos. (d, e) Mean peak intensity and mean overall F-actin intensity along cell contour for control and mys −/− embryos. (f) Mean density of medial actin bundles within the cell measured as bundles per micrometer squared for control and mys −/− embryos. (g) Mean area of actin bundles in control and mys −/− embryos. (h) Mean overall F-actin staining intensity within cells from control and mys −/− embryos (three embryos, 8–11 cells). Error bars indicate SEM. *p < 0.05, **p < 0.001.
Cell–ECM adhesions, AS cell oscillatory behaviors, and force transmission are perturbed when cell–cell adhesion is reduced
Based on these findings and previous work from our group (Goodwin et al., 2016), precise levels of both cell–ECM and cell–cell adhesion appear to be critical in regulating cell oscillatory behavior and force transmission during DC. Loss of integrin-mediated adhesion resulted in irregular distribution of cadherin, including patches of increased concentration of cell–cell adhesion that led to irregular distribution of actin networks. Given that abnormal distributions of E-cadherin were associated with defective closure in mys −/− mutants, we asked whether depletion of cadherin-mediated adhesion would affect DC in ways similar to those observed when cell–ECM adhesion was misregulated. To address this question, we genetically reduced cadherin-mediated adhesion using a null mutation in the gene shotgun (shgR69b), which encodes the Drosophila E-cadherin gene (Figure 4). We found that in shgR69b mutant embryos, DC is severely disrupted and occurs at a slower rate than in wild-type controls (Figure 4, a and b). Of interest, we also found that cell–ECM adhesion is increased and stabilized when cell–cell adhesion was lost. Specifically, when GFP-tagged talin—a cytoplasmic linker protein that binds integrins and actin—was used as a marker to label FALS in shgR69 embryos, a noticeable increase in their area, intensity, and density was observed (Figure 4, c–f). Moreover, tracking FALS in the AS of shgR69b mutant embryos during cell oscillations showed that they moved with lower speed and had a lower effective diffusion constant (Figure 4, g and h). Our results thus far show that the distribution and stability of cadherin-mediated adhesions was disrupted when integrin-based cell–ECM adhesion was compromised, and conversely that the density and dynamics of cell–ECM adhesion in FALS was abnormal upon disruption of cell–cell adhesion. Of importance, disruption of either cadherin-based cell–cell or integrin-based cell–ECM adhesion affected DC. This suggests that interdependence between cell–cell and cell–ECM adhesion in vivo may play an important morphogenetic role in this context.
FIGURE 4:
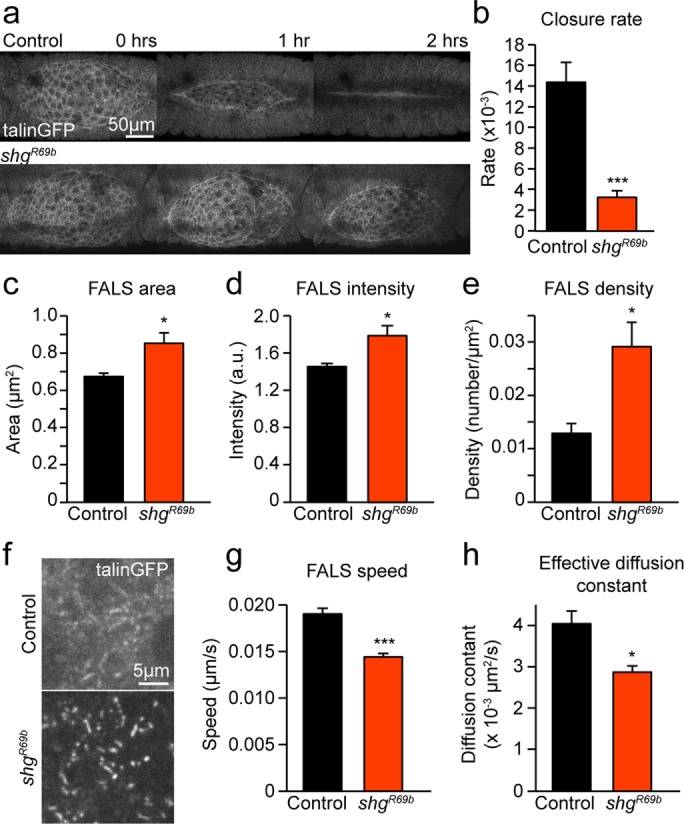
Loss of cell–cell adhesion causes DC defects and an increase in the amount and stability of cell–ECM adhesions. (a) Sample z-projected images of control and shgR69b embryos expressing talin-GFP at 1-h intervals during DC. (b) Mean rate of dorsal closure in control and shgR69b embryos (five embryos). (c–e) FALS area (c), intensity (normalized to cytoplasmic intensity (d), and density (e) in control and shgR69b embryos (seven or eight embryos, 242–503 FALS). (f) Sample z-projected images of the basal domain of AS cells showing FALS in control and shgR69b embryos. (g, h) Mean speed (g) and effective diffusion constant (h) of FALS movement in control and shgR69b embryos (four to seven embryos, 34–58 FALS). Error bars indicate SEM. *p < 0.05, ***p < 0.0001.
To explore further the possible functional and mechanistic consequences of interdependence between cadherin-based cell–cell and integrin-based cell–ECM adhesion in the AS, we studied cell behaviors and tissue properties that require precise regulation of cell–ECM adhesion in shgR69b mutant embryos. On the basis of our previous findings, we hypothesized that the increase in the area and density of cell–ECM adhesion we observe in the AS of shgR69b mutants would result in reduced cell deformation (Goodwin et al., 2016). Reduced cell deformation would, in turn, give rise to changes in the oscillatory behavior of AS cells, as well as abnormal tissue responses to recoil after laser ablation (Figure 5). Consistent with this hypothesis, we observed severe impairment in cell deformation in shgR69b mutants: the amplitude and period of oscillations and the speed of the cell centroid were all reduced (Figure 5, a–d). Tissue biomechanical properties were analyzed in shgR69b mutant embryos using laser ablation experiments (Figure 5, e–g). In these experiments, a laser pulse penetrates and cuts the lateral membrane between two cells; in the case of AS cells, which are very thin, the cut likely spans most of the apical–basal axis of lateral membranes. Initial recoil speed—a general indicator for tension experienced across cell membranes—was not different between shgR69b embryos and controls (Figure 5, e and f). However, recoil speed also depends on local tissue biomechanical properties, including viscosity and stiffness; if local biomechanical properties differ between experimental groups, recoil velocity may not accurately reflect differences in tension (Hutson et al., 2003). Using a viscoelastic model to fit the displacement of junctions over time after ablation, we can infer the ratio of stiffness to viscosity, known as the relaxation time (Fernandez-Gonzalez et al., 2009). We find that relaxation time is significantly reduced in shgR69b mutant embryos, indicating that movement of junctions away from the ablation site ceases earlier than in controls (Figure 5, e and g). Overall these experiments reveal that tissue biomechanical properties are altered in shgR69b embryos in a manner reminiscent of but not identical to that observed in embryos with increased cell–ECM adhesion.
FIGURE 5:

Cell oscillatory behavior and force transmission are suppressed in shgR69b mutants. (a) Temporal overlay of cell contours 20 s apart from time-lapse movies of control and shgR69b embryos during early DC. (b–d) Mean amplitude (b) and period (c) of apical area oscillations and mean speed of apical cell centroid movement (d) for control and shgR69b embryos (four embryos, 21–25 cells). (e) Sample images of control and shgR69b embryos before and after laser ablation. Yellow line indicates cut location, and white arrows indicate tracked first-neighbor junctions. Junction separation at t = 0 (dt=0 s) and 15 s (dt=15 s) are indicated to help visually assess differences in separation over time between samples. (f, g) Recoil velocity (f) and relaxation time (g) of first-neighbor junctions after laser ablation in control and shgR69b embryos (13–24 cuts). (h) Spatial decay of maximum displacement in control and shgR69b embryos measured by fitting the maximum displacement of first-neighbor (1st nbr), second-neighbor (2nd nbr), and background (Bkgd) junctions to an exponential. R2 values and overall spatial decay coefficient (k) are indicated on plots. (i) Mean spatial decay coefficient in controls and shgR69b embryos. Error bars indicate SEM (unless otherwise indicated). *p < 0.05, ***p < 0.0001.
Our recent work showed that higher cell–ECM adhesion resulted in reduced force transmission between AS cells (Goodwin et al., 2016). Therefore we used a methodology we previously used to study force transmission across AS cells by analyzing broader tissue responses to laser ablation in control and mutant embryos (Fischer et al., 2014; Goodwin et al., 2016). To this end, we tracked the movement of first-neighbor junctions (immediately adjacent to the cut) and second-neighbor junctions (two cell segments away from the cut), as well as background movement of junctions (at least three cell segments away from the cut). If we plot the maximum displacement of each of these junctions as a function of their distance from the cut, we can fit these data to an exponential function and estimate the spatial decay of recoil effects. We find that the spatial decay coefficient (k) is increased in shgR69b embryos, indicating that recoil effects are more quickly dissipated as we move away from the ablation site (Figure 5, h and i). Overall this suggests that increased cell–ECM adhesion upon loss of cadherin-based cell–cell adhesion has important functional consequences for the efficiency of force transmission across AS cells. Specifically, the increase in cell–ECM adhesion in cadherin-deficient embryos could account for the defects in cell area oscillations and constriction of the AS, which contribute to the defective DC we observed in shgR69b mutants.
DISCUSSION
Using genetic manipulations and quantitative analysis, we showed that perturbations to cell–ECM or cell–cell adhesion alter cell behavior and tissue biomechanical properties in the AS, leading to defective DC. Of importance, we found that depleting either cell–cell or cell–ECM adhesion led to changes in the recruitment and stability of the other type of adhesion. When cell–ECM adhesion was reduced, cadherin localization along the apical membrane was highly irregular, and cadherin stability was altered. Further, we found that cortical and medial actin was unevenly distributed across the cell and that myosin flow speed was reduced when cell–ECM adhesion was depleted. Similarly, when cell–cell adhesion was reduced, recruitment of talin to cell–ECM adhesions was increased, yielding larger, more stable FALS. As a result, cell deformation and force transmission across AS cells were limited, preventing proper progression of DC. These results suggest that cooperation or interdependence between cell–cell and cell–ECM adhesions may play a role in regulating DC.
As DC progresses, actomyosin-mediated pulsatile contraction of cells results in incremental decreases in apical cell area. Changes in the stereotypic pattern of contraction rates of AS cells have been reported in mys zygotic embryos (Gorfinkiel et al., 2009). Under wild-type conditions, cells on the periphery of the AS experience increased contraction compared with central cells, and in mys embryos, this relationship is reversed (Gorfinkiel et al., 2009). Moreover, in mys embryos, the contraction rate of central cells was increased compared with central cells of wild-type embryos (Gorfinkiel et al., 2009). These earlier observations are consistent with our data, specifically, our analysis of myosin localization and dynamics. We found that in central AS cells, myosin intensity increased and its flow speed decreased during the slow phase of contraction. These changes in myosin localization and activity may explain the increased contraction rates described previously (Gorfinkiel et al., 2009) and, consequently, the reduction in apical area we observed in AS cells of mys −/− embryos.
Myosin localization and dynamics can be regulated by cell–cell adhesions in vivo. In the Drosophila germband, asymmetries in E-cadherin distribution can direct myosin flows by providing an unbalanced mechanical landscape across the cell (Levayer and Lecuit, 2013). Uniformly high cadherin levels lead to increased tension and a more balanced actomyosin network across the cell, thus reducing myosin flow speed (Levayer and Lecuit, 2013). In the AS, we observed that loss of cell–ECM adhesion led to both reduced myosin flow speed and aberrant localization of cell–cell adhesions. E-cadherin appeared to be clustered together in bright peaks instead of being evenly distributed along the cell membrane. Although the phenotypes we observe in mys mutants cannot be explained simply by the framework provided by the germband model, it is possible to modify this framework to propose a model that fits our observations. For example, although E-cadherin accumulation was irregular in the AS of mys mutants, it was not planar polarized and would therefore not generate the imbalances required to support high myosin flow. Instead, the irregular accumulation of E-cadherin may lead to an overall increase in tension across the network, leading to more balanced mechanical tension and affecting myosin flow, as described previously (Levayer and Lecuit, 2013).
Adherens junctions are remodeled in response to changing mechanical force. Increased magnitude of the “tugging” force exerted by a pair of adhering cells on each other results in increased size of cell–cell junctions (Liu et al., 2010). In embryos lacking cell–ECM adhesion, force transmission across the apical plane of cells is increased (Goodwin et al., 2016), which may produce a “tugging” force, leading to larger clusters of cadherin-based adhesion. Further, this aberrant distribution of cell–cell adhesions could lead to the disorganization of actin networks observed in mys −/− embryos by providing spatial or mechanical cues. In cell culture, mechanotransduction via cadherins can trigger changes in actomyosin networks and contractility (Muhamed et al., 2016). Cadherin mechanotransduction requires coupling to the actin cytoskeleton via linker proteins, including α-catenin (Yonemura et al., 2010). Recent work in Drosophila showed that α-catenin is required for actomyosin pulses and stabilization of E-cadherin at the membrane in the AS, demonstrating the importance of this coupling to actin in vivo (Jurado et al., 2016). Here we see that loss of basal cell–ECM adhesion (and the resultant up-regulation of apical force transmission; Goodwin et al., 2016) leads to changes in cadherin localization, actin organization, and myosin dynamics. Similar phenotypes were observed in the AS upon disruption of Crumbs, a protein involved in epithelial polarity; loss of normal Crumbs function disrupted adherens junctions, leading to abnormally high myosin activity and failure to complete DC (Flores-Benitez and Knust, 2015). Taken together, these studies and the results presented here highlight the importance of proper linkage between adherens junctions and actomyosin networks in regulating cell behaviors required for DC.
Cross-talk between cell–cell and cell–ECM adhesions has been described in many contexts both in vivo and in cell culture (Borghi et al., 2010; Julich et al., 2015; Yamamoto et al., 2015; Coburn et al., 2016). The interplay between these two types of adhesions can affect their localization, the positioning or structure of actomyosin networks, and the mechanical landscape of the cell (Mui et al., 2016). Here we demonstrate the existence of interdependence between cell–cell and cell–ECM adhesions in the AS. Loss of integrin function led to changes in cadherin localization and stability. Similarly, loss of cadherin-mediated adhesion increased recruitment of cell–ECM adhesions in the form of larger and more stable FALS. In embryos with increased cell–ECM adhesion and larger FALS, cell deformation is hindered; we see a similar phenotype in embryos lacking cell–cell adhesions, suggesting that FALS may be resisting apical deformations in this context as well. Further, the loss of cadherin would reduce coupling between apicomedial actomyosin networks and the cell membrane, possibly preventing apical shape changes, leading to a much more severe defect in cell oscillations than observed by specifically increasing cell–ECM adhesion.
Depletion of cell–cell adhesion and the resultant increase in cell–ECM adhesion at FALS also affected tissue biomechanical properties as probed by laser ablation experiments. Recoil velocity was unchanged, but relaxation time of junctions was significantly reduced; stated differently, junctions ceased moving away from each other after laser-induced release of tension sooner in shgR69b embryos. Shorter relaxation time suggests that either stiffness is reduced or viscosity is increased (Fernandez-Gonzalez et al., 2009). Loss of cadherin-mediated cell–cell adhesions from the cell membrane could result in a more easily deformable (and less stiff) apical membrane. In addition, the observed changes to cell–ECM adhesion seen in shgR69b mutants could affect the behavior of junctions after ablation. We previously showed that recoil can be resisted basally by FALS and that mutations that specifically increased the size and density of FALS resulted in reduced recoil velocity and force transmission across the apical plane of cells (Goodwin et al., 2016). Of interest, embryos with increased cell–ECM adhesions showed no change in relaxation time, likely because perturbations to basal adhesions do not affect the viscosity or stiffness of the apical part of the cell. In shgR69b embryos, loss of cadherin-mediated adhesion would undoubtedly affect apical biomechanical properties, resulting in reduced relaxation time. Further, reduced force transmission in the AS of shgR69b embryos is likely to be, at least in part, a direct consequence of reduced adhesion between adjacent cells, which is required to mechanically couple them. However, the increase in size and density of FALS observed in shgR69b embryos suggests that increased cell–ECM adhesion underlies, at least in part, the shgR69b mutant phenotype. The similarities observed between embryos with increased cell–ECM adhesion and those with decreased cell–cell adhesion suggest an important role for regulatory cross-talk between the two types of adhesive networks during DC. Whether the adhesion interdependence in the AS reported here involves direct interactions or functional communications between signaling pathways and adhesion complexes is unknown. Future work should focus on elucidating the molecular and/or physical mechanisms that may link cell–cell and cell–ECM adhesion during DC; for example, RhoA signaling, which we previously showed regulates FALS morphology (Goodwin et al., 2016), may also regulate adherens junctions and adhesion interdependence in the AS. Further, simultaneous genetic manipulation of cell–cell and cell–ECM adhesion may help to uncover the mechanisms of adhesion interdependence and the exact cellular activities this interdependence regulates.
Given their functional similarities and the requirement for coordinated adhesion in driving dynamic cellular behaviors during morphogenesis, it is unsurprising that interdependence exists between the integrin and cadherin adhesion complexes. In fact, evidence for such interdependence is extensive in diverse settings both in vitro and in vivo (Weber et al., 2011; Burute and Thery, 2012; McMillen and Holley, 2015). However, the precise biological roles of cross-talk between cadherin- and integrin-mediated adhesion remain somewhat obscure, perhaps because such interdependence may be context specific (reviewed in McMillen and Holley, 2015). For example, work using murine cancer cells found that engagement of integrin adhesion receptors specifically up-regulates cadherin-based cell–cell adhesion (Martinez-Rico et al., 2010). When cells were incubated with ECM-coated beads, more force was required to separate cell doublets as than when cells were exposed to poly-l-lysine–coated beads. Of importance, this up-regulation was found to depend on actomyosin activity (Martinez-Rico et al., 2010). Other studies found an opposite relationship between integrin- and cadherin-based adhesions. When the same murine cancer cell line was plated on micropatterned fibronectin substrata designed to modulate integrin adhesion, an increase in cell–ECM adhesion resulted in delayed formation and decreased strength of cell–cell attachment (Al-Kilani et al., 2011). There are a number of possible technical explanations for such inconsistencies: differences between adhesions formed by cells in suspension versus in two-dimensional (2D) culture, methods of measuring strength of cell–cell adhesions, and stiffness of substrate (McMillen and Holley, 2015). This discrepancy serves to highlight the obstacles to understanding the interdependence between integrin and cadherin signaling and supports the notion that it is likely dependent on biophysical context. It is therefore imperative to try to understand cross-talk between cell–ECM and cell–cell adhesion in vivo (McMillen and Holley, 2015). The work we present here illustrates how the use of genetic and biomechanical approaches in the context of animal morphogenesis can provide insight into the biological role of this interdependence. Moreover, the robust phenotypes we uncover in our studies provide strong evidence that interdependence between cell–ECM and cell–cell adhesion is an essential component of tissue morphogenesis.
MATERIALS AND METHODS
Fly stocks and genetics
The βPS-integrin–null mutants were generated using the dominant female sterile germline clone technique (Chou and Perrimon, 1996) in order to remove both maternal and zygotic contributions of integrin. Virgins of mysXG43, FRT101 were crossed to males of ywhsFlp,OvoD1, FRT101 (1813; Bloomington Drosophila Stock Center), and the larval progeny were subjected to a heat shock regime to generate a mys mosaic germline. Virgins were then crossed to dfd-GMR-nvYFP males. Mutant embryos were identified by lack of the fluorescent marker. Embryos carrying the fluorescent marker (i.e., heterozygous mys/+ siblings) were used as controls. For live-imaging experiments, ubi-DE-Cadherin-GFP (Oda and Tsukita, 2001) and sqh-mCherry (Martin et al., 2009) were zygotically provided.
The shgR69 line (Godt and Tepass, 1998; a gift of Uli Tepass, University of Toronto) was used to generate virgins of the genotype talinGFP;shgR69b,FRT42D/CyO,dfd-GMR-nvYFP, which were crossed to males of the genotype w-;shgR69b,FRT42D/CyO,dfd-GMR-nvYFP. Embryos lacking the balancer were used for analyses. Wild-type talin-GFP embryos were used for controls, as defects in heterozygous shgR69b embryos were as severe as in homozygous ones.
Confocal immunofluorescence and live imaging
Embryos were dechorionated in 50% bleach for ∼4 min and staged according to the criteria of Ellis et al. (2013). For live imaging, embryos were aligned and glued to a coverslip using embryo glue (Scotch double-sided tape dissolved in heptane), dorsal side down. Coverslips were mounted in halocarbon oil (Sigma-Aldrich) on glass slides with a coverslip bridge to prevent compression of the embryos. For fixed imaging, embryos were fixed in 4% paraformaldehyde according to standard protocols. The following antibodies were used in our analysis: mouse monoclonal anti–βPS-integrin (1:50; Developmental Studies Hybridoma Bank), rat monoclonal anti–DE-cadherin (1:40; Developmental Studies Hybridoma Bank), and rhodamine-conjugated phalloidin (1:400; Invitrogen). Fluorescently conjugated Alexa Fluor 488, Cy3, and Cy5 secondary antibodies were used at 1:400 dilution (Molecular Probes).
Images were collected using an Olympus FV1000 inverted confocal microscope and an UplanFL N 40×/1.30 numerical aperture (NA) oil objective or a UplanSApo 60×/1.35 NA objective, or a Zeiss Axiovert 200 M spinning-disk confocal microscope using a 63×/1.40 NA or 100×/1.45 NA objective. All images were acquired maintaining consistent exposure time, laser power, gain, and offset settings between control and experiment embryos to allow for direct comparison. For movies of cell oscillations, myosin flows, and/or FALS movement, five to ten 1-μm confocal sections were collected at 20-s intervals for a 20-min time period. For movies of DC, 10 to 20 2 μm-confocal sections were collected at 5-min intervals for a 5-h time period. At least five movies were taken of each genotype. For fixed images of cells, z-stacks were assembled from ten 0.5-μm confocal sections. Animations were assembled and processed using ImageJ (National Institutes of Health, Bethesda, MD).
FRAP
Stage 14 embryos were collected and mounted for live imaging as described earlier. FRAP analysis was performed at room temperature. Photobleaching was performed using a 473-nm laser at 10% power with the Tornado scanning tool (Olympus) for 2 s at 100 ms/pixel. Fluorescence recovery was recorded over 13 min at rate of 1 frame every 4 s. To control for drift of embryos, we selected multiple regions of interest (ROIs) in nonphotobleached regions; we used only samples for which intensities within control ROIs remained steady throughout the FRAP experiment. The mobile fraction and recovery time were computed in MatLab, and statistical tests were performed using Prism software.
Time-lapse image analysis: closure time, cell oscillations, and myosin flows
In all time-lapse analyses, z-projected images were used for each time point to encompass large portions of the curved surface of the AS. To measure AS area over time, AS outlines were traced manually using ImageJ and processed in MatLab. Closure curves were then fitted to the exponential A(t) = A0e-kt, where A is area, A0 is initial area, k is the rate of closure, and t is time.
Analysis of cell oscillations was done using custom tools generated in MatLab. Cell outlines were generated from time-lapse movies either semiautomatically using SIESTA (Fernandez-Gonzalez and Zallen, 2011) or manually using ImageJ. These outlines were then processed in MatLab to calculate cell area and centroid (geometric center of the cell outline) over time. Cell area curves were detrended to obtain measurements of the amplitude and period of cell oscillations. Centroid speed was measured as the mean change in position of the centroid between frames divided by the time between frames. Myosin intensity was computed as the mean fluorescence intensity within the cell contour.
Particle image velocimetry (PIV) was performed using custom-written MatLab scripts based on the approach described in Levayer and Lecuit (2013) in order to measure movement of fluorescently tagged myosin. Images were divided into 16 × 16 pixel (2.5 × 2.5 μm) interrogation windows overlapping by 75%. The 2D cross-correlation of each window for each frame over the duration of the movie was used to calculate the direction and magnitude of intensity movement. Cell outlines were calculated as described and used to isolate PIV vectors corresponding to individual cells. The magnitude of the vectors was used to compute mean instantaneous speed. The mean for a cell was defined as the mean of the speeds described by all vectors within the cell.
Quantification of cadherin and actin localization
Measurement of cadherin and cortical actin intensity along cell edges was done using custom MatLab scripts using z-projected images of AS cells spanning the apical–basal axis of the cell. The script used a user-generated cell outline and refined it using a series of morphological image processing steps to ensure accuracy and remove user bias. The resultant outline coordinates were used to determine cadherin or actin intensity at each point along the outline. For each point, pixel brightness values were determined for a square region of 12 × 12 pixels (0.8 × 0.8 μm) around the point. Pixels with brightness greater than one SD of the mean brightness (computed from the entire image) were included in intensity calculation. The mean of all included pixels was used as the intensity value for that point along the outline. The resultant curve of intensity versus position along the cell outline was used to compute the magnitude and spacing of intensity peaks. Peaks greater than one SD above the mean intensity of the entire cell outline were included. Peak density was computed as the number of peaks divided by the cell perimeter. To quantify the size and number of medial actin bundles, images were filtered using a difference-of-Gaussians approach and thresholded to identify and count bright objects within the cell. The size of these identified objects was used to measure actin bundle size, and the number within a cell divided by the cell area was used to compute bundle density (Supplemental Figure S1).
FALS detection and tracking
We developed automatic image segmentation and analysis tools in MatLab to measure FALS area and intensity. Single-slice images of the basal portion of AS cells were filtered using a difference-of-Gaussians approach. The filtered images were thresholded and applied as masks to the original images to identify FALS and measure area and pixel intensity. FALS density was determined by counting the number of FALS per image and dividing by image area. FALS were tracked manually in ImageJ. Speed was defined as the change in position divided by the time between frames. Mean square displacement (MSD) analyses of FALS were performed to quantify the movement of FALS as a result of disordered pulsatile contractions of AS cells. MSD curves were computed as described in Suraneni et al. (2012) and fitted to the general formula MSD(τ) = 4Dτ, where D is the effective diffusion constant.
Laser ablation and recoil analysis
Laser ablation experiments were carried out using a Revolution XD spinning-disk confocal microscope equipped with an iXon Ultra 897 camera (Andor) and a 60×/NA 1.35 oil immersion lens (Olympus). Ablation was induced using a pulsed Micropoint N2 laser (Andor) tuned to 365 nm. Z-stacks were acquired every 3 s for up to 60 s after ablation. Cuts were performed in embryos from timed cages at the onset of the slow phase of DC as judged by the shape of the canthi (for a description of the phases of DC, see Gorfinkiel et al., 2009). Movement of first- and second-neighbor junctions was measured using SIESTA (Fernandez-Gonzalez and Zallen, 2011). First neighbors are defined as junctions immediately adjacent to the cut, and second neighbors are the two junctions next to each first neighbor. Background movement was measured by tracking junctions at least three membrane segments away from the cut.
Recoil velocity was determined by the displacement of junctions immediately after the cut. Maximum displacement was measured as the maximum distance from each junction to the midpoint between first-neighbor junctions immediately before the cut. Spatial decay was measured by fitting the maximum displacement as a function of the junction’s distance from the midpoint before ablation to an exponential function D = cekd, where D is maximum displacement, k is the spatial decay coefficient, d is distance from the cut, and c is a constant. To compare decay rate between mutant and control groups, we randomly resampled the data set with replacement and fit again to obtain new estimates for k. This was repeated 1000 times to obtain a distribution of values for k that could be compared between experimental groups using a two-sample t test.
To estimate mechanical properties of AS cells, junction retraction was fitted using a viscoelastic model in which membrane segments are approximated as a spring and dashpot in parallel, described by the equation L(t) = D(1 − et/τ), where L is the change in distance between first-neighbor junctions, D is proportional to tensile force, t is time, and τ is the relaxation time, equal to the ratio of viscosity to elasticity (Fernandez-Gonzalez et al., 2009).
Supplementary Material
Acknowledgments
This study was supported by a CIHR Operating Grant to G.T. (MOP-285391) and a grant from the Canada Foundation for Innovation (30279) to R.F.G. T.Z.C. was supported by an Ontario Trillium Scholarship and a Delta Kappa Gamma Society International World Fellowship. We thank Uli Tepass and the Bloomington Drosophila Stock Center for fly lines.
Abbreviations used:
- AS
amnioserosa
- DC
dorsal closure
- ECM
extracellular matrix
- FALS
focal adhesion–like structures
- FRAP
fluorescence recovery after photobleaching
- PIV
particle image velocimetry.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E17-01-0033) on March 22, 2017.
REFERENCES
- Al-Kilani A, de Freitas O, Dufour S, Gallet F. Negative feedback from integrins to cadherins: a micromechanical study. Biophys J. 2011;101:336–344. doi: 10.1016/j.bpj.2011.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanchard GB, Murugesu S, Adams RJ, Martinez-Arias A, Gorfinkiel N. Cytoskeletal dynamics and supracellular organisation of cell shape fluctuations during dorsal closure. Development. 2010;137:2743–2752. doi: 10.1242/dev.045872. [DOI] [PubMed] [Google Scholar]
- Borghi N, Lowndes M, Maruthamuthu V, Gardel ML, Nelson WJ. Regulation of cell motile behavior by crosstalk between cadherin- and integrin-mediated adhesions. Proc Natl Acad Sci USA. 2010;107:13324–13329. doi: 10.1073/pnas.1002662107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burute M, Thery M. Spatial segregation between cell-cell and cell-matrix adhesions. Curr Opin Cell Biol. 2012;24:628–636. doi: 10.1016/j.ceb.2012.07.003. [DOI] [PubMed] [Google Scholar]
- Chou TB, Perrimon N. The autosomal FLP-DFS technique for generating germline mosaics in Drosophila melanogaster. Genetics. 1996;144:1673–1679. doi: 10.1093/genetics/144.4.1673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coburn L, Lopez H, Caldwell BJ, Moussa E, Yap C, Priya R, Noppe A, Roberts AP, Lobaskin V, Yap AS, et al. Contact inhibition of locomotion and mechanical cross-talk between cell-cell and cell-substrate adhesion determine the pattern of junctional tension in epithelial cell aggregates. Mol Biol Cell. 2016;27:3436–3448. doi: 10.1091/mbc.E16-04-0226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ducuing A, Vincent S. The actin cable is dispensable in directing dorsal closure dynamics but neutralizes mechanical stress to prevent scarring in the Drosophila embryo. Nat Cell Biol. 2016;18:1149–1160. doi: 10.1038/ncb3421. [DOI] [PubMed] [Google Scholar]
- Duque J, Gorfinkiel N. Integration of actomyosin contractility with cell-cell adhesion during dorsal closure. Development. 2016;143:4676–4686. doi: 10.1242/dev.136127. [DOI] [PubMed] [Google Scholar]
- Ellis SJ, Goult BT, Fairchild MJ, Harris NJ, Long J, Lobo P, Czerniecki S, Van Petegem F, Schock F, Peifer M, et al. Talin autoinhibition is required for morphogenesis. Cur Biol. 2013;23:1825–1833. doi: 10.1016/j.cub.2013.07.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Gonzalez R, Simoes Sde M, Roper JC, Eaton S, Zallen JA. Myosin II dynamics are regulated by tension in intercalating cells. Dev Cell. 2009;17:736–743. doi: 10.1016/j.devcel.2009.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Gonzalez R, Zallen JA. Oscillatory behaviors and hierarchical assembly of contractile structures in intercalating cells. Phys Biol. 2011;8:045005. doi: 10.1088/1478-3975/8/4/045005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer SC, Blanchard GB, Duque J, Adams RJ, Arias AM, Guest SD, Gorfinkiel N. Contractile and mechanical properties of epithelia with perturbed actomyosin dynamics. PLoS One. 2014;9:e95695. doi: 10.1371/journal.pone.0095695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flores-Benitez D, Knust E. Crumbs is an essential regulator of cytoskeletal dynamics and cell-cell adhesion during dorsal closure in Drosophila. Elife. 2015;4:e07398. doi: 10.7554/eLife.07398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geiger B, Spatz JP, Bershadsky AD. Environmental sensing through focal adhesions. Nat Rev Mol Cell Biol. 2009;10:21–33. doi: 10.1038/nrm2593. [DOI] [PubMed] [Google Scholar]
- Godt D, Tepass U. Drosophila oocyte localization is mediated by differential cadherin-based adhesion. Nature. 1998;395:387–391. doi: 10.1038/26493. [DOI] [PubMed] [Google Scholar]
- Goodwin K, Ellis SJ, Lostchuck E, Zulueta-Coarasa T, Fernandez-Gonzalez R, Tanentzapf G. Basal cell-extracellular matrix adhesion regulates force transmission during tissue morphogenesis. Dev Cell. 2016;39:611–625. doi: 10.1016/j.devcel.2016.11.003. [DOI] [PubMed] [Google Scholar]
- Gorfinkiel N, Arias AM. Requirements for adherens junction components in the interaction between epithelial tissues during dorsal closure in Drosophila. J Cell Sci. 2007;120:3289–3298. doi: 10.1242/jcs.010850. [DOI] [PubMed] [Google Scholar]
- Gorfinkiel N, Blanchard GB, Adams RJ, Martinez Arias A. Mechanical control of global cell behaviour during dorsal closure in Drosophila. Development. 2009;136:1889–1898. doi: 10.1242/dev.030866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutson MS, Tokutake Y, Chang MS, Bloor JW, Venakides S, Kiehart DP, Edwards GS. Forces for morphogenesis investigated with laser microsurgery and quantitative modeling. Science. 2003;300:145–149. doi: 10.1126/science.1079552. [DOI] [PubMed] [Google Scholar]
- Hynes RO. Integrins: bidirectional, allosteric signaling machines. Cell. 2002;110:673–687. doi: 10.1016/s0092-8674(02)00971-6. [DOI] [PubMed] [Google Scholar]
- Jacinto A, Wood W, Balayo T, Turmaine M, Martinez-Arias A, Martin P. Dynamic actin-based epithelial adhesion and cell matching during Drosophila dorsal closure. Curr Biol. 2000;10:1420–1426. doi: 10.1016/s0960-9822(00)00796-x. [DOI] [PubMed] [Google Scholar]
- Julich D, Cobb G, Melo AM, McMillen P, Lawton AK, Mochrie SG, Rhoades E, Holley SA. Cross-scale integrin regulation organizes ECM and tissue topology. Dev Cell. 2015;34:33–44. doi: 10.1016/j.devcel.2015.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurado J, de Navascues J, Gorfinkiel N. alpha-Catenin stabilises cadherin-catenin complexes and modulates actomyosin dynamics to allow pulsatile apical contraction. J Cell Sci. 2016;129:4496–4508. doi: 10.1242/jcs.193268. [DOI] [PubMed] [Google Scholar]
- Leckband DE, de Rooij J. Cadherin adhesion and mechanotransduction. Annu Rev Cell Dev Biol. 2014;30:291–315. doi: 10.1146/annurev-cellbio-100913-013212. [DOI] [PubMed] [Google Scholar]
- Lecuit T, Yap AS. E-cadherin junctions as active mechanical integrators in tissue dynamics. Nat Cell Biol. 2015;17:533–539. doi: 10.1038/ncb3136. [DOI] [PubMed] [Google Scholar]
- Levayer R, Lecuit T. Oscillation and polarity of E-cadherin asymmetries control actomyosin flow patterns during morphogenesis. Dev Cell. 2013;26:162–175. doi: 10.1016/j.devcel.2013.06.020. [DOI] [PubMed] [Google Scholar]
- Levayer R, Pelissier-Monier A, Lecuit T. Spatial regulation of Dia and Myosin-II by RhoGEF2 controls initiation of E-cadherin endocytosis during epithelial morphogenesis. Nat Cell Biol. 2011;13:529–540. doi: 10.1038/ncb2224. [DOI] [PubMed] [Google Scholar]
- Liu Z, Tan JL, Cohen DM, Yang MT, Sniadecki NJ, Ruiz SA, Nelson CM, Chen CS. Mechanical tugging force regulates the size of cell-cell junctions. Proc Natl Acad Sci USA. 2010;107:9944–9949. doi: 10.1073/pnas.0914547107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin AC, Kaschube M, Wieschaus EF. Pulsed contractions of an actin-myosin network drive apical constriction. Nature. 2009;457:495–499. doi: 10.1038/nature07522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Rico C, Pincet F, Thiery JP, Dufour S. Integrins stimulate E-cadherin-mediated intercellular adhesion by regulating Src-kinase activation and actomyosin contractility. J Cell Sci. 2010;123:712–722. doi: 10.1242/jcs.047878. [DOI] [PubMed] [Google Scholar]
- McMillen P, Holley SA. Integration of cell-cell and cell-ECM adhesion in vertebrate morphogenesis. Curr Opin Cell Biol. 2015;36:48–53. doi: 10.1016/j.ceb.2015.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muhamed I, Wu J, Sehgal P, Kong X, Tajik A, Wang N, Leckband DE. E-cadherin-mediated force transduction signals regulate global cell mechanics. J Cell Sci. 2016;129:1843–1854. doi: 10.1242/jcs.185447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mui KL, Chen CS, Assoian RK. The mechanical regulation of integrin-cadherin crosstalk organizes cells, signaling and forces. J Cell Sci. 2016;129:1093–1100. doi: 10.1242/jcs.183699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narasimha M, Brown NH. Novel functions for integrins in epithelial morphogenesis. Curr Biol. 2004;14:381–385. doi: 10.1016/j.cub.2004.02.033. [DOI] [PubMed] [Google Scholar]
- Oda H, Tsukita S. Real-time imaging of cell-cell adherens junctions reveals that Drosophila mesoderm invagination begins with two phases of apical constriction of cells. J Cell Sci. 2001;114:493–501. doi: 10.1242/jcs.114.3.493. [DOI] [PubMed] [Google Scholar]
- Pasakarnis L, Frei E, Caussinus E, Affolter M, Brunner D. Amnioserosa cell constriction but not epidermal actin cable tension autonomously drives dorsal closure. Nat Cell Biol. 2016;18:1161–1172. doi: 10.1038/ncb3420. [DOI] [PubMed] [Google Scholar]
- Pokutta S, Weis WI. Structure and mechanism of cadherins and catenins in cell-cell contacts. Annu Rev Cell Dev Biol. 2007;23:237–261. doi: 10.1146/annurev.cellbio.22.010305.104241. [DOI] [PubMed] [Google Scholar]
- Roper K ( Integration of cell-cell adhesion and contractile actomyosin activity during morphogenesis. Curr Top Dev Biol. 2015;112:103–127. doi: 10.1016/bs.ctdb.2014.11.017. [DOI] [PubMed] [Google Scholar]
- Saias L, Swoger J, D’Angelo A, Hayes P, Colombelli J, Sharpe J, Salbreux G, Solon J. Decrease in cell volume generates contractile forces driving dorsal closure. Dev Cell. 2015;33:611–621. doi: 10.1016/j.devcel.2015.03.016. [DOI] [PubMed] [Google Scholar]
- Solon J, Kaya-Copur A, Colombelli J, Brunner D. Pulsed forces timed by a ratchet-like mechanism drive directed tissue movement during dorsal closure. Cell. 2009;137:1331–1342. doi: 10.1016/j.cell.2009.03.050. [DOI] [PubMed] [Google Scholar]
- Sun Z, Guo SS, Fassler R. Integrin-mediated mechanotransduction. J Cell Biol. 2016;215:445–456. doi: 10.1083/jcb.201609037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suraneni P, Rubinstein B, Unruh JR, Durnin M, Hanein D, Li R. The Arp2/3 complex is required for lamellipodia extension and directional fibroblast cell migration. J Cell Biol. 2012;197:239–251. doi: 10.1083/jcb.201112113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyama Y, Peralta XG, Wells AR, Kiehart DP, Edwards GS. Apoptotic force and tissue dynamics during Drosophila embryogenesis. Science. 2008;321:1683–1686. doi: 10.1126/science.1157052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber GF, Bjerke MA, DeSimone DW. Integrins and cadherins join forces to form adhesive networks. J Cell Sci. 2011;124:1183–1193. doi: 10.1242/jcs.064618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wells AR, Zou RS, Tulu US, Sokolow AC, Crawford JM, Edwards GS, Kiehart DP. Complete canthi removal reveals that forces from the amnioserosa alone are sufficient to drive dorsal closure in Drosophila. Mol Biol Cell. 2014;25:3552–3568. doi: 10.1091/mbc.E14-07-1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto H, Ehling M, Kato K, Kanai K, van Lessen M, Frye M, Zeuschner D, Nakayama M, Vestweber D, Adams RH. Integrin beta1 controls VE-cadherin localization and blood vessel stability. Nat Commun. 2015;6:6429. doi: 10.1038/ncomms7429. [DOI] [PubMed] [Google Scholar]
- Yonemura S, Wada Y, Watanabe T, Nagafuchi A, Shibata M. alpha-Catenin as a tension transducer that induces adherens junction development. Nat Cell Biol. 2010;12:533–542. doi: 10.1038/ncb2055. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.