

Abstract
Resistance to angiotensin II (Ang II)-induced hypertension in T-cell deficient male mice with a targeted mutation in the recombination activating gene-1 (Rag1) on the C57BL/6J background (B6.Rag1−/−-M), which was reported by five independent laboratories including ours prior to 2015, has been lost. In mice purchased from Jackson Laboratory in 2015 and 2016, the time course and magnitude increase in mean arterial pressure (MAP) induced by two weeks of Ang II infusion at 490 ng/kg/min was identical between B6.Rag1−/−-M and wildtype littermates (B6.WT-M). Moreover, there were no differences in the time course or magnitude increase in MAP at the lowest dose of Ang II (200 ng/kg/min) that increased MAP. This loss in Ang II resistance is independent of T-cells. Angiotensin-type1-receptor (AT1R) binding was 1.4-fold higher in glomeruli isolated from recently purchased B6.Rag1−/−-M suggesting an increase in renal AT1R activity masks the blood pressure protection afforded by the lack of T-cells. The phenotypic change in B6.Rag1−/−-M has implications for investigators using this strain to study mechanisms of T-cell modulation of Ang II-dependent blood pressure control. These findings also serve as a reminder that the universal drive for genetic variation occurs in all animals including inbred mouse strains and that spontaneous mutations leading to phenotypic change can compromise experimental reproducibility over time and place. Lastly, these observations illustrate the importance of including experimental details regarding the location and time period over which animals are bred in publications involving animal studies to promote rigor and reproducibility in the scientific literature.
Keywords: backcross, genetic contamination, genetic drift, genetic stability, knockout, inbred mice, spontaneous mutation
The Rag1 gene codes for an enzyme that plays a critical role in the formation of the T-cell receptor and this protein is required for generating mature T and B cells1. In 2007, David Harrison’s laboratory, then at Emory, published that the increase in systolic blood pressure (SBP) induced by Ang II infusion was greater in male wild type mice (WT-M) compared to male Rag1 knockout mice (Rag1−/−-M) on the C57BL/6J background strain purchased from Jackson Laboratory2. Two weeks of Ang II infusion at 490 ng/kg/min caused a 41±3 mm Hg increase in SBP in WT-M whereas the SBP increased by only 21±1 mm Hg in Rag1−/−-M.
Rag1−/−-M resistance to Ang II-induced hypertension was replicated by several independent laboratories including us at Georgetown University, Washington, DC3, and investigators at the University of Kentucky4, Louisiana State University, Shreveport5 and at the University of Arizona, Tucson6 regardless of how blood pressure was measured including by tail cuff2, 4, 6, a pressure transducer via an implanted femoral artery catheter5 or by telemetry3. All of these studies were conducted on male B6.129S7-Rag1tm1Mom/J (B6.Rag1−/−-M purchased from Jackson Laboratory prior to 2015. Here we report on the loss of this phenotype. B6.Rag1−/−-M purchased in 2015 and 2016 are no longer resistant to Ang II-induced increases in blood pressure. We also discuss the implications for investigators using the B6.Rag1−/−-M to study immune modulation of blood pressure.
Methods
Mice
Source
Jackson Lab reports that the Rag1tm1Mom mutant strain was developed by Dr. Peter Mombaerts at the Massachusetts Institute of Technology7. Homologous recombination of the targeting vector with the Pgk-neo marker resulted in a 5′ deletion of 1,356 bp in the coding sequence in the 129S7/SvEvBrd-Hprt+-derived AB1 embryonic stem cell line. Backcrossing mice carrying the Rag1tm1Mom mutation a minimum of ten times to C57BL/6J inbred mice resulted in the generation of B6.Rag1tm1Mom (B6.Rag1−/−). These mice and their WT littermates were purchased from Jackson Laboratory (catalog #002216) at 10 weeks of age from March 2015 to October 2016.
Diet
At Jackson Labs, the B6.Rag1−/−-M and B6.WT were maintained on LabDiet 5K52 (22% protein; 16% fat; 62% carbohydrates). According to Jackson Labs, there have been no changes to the formulation of this diet since the life of this mouse line. Once these mice arrived at Georgetown University, they were maintained on LabDiet 5053 (24% protein; 13% fat; 62% carbohydrates). Both of these diets were purchased from LabDiet (St. Louis, MO).
Housing
Both at Jackson Labs and Georgetown University, the B6.Rag1−/−-M and B6.WT littermates were maintained in a pathogen-free barrier facility. Ivanov et al.8 reported that mice from Jackson Labs are free of segmented filamentous bacteria (SFB), which influence Th17 cells and subsequent studies have confirmed this finding9. Jackson Labs reports that in 2013, they tested all of their foundation stocks and animal rooms for SFB and failed to find any positive results for SFB. Since September 2015, they regularly test their mouse rooms for SFB and also have yet to find any positive results. Thus, it is unlikely that there was any difference in the SFB status of recently purchased B6.Rag1−/−-M and those purchased prior to 2015.
All experimental methods were approved by the Georgetown University Animal Care and Use Committee.
Genotyping
Genomic DNA was extracted from mouse tails, as described previously10. PCR was used to genotype B6.Rag1−/−7. The B6.WT forward primer (oIMR1746) was 5′- GAG GTT CCG CTA CGA CTC TG-3′. The B6.Rag1−/− forward primer (oLMR8162) was 5′- TGG ATG TGG AAT GTG TGC GAG-3′. The common reverse primer (oLMR3104) was 5′- CCG GAC AAG TTT TTC ATC GT-3′. PCR cycling was as follows: 94°C for 2 min then 35 cycles of 94°C for 30 sec, 58°C for 45 sec, 72°C for 45 sec, and 1 elongation cycle of 72°C for 2 min. The B6.WT and disrupted alleles in the B6.Rag1−/− generated amplicons of 474 bp and 530 bp, respectively.
Mean Arterial Pressure (MAP) and Heart Rate (HR) Measurements
TA11PA-C10 radiotransmitters (Data Sciences Int., St. Paul, MN) were implanted into B6.WT-M and B6.Rag1−/−-M as we previously described11. Recording began on the 5–7th day after transmitter implantation. Recordings were taken at 30 second intervals every 10 minutes from 6 pm to 6 am and presented as daily averages using a Data Acquisition and Analysis System (Data Sciences Int.).
Angiotensin II (Ang II) Infusion
After recording a stable basal MAP for at least 3 days, Alzet osmotic minipumps (model #1002; 100 μl reservoir vol; 0.25 μl/h; Durect Corporation, Cupertino, CA) filled with Ang II (Sigma, Saint Louis, MO; #A9525) dissolved in saline were implanted under isoflurane anesthesia. After two weeks of Ang II infusion at 200 ng/kg/min, pumps were removed and exchanged with new pumps infusing Ang II at 490 ng/kg/min. A subset of the mice received a pump with the Ang II dose of 75 ng/kg/min. At the termination of the experiment, mice were euthanized and the spleens were collected along with whole blood.
Isolation from Peripheral Blood Mononuclear Cells (PBMC) and T-cells from Spleen
PBMC were isolated from whole blood using red blood cell lysis buffer and CD3+ T-cells were isolated from spleen by negative magnetic sorting (Pan T-cell Isolation Kit II, Miltenyi, Auburn, CA) from single cell suspensions of splenocytes, as we previously described2.
Isolation of Glomeruli
Glomeruli were isolated from whole kidneys as we described previously12 with the following modifications: mesh sizes for mouse kidneys were 125 μm (mesh size #120) for initial sieving followed by two stacks of sieves with 150 μm (mesh size #100) on the top and 63 μm (sieve size #230) on the bottom. The glomeruli were collected in a glass beaker from the bottom mesh by rinsing the mesh with ice-cold phosphate buffered saline (PBS) in the presence of protease Inhibitors (Final concentrations: 0.2 μM phenylmethane-sulfonyl fluoride (PMSF); 1 μg/mL Leupeptin; 2 μg/mL Antipain; 1 unit/L Aprotinin). The glomeruli were then centrifuged in 50 mL tubes at 3,000 rpm for 15 min. The supernatant was removed by vacuum and the pellet was washed with PBS and recentrifuged in a 1.5 mL microcentrifuge tube at 12,000 rpm for 10 min. The final pellet was resuspended in homogenization buffer (50 mM TRIS-HCl, 1 mM EDTA buffer, pH 7.4) supplemented with protease inhibitors (Final concentrations: 0.2 μM phenylmethane-sulfonyl fluoride (PMSF); 1 μg/mL Leupeptin; 2 μg/mL Antipain; 1 unit/L Aprotinin). Protein concentrations of isolated glomeruli were determined by the Bradford method using bovine serum albumin as the standard (Bio-Rad Laboratories, Richmond, CA, USA).
125I-[Sar1,Ile8]Ang II Binding to Glomeruli
125I-Sar1,Ile8 Ang II specific binding to glomerular membranes (10 μg protein/tube) was determined at a concentration of 0.4 nM (approximately 400,000 cpm/300 μl) in the presence of 10 μM PD123319 (to block AT2 receptor binding of 125I-Sar1,Ile8 Ang II) and the presence or absence of 3 μM Ang II to define specific, Ang II displaceable 125I-Sar1,Ile8 Ang II binding to glomerular membrane AT1 receptors.
Statistical Analysis
Prism 7.0 (GraphPad Software, La Jolla, CA) was used to analyze all data. The data are expressed as the mean plus the standard error of the mean (SEM). Unpaired Student’s t-test was used to analyze differences between groups. Two-way analysis of variance (ANOVA) was used to analyze differences in the effects of Ang II between mouse strains. The significance threshold was defined as 0.05.
Literature Search
Searches of OVID and PubMed were conducted in journals through January 3, 2017. All languages were included and no start date was imposed in the search. Two separate searches were performed before Boolean logic was applied:
Recombination activating gene OR Rag
Blood pressure OR arterial pressure OR systolic pressure OR hypertension
Boolean logic: (1) AND (2). Two reviewers (AVP & KS) independently performed the search and analyzed the results.
Results
Genotyping
PCR of the Rag1−/− gene showed that DNA extracted from B6.Rag1−/−-M purchased between March 2014 and October 2016 had an additional 56 bp compared to their B6.WT-M littermates, which is identical to the genotyping results from B6.Rag1−/−-M purchased between February 2011 and September 2012 with which we reported sex-specific T- cell regulation of Ang II-dependent hypertension3 (Figure 1).
Figure 1.
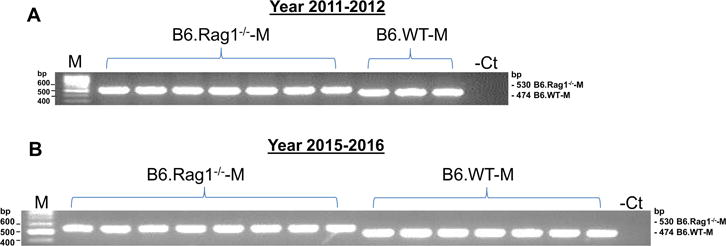
Genotyping of male B6.WT and B6.Rag1−/−. Shown are representative examples of gels of amplicons generated by PCR from RNA isolated from B6.WT-M and B6.Rag1−/−-M purchased from Jackson Laboratory in 2011–2012 (A) and 2015–2016 (B).
Body and Spleen Weight
B6.Rag1−/−-M had body and spleen weights that were similar to our previous findings3 (Figure 2). Whereas no differences in body weight were observed between B6.Rag1−/−-M and B6.WT-M, B6.Rag1−/−-M spleens were less than 25% of B6.WT-M spleen weights.
Figure 2.
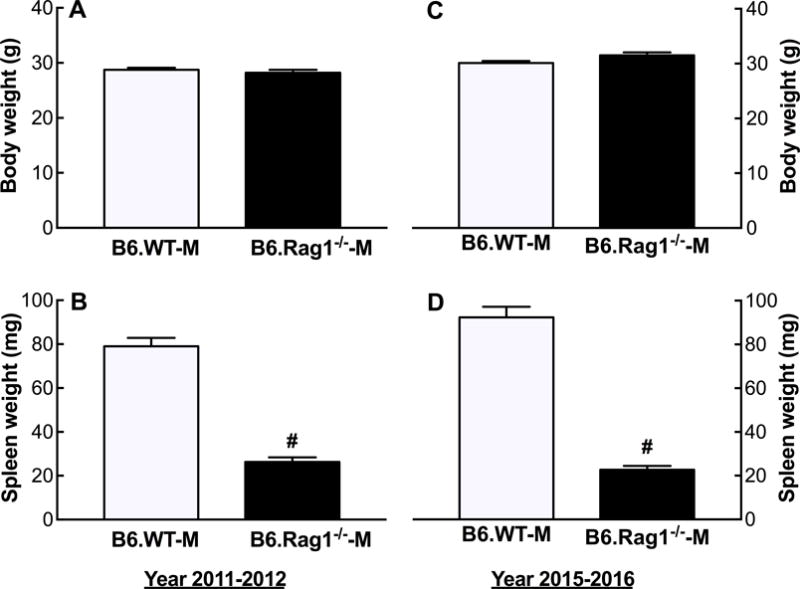
Body and spleen weights of male B6.WT and B6.Rag1−/− mice. Shown are the mean + SEM of the body (A,C) and spleen weights (B,D) determined in B6.WT-M and B6.Rag1−/−-M purchased from Jackson Laboratory in 2011–2012 (A,B) and 2015–2016 (C,D). #P<0.001 vs. B6.WT-M, same year; n=12–15.
MAP & HR
At 75 ng/kg/min, two weeks of Ang II infusion did not significantly increase MAP in B6.WT-M [MAP(mm Hg): Basal, 114 ± 5.5 vs Ang II, 121 ± 4.4; ns] or in B6.Rag1−/−-M [MAP(mm Hg): Basal, 117 ± 1.8 vs Ang II, 123 ± 4.8; ns]. At 200 ng/kg/min, Ang II infusion gradually increased MAP over two weeks in both B6.WT-M and B6.Rag1−/−-M with the peak MAP response occurring at 12 days (Figure 3A). No differences in the time course or peak response were found between B6.WT-M and B6.Rag1−/−-M (P= 0.35; Two-way ANOVA: Factors of strain & time). At 490 ng/kg/min, Ang II infusion rapidly increased MAP in both B6.WT-M and B6.Rag1−/−-M with the peak response occurring at 9–12 days. No differences in the time course or peak response were found between B6.WT-M and B6.Rag1−/−-M (P= 0.82; Two-way ANOVA: Factors of strain & time). No differences in the time course or magnitude changes in HR after infusion of Ang II were observed at the 200 and 490 doses in either strain (Figure 3B).
Figure 3.
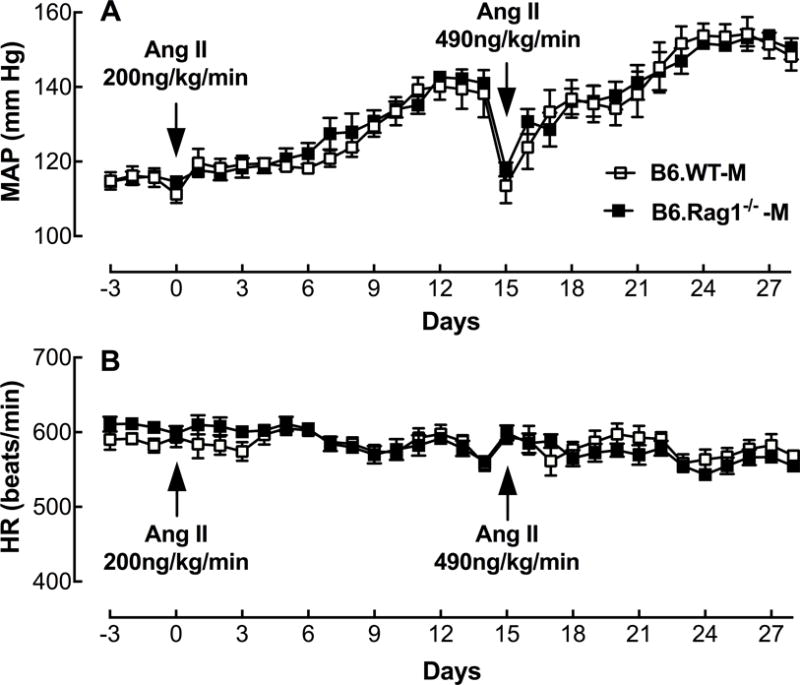
Effect of Ang II dose on MAP in male B6.WT and B6.Rag1−/− purchased from Jackson Laboratory in 2015–2016. Shown are the mean ± SEM of MAP (A) and HR (B) in B6.WT-M (opened square) and B6.Rag1−/−-M (closed square) infused with Ang II at 200 ng/kg/min (n=7–8) and 490 ng/kg/min (n=6–8).
T-cells
Rag1−/−-M purchased in 2015 and 2016 were T-cell deficient. The number of T cells in the spleen were less than 3% of B6.WT-M, similar to the mice purchased in 2011 and 2012 with which we reported sex-specific T-cell regulation of Ang II-dependent hypertension3 (Figure 4).
Figure 4.
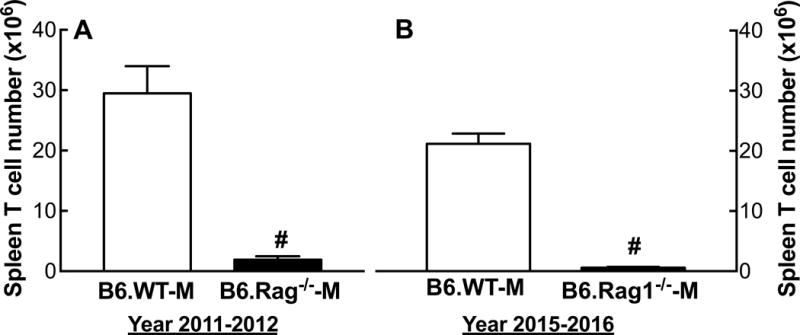
Spleen T-cell numbers in male B6.WT and B6.Rag1−/−-M. Shown are the mean + SEM of the total CD3+ T-cell count isolated from whole spleen in B6.WT-M and B6.Rag1−/−-M purchased from Jackson Laboratory in 2011–2012 (A) and 2015–2016 (B). #P<0.001 vs. B6.WT-M, same year; n=12–17.
125I-[Sar1,Ile8]Ang II Binding
There were no detectable differences in specific binding of 125I-[Sar1,Ile8]Ang II to glomeruli in in B6.WT-M purchased recently and prior to 2015 (Figure 5). In contrast, there was 1.4-fold higher 125I-[Sar1,Ile8]Ang II binding in glomeruli isolated from recently purchased B6.Rag1−/−-M compared to mice purchased prior to 2015.
Figure 5.
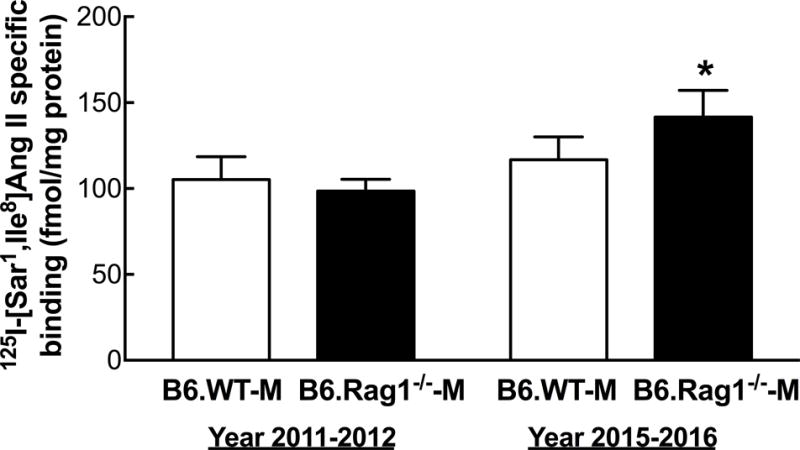
AT1R specific binding in male B6.WT and B6.Rag1−/−-M. Shown are the mean + SEM of 125I-[Sar1,Ile8]-Ang II specific binding to isolated glomeruli from B6.WT-M and B6.Rag1−/−-M purchased from Jackson Laboratory in 2011–2012 and 2015–2016. *P<0.02 vs 2011–2012, same strain; n=5–8.
Systematic Review
Through a systematic literature search, 17 unique records were identified that cited use of Rag1−/− and measurement of blood pressure. All were screened. Sixteen publications included original data and of these (Table 1), 10 included blood pressure measurements after Ang II infusion in B6.Rag1−/−-M. Seven publications included comparative blood pressure measurements in B6.WT-M littermates.
Table 1.
Reports comparing Ang II-induced increases in blood pressure in B6.Rag1−/−-M, B6.WT-M & CD3➔B6.Rag1−/−-M. All blood pressure studies were conducted in conscious B6.Rag1−/−-M or B6.WT-M purchased from Jackson Laboratory using osmotic minipumps to deliver Ang II.
| Author | Year | Ang II dose (ng/kg/min) |
Blood Pressure (mm Hg) | ||
|---|---|---|---|---|---|
| B6.Rag1−/−-M ΔAngII-Basal |
B6.WT-M ΔAngII-Basal |
CD3➔B6.Rag1−/−-M Δ Ang II-Basal |
|||
| Guzik et al.2 | 2007 | 490 | SBP (tail cuff) Basal: 118±4 Ang II: 139±5 Δ21 |
SBP (tail cuff) Basal: 118±2 Ang II: 159±5 Δ41 |
SBP (tail cuff) Basal: 107±3 Ang II: 154±5 Δ47 |
| Uchida et al.4 | 2010 | 1000 | SBP (tail cuff) Basal: 113±2 Ang II: 144±4 Δ31 |
SBP (tail cuff) Basal: 112±2 Ang II: 160±4 Δ48 |
|
| Vital et al.5 | 2010 | 2000 | MAP (indwelling catheter) Basal: 111±3 Ang II: 115±3 Δ5 |
MAP (indwelling catheter) Basal: 111±3 Ang II: 126±2 Δ15 |
|
| Senchenkova et al.14 | 2011 | 1000 | SBP (tail cuff) Ang II: 159±9 Δ52* |
SBP (tail cuff) Basal: 107±2 Ang II: 154±9 Δ47 |
SBP (tail cuff) Ang II: 173±5 Δ66* |
| Ji et al.3 | 2014 | 200 490 1000 |
MAP (telemetry) Basal: 111±1.4 Ang II: 200: 116±8 Δ5 490: 122±2 Δ16 1000:145±15 Δ34 |
MAP (telemetry) Basal: 114±1.3 Ang II: 200: 142±7 Δ28 490: 143±3 Δ29 1000:146±4 Δ32 |
MAP (telemetry) Basal: 112±1 Ang II: 141±3 Δ29 |
| Pollow et al.6 | 2014 | 490 | SBP (tail cuff) Basal: 101±3 Ang II: 123±5 Δ22 |
SBP (tail cuff) Basal: 103±3 Ang II: 141±7 Δ38±7 |
|
| Trott et al.13 | 2014 | 490 | SBP (tail cuff) Basal: 119±2 Ang II: 143±2 Δ24 |
SBP (tail cuff) Basal: 119±2 Ang II: 178±7 Δ61 |
SBP (tail cuff) Basal: 119±3 Ang II: 173±5 Δ66 |
| Wu et al.22 | 2014 | 490 | SBP (telemetry) Basal: 123±4 Ang II: 139±4 Δ16 |
SBP (telemetry) Basal: 120±4 Ang II: 170±3 Δ50 |
SBP (telemetry) Basal: 120±4 Ang II: 178±8 Δ58 |
| Mian et al.10 | 2016 | 490 | SBP (telemetry) Basal: 124±1 Ang II: 161±4 Δ37 |
SBP (telemetry) Basal: 121±4 Ang II: 160±5 Δ40 |
|
Calculated from B.6.WT-M basal SBP.
Of the 3 that did not include comparative measurements in B6.WT, 2 papers6, 13 compared B6.Rag1−/−-M blood pressure measurements with B6.Rag1−/−-M after reconstituting the B6.WT CD3+ T-cell population by adoptive transfer (CD3➔B6.Rag1−/−-M) (Table 1). Five papers compared Ang II-induced increases in blood pressure in B6.Rag1−/−-M with both B6.WT-M and CD3➔B6.Rag1−/−-M (Table 1). One paper14 reported that adoptive transfer of B6.WT CD3+ T-cells into B6.Rag1−/− restored the hypertensive response to Ang II to a level observed in Ang II-infused B6.WT; however, this data was not shown.
Discussion
The main finding of this report is that compared to their male B6.WT littermates, B6.Rag1−/−-M purchased from Jackson Laboratory in 2015 and 2016 were no longer resistant to Ang II-induced hypertension (Figure 3). This finding is in stark contrast to our previous results3 and those of four other independent laboratories including Drs. DG Harrison2, 15, 16, A Daugherty & L. Cassis4, DN Granger5, and HL Brooks & M Hay6 (Table 1).
Our new data suggests that a genetic drift has occurred in the Jackson Laboratory B6.129S7-Rag1tm1Mom/J mouse strain (catalog #002216), which has led to a loss of resistance to Ang II-induced hypertension. This change in phenotype is independent of T-cell deficiency since genotyping of these recently purchased B6.Rag1−/−-M demonstrated that the Rag1 gene remained disrupted (Figure 1). Furthermore, the markedly reduced spleen weights (Figure 2), minimal T-cell count in Rag1−/−-M spleens (Figure 4) and low numbers of PBMC confirms that these mice were deficient in their ability to produce mature T-cells.
A recently published study by Mian et al.13 at McGill University supports our current observation. Using tail cuff assessments of SBP, they showed that two weeks of Ang II infusion at 490 ng/kg/min in the B6.Rag1−/−-M caused a 41 mm Hg increase in SBP (Table 1). This increase in SBP was markedly larger than previous reports using the tail cuff method including studies by Guzik et al.2 (Δ21 mm Hg), Trott16 (Δ24 mm Hg) and Pollow et al.6 (Δ22 mm Hg) (Table 1).
Mian et al.13 did not compare the magnitude of SBP increases induced by Ang II infusion in B6.Rag1−/−-M to B6.WT-M littermates; however, they did compare their data with Ang II infusion results in B6.Rag1−/−-M beginning 2 weeks after adoptive transfer of B6.WT CD3+ T-cells and found no differences in Ang II-induced increases in SBP between B6.WT-M and T-cell reconstituted B6.Rag1−/−-M. In contrast, previous studies from four independent laboratories conducted on B6.Rag1−/−-M purchased from Jackson Laboratory prior to 20152–6 have shown that reconstitution of T-cells into B6.Rag1−/−-M restores the blood pressure sensitivity to Ang II to B6.WT-M levels.
Only one study published before 2016 reported no differences in the magnitude of hypertension induced by Ang II between B6.WT-M and B6.Rag1−/−-M17; however, this study infused a high dose of Ang II (1000 ng/kg/min). We previously showed that Rag1−/−-M resistance to Ang II-induced increases in blood pressure is dependent on the dose of Ang II3. While we showed that blood pressure in B6.Rag1−/−-M purchased in 2011 and 2012 is resistant to elevation by slow pressor doses of Ang II (200 and 490 ng/kg/min), we observed no differences in blood pressure elevation at the 1000 ng/kg/min Ang II dose3. In contrast, in the mice purchased in 2015 and 2016, no differences in the elevation of MAP were observed between B6.WT-M and B6.Rag1−/−-M at the lowest dose of Ang II (200 ng/kg/min) that caused an increase in MAP (Figure 3A). Note, that at 75 ng/kg/min, Ang II did not significantly increase MAP in either strain.
In an effort to improve rigor and reproducibility of scientific research, the National Institutes of Health now requires grant applicants to report experimental details to ensure full transparency in interpreting results (https://grants.nih.gov/reproducibility/index.htm). A section on the authentication of key biological resources is also now required in all grant applications. Our findings indicate that in addition to using the standardized genetic nomenclature system to accurately describe mouse genes, mutations and strains (http://www.informatics.jax.org), experimental details of animal studies should also include the source, location and time period over which animals are bred.
Diet is also important to report especially since new studies suggest the microbiota can influence the blood pressure sensitivity to Ang II. Karbach et al.18 recently showed that blood pressure responses to Ang II were attenuated in germ free animals compared to WT-M and studies such as from Desvarieux et al.19 and Chen et al.20 have shown that infection is associated with hypertension. Thus, reporting SFB status (negative in this case) is also essential. It is unlikely, however, that differences in infection contributed to the loss in phenotype since if anything, we have become more proficient at animal handling with time and our facility has become cleaner rather than dirtier. Other important information to include in ‘Methods’ sections of manuscripts is the age of the breeders since the age of the dam and potentially the sire, can impact the developmental programming of hypertension21.
The two sources of genetic variation in inbred mouse strains are genetic contamination and genetic drift. Genetic contamination is the accidental intercrossing of mouse strains as a result of animal management errors. Jackson Laboratory, one of the major suppliers worldwide of inbred mouse strains, has a genetic quality control program (Genetic Stability Program) developed from over 85 years of experience that addresses this potential problem22. Thus, it is unlikely that the B6.Rag1−/−-M loss of resistance to the Ang II pressor effect is due to an accidental intercrossing especially since we consistently observed this loss over six separate purchases from Jackson Laboratory between March 2015 and October 2016.
The finding that there is a 1.4-fold higher level of AT1R binding to glomeruli in B6.Rag1−/−-M purchased recently compared to B6.Rag1−/−-M purchased prior to 2015 (Figure 5), suggests that the loss of the Ang II resistant blood pressure phenotype in the B6.Rag1−/−-M mouse could be due to spontaneous mutations that directly increased renal mechanisms involving the AT1R by which Ang II increases blood pressure. Thus, these putative mutations may mask the mechanisms by which the absence of T cells protects the mice from Ang II-induced increases in blood pressure. Alternatively, the differences in mice purchased recently from those purchased prior to 2015 may be a result of dilution of unrelated 129S genome pieces left over from the original backcrosses.
While inbred mouse strains are remarkably stable, genetic drift can occur through spontaneous and ‘quiet’ mutations that can become fixed in mouse colonies23. Thus, this loss in resistance to the pressor effects of Ang II could be due to a spontaneous mutation(s) that makes the T-cell deficient B6.Rag1−/−-M no longer a model of Ang II-resistant hypertension. While these findings may call into question the physiological importance of T-cells in Ang II-dependent hypertension, a study by Batchu et al.24 using the same Jackson Laboratory strain of B6.Rag1−/−-M showed that B6.Rag1−/−-M were resistant to DOCA-salt-induced hypertension. The SBP increased by only 4 mm Hg in B6.Rag1−/−-M compared to 21 mm Hg in B6.WT-M or to 19 mm Hg in CD3➔B6.Rag1-M. Taken together, these findings raise the possibility that T-cell modulation of blood pressure may be more dependent on sodium than Ang II in B6.Rag1−/−-M.
The SS-Rag1em1Mcwi rat (SS.Rag1−/−) is a model of T-cell deficiency on a Dahl salt-sensitive background25. Mattson et al.25 demonstrated these rats are less susceptible to the development of salt-sensitive hypertension. Twenty one days of a high sodium diet (4%NaCl) resulted in a 30 mmHg increase in MAP in the SS.Rag1−/−-M which was much lower than the 50 mmHg increase observed in male WT littermates. A year later in 2014, this laboratory showed similar results in a second Dahl salt-sensitive rat model of functional T-cell deficiency, the SS-Cd247em1Mcwi rat (SS.CD247−/−). The CD247 gene codes for the CD3 ζ chain, which is required for T-cell signaling. Three weeks of salt loading increased MAP by only 12 mmHg in the SS.CD247−/−-M compared to 32 mmHg in male WT littermates26. Both these studies suggest that in the absence of functional T-cells, male Dahl salt-sensitive rats are less susceptible to developing salt-sensitive hypertension. Determining if these rats are also resistant to Ang II-induced increases in blood pressure could provide additional insight into the role of T-cells in Ang II-dependent hypertension.
Even if the data on T-cell modulation of Ang II-dependent hypertension is questionable, there is strong evidence that T-cells modulate other important physiologic processes mediated by Ang II. Mian et al.13 showed that adoptive transfer of T regulatory cells protected T cell reconstituted male B6.Rag1 deficient mice (CD3➔ B6.Rag1−/−-M) from Ang II-induced endothelial dysfunction even though no differences in SBP were observed.
Whatever the case, accidental discovery of new phenotypes arising from quiet mutations can ultimately yield important insights for researchers23. For example, investigators discovered that a colony of C57BL/6 maintained at the Dunn School of Pathology in Oxford, UK had nerves that were resistant to degeneration27. This led to the identification of the Wallerian degeneration slow (Wld) mutation28 and now the C57BL/6OlaHsd-Wld is a widely studied model that has provided valuable insights into neural degeneration. Thus, investigating how the B6.Rag1−/−-M mouse has lost its resistance to Ang II-dependent hypertension may also yield important information for those investigators studying the role of T-cells in hypertension.
Perspectives.
The most important aspect of this research is that T-cell deficient Rag1−/− male mice on the C57BL/6J background (B6.Rag1−/−-M) purchased from Jackson Laboratory as of 2015 are no longer a model of resistance to Ang II-induced hypertension. This phenotypic change impacts the design of studies using this strain to study mechanisms of T-cell modulation of Ang II-dependent blood pressure control. Spontaneous mutations leading to phenotypic change is common in animals including inbred mouse strains as a result of the universal drive to increase genetic variation. Therefore, experimental details regarding the location and time period over which animals are bred should be reported in scientific method descriptions to promote rigor and reproducibility in the scientific literature.
Novelty and Significance.
What Is New?
The previously reported phenotype of resistance to Ang II-induced hypertension in the B6.129S7-Rag1tm1Mom/J Jackson Laboratory mouse has been lost. The time course and magnitude increase in MAP induced by Ang II infusion at 200 and 490 ng/kg/min was identical between B6.WT-M and B6.Rag1−/−-M in mice purchased in 2015 and 2016.
This loss of resistance to Ang II-induced hypertension is independent of T-cells; B6.Rag1−/−-M remain T-cell deficient.
Glomerular AT1R specific binding is 1.4-fold higher in recently purchased B6.Rag1−/−-M compared to mice purchased prior to 2015.
What Is Relevant?
Investigators using male B6.129S7-Rag1tm1Mom/J to study T-cell modulation of Ang II-dependent mechanisms of blood pressure control need to be aware of this phenotypic change in the Jackson Laboratory strain to avoid loss of time, waste of expensive resources and the uselessness of uninterpretable results.
Summary.
These findings serve as an important reminder to investigators that the universal drive for genetic variation occurs in all animals including inbred mouse strains and that spontaneous mutations can lead to phenotypic change, which can compromise experimental reproducibility over time and location.
Acknowledgments
Sources of Funding: This work was supported by grants from the National Institutes of Health: TL1-TR001431 (AVP), UL1-TR001409 (KS) and R01-HL119380 (KS, HJ).
Footnotes
Disclosures: None.
References
- 1.Jones JM, Gellert M. The taming of a transposon: V(D)J recombination and the immune system. Immunol Rev. 2004;200:233–248. doi: 10.1111/j.0105-2896.2004.00168.x. [DOI] [PubMed] [Google Scholar]
- 2.Guzik TJ, Hoch NE, Brown KA, McCann LA, Rahman A, Dikalov S, Goronzy J, Weyand C, Harrison DG. Role of the T cell in the genesis of angiotensin II induced hypertension and vascular dysfunction. J Exp Med. 2007;204:2449–2460. doi: 10.1084/jem.20070657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ji H, Zheng W, Li X, Liu J, Wu X, Zhang MA, Umans JG, Hay M, Speth RC, Dunn SE, Sandberg K. Sex-specific T-cell regulation of angiotensin II-dependent hypertension. Hypertension. 2014;64:573–582. doi: 10.1161/HYPERTENSIONAHA.114.03663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Uchida HA, Kristo F, Rateri DL, Lu H, Charnigo R, Cassis LA, Daugherty A. Total lymphocyte deficiency attenuates Ang II-induced atherosclerosis in males but not abdominal aortic aneurysms in apoE deficient mice. Atherosclerosis. 2010;211:399–403. doi: 10.1016/j.atherosclerosis.2010.02.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vital SA, Terao S, Nagai M, Granger DN. Mechanisms underlying the cerebral microvascular responses to angiotensin II-induced hypertension. Microcirculation. 2010;17:641–649. doi: 10.1111/j.1549-8719.2010.00060.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pollow DP, Uhrlaub J, Romero-Aleshire MJ, Sandberg K, Nikolich-Zugich J, Brooks HL, Hay M. Sex differences in T-lymphocyte tissue infiltration and development of angiotensin II hypertension. Hypertension. 2014;64:384–390. doi: 10.1161/HYPERTENSIONAHA.114.03581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mombaerts P, Lacomini J, Johnson RS, Herrup K, Tonegawa S, Papaioannou VE. RAG-1-deficient mice have no mature B and T lymphocytes. Cell. 1992;68:869–877. doi: 10.1016/0092-8674(92)90030-g. [DOI] [PubMed] [Google Scholar]
- 8.Ivanov, Atarashi K, Manel N, et al. Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell. 2009;139:485–498. doi: 10.1016/j.cell.2009.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Celaj S, Gleeson MW, Deng J, O’Toole GA, Hampton TH, Toft MF, Morrison HG, Sogin ML, Putra J, Suriawinata AA, Gorham JD. The microbiota regulates susceptibility to Fas-mediated acute hepatic injury. Lab Invest. 2014;94:938–949. doi: 10.1038/labinvest.2014.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Miller SA, Dykes DD, Polesky HF. A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res. 1988;16:1215. doi: 10.1093/nar/16.3.1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ji H, Zheng W, Wu X, Liu J, Ecelbarger CM, Watkins R, Arnold AP, Sandberg K. Sex chromosome effects unmasked in angiotensin II-induced hypertension. Hypertension. 2010;55:1275–1282. doi: 10.1161/HYPERTENSIONAHA.109.144949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zheng W, Ji H, Szabo Z, Brown PR, Yoo SE, Sandberg K. Coordinate regulation of canine glomeruli and adrenal angiotensin receptors by dietary sodium manipulation. Kidney Int. 2001;59:1881–1890. doi: 10.1046/j.1523-1755.2001.0590051881.x. [DOI] [PubMed] [Google Scholar]
- 13.Mian MO, Barhoumi T, Briet M, Paradis P, Schiffrin EL. Deficiency of T-regulatory cells exaggerates angiotensin II-induced microvascular injury by enhancing immune responses. J Hypertens. 2016;34:97–108. doi: 10.1097/HJH.0000000000000761. [DOI] [PubMed] [Google Scholar]
- 14.Hoch NE, Guzik TJ, Chen W, Deans T, Maalouf SA, Gratze P, Weyand C, Harrison DG. Regulation of T-cell function by endogenously produced angiotensin II. Am J Physiol Regul Integr Comp Physiol. 2009;296:R208–216. doi: 10.1152/ajpregu.90521.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wu J, Saleh MA, Kirabo A, et al. Immune activation caused by vascular oxidation promotes fibrosis and hypertension. J Clin Invest. 2016;126:50–67. doi: 10.1172/JCI80761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Trott DW, Thabet SR, Kirabo A, Saleh MA, Itani H, Norlander AE, Wu J, Goldstein A, Arendshorst WJ, Madhur MS, Chen W, Li CI, Shyr Y, Harrison DG. Oligoclonal CD8+ T cells play a critical role in the development of hypertension. Hypertension. 2014;64:1108–1115. doi: 10.1161/HYPERTENSIONAHA.114.04147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Senchenkova EY, Russell J, Kurmaeva E, Ostanin D, Granger DN. Role of T lymphocytes in angiotensin II-mediated microvascular thrombosis. Hypertension. 2011;58:959–965. doi: 10.1161/HYPERTENSIONAHA.111.173856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Karbach SH, Schonfelder T, Brandao I, et al. Gut microbiota promote angiotensin II-induced arterial hypertension and vascular dysfunction. J Am Heart Assoc. 2016;5:e003698. doi: 10.1161/JAHA.116.003698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Desvarieux M, Demmer RT, Jacobs DR, Jr, Rundek T, Boden-Albala B, Sacco RL, Papapanou PN. Periodontal bacteria and hypertension: The oral infections and vascular disease epidemiology study (INVEST) J Hypertens. 2010;28:1413–1421. doi: 10.1097/HJH.0b013e328338cd36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen LW, Chien CY, Yang KJ, Kuo SF, Chen CH, Chien RN. Helicobacter pylori infection increases insulin resistance and metabolic syndrome in residents younger than 50 years old: A community-based study. PLoS One. 2015;10:e0128671. doi: 10.1371/journal.pone.0128671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Alexander BT, Dasinger JH, Intapad S. Fetal programming and cardiovascular pathology. Compr Physiol. 2015;5:997–1025. doi: 10.1002/cphy.c140036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Taft RA, Davisson M, Wiles MV. Know thy mouse. Trends Genet. 2006;22:649–653. doi: 10.1016/j.tig.2006.09.010. [DOI] [PubMed] [Google Scholar]
- 23.Stevens JC, Banks GT, Festing MF, Fisher EM. Quiet mutations in inbred strains of mice. Trends Mol Med. 2007;13:512–519. doi: 10.1016/j.molmed.2007.10.001. [DOI] [PubMed] [Google Scholar]
- 24.Batchu SN, Hughson A, Wadosky KM, Morrell CN, Fowell DJ, Korshunov VA. Role of axl in T-lymphocyte survival in salt-dependent hypertension. Arterioscler Thromb Vasc Biol. 2016;36:1638–1646. doi: 10.1161/ATVBAHA.116.307848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mattson DL, Lund H, Guo C, Rudemiller N, Geurts AM, Jacob H. Genetic mutation of recombination activating gene 1 in Dahl salt-sensitive rats attenuates hypertension and renal damage. Am J Physiol Regul Integr Comp Physiol. 2013;304:R407–414. doi: 10.1152/ajpregu.00304.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rudemiller N, Lund H, Jacob HJ, Geurts AM, Mattson DL, PhysGen Knockout Program CD247 modulates blood pressure by altering T-lymphocyte infiltration in the kidney. Hypertension. 2014;63:559–564. doi: 10.1161/HYPERTENSIONAHA.113.02191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lunn ER, Perry VH, Brown MC, Rosen H, Gordon S. Absence of wallerian degeneration does not hinder regeneration in peripheral nerve. Eur J Neurosci. 1989;1:27–33. doi: 10.1111/j.1460-9568.1989.tb00771.x. [DOI] [PubMed] [Google Scholar]
- 28.Gagnon LH, Longo-Guess CM, Berryman M, Shin JB, Saylor KW, Yu H, Gillespie PG, Johnson KR. The chloride intracellular channel protein CLIC5 is expressed at high levels in hair cell stereocilia and is essential for normal inner ear function. J Neurosci. 2006;26:10188–10198. doi: 10.1523/JNEUROSCI.2166-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]