

Abstract
To many of us in the field, working on matrix metalloproteinases (MMPs) has felt like riding a roller coaster, traveling through times of both excitement and despair. I was fortunate to join the ride when it was a mere carousel of three activities thought to target the proteins that comprise the extracellular matrix (ECM). New technologies brought the thrills of discovery as we uncovered specific proteinase genes and defined specialized activities in different cellular processes. The MMPs and the sister families of “a disintegrin and metalloproteinase” (ADAMs), ADAMs with thrombospondin domains (ADAM-TS), and Astacins are now recognized as key signaling “scissors” that drive rapid changes in a plethora of cellular pathways. My many excellent colleagues and collaborators and I were enthused to contribute to the early development of the field and continue to be amazed at its growth and sophistication. In contrast, the hype and failure of early inhibitor discovery have dogged our standing with the pharmaceutical industry and grant-giving bodies. However, the true believers have kept going, and knowledge of particular functions of MMPs and their contributions to disease progression has progressed. Recognition of the strategic importance of proteinase function should inspire more work harnessing new technologies such as imaging, proteomics, and gene editing to generate a more precise understanding of individual situations. New approaches to inhibitor design and assessment are possible, and the consequent ability to precisely abrogate specific MMP activity could contribute to the fight against a number of pathologies with unmet needs. What a ride it could be!
Keywords: ADAMTS, extracellular matrix, matrix metalloproteinase (MMP), proteinase, tissue inhibitor of metalloproteinase (TIMP)
Introduction
Inspired by the newly emerging understanding of DNA structure and function in the early 1960s, I abandoned my childhood wish to become a farmer and decided to enroll in a B. Sc. course in Biochemistry at Birmingham University. I then took a brief sojourn doing service for Voluntary Service Overseas (VSO) as a biology teacher in Montserrat, West Indies, which was the closest to the modern equivalent of a “gap year.” The VSO scheme does not allow entrants a choice of location, and my vision of a jungle school in Asia was supplanted by a placement on an exotic island teaching children a British school curriculum! I returned to Birmingham to undertake a Ph. D. with Deryck Walker in the Biochemistry department. Here I became interested in malic enzyme function and followed up my Ph. D. with a brief postdoctoral post with Nick Kuhn working on lactose synthetase. I was lucky enough to gain a Royal Society/NATO Fellowship to go to the laboratory of the Nobel Laureate Feodor Lynen in the then new Max Planck Institute at Martinsried, Munich. I pursued Prof Lynen's interest in multi-enzyme complexes and simply enjoyed the academic freedom of research on topics that attracted one's curiosity, with the possible objective of becoming a university teacher in the long run. For my return to the UK, I opted to join John J. Reynolds' group at the Strangeways Research Laboratory in Cambridge where I could continue to pursue my fascination with enzymes, but this time in the form of proteinases.
“The Strangeways” was originally established in 1911 and became a mecca for the development of tissue culture techniques under the directorship of Dr. T. S. P. Strangeways and later Dame Honor Fell (1) (Fig. 1). Although situated in the university town of Cambridge UK, Strangeways remained independent of the University until 1997 and in the early years played host to many young scientists with independent ideas (1). Later, under the directorship of John Dingle, research into “connective tissue diseases” became the major thrust of the laboratory (Fig. 2). I really wanted to continue to work on enzyme biochemistry but first had to overcome the huge culture shock and learning curve of coming into what was essentially a laboratory focused on basic aspects of arthritis research. My challenge was to study the destructive processes underlying arthritic diseases at a molecular level with minimal protein resources. Strangeways offered a unique environment in the UK where one could really pursue one's own scientific interests within a very broad framework. It was a great fortune for me that the eminent proteinase biochemist Alan Barrett was already guiding proteinase biochemistry at the Strangeways and, as a consequence, a host of exciting American visitors were coming through the door with stimulating ideas and approaches. The current dogma at that time, originally developed by researchers in the USA, was that secreted metal-dependent proteinases, later to be renamed as MMPs,2 were responsible for the cleavage of extracellular matrix proteins and could be considered as the “arbiters of destruction” in arthritis and cancer. Zena Werb and Ted Harris had both been working with John Reynolds and had recently departed, leaving a wealth of questions to pursue. So I stepped onto the MMP roller coaster (Fig. 3), initially as a postdoctoral fellow and later as the joint Head of Department and an Arthritis Research Campaign Senior Fellow. From then on, the MMP story, with all its thrills and spills, is a reflection of how an international community of researchers can function as one big family, all pulling together to unravel the mysteries of what started as enigmatic activities with no initial understanding of their fundamental nature or mechanisms of action.
Figure 1.

Strangeways Research Laboratory, Cambridge UK. The Strangeways Research Laboratory was founded by T. S. P. Strangeways (left inset) in 1905 as the Cambridge Research Hospital to promote the scientific investigation of joint disease. The building shown was opened in 1912 and was renamed in 1928 when Dame Honor Fell (right inset) became Chief of the Laboratory. “The Strangeways” was the home of tissue culture developments in the UK, and many famous scientists of the time visited and worked there (1). It remained independent of Cambridge University until 1997, albeit with extensive collaborative links.
Figure 2.

Strangeways Research Laboratory in 1980 under the directorship of John Dingle. The Laboratory was largely supported by the Medical Research Council, the Empire Rheumatism Council (renamed Arthritis Research Council and now Arthritis Research UK), and the Wellcome Trust (1). The author is 2nd from the left in the 2nd row from the front. John Reynolds is at 3rd from the left, and John Dingle is at 10th from the left in the front row. Alan Barrett is 5th from the right in the front row.
Figure 3.

John Reynolds' Cell Physiology group at Strangeways. John is at the right, in the back row. The author is seated at the far left.
A brief early history
The MMP story started in the early 1960s with the discovery of collagenolytic activities and their characterization as metal-dependent activities, functioning at neutral pH in extracts of uterine tissues (2) and then in cultured tissues (tadpole gut, gill, and intestine and mammalian uterus, bone, and skin wounds) (3, 4). Over the next decade, biochemical studies were focused on activities isolated from cultured human skin, involuting uterus, and rheumatoid synovium that secreted fairly good amounts of the activity designated as “collagenase” (5–7). At Strangeways, John Reynolds' interests were largely focused on collagen turnover in growing bones. To carry out fundamental biochemical evaluations, our work required the laborious culture of young rabbit calvariae as a source of the matrix-degrading enzymes. We identified both a collagenolytic activity and a protein inhibitor (8) and subsequently two further metal-dependent activities degrading gelatin and proteoglycans, respectively, all of which were secreted into the culture medium (9). Reports from around the world showed that comparable activities were produced in the cultures of many tissues, and eventually these were all grouped together using the MMP nomenclature (for more history of the field, see Ref.10 and further discussion below).
There were also indications that these activities were regulated by the secreted protein inhibitor we had discovered. Tim Cawston led the Strangeways' purification of the inhibitor from rabbit bone cultures to confirm that one protein could be responsible for inhibiting all the metal-dependent matrix-degrading activities isolated from the same system, and it was tentatively named tissue inhibitor of metalloproteinases (TIMP) (11). Meanwhile a collagenase inhibitor was identified from skin in 1979 and purified from fibroblast cultures in 1983 (12). Subsequently, three further related TIMPs were identified from their cDNAs, isolated, and characterized (reviewed in Ref. 13). The question of the nature of the observed latency of the secreted forms of metalloproteinases occupied many of the laboratories over the next few years (14). In 1978, Stricklin et al. (15) had shown that purified latent collagenase could be activated by trypsin treatment, and we and others had found that treatment with organomercurials could also activate the rabbit bone enzyme (16). It was proposed that these might act to dissociate a complex of collagenase and the natural inhibitor, but this was later dismissed as it became evident that the matrix-degrading metalloenzymes were secreted in an N-terminally extended pro form and that propeptide cleavage was needed to generate the full activity. Looking back, it is amazing how much progress we were able to make given the very small amounts of crude activities available to many researchers.
It is now known that most proteolytic enzymes, not just the MMPs, are synthesized as inactive zymogens and activation by proteolytic removal of a propeptide is a key regulatory step. In the case of the MMPs, the N-terminal propeptide interacts with the active-site cleft, mediated in part by a cysteine sulfhydryl that interacts with the catalytic zinc ion (17). In subsequent studies, it became clear that the activation process for latent (pro)MMPs could be viewed as a form of proteolytic cascade with plasmin (generated extracellularly from plasminogen by the ubiquitous proteinase urokinase-like plasminogen activator) being a key player. Other proteinases, including some MMPs, can also act as extracellular activators, and even autolytic activation can occur. More recently, the discovery of the action of furin as an intracellular activator of some MMPs, as well as ADAM and ADAM-TS, has contributed to the appreciation of the complexity of the regulatory processes for extracellular proteolysis (Fig. 4) (18, 19).
Figure 4.

Activation of the secreted pro forms of the MMPs is thought to be due to the action of a number of other cellular proteinases, including plasmin and MMPs, as well as self-activation mechanisms. The membrane-type MMPs are activated by furin during secretion (18, 19).
In the 1980s, cDNA cloning became widespread, and the subsequent advent of recombinant protein production techniques, which were seminal to our efforts at Strangeways, was aided by our key collaboration with Andy Docherty and colleagues at the nascent UK biotechnology company Celltech. Initially, we were able to clone and express TIMP1, confirming its similarity to the inhibitor found in many tissue and cell cultures (20). Collagenase and the proteoglycan-degrading proteinase were also cloned, and the zymogenic nature of both enzymes, as well as the ability of stromelysin to act as a procollagenase activator, as proposed by other laboratories, was confirmed. In 1986, Okada et al. (21) proposed the nomenclature of MMPs, with collagenase being MMP1, gelatinase being MMP2, and proteoglycanase (already renamed stromelysin) being MMP3. Subsequently, ever more MMPs (23 in humans) were being discovered by the collective cloning efforts of different laboratories around the world (and a few were laid to rest!). The roller coaster soared higher! The numerical MMP nomenclature was adopted by the International Union of Biochemistry and Molecular Biology, and the concept of the MMP “family” was established, being characterized by a three-histidine zinc-binding catalytic motif (His-Glu-Xaa-Xaa-His + His) and a conserved methionine (Met) following the active-site residues. A glutamate residue within the catalytic motif activates a zinc-bound H2O molecule, providing the nucleophile that cleaves peptide bonds.
Subsequently, it was recognized that metalloproteinase families with very similar active-site motifs, but vastly differing constituent domains, existed and could be grouped into a clan termed the “Metzincins” (22) (http://merops.sanger.ac.uk). The families included the ADAMs, the ADAM-TS, and the human Astacins (Ref. 23 and references therein, and Refs. 24–26) (Fig. 5). The clan members are all largely secreted and play substantial roles in the pericellular activities of many cell types. Below I will describe the problems generated by the existence of over 60 proteinases with vastly different functions, but highly similar active-site residues and catalytic mechanisms, when addressing the issue of inhibitor design!
Figure 5.

MMPs, also named Matrixins, are a family within the clan of metallo-endopeptidases called the Metzincins (18, 23, 33). Other Metzincin families that largely function extracellularly include the ADAMs, the ADAM-TS, and the Astacins, including the meprins and bone morphogenetic protein 1 (BMP1). The MMPs share a common domain structure: the pre-domain that contains a signal peptide responsible for secretion; the pro-domain that keeps the enzyme inactive by an interaction between a cysteine residue and the Zn2+ ion group from the catalytic domain; and the hemopexin-like C-terminal domain, which is linked to the catalytic domain by a flexible hinge region. MMP7 and MMP26 lack the hinge region and the hemopexin domain. MMP2 and MMP9 contain a fibronectin type II motif inserted into the catalytic site, and MT-MMPs have a transmembrane domain or a glycosylphosphatidylinositol (GPI) anchor at the C terminus. MMP23 has unique features: the N-terminal signal anchor that targets MMP23 to the cell membrane, a cysteine array, and an immunoglobulin-like domain. Figure from Vandenbrouke and Libert (30). Reprinted by permission from Macmillan Publishers Ltd.: Nature Drug Discovery 13, 904–927 copyright (2014).
One of our major contributions at that time was to examine the domain structure and function of a number of the MMP family members (Fig. 5), as well as the TIMPs. This was largely effected by the expression of domain deletion mutants and point mutagenesis, coupled with enzyme kinetic assessment of MMP functions, including TIMP binding. Seminal to the work carried out at Strangeways was the collaboration with the kinetics expert Frances Willenbrock, who was also at Celltech at that time.
As one example of the value of a kinetic approach that supported the cell biology studies of many laboratories, including ours, we investigated potential physiological activation mechanisms for proMMP2. Previous results had implicated a membrane-mediated process in the activation of secreted proMMP2 by stimulated cells, and had led to the identification of the membrane-type MMPs (MT-MMPs) as potential mediators of the activation process. It had also been observed that activation of proMMP2 at the cell surface required its C-terminal domain and that this allowed cell membrane association of MMP2. Further, both MMP14 (MT1-MMP) expressed at the surface of cells and proMMP2 were associated with TIMP2. However, it was not known how MMP14 activated proMMP2 and what role the inhibitor TIMP2 played in the process. We embarked on a study of proMMP2 activation by MMP14 and the role of TIMP2 using recombinant enzymes and mutagenesis strategies and addressed the questions of the precise kinetics and concentration dependence of their interactions. We assessed the role of the C-terminal domain of proMMP2 by the use of C-terminal domain mutants to define the role of interactions of proMMP2 and MMP14 in the binding of TIMP2 and in the cell-associated activation of proMMP2. TIMP2 was shown to be essential for the activation process by binding to proMMP2 through interactions between the C-terminal domain of the enzyme and the three C-terminal loops and the charged tail of the inhibitor. Soluble constructs of MMP14 were used to demonstrate that binding with TIMP2 occurs primarily through N-terminal domain interactions, leaving the C-terminal domain free for interactions with proMMP2. We also found that an initial cleavage of proMMP2 by MMP14 led to subsequent MMP2 self-cleavage to generate the fully active form of MMP2. Further, the rate of autolytic activation of proMMP2 initiated by MMP14 cleavage could be potentiated by concentration of the proenzyme by binding to heparin (mimicking the potential role of cell-surface heparan sulfate proteoglycans (HSPGs)). Together, the data supported the concept that TIMP2 forms a receptor with MMP14 that regulates the concentration and efficient generation of functionally active MMP2, and that cell-surface molecules such as HSPGs may modulate activation (Fig. 6) (27).
Figure 6.

The model proposed for the activation of proMMP2 by MT1 MMP (MMP14) by the formation of an MT1 MMP-TIMP2 “receptor,” based on cell studies and kinetic analyses of MMP and TIMP2 mutants. This requires activated MT1 MMP to exist at the cell surface in the form of oligomers, minimally dimers. Formation of a complex with TIMP2 confined to N-domain interactions of both partners leaves the C-domain of TIMP2 free to bind the hemopexin domain of proMMP-2, presenting the prodomain of the latter for cleavage by the uninhibited molecule of MT1 MMP (27). This figure was originally published in Murphy et al. (2003) Role of TIMPs (tissue inhibitors of metalloproteinases) in pericellular proteolysis: the specificity is in the detail. Biochem. Soc. Symp. 70, 65–80 (27).
Development of inhibitors: Bumps in the ride
The early observations on the function of MMPs emphasized their ability to degrade components of the extracellular matrix (ECM). As secreted proteinases, functional at neutral pH, and with an extracellular zymogen activation mechanism, they were regarded as key orchestrators of ECM turnover in degradative diseases such as arthritis and in cell migration events driving tumor invasion and metastasis. Thus, in the 1990s, enthusiasm for targeting the MMPs soared in the pharmaceutical industry in their drive to develop potential therapies. Although we didn't have any substantial structural information on the MMPs in those early years, it seemed chemically relatively straightforward to target activities with a Zn2+ ion at the heart of the catalytic mechanism. Thus, scientists rapidly started making inhibitors, which largely consisted of simple peptide derivatives that contained a zinc-chelating moiety and would interact with residues in the active-site cleft. At least 50 of these were tested in animal models, with promising results, and quickly moved into clinical trials using patients with many different forms of cancer (28–30). Our roller coaster plummeted as all the trials failed, or presented side effects (31). Prolonged administration of inhibitors in patient treatment protocols during clinical trials was associated with musculo-skeletal problems, notably forms of tendinitis. This resulted in the use of lower, often inadequate, MMP inhibitor doses in subsequent trials. Trials were conducted in patients with end-stage disease with heroic end-point criteria (32). This explains the discrepancies in outcomes when compared with the mouse models conducted in early stages of disease that had demonstrated the effectiveness of MMP inhibitors. Unfortunately, these results have continued to blight the concept of MMP/metalloproteinase inhibition as a therapeutic approach, even today.
What went wrong? We now appreciate that the problems of these early attempts were manyfold: at that stage, it was not known that there are 23 human MMPs and that they also function to control cell processes as diverse as the functions of cytokines, chemokines, antimicrobial peptides, and cell-surface proteins (23). Consequently, MMP proteolytic activities modulate critical processes related to immunity, inflammation, and cell transformation, to name but a few, in addition to ECM remodeling. Such activities relate to both physiological as well as pathological processes. Further, the extent of the metalloproteinase families with a similar active-site structure within the Metzincin clan was not known, meaning that we couldn't appreciate the multiple outcomes a single inhibitor might effect. Indeed, subsequent research has shown that the inhibitors developed were frequently active against many members of the constituent families of the Metzincins.
Basic science catches up
Studies in subsequent decades, notably using individual MMP gene ablation models in mice (33), coupled with sophisticated proteomic technologies (34, 35), have given us an improved understanding of the multifarious roles of individual MMPs in physiological events as well as in disease. Interestingly, analyses of genetically engineered mice have shown that most MMPs have no, or expendable, roles in embryogenesis. This is surprising given the original concept that these enzymes orchestrated ECM remodeling, and is indicative of the need to understand how other proteinase families may have specific roles in developmental remodeling (and how they may even get reactivated in disease to work in concert with the MMPs). Turnover of ECM components is effected by some MMPs to control processes in stem cell biology, muscle biology, central nervous system homeostasis and disease, angiogenesis, tissue repair in skin and liver, inflammation, vascular disease, destructive lung disease, and some cancers. For instance, the collagenolytic enzymes MMP13, MMP14 (MT1-MMP), and MMP16 (MT3-MMP) are involved in the remodeling of the collagen matrix in bone and elsewhere (36, 37). Thanks to the information derived from a number of parallel studies, it has emerged that many MMPs act beyond the ECM with roles in the modulation of effectors within signaling pathways associated with inflammation and immunity. Notably, the generation and analysis of engineered mouse models causing gain or loss of individual MMP gene function have led to the discovery of a number of such novel functions as well as several clues about the causal relationships between MMP deregulation and the development of different human diseases. The identification of MMP polymorphisms and mutations involved in human diseases has also contributed to the growing picture of MMP function, although there have been some discrepancies in the effects of MMP modulation between mice and humans. Because many MMP-null mice had no overt phenotypic alterations, only further analyses of disease models activating specific cellular responses gave some insights into the contribution of MMPs (33). Similarly, MMP overexpression models gave useful guides to their activities in vivo (33). Indeed, the use of transgenic mouse models, instead of simply interrogating existing mouse biology, has proved to be seminal to our understanding of the significance of MMPs in disease. Interestingly, some studies have shown that MMPs can have beneficial effects in disease resolution (33, 38).
What we've learned from this body of work is that MMPs regulate cellular functions through tightly controlled processing of a variety of signaling events and interact with regulatory elements such as inhibitors and other proteinase pathways; the combined network of these interactions and elements has been termed the “proteinase web” (38). This interconnected structure makes it difficult to directly interpret all the data from transgenic mouse models where one factor is modulated. The advent of proteomic approaches (for proteinases, termed “degradomics”) has allowed researchers to dig deeper into the interactions between MMP function and signaling and proteolytic cascades (35). Mass spectrometry (MS)-based studies have evolved from the initial identification and quantitation of MMPs and TIMPs to the identification of substrate cleavage sites and eventually MMP activities in cells and tissues. A library of metalloproteinase-directed probes with complementary target selectivity was developed, and the labeling profiles in tissues and cells were analyzed by MS techniques (39). Further, a new generation of methods for the system-wide discovery of proteinase substrates that exploited the generation of new N and C termini upon proteolysis was extended to the determination of the cleavage sites of MMPs (40).
Moving on
For us, the realization that the MMPs and other Metzincins had so many potential roles in cellular interactions with the external microenvironment was at first rather daunting. However, we found our niche, undertaking biochemical studies of the ADAMs in collaboration with Andy Docherty and colleagues. We were in our element expressing, purifying, and characterizing these complex multi-domain proteinases. We were excited to discover that some were regulated by the TIMPs, and became engrossed in the mechanistic details of these interactions using recombinant proteins and serial mutagenesis strategies. In the midst of this happy time, we were dismayed at the closure of Strangeways following the retirement of the Director. My group moved to the School of Biological Sciences at the University of East Anglia (UEA), where I became the Professor of Cell Biology. At UEA, we decided to focus on membrane-associated Metzincins such as MMP14 and some of the most abundant ADAMs, including ADAM9, ADAM10, and ADAM17. The premise here was that these proteinases could be more closely regulated by trafficking activities of the cell and could rapidly effect seminal cellular responses as necessary. Further, deciphering the role of their extra-catalytic domains in enzyme function would provide a treasure trove of biochemical studies and should lead to new concepts in proteinase inhibition.
With our new focus on membrane-associated Metzincins and the functions of their extra-catalytic domains, we embarked on the development of antibodies that were sufficiently potent and specific to attain activity in animal models of disease. In the case of the membrane-associated proteinases, this can be challenging as the enzyme/substrate conformations are likely to differ markedly from their solution structures, necessitating screening in cell-based assays. In 2002, my group moved back to Cambridge, this time to the Department of Oncology at the University of Cambridge, initially housed in the Cambridge Institute for Medical Research and finally in a newly built Institute supported by Cancer Research UK (now called the Cancer Research UK Cambridge Institute), and we were fortunate to team up with groups developing sophisticated phage display technologies to produce potent antibodies. We succeeded in producing inhibitory antibodies to MMP14 that prevent collagen degradation at the cell surface. These targeted the hemopexin domain, or the catalytic domain outside the active-site cleft (41, 42). In other work, a cross domain antibody to ADAM17, binding both an extra-active-site cleft site on the catalytic domain and the cysteine-rich domain that we developed, was similarly active in vivo (43), allowing us to develop new concepts about metalloproteinase interactions with some of the growth factors that modulate tumorigenesis.
Outlook for the future: Looking down the track
With these and other steps forward, the roller coaster has taken off again, but cautious brakes are now being applied. The outcomes of the lack of efficacy of the early phase inhibitors had a very marked effect on attitudes toward the funding of academic research concerning MMPs and the other Metzincins, as well as concerning the efforts within the pharmaceutical industry. This is an opportunity lost in many ways, although the more recent discoveries of the academic community are slowly changing attitudes about this fascinating but challenging group of enzymes. With the advent of more precise definitions of the roles of different metalloproteinases, we can begin to understand both the negative (anti-target) as well as the positive effects of their inhibition and to target inhibitors more appropriately (38, 44). With current structural knowledge, the design of specific inhibitors of MMPs is possible, although it remains challenged by the remarkable similarities in the structure of the active-site clefts, which is thought to have resulted from extensive gene duplication. It may be useful to note that MMPs have a notable predilection to cleave at hydrophobic residues that would normally be buried in large protein substrates. The roles for specific conformational alterations, i.e. enhancement of substrate availability, may be key features of the regulation of MMP activities. In the case of ECM substrates, such changes could be effected by changes in ECM protein interactions due to mechanical, proteolytic, or other effectors. Hence, very subtle and specific microenvironment changes that vary according to the cellular situation need to be considered, in itself a technological challenge. Based in part on the original studies on MMP domain functions, much recent interest has focused on the concept of “exo-sites” of the MMPs that define specific proteolytic functions and the potential to target these alternative sites in the development of inhibitors. Several groups, including ours (see above), have used antibodies targeted at exosites, which supports the notion that specificity of action can be achieved by such approaches (41–43, 45, 46).
Since the early work on cloning and identifying MMPs (and their Metzincin relatives), we've uncovered many questions regarding their regulation at the gene and post-translational levels (47–49) (Fig. 7). Defining these mechanisms is itself a remarkable story because these enzymes are very tightly regulated beyond synthesis, through secretion, activation, ECM, and cell-surface sequestration and endocytic processes. Even substrate availability questions arise at a number of levels, ranging from delivery to the pericellular environment to conformational modulation, or simple masking by other proteins or glycosaminoglycans, all of which may determine proteolytic rates (48–50).
Figure 7.
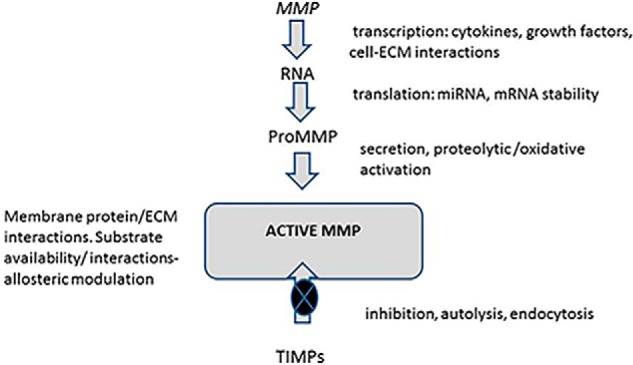
Matrix metalloproteinase regulation. MMP genes are regulated at the transcriptional level by cytokines and the action of other cell factors, including cytokines and growth factors. Changes in interactions of cell receptors with the environment (ECM binding, mechanical tension, etc.) also modulate transcriptional rates. Epigenetic mechanisms and factors governing RNA stability also play substantial regulatory roles in many cases. Post-translational regulation of MMP and TIMP expression are known to be modulated by factors such as microRNAs (miRNAs). MMP protein secretion, proteolytic activation (including the presence of activating proteinases), and sequestration either at the cell membrane or within the pericellular ECM are all documented levels of regulation (47–50). Substrate conformation and/or availability are also thought to be important factors determining the extents of MMP action. The major inhibitors are the secreted TIMPs within the context of pericellular proteolysis. More recently, substantial knowledge has also been accrued on endocytic mechanisms that remove both MMPs and MMP-TIMP complexes from the pericellular environment for intracellular destruction. MT-MMPs may also be recycled to different cell membrane sites (50).
The metalloproteinase community has seen it all: the thrills and spills of new discoveries and unfortunate failures. Throughout it all, we've been resilient, and the current sophistication of research in the field is breathtaking (see, for example, Ref. 23 and references therein). However, a major shift in perception of what MMPs and other Metzincins can do is now needed for the greater biomedical community. Without a doubt, there are still many difficulties in studying proteinases with overlapping substrates and structures. The need for the painstaking application of high-throughput, high-content genomic and proteomic analyses for each individual proteinase and pathophysiological scenario has not gone away. Many substrates and functions are still not known. However, the opportunities are there: validation of any newly identified activities could be performed in specifically designed animal models. System-wide knowledge of MMPs and their place in the proteinase web could pave the way for new therapeutic approaches (40). The need to develop inhibitors that target disease-related MMPs while sparing those deigned to be “anti-targets” (although note that these can vary in different pathologies!) is a worthwhile goal that could present exciting new therapeutic opportunities.
MMP/Metzincin inhibitor development has entered a more promising era, thanks in part to high-throughput techniques. Small molecules targeting the active-site cleft now largely steer clear of the chelation of zinc. The use of X-ray crystallography and NMR methods, combined with computational methods, has enabled the modeling of drug-protein interactions with inhibitors of MMPs that bind to sites within the catalytic domain outside the catalytic site. This makes it possible to design compounds with greater potency and selectivity; indeed, inhibitors with up to three orders of magnitude stronger inhibition capacity of target MMPs when compared with non-target MMPs have been developed (30). A molecular understanding of the interactions of MMPs with substrates and other protein partners can yield important information for the design of inhibitors. Such strategies target motifs away from the catalytic cleft and in the other non-catalytic domains of MMPs (45, 46), similar to the antibody approach that we and others have investigated. The reassessment of MMPs as molecular targets for therapeutic intervention in disease alongside the development of more specific inhibitors would now be timely. Issues of stability and bioavailability of new inhibitors also need to be addressed (51).
One of the most important hurdles when evaluating enzyme inhibitors as therapeutics is the need to have reliable screening tools to follow the abrogation of enzyme function. This was a major problem for the MMP field due to the massive overlap in enzyme functions as well as the presence of really low levels of functional proteins. Fortunately, recent years have seen tremendous advances in MMP imaging techniques for both in vitro and in vivo applications. Currently available imaging probes based on optical imaging, positron emission tomography, fluorescence resonance energy transfer, single-photon emission computed tomography (SPECT), magnetic resonance imaging, and photoacoustic modalities are now under evaluation. Various MMP-activated optical probes have also recently been developed for in vivo MMP imaging, but the use of fluorescent, synthetic, low-molecular-mass MMP inhibitors has also been suggested. These optical probes are valuable tools to study localization of MMP activity but largely lack true specificity (52–54). Further, finding the best way to construct and deliver these types of agents that can be integrated into current clinical facilities (e.g. PET/SPECT- or MRI-based rather than optical) is still challenging.
Overall our increased knowledge of MMP structure and function has opened up new opportunities for the design of innovative agents with the goal of fighting human disease. From humble beginnings, the field of MMPs in disease roared into life too soon. We were running before we could walk. With careful resetting of the goals, the MMP world has made important strides in defining how these enzymes play roles in both physiology and pathology and the delicate balances involved. The knowledge that many MMPs have significant roles in diverse immune and inflammation processes rather than in the turnover of the ECM has been key to the current and future strategies. Attempts to inhibit metalloproteinases in human diseases thus require continuing appraisal of their biological roles and cautious evaluation of potential new therapeutic opportunities.
Acknowledgments
I am exceedingly grateful to my colleagues and students for working so hard and sharing the thrill of discovery with me. I am also grateful to all in the MMP community across the world for sharing the excitements and disappointments of research in this field and for such valuable collaborations. I thank the Medical Research Council, UK, the Wellcome Trust, Arthritis Research UK, Cancer Research UK and the European Union Framework Programmes for supporting our research from 1975 to 2014. Thanks to Hideaki Nagase for critical comments on the manuscript.
The author declares that she has no conflicts of interest with the contents of this article.
- MMP
- matrix metalloproteinase
- MT-MMP
- membrane-type MMPs
- ADAM
- a disintegrin and metalloproteinase
- ADAM-TS
- ADAM with thrombospondin domain
- TIMP
- tissue inhibitor of metalloproteinases
- ECM
- extracellular matrix.
References
- 1. Hall L. A. (1996) The Strangeways Research Laboratory: archives in the Contemporary Medical Archives Centre. Med. Hist. 40, 231–238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Woessner J. F., Jr. (1962) Catabolism of collagen and non-collagen protein in the rat uterus during post-partum involution. Biochem. J. 83, 304–314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Gross J., and Lapiere C. M. (1962) Collagenolytic activity in amphibian tissues: a tissue culture assay. Proc. Natl. Acad. Sci. U.S.A. 48, 1014–1022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Nagai Y., Lapiere C. M., and Gross J. (1966) Tadpole collagenase: preparation and purification. Biochemistry 5, 3123–3130 [DOI] [PubMed] [Google Scholar]
- 5. Evanson J. M., Jeffrey J. J., and Krane S. M. (1967) Human collagenase: identification and characterization of an enzyme from rheumatoid synovium in culture. Science 158, 499–502 [DOI] [PubMed] [Google Scholar]
- 6. Eisen A. Z., Jeffrey J. J., and Gross J. (1968) Human skin collagenase: isolation and mechanism of attack on the collagen molecule. Biochim. Biophys. Acta 151, 637–645 [DOI] [PubMed] [Google Scholar]
- 7. Jeffrey J. J., and Gross J. (1970) Collagenase from rat uterus: isolation and partial characterization. Biochemistry 9, 268–273 [DOI] [PubMed] [Google Scholar]
- 8. Sellers A., and Reynolds J. J. (1977) Identification and partial characterization of an inhibitor of collagenase from rabbit bone. Biochem. J. 167, 353–360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sellers A., Reynolds J. J., and Meikle M. C. (1978) Neutral metallo-proteinases of rabbit bone: separation in latent forms of distinct enzymes that when activated degrade collagen, gelatin and proteoglycans. Biochem. J. 171, 493–496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Iyer R. P., Patterson N. L., Fields G. B., and Lindsey M. L. (2012) The history of matrix metalloproteinases: milestones, myths, and misperceptions. Am. J. Physiol. Heart Circ. Physiol. 303, H919–H30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Cawston T. E., Galloway W. A., Mercer E., Murphy G., and Reynolds J. J. (1981) Purification of rabbit bone inhibitor of collagenase. Biochem. J. 195, 159–165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Stricklin G. P., and Welgus H. G. (1983) Human skin fibroblast collagenase inhibitor: purification and biochemical characterization. J. Biol. Chem. 258, 12252–12258 [PubMed] [Google Scholar]
- 13. Brew K., and Nagase H. (2010) The tissue inhibitors of metalloproteinases (TIMPs): an ancient family with structural and functional diversity. Biochim. Biophys. Acta 1803, 55–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Harper E., Bloch K. J., and Gross J. (1971) The zymogen of tadpole collagenase. Biochemistry 10, 3035–3041 [DOI] [PubMed] [Google Scholar]
- 15. Stricklin G. P., Eisen A. Z., Bauer E. A., and Jeffrey J. J. (1978) Human skin fibroblast collagenase: chemical properties of precursor and active forms. Biochemistry 17, 2331–2337 [DOI] [PubMed] [Google Scholar]
- 16. Sellers A., Cartwright E., Murphy G., and Reynolds J. J. (1977) Evidence that latent collagenases are enzyme-inhibitor complexes. Biochem. J. 163, 303–307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Van Wart H. E., and Birkedal-Hansen H. (1990) The cysteine switch: a principle of regulation of metalloproteinase activity with potential applicability to the entire matrix metalloproteinase gene family. Proc. Natl. Acad. Sci. U.S.A. 87, 5578–5582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Nagase H., Visse R., and Murphy G. (2006) Structure and function of matrix metalloproteinases and TIMPs. Cardiovasc. Res. 69, 562–573 [DOI] [PubMed] [Google Scholar]
- 19. Ra H. J., and Parks W. C. (2007) Control of matrix metalloproteinase catalytic activity. Matrix Biol. 26, 587–596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Docherty A. J., Lyons A., Smith B. J., Wright E. M., Stephens P. E., Harris T. J., Murphy G., and Reynolds J. J. (1985) Sequence of human tissue inhibitor of metalloproteinases and its identity to erythroid-potentiating activity. Nature 318, 66–69 [DOI] [PubMed] [Google Scholar]
- 21. Okada Y., Nagase H., and Harris E. D. Jr. (1986) A metalloproteinase from human rheumatoid synovial fibroblasts that digests connective tissue matrix components: purification and characterization. J. Biol. Chem. 261, 14245–14255 [PubMed] [Google Scholar]
- 22. Rawlings N. D., Barrett A. J., and Finn R. (2016)Twenty years of the MEROPS database of proteolytic enzymes, their substrates and inhibitors. Nucleic Acids Res. 44(D1), D343–D350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Apte S. S., and Parks W. C. (2015) Metalloproteinases: a parade of functions in matrix biology and an outlook for the future. Matrix Biol. 44–46, 1–6 [DOI] [PubMed] [Google Scholar]
- 24. Kelwick R., Desanlis I., Wheeler G. N., and Edwards D. R. (2015) The ADAMTS (A Disintegrin and Metalloproteinase with Thrombospondin motifs) family. Genome Biol. 16, 113–129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Murphy G. (2008) The ADAMs: signalling scissors in the tumour microenvironment. Nat. Rev. Cancer 8, 929–941 [DOI] [PubMed] [Google Scholar]
- 26. Sterchi E. E., Stöcker W., and Bond J. S. (2008) Meprins, membrane-bound and secreted astacin metalloproteinases. Mol. Aspects Med. 29, 309–328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Murphy G., Knäuper V., Lee M.-H., Amour A., Worley J. R., Hutton M., Atkinson S. A., Rapti M., and Williamson R. (2003) Role of TIMPs (tissue inhibitors of metalloproteinases) in pericellular proteolysis: the specificity is in the detail. Biochem. Soc. Symp. 70, 65–80 [DOI] [PubMed] [Google Scholar]
- 28. Brown P. D., and Giavazzi R. (1995) Matrix metalloproteinase inhibition: a review of anti-tumour activity. Ann. Oncol. 6, 967–974 [DOI] [PubMed] [Google Scholar]
- 29. Watson S. A., Morris T. M., Parsons S. L., Steele R. J., and Brown P. D. (1996) Therapeutic effect of the matrix metalloproteinase inhibitor, batimastat, in a human colorectal cancer ascites model. Br. J. Cancer 74, 1354–1358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Vandenbroucke R. E., and Libert C. (2014) Is there new hope for therapeutic matrix metalloproteinase inhibition? Nat. Rev. Drug Discov. 13, 904–927 [DOI] [PubMed] [Google Scholar]
- 31. Pavlaki M., and Zucker S. (2003) Matrix metalloproteinase inhibitors (MMPIs): the beginning of phase I or the termination of phase III clinical trials. Cancer Metastasis Rev. 22, 177–203 [DOI] [PubMed] [Google Scholar]
- 32. Zucker S., Cao J., and Chen W. T. (2000) Critical appraisal of the use of matrix metalloproteinase inhibitors in cancer treatment. Oncogene 19, 6642–6650 [DOI] [PubMed] [Google Scholar]
- 33. Fanjul-Fernández M., Folgueras A. R., Cabrera S., and López-Otín C. (2010) Matrix metalloproteinases: evolution, gene regulation and functional analysis in mouse models. Biochim. Biophys. Acta 1803, 3–19 [DOI] [PubMed] [Google Scholar]
- 34. Butler G. S., and Overall C. M. (2009) Updated biological roles for matrix metalloproteinases and new “intracellular” substrates revealed by degradomics. Biochemistry 48, 10830–10845 [DOI] [PubMed] [Google Scholar]
- 35. Schlage P., and auf dem Keller U. (2015) Proteomic approaches to uncover MMP function. Matrix Biol. 44–46, 232–238 [DOI] [PubMed] [Google Scholar]
- 36. Shi J., Son M. Y., Yamada S., Szabova L., Kahan S., Chrysovergis K., Wolf L., Surmak A., and Holmbeck K. (2008) Membrane-type MMPs enable extracellular matrix permissiveness and mesenchymal cell proliferation during embryogenesis. Dev. Biol. 313, 196–209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Aiken A., and Khokha R. (2010) Unraveling metalloproteinase function in skeletal biology and disease using genetically altered mice. Biochim. Biophys. Acta 1803, 121–132 [DOI] [PubMed] [Google Scholar]
- 38. Noël A., Gutiérrez-Fernández A., Sounni N. E., Behrendt N., Maquoi E., Lund I. K., Cal S., Hoyer-Hansen G., and López-Otín C. (2012) New and paradoxical roles of matrix metalloproteinases in the tumor microenvironment. Front Pharmacol. 3, 140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Sieber S. A., Niessen S., Hoover H. S., and Cravatt B. (2006) Proteomic profiling of metalloproteinase activities with cocktails of active-site probes. Nat. Chem. Biol. 2, 274–281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Rodríguez D., Morrison C. J., and Overall C. M. (2010) Matrix metalloproteinases: what do they not do? New substrates and biological roles identified by murine models and proteomics. Biochim. Biophys. Acta 1803, 39–54 [DOI] [PubMed] [Google Scholar]
- 41. Basu B., Correa de Sampaio P., Mohammed H., Fogarasi M., Corrie P., Watkins N. A., Smethurst P. A., English W. R., Ouwehand W. H., and Murphy G. (2012) Inhibition of MT1-MMP activity using functional antibody fragments selected against its hemopexin domain. Int. J. Biochem. Cell Biol. 44, 393–403 [DOI] [PubMed] [Google Scholar]
- 42. Botkjaer K. A., Kwok H. F., Terp M. G., Karatt-Vellatt A., Santamaria S., McCafferty J., Andreasen P. A., Itoh Y., Ditzel H. J., and Murphy G. (2016) Development of a specific affinity-matured exosite inhibitor to MT1-MMP that efficiently inhibits tumor cell invasion in vitro and metastasis in vivo. Oncotarget 7, 16773–16792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Tape C. J., Willems S. H., Dombernowsky S. L., Stanley P. L., Fogarasi M., Ouwehand W., McCafferty J., and Murphy G. (2011) Cross-domain inhibition of TACE ectodomain. Proc. Natl. Acad. Sci. U.S.A. 108, 5578–5583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Dufour A., and Overall C. M. (2013) Missing the target: matrix metalloproteinase antitargets in inflammation and cancer. Trends Pharmacol. Sci. 34, 233–242 [DOI] [PubMed] [Google Scholar]
- 45. Sela-Passwell N., Rosenblum G., Shoham T., and Sagi I. (2010) Structural and functional bases for allosteric control of MMP activities: Can it pave the path for selective inhibition? Biochim. Biophys. Acta 1803, 29–38 [DOI] [PubMed] [Google Scholar]
- 46. Fields G. B. (2015) New strategies for targeting matrix metalloproteinases. Matrix Biol. 44–46, 239–246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Clark I. M., Swingler T. E., Sampieri C. L., and Edwards D. R. (2008) The regulation of matrix metalloproteinases and their inhibitors. Int. J. Biochem. Cell Biol. 40, 1362–1378 [DOI] [PubMed] [Google Scholar]
- 48. Löffek S., Schilling O., and Franzke C.-W. (2011) Biological role of matrix metalloproteinases: a critical balance. Eur. Respir. J. 38, 191–208 [DOI] [PubMed] [Google Scholar]
- 49. Murphy G., and Nagase H. (2011) Localizing matrix metalloproteinase activities in the pericellular environment. FEBS J. 278, 2–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Yamamoto K., Murphy G., and Troeberg L. (2015) Extracellular regulation of metalloproteinases. Matrix Biol. 44–46, 255–263 [DOI] [PubMed] [Google Scholar]
- 51. Amar S., and Fields G. B. (2015) Potential clinical implications of recent matrix metalloproteinase inhibitor design strategies. Expert Rev. Proteomics 12, 445–447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Shay G., Lynch C. C., and Fingleton B. (2015) Moving targets: emerging roles for MMPs in cancer progression and metastasis. Matrix Biol. 44–46, 200–206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Waschkau B., Faust A., Schäfers M., and Bremer C. (2013) Performance of a new fluorescence-labeled MMP inhibitor to image tumor MMP activity in vivo in comparison to an MMP-activatable probe. Contrast Media Mol. Imaging 8, 1–11 [DOI] [PubMed] [Google Scholar]
- 54. Matusiak N., van Waarde A., Bischoff R., Oltenfreiter R., van de Wiele C., Dierckx R. A., and Elsinga P. H. (2013) Probes for non-invasive matrix metalloproteinase-targeted imaging with PET and SPECT. Curr. Pharm. Des. 19, 4647–4672 [DOI] [PubMed] [Google Scholar]