

Abstract
Hydrogen sulfide (H2S) is produced endogenously in vivo and has multiple effects on signaling pathways and cell function. Mitochondria can be both an H2S source and sink, and many of the biological effects of H2S relate to its interactions with mitochondria. However, the significance of mitochondrial H2S is uncertain, in part due to the difficulty of assessing changes in its concentration in vivo. Although a number of fluorescent H2S probes have been developed these are best suited to cells in culture and cannot be used in vivo. To address this unmet need we have developed a mitochondria-targeted H2S probe, MitoA, which can be used to assess relative changes in mitochondrial H2S levels in vivo. MitoA comprises a lipophilic triphenylphosphonium (TPP) cation coupled to an aryl azide. The TPP cation leads to the accumulation of MitoA inside mitochondria within tissues in vivo. There, the aryl azido group reacts with H2S to form an aryl amine (MitoN). The extent of conversion of MitoA to MitoN thus gives an indication of the levels of mitochondrial H2S in vivo. Both compounds can be detected sensitively by liquid chromatography tandem mass spectrometry (LC-MS/MS) analysis of the tissues, and quantified relative to deuterated internal standards. Here we describe the synthesis and characterization of MitoA and show that it can be used to assess changes in mitochondrial H2S levels in vivo. As a proof of principle we used MitoA to show that H2S levels increase in vivo during myocardial ischemia.
Keywords: chemical biology, hydrogen sulfide, hypoxia, mass spectrometry (MS), mitochondria, analytical chemistry, chemical biology, energy metabolism, mitochondria, hypoxia, hydrogen sulfide
Introduction
There is considerable interest in the biological roles of hydrogen sulfide (H2S), both as an endogenously produced modulator of mammalian metabolism and for its potential biomedical effects when generated pharmacologically (1–3). Changes in endogenous H2S levels have been claimed to impact on a diverse range of physiological processes, including neuronal function, blood pressure, angiogenesis, oxygen sensing, inflammation, and mitochondrial energy production (4–16), and exposure to H2S can induce a suspended animation-like state in mice (17), but not in larger animals (18, 19).
Within mammals there are three main enzymatic sources of H2S: cystathionine β-synthase and cystathionine γ-lyase in the cytosol, and mitochondrial 3-mercaptopyruvate sulfurtransferase in conjunction with cysteine aminotransferase (Fig. 1A) (20). Once generated, H2S can be oxidized by sulfide:quinone oxidoreductase (SQR)4 in the mitochondrial inner membrane, passing electrons to the CoQ pool, and in parallel generating glutathione persulfide in the matrix, which is further metabolized to thiosulfate/sulfite/sulfate in the mitochondrial matrix and intermembrane space (21, 22) (Fig. 1A).
Figure 1.

Overview of endogenous H2S production and rationale for its detection by MitoA. A, overview of H2S metabolism (sulfur-containing compounds in bold). CBS, cystathionine β-synthase; CSE, cystathionine β-lyase; CAT, cysteine aminotransferase; GSSH, glutathione persulfide; 3-MP, 3-mercaptopyruvate; 3-MST, 3-mercaptopyruvate sulfurtransferase. B, reaction of MitoA with H2S to form MitoN. C, model of uptake of MitoA into mitochondria in vivo, followed by its conversion to MitoN upon reaction with H2S. The subsequent extraction of MitoA and MitoN from the tissue and analysis by LC-MS/MS enables changes in the levels of H2S in vivo to be inferred.
The biological activity of H2S arises from four interrelated processes: protein persulfidation, binding to metalloprotein centers (most notably, inhibition of cytochrome oxidase), interaction with NO signaling pathways, and as an antioxidant (14, 20). Protein persulfidation is a recently emerging mode of reversible posttranslational modification (PTM) of protein thiols in which H2S can react with a disulfide or a sulfenic acid, but not unmodified protein thiols, to form a persulfidated protein thiol (PrSSH), which can be reduced back to a thiol by the glutathione or thioredoxin systems (23, 24). This reversible PTM can in principle modify protein activity in a similar way to other PTMs, or can act as a releasable pool of H2S; however, the extent and physiological significance of protein persulfidation is still emerging. H2S can also inhibit cytochrome oxidase (25), which is why high concentrations of H2S are toxic, but whether these interactions with cytochrome oxidase or other heme proteins are of physiological or pharmacological importance is unclear. H2S signaling is intimately interwoven with that of NO, due to the formation of HSNO and other intermediates, and it is likely that protein modification by persulfidation and S-nitrosation are facets of a general signaling pathway (26, 27). Finally, H2S may also contribute to antioxidant defenses, by reacting directly with oxidants, or indirectly through its interactions with protein thiols (20, 28).
Abnormal H2S levels have been reported in a range of pathologies including Down syndrome (29), diabetes (30), liver cirrhosis, and in ethylmalonic acid encephalopathy, which occurs due to mutation to ETHE1 (31). However, the factors that modify H2S levels in vivo under normal and pathological conditions are obscure. In addition, the contribution of H2S generation to the efficacy of pharmacological agents that release H2S is uncertain. Therefore, understanding how the levels of H2S change in vivo is vital to understanding its roles in physiology, pathology, and pharmacology.
There are a number of methods to assess H2S in cells using fluorescent probes that allow real-time detection of changes in H2S levels (32–40). Although these methods can be applied to the surfaces of animals (41), and to blood ex vivo (42), they cannot be used in whole animals in vivo. Consequently, there are considerable uncertainties about how H2S tissue levels change in response to physiological, pathological, or pharmacological events. These uncertainties are a major impediment to better understanding the biological roles of H2S and its downstream targets.
To overcome this obstacle, we have developed MitoA, a mitochondria-targeted mass spectrometry probe for H2S detection in vivo (Fig. 1B). The probe comprises an H2S-sensitive aryl azide moiety coupled to the lipophilic triphenylphosphonium (TPP) cation (Fig. 1B). The TPP cation targets a wide range of bioactive and probe compounds to mitochondria in vivo in response to the mitochondrial membrane potential (43, 44), and thus should lead to the rapid uptake of MitoA into mitochondria in vivo. Within the mitochondrial matrix the aryl azide moiety is intended to react with H2S to form the aryl amine, MitoN (Fig. 1B). This chemistry is based on that of widely used H2S-sensitive fluorescent probes, which rely on the reaction of a non-fluorescent aryl azide to give a fluorescent aryl amine (33, 37, 45, 46). The reaction occurs by initial nucleophilic attack of HS− on the electrophilic terminal nitrogen of the aryl azide, followed by a rate-determining reaction with a second HS−, forming HS2−, N2, and the aryl amine (37) (Fig. 1B). Thus, MitoA can be injected into animals and the extent of generation of the exomarker MitoN will depend on the levels of H2S within mitochondria in vivo. Similar exomarker approaches have been used in vivo to assess hydrogen peroxide using MitoB (47, 48), and glyoxals through MitoG (49, 50). To analyze MitoA and its conversion to MitoN, both are extracted from animal tissue and then analyzed by LC-MS/MS relative to deuterated internal standards (Fig. 1C). Because MitoA is mainly present within mitochondria in vivo, it will report on the local concentration of H2S within mitochondria. However, because H2S diffuses rapidly across cellular membranes, H2S levels in the mitochondria are likely to reflect or influence overall changes in H2S within cells in vivo. Here we report the development of MitoA and show that it can be used to assess changes in H2S in vivo.
Results and discussion
Reaction of MitoA with H2S to form MitoN
We first determined whether MitoA reacted with H2S to form MitoN by measuring the effect of H2S on the UV absorption spectra of MitoA over time (Fig. 2A). Addition of NaHS, which rapidly generates H2S (pKa = 6.8 (51)), led to the gradual conversion of MitoA to MitoN. RP-HPLC analysis of MitoA following overnight incubation with excess NaHS showed its complete conversion to MitoN (Fig. 2B). We could readily detect MitoA and MitoN by mass spectrometry (MS) and their spectra matched those predicted from the natural isotope distribution (supplemental Fig. S2). There was decomposition of MitoA ([C28H26N4OP]+; predicted m/z 465.18) by neutral loss of N2 to form the nitrene ([C28H26N2OP]+; predicted m/z 437.17), the extent of which depended on MS conditions (Fig. 2C). MS analysis of MitoA after overnight incubation with excess NaHS showed formation of MitoN ([C28H29N2OP]+; predicted m/z 439.19) further confirming that MitoA reacts with H2S to form only MitoN (Fig. 2C); under these MS conditions only the nitrene product of MitoA was found. When the reaction of MitoA with H2S was monitored continuously using time-resolved MS, where both MitoA and its nitrene product were observed, the gradual loss of MitoA and the accumulation of MitoN were seen over time, consistent with MitoA reacting selectively with H2S to generate MitoN (Fig. 2D).
Figure 2.

Reaction of MitoA with H2S to form MitoN. A, UV-visual absorbance spectra of MitoA reacting with NaHS. MitoA (50 μm) in KCl buffer was mixed with 5 mm NaHS and spectra were taken every 15 min. The inset shows spectra of pure MitoA and MitoN (25 μm each). B, RP-HPLC analysis of MitoA, MitoN, and its reactivity with H2S. Standards of 10 nmol of MitoA (upper trace) or MitoN (middle trace) were separated by RP-HPLC. For the lower trace MitoA (100 μm) in KCl buffer was incubated overnight with 1 mm NaHS at room temperature. Then 5 nmol eq of starting MitoA was analyzed by RP-HPLC. Spiking the NaHS-treated MitoA with MitoN increased the MitoN peak (data not shown). C, mass spectra of MitoA, MitoN, or MitoA incubated overnight with NaHS. Mass spectra were taken of MitoA (100 μm), MitoN (100 μm), or MitoA (100 μm) incubated with 1 mm NaHS in KCl buffer, pH 7.2, overnight at room temperature. Samples were diluted in 20% ACN, 0.1% FA and analyzed within 5 h. Predicted m/z: MitoA, [C28H26N4OP]+ = 465.18; nitrene from neutral loss of N2, [C28H26N2OP]+ = 437.17; MitoN, [C28H29N2OP]+ = 439.19. The MS of MitoA incubated at room temperature overnight was the same as that for freshly prepared MitoA (data not shown). D, time-resolved analysis of the reaction of MitoA with H2S. MitoA (100 μm) was mixed with 2.5 mm H2S in 300 mm ammonium carbonate, pH 7.4, and sprayed continuously over 45 min into an ultra high resolution mass spectrometer (maXis Bruker Daltonics). Spectra at selected time points are shown. Asterisk shows the nitrene formed from MitoA by the neutral loss of N2.
The interaction of MitoA with other potential reactive species that it may encounter in vivo was assessed by RP-HPLC (Table 1). None of the tested compounds reacted to any significant extent with MitoA to form MitoN (Table 1): the greatest conversion of MitoA to MitoN was 7.9% and required incubation with 10 mm glutathione for 24 h. A significant potential confounder for the selectivity of MitoA for H2S is the possibility of its reacting with low molecular weight and protein persulfides. Therefore we next assessed the direct reactivity of MitoA with NAP-SSH, a low molecular weight persulfide derivative of N-acetylpenicillamine (NAP) (24, 52). A complication of analyzing persulfides is that they spontaneously release H2S, making it difficult to separate the reaction of MitoA with H2S from that with the persulfide itself (24). To address this, we compared the reaction of MitoA with NAP-SSH to that of MitoA with the same concentration of H2S (supplemental Fig. S3). These experiments showed the expected reaction of MitoA with H2S with formation of MitoN, whereas under the same conditions the reaction of MitoA with NAP-SSH to form MitoN was negligible (supplemental Fig. S3). Furthermore, the incubation of 5 μm persulfidated human serum albumin with 10 μm MitoA led to negligible (<7%) consumption of MitoA over 4 h at room temperature (data not shown), most probably due to spontaneous decay of persulfides to release H2S (53).
Table 1.
MitoA (10 μm) was incubated with each substance for the indicated time and temperature in KCl buffer and analyzed by RP-HPLC
Dimethyl trisulfide was dissolved in 70% EtOH. For NO, DetaNONOate (50 μm) was disolved in KCl buffer, which had been deoxygenated by bubbling with argon for 30 min. Superoxide was generated using 5 milliunits/ml of xanthine oxidase and 1 mm hypoxanthine in KCl buffer and its production quantified as the SOD-sensitive reduction of ferricytochrome c. Reactivity = (MitoN × 100)/(MitoN + MitoA).
| Concentration | Incubation condition | Reactivity | |
|---|---|---|---|
| % | |||
| H2S | 100 μm | 4 h, RTa | 94 |
| Glutathione | 5 mm | 24 h, 37 °C | 7.3 |
| 5 mm | 4 h, 37 °C | 2.2 | |
| 10 mm | 24 h, 37 °C | 7.9 | |
| 10 mm | 4 h, 37 °C | 4.6 | |
| Cysteine | 2 mm | 4 h, RT | <LODb |
| Dimethyl trisulfide | 100 μm | 4 h, RT | <LOD |
| Lipoic acid | 100 μm | 4 h, RT | <LOD |
| Dihydrolipoic acid | 100 μm | 4 h, RT | <LOD |
| Na2S2O3 | 100 μm | 4 h, RT | <LOD |
| Na2S2O4 | 100 μm | 4 h, RT | <LOD |
| NADH | 5 mm | 4 h, RT | <LOD |
| NADPH | 5 mm | 4 h, RT | <LOD |
| NaSCN | 100 μm | 4 h, RT | <LOD |
| NaNO2 | 100 μm | 4 h, RT | <LOD |
| NO | 50 μm | 4 h, RT | <LOD |
| ONOO− | 100 μm | 4 h, RT | 1.25 |
| H2O2 | 100 μm | 4 h, RT | <LOD |
| t-BuOOH | 100 μm | 4 h, RT | <LOD |
| O2˙̄ | 1.79 nmol of cytochrome c/min | 4 h, RT | <LOD |
| HOCl | 100 μm | 4 h, RT | <LOD |
a RT, room temperature.
b LOD, limit of detection.
We next measured the rate of reaction of MitoA with H2S from the time-dependent disappearance of the MitoA peak at ∼270 nm and the appearance of a new peak at 306 nm due to MitoN (Fig. 3A). The absorption maxima for the non-TPP aromatic rings were identified using 4-azido-N-(hex-1-yl)benzamide and 4-amino-N-(hex-1-yl)benzamide, which are analogues of MitoA and MitoN, respectively, but without the TPP targeting group. We then incubated MitoA with known concentrations of H2S and measured the formation of MitoN at 306 nm (Fig. 3B). From these we were able to plot the observed initial rate constant (kobs) for MitoN formation against the concentration of H2S and calculate a rate constant: 0.16 ± 0.03 m−1 s−1 at 22 °C (Fig. 3C). This compares with literature rates for the reaction of H2S with phenyl azides: 0.12 m−1 s−1 at 10 °C and 0.95 m−1 s−1 at 25 °C (37). One concern about the use of MitoA is that its reaction consumes H2S and generates H2S2 (37), thereby potentially disrupting H2S signaling pathways and altering protein persulfidation. Hence the slow reaction of MitoA with H2S is advantageous because it allows selective detection, while also minimizing disruption to endogenous H2S signaling pathways. Together these data indicate that, in agreement with other aryl azides (45, 46), MitoA reacts selectively with H2S and therefore that the conversion of MitoA to MitoN can be used to assess H2S in vivo.
Figure 3.
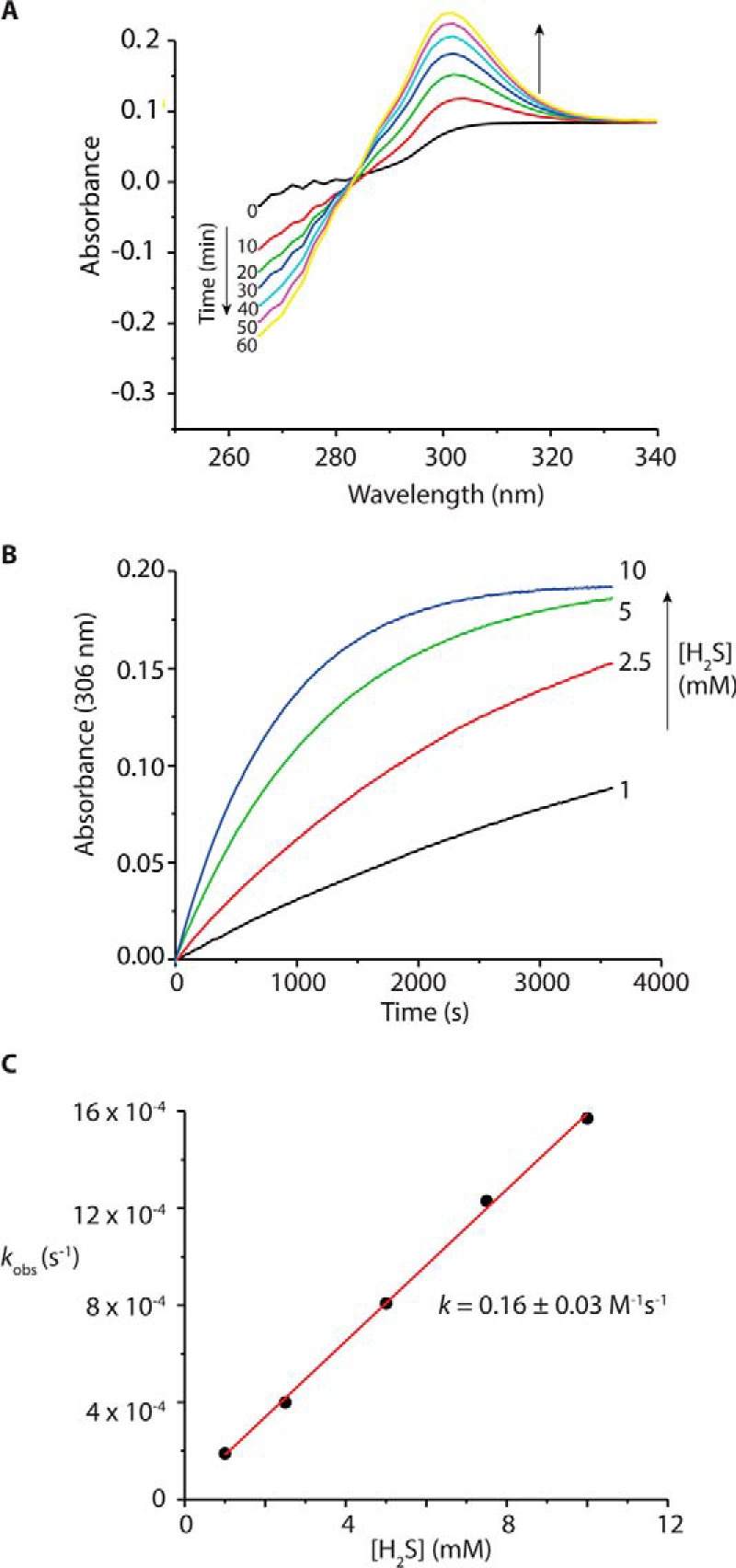
Rate of reaction of MitoA with H2S to form MitoN. A, differential time-resolved spectra where 40 μm MitoA was mixed with 2.5 mm H2S, with MitoA serving as the blank. The spectra illustrate the time-dependent disappearance of the MitoA peak at ∼270 nm and the appearance of a new peak assigned to MitoN at ∼306 nm. B, kinetic traces at 306 nm obtained for the reaction of 40 μm MitoA with increasing concentrations of H2S, the absorbance was observed over 6 min at 306 nm using a Hewlett Packard 8452A Diode Array Spectrophotometer under pseudo-first order conditions. C, plot of kobs versus [H2S]. The experiments were performed in 300 mm potassium phosphate buffer, pH 7.4, at 22 °C in triplicates.
Analysis of MitoA and MitoN by LC-MS/MS
To be useful as an in vivo mass spectrometric probe MitoA and MitoN have to be extracted from tissues and analyzed by LC-MS/MS, relative to deuterated internal standards (IS) (47–49). To establish the LC-MS/MS assay we first determined fragmentation conditions that enabled sensitive detection of MitoA and MitoN, and their deuterated internal standards (Fig. 4A). For the analysis of MitoA we took into account the neutral loss of N2 from MitoA to generate the nitrene, which under these conditions was complete upon entrance to the first mass spectrometer quadrupole. Using the transitions shown in Fig. 4A we could detect MitoA and MitoN, and their deuterated IS, selectively by LC-MS/MS (Fig. 4B). Hence we were able to generate standard curves for MitoA and MitoN down to picomole levels (Fig. 4C).
Figure 4.

Detection of MitoA and MitoN by LC-MS/MS. A, the transitions used for analysis of MitoA and MitoN, and their deuterated derivatives, are shown. B, typical chromatograms showing the m/z transitions measured simultaneously for 50 pmol of d15-MitoA, MitoA, d15-MitoN, or MitoN. Each trace is normalized to the highest total ion count peak within that trace. C, typical standard curves for MitoA and MitoN detection by LC-MS/MS. Samples were prepared in 20% ACN, 0.1% FA and contained known MitoA or MitoN concentrations and their deuterated internal standards. Each point is mean ± range of duplicate measurements.
A potential concern for the LC-MS/MS analysis is that MitoA might react with H2S generated upon homogenization and extraction of tissues, for example, by release of labile sulfur from iron-sulfur centers. The possibility of H2S release was minimized by omitting acid during tissue extraction. Even so, to assess whether an artifactual reaction with released H2S could occur, we incubated MitoA in tissue extraction medium supplemented with H2S for up to 4 h, and then measured MitoN formation by LC-MS/MS. As expected, this showed some conversion of MitoA to MitoN, but with a very small (∼0.02) increase in the MitoN/MitoA ratio (supplemental Fig. S4). Considering the high H2S concentration added, and that the incubation time of 4 h was far longer than that used during conventional extraction, we conclude that the artifactual conversion of MitoA to MitoN during tissue extraction is negligible. In any case such a potential artifact would be equal for all tissue samples and therefore would not affect comparisons. To conclude, we have established a sensitive LC-MS/MS assay for H2S that can be applied to tissues.
Uptake of MitoA and MitoN by mitochondria and cells
To be useful for the analysis of H2S in vivo, MitoA must be taken up into mitochondria in response to the membrane potential. To see if this occurred we next assessed the accumulation of MitoA and MitoN by isolated mitochondria using a triphenylphosphonium (TPP)-selective electrode (54) (Fig. 5, A and B). Both probes accumulated inside mitochondria ∼650- to ∼1,000-fold in response to the membrane potential and this uptake was reversed by abolishing the mitochondrial membrane potential with the uncoupler FCCP. Therefore, MitoN and MitoA are taken up by energized mitochondria as expected for TPP compounds. To determine whether MitoA was accumulated within cells we first established that the toxicity of MitoA or MitoN to cells in vitro was negligible at concentrations up to 12.5 μm, determined by following the effects of the compounds on cell proliferation (supplemental Fig. S5). To assess the uptake of MitoA into cells, we incubated HCT116 cells with MitoA and measured the amounts taken up by the cells over time by LC-MS/MS (Fig. 5C). This showed that MitoA was taken up into cells over time, coming to a steady state after ∼1 h. Over this time the amount of MitoN generated was small (Fig. 5C), consequently the MitoN/MitoA ratio stayed low and constant throughout the incubation (Fig. 5D). Therefore MitoA is taken up by cells and, at least under these conditions, there was low MitoN formation.
Figure 5.
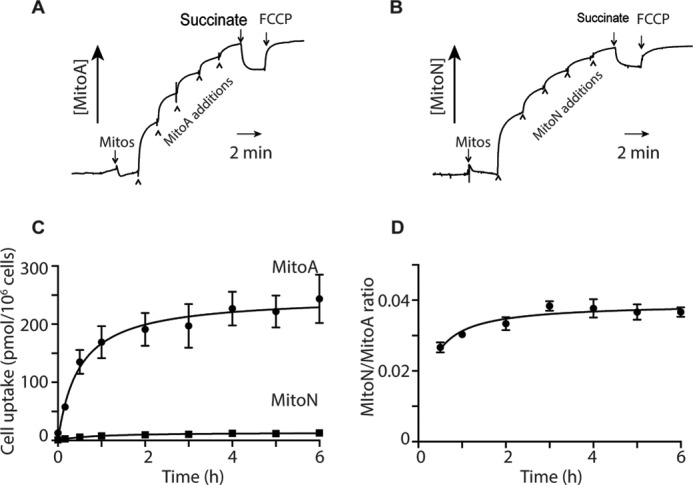
Accumulation of MitoA and MitoN by isolated mitochondria and cells. The uptake of MitoA (A) and MitoN (B) was examined in isolated mitochondria (2 mg of protein/ml) incubated in KCl medium supplemented with 4 μg/ml of rotenone and 100 nm nigericin using a TPP-selective electrode. The compounds were added in 1 μm steps, followed by 10 mm succinate and 500 nm FCCP. C, MitoA uptake by HCT116 cells. HCT116 cells were plated at 250,000 cells/well (∼27,000 cells/cm2) in 6-well plates overnight and then incubated with 10 μm MitoA in DMEM containing 10% FBS and antibiotics. At various times 1 ml of supernatant was removed for analysis, the rest was discarded, and the cells were rinsed with 1 ml of PBS and collected by scraping into 0.5 ml of PBS and pelleted by centrifugation (16,000 × g, 3 min, room temperature). Cell pellets were snap frozen and stored at −20 °C. Compounds were extracted and quantified by LC-MS/MS. Results are mean ± S.E. for n = 3. MitoA levels in the supernatant did not change over 6 h (data not shown). D, for the experiment in C, the level of MitoN in the cell pellets were assessed in parallel with MitoA and the MitoN/MitoA ratio is shown.
Reactivity and stability of MitoA in tissue homogenates
MitoA is designed to assess H2S in tissues in vivo. To determine whether there were any enzymatic processes in tissues that could convert MitoA to MitoN in the absence of H2S, we incubated MitoA with liver homogenates and measured MitoN formation (Fig. 6A). Under these conditions enzymes are available to react with MitoA, but the lack of tissue and cell architecture means that any H2S generated will diffuse away. Incubation of MitoA with a liver homogenate led to a negligible MitoN formation (Fig. 6A) and no increase in the MitoN/MitoA ratio (Fig. 6B), even though the reaction proceeded readily when H2S was added (Fig. 6, A and B). Similarly, incubation of MitoN with a liver homogenate in the presence or absence of H2S led to very little loss of the compound (Fig. 6C). Together these data suggest that MitoA conversion to MitoN in tissues is negligible in the absence of H2S, and that once formed MitoN is stable.
Figure 6.
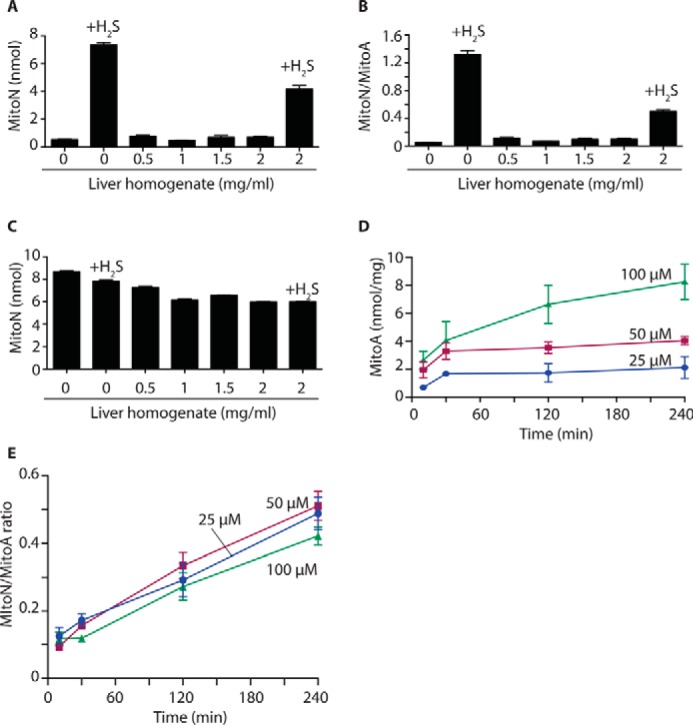
MitoA and MitoN metabolism in the liver ex vivo. A and B, MitoA metabolism by a liver homogenate. MitoA (10 μm) was incubated with various amounts of liver homogenate, or with liver homogenate and NaHS (25 μm), for 2 h at 37 °C. Samples were then analyzed by LC-MS/MS to quantify MitoA (data not shown), MitoN (A), and the MitoN/MitoA ratio (B). Data are mean ± S.E., n = 3. C, MitoN metabolism by a liver homogenate. MitoN (10 μm) was incubated with various amounts of liver homogenate, or with liver homogenate and NaHS (25 μm), for 2 h at 37 °C. Samples were then analyzed by LC-MS/MS for MitoN. Data are mean ± S.E., n = 3. D, uptake of MitoA by liver sections. Rat liver sections of ∼20 to 50 mg wet weight were incubated with MitoA in University of Wisconsin solution at 4 °C. After incubation, the tissue pieces were rinsed in PBS, dried, and snap frozen prior to quantification of MitoA and MitoN by LC-MS/MS. Data show the accumulation of MitoA over time and are mean ± S.E., n = 3. E, MitoN/MitoA ratio in liver sections. The MitoN/MitoA ratio was determined over time in liver sections as described in D.
Response of MitoA to H2S in ischemic liver sections
The next step was to see how MitoA responded to an increase in endogenous H2S within a tissue. To assess this we incubated liver tissue sections with various concentrations of MitoA at 4 °C, designed to mimic conditions during organ storage for transplantation (55, 56). MitoA was taken up into the tissue sections over time (Fig. 6D). Under these conditions the tissue will be largely ischemic, as there is no perfusion and O2 will be depleted by the oxidation of endogenous substrates. The lack of O2 will block the activity of the mitochondrial SQR, which is the major way in which H2S is degraded in vivo (Fig. 1A). It is known that when the activity of the SQR is decreased by depletion of the mitochondrial CoQ pool, that there is an accumulation of H2S within mice in vivo (57). Thus, under ischemic conditions the inhibition of SQR is expected to increase H2S levels. Furthermore, H2S production by the mitochondrial 3-mercaptopyruvate sulfurtransferase enzyme is driven by the reduced form of thioredoxin, which will also increase in concentration upon ischemia (58). Therefore it is likely that there will be an increase in H2S levels in these liver sections upon ischemia.
To see if this increase in H2S led to MitoN formation in the ischemic liver sections, we measured MitoN levels over time and expressed these relative to MitoA. This showed an increase in the MitoN/MitoA ratio over time (Fig. 6E), consistent with increased H2S formation in ischemic organs over time. Importantly, the MitoN/MitoA ratios at a given time point were similar for all initial MitoA concentrations used. Thus the ratiometric analysis of MitoN relative to MitoA corrects for differences in MitoA content and suggests that the MitoA/MitoN ratio is a robust measure of changes in H2S levels.
Uptake and metabolism of MitoA and MitoN within tissues in vivo
Previously developed exomarkers such as MitoB were administered to mice as a single intravenous (i.v.) bolus injection (47). This approach enabled the compounds to be very rapidly accumulated within tissues in vivo, after which they are gradually lost over the following hours. This protocol gives sufficient time for the accumulated compound (e.g. MitoB) to react with its target (e.g. H2O2) to form an exomarker (e.g. MitoP). To see if MitoA could also be used in this way, we assessed MitoA distribution within mouse tissues following a single i.v. injection (Fig. 7, A and B). This showed rapid clearance of MitoA from the blood and its accumulation into the heart and liver, but not the brain, as expected for TPP compounds in mice (59, 60) (Fig. 7A). The kidney accumulated more MitoA than the other tissues, presumably due to direct removal of the initial bolus from the circulation, as well as redistribution of MitoA from other tissues over time (Fig. 7B). MitoA was gradually lost from the tissues over time (Fig. 7, A and B), consistent with other TPP compounds (59). These findings suggest that MitoA is accumulated rapidly by tissues and is retained long enough to respond to local levels of H2S.
Figure 7.

Uptake and metabolism of MitoA in vivo. A and B, mice were injected with 50 nmol of MitoA and tissues were collected, extracted, and analyzed by LC-MS/MS. Results are mean ± S.E., n = 3. C, MitoA (50 nmol) was administered to mice. After 60 min the liver was collected, extracted, and prepared as for mass spectrometry (LC-MS/MS) but omitting the internal standards and analyzed by mass spectrometry by direct infusion at 5 μl/min. C, i, shows the mass spectrum of the liver extract. ii, shows the MS/MS spectrum of the liver extract assessed for MitoA (437 > 183). The inset shows the MS/MS spectrum for pure MitoA. iii, shows the MS/MS spectrum of the liver extract assessed for MitoN (439 > 183). The inset shows the MS/MS spectrum for pure MitoN. iv, shows the precursor scan of the liver extract assessed for precursor ions that generate a product ion of m/z 183. The inset shows the precursor ion scan for pure MitoA. D, MitoN (50 nmol) was administered to mice. After 60 min the liver was collected, extracted into solvent, and prepared as for mass spectrometry, but omitting the internal standards, and analyzed by MS by direct infusion at 5 μl/min. D, i, shows the mass spectrum of the liver extract. ii, shows the MS/MS spectrum of the liver extract assessed for MitoN (439 > 183). The inset shows the MS/MS spectrum for pure MitoN. iii, shows the precursor scan of the liver extract assessed for precursor ions that generate a product ion of m/z 183. The inset shows the precursor ion scan for pure MitoN. For all spectra the maximum ion counts are indicated on the spectra and all ion counts are expressed as % of the maximum.
To report on H2S levels within tissues in vivo MitoA should only be converted to MitoN by reaction with H2S, with minimal side reactions. To assess this we injected MitoA into mice and 1 h later isolated the liver. The liver was chosen because it readily accumulates MitoA (Fig. 7A) and has extensive xenobiotic metabolism. Thus, if MitoA is biotransformed to compounds other than MitoN in vivo the liver should demonstrate this biotransformation. Extraction and MS analysis of the liver showed a large number of compounds (Fig. 7C, i). Analysis by MS/MS at the transition diagnostic for MitoA (437 > 183) showed the presence of MitoA (Fig. 7C, ii). Similarly, analysis at the transition diagnostic for MitoN (439 > 183) showed some MitoN formation in vivo (Fig. 7C, iii). These mass spectra were identical to those of pure MitoA and MitoN (Fig. 7C, ii and iii, insets). To see if MitoA and MitoN were metabolized in the liver to other TPP containing molecules, we next carried out a precursor scan of the extract to identify all compounds that fragmented to a product with m/z 183, diagnostic of a TPP-containing compound (47, 61). In the liver extract the only precursor molecules that gave a product ion of m/z 183 corresponded to the nitrene product of MitoA upon neutral loss of N2 (m/z 437), MitoN (m/z 439), with a small amount of intact MitoA (m/z 465) (Fig. 7C, iv). This precursor ion pattern was similar to that of pure MitoA (Fig. 7C, iv, inset).
Another potential concern is whether MitoN could be further metabolized in vivo, following its formation from MitoA. To assess this we injected MitoN into mice and 1 h later isolated the liver and analyzed this extract by MS analysis (Fig. 7D, i). Analysis by MS/MS at the transition diagnostic for MitoN (439 > 183) showed the presence of MitoN (Fig. 7D, ii), and the spectrum was identical to that of pure MitoN (Fig. 7D, ii, inset). To see if MitoN was metabolized within the liver in vivo, we next carried out a precursor scan of the extract to identify all compounds that fragmented to a product with m/z 183, diagnostic of a TPP containing compound (47, 61). In the liver homogenate the only parent molecules that gave a daughter ion of m/z 183 corresponded to MitoN (m/z 439), with a small peak at 455 (Fig. 7D, iii). This precursor ion pattern was similar to that of pure MitoN (Fig. 7D, iii, inset). The precursor ion at 455 is a +16 shift from MitoN and may represent oxidation of MitoN by addition of an oxygen atom. Even so, it suggests that once formed MitoN is relatively stable and that any further metabolism of MitoN is unlikely to affect the analysis of H2S in vivo.
Use of MitoA to analyze H2S in the ischemic heart in vivo
The increase in the MitoN/MitoA ratio in the ischemic liver (Fig. 6E) is consistent with an increase in H2S in tissue during ischemia. To extend this, we next focused on the heart as its metabolism changes dramatically upon ischemia (62, 63). As a first step to see whether there was any accumulation of H2S in the ischemic heart we administered MitoA to mice, isolated the heart, and stored it under ischemic conditions mimicking those used for organ storage during transplantation (Fig. 8A). The MitoN/MitoA ratio increased after 240 min ischemia, consistent with elevated H2S during the ischemia inherent in organ storage.
Figure 8.

Assessment of H2S in vivo using MitoA. A, MitoA in the heart during storage ex vivo. MitoA (50 nmol) was administered to mice via tail vein injection. The heart was removed after ∼12 min of warm ischemia. The hearts were then either snap frozen or stored for 240 min on ice and then snap frozen. Samples were analyzed by LC-MS/MS for MitoA and MitoN, and the MitoN/MitoA ratio was calculated. Results are mean ± S.E., n = 3. *, p < 0.05 by Student's two-tailed t test. B, protocol for the LAD artery occlusion experiments. C, mice were injected with 50 nmol of MitoA prior to ischemia of the left ventricle induced by occlusion of the LAD artery. At the end of the protocol the hearts were separated into right (normoxic) and left (normoxic or ischemic) ventricles, snap frozen, and analyzed for MitoA and MitoN content by LC-MS/MS, enabling the MitoN/MitoA ratio to be calculated. Data are mean ± S.E., n = 3. *, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001, by two-way analysis of variance followed by post hoc Bonferroni test.
To explore further how H2S levels respond to ischemia we next used the left anterior descending (LAD) model of acute myocardial infarction. MitoA was injected into mice and then cardiac ischemia was induced by closing off the LAD artery, thus subjecting a large section of the left ventricle to ischemia. In previous work we have shown that when the occlusion to the LAD artery was removed after 30 min ischemia and the tissue reperfused with oxygenated blood, there was extensive ischemia-reperfusion injury (63). This model thereby enables us to use MitoA to assess variations of H2S levels during both ischemia and reperfusion. In comparing the formation of MitoN through these changes it is important that MitoA is present in the tissue for the same duration. To ensure this, we injected MitoA at various time points before or after initiation of ischemia (Fig. 8B). Importantly, this protocol meant that MitoA was exposed to a range of times of ischemia and reperfusion, whereas always being present in the tissue for 40 min, thereby facilitating comparison. In the LAD model only the left ventricle is exposed to ischemia and reperfusion, however, as it was unclear what effect ischemia in the left ventricle would have on the right ventricle from the same mouse, the right ventricle from the same mouse cannot act as a control. Therefore the left and right ventricles from mice that underwent ischemia were compared with the appropriate ventricle from MitoA-treated normoxic mice.
When MitoA was injected into control mice in which the hearts were not exposed to ischemia or reperfusion, analysis of the right and left ventricles 40 min later showed a low MitoN/MitoA ratio in both ventricles (Fig. 8C). When MitoA was injected before reperfusion, the MitoN/MitoA ratio was increased compared with normoxic hearts, presumably as result of H2S present in the tissue before reperfusion. However, administration 5 min after reperfusion resulted in the MitoN/MitoA ratio similar to normoxic hearts. Furthermore, during ischemia there was a dramatic increase in the MitoN/MitoA ratio in the left ventricle, but not in the right (Fig. 8C). The MitoN/MitoA ratio increased the longer the duration of ischemia to which the left ventricle was exposed (Fig. 8C). Comparison between the ischemic and non-ischemic zones of the heart showed that the H2S increase was local to the ischemic tissue area, and did not diffuse to surrounding tissue of the same organ. Together, these findings are consistent with an increase in H2S during ischemia that is rapidly reversed once oxygen is reintroduced into the tissue upon reperfusion. Furthermore, these findings indicate that MitoA can be used to assess changes in H2S in vivo.
Conclusions
Here we have applied the exomarker approach to the analysis of H2S in vivo. To do this we used the well established chemistry of aryl azides and their conversion to an amine on reaction with H2S (33, 36, 46). The combination of this chemistry with mitochondria targeting by the TPP moiety led to its rapid uptake into cells in vivo where it is predicted to be mainly located in the mitochondria. There, MitoA should react with H2S to form the diagnostic exomarker MitoN. The chemical selectivity for H2S of the MitoA probe was clear, and it did not react with other potential factors found in biological environments. Most importantly, it did not react directly with persulfides, suggesting that in vivo it reports on free H2S independently of any changes in the levels of protein or low molecular weight persulfides.
The most important aspect of the development of MitoA is that it enables the assessment of changes in H2S in vivo. This exomarker approach has previously been used to infer levels of evanescent species such as H2O2 in a range of living organisms, where assessments ex vivo or in isolated cells would have introduced confounding factors making the data hard to interpret. The experimental constraints are similar with assessment of free H2S in tissues, and are in fact exacerbated due to the rapid diffusion of H2S from in vitro systems, such as cell culture where H2S rapidly disperses into the large amount of medium above a cell monolayer and is then lost into the head space. In contrast, in tissues the situation is quite different, where the local architecture can enable a large build up of H2S. Consequently, measurements of H2S levels within tissues in vivo under normal and pathological conditions are essential to infer the roles of H2S in vivo.
As a proof of principle to test MitoA in vivo, we assessed changes in H2S in tissues undergoing ischemia/hypoxia. Ischemia is expected to lead to an increase in H2S due to the inhibition of H2S degradation by SQR that occurs in the absence of O2. An advantage of this model to test the efficacy of MitoA was that there was no intervention with an exogenous agent or H2S donor, thus it indicates that MitoA can respond to H2S levels that may arise in vivo under pathological conditions. These findings also demonstrated that there were dramatic changes in H2S between the ischemic and normoxic regions of the heart, and that the H2S levels rapidly returned to normal upon reperfusion with oxygenated blood. None of these assessments would have been possible prior to the development of MitoA. Interestingly, as well as enabling the testing of MitoA, these findings suggested that H2S levels may be a significant aspect of how tissues respond to ischemia and these changes in H2S will be explored further as a potential ischemic signaling pathway.
There are a few limitations to the exomarker approach. Because the ratio of MitoN to MitoA can only be determined from tissue samples ex vivo, each time point requires killing a mouse. Furthermore, the analysis is inevitably an average of many different cells and mitochondria. Consequently, complementary techniques are required to assess changes in individual cells and in real time. Another caveat is that many of the effects of altering H2S metabolism are likely to be caused by changes in protein persulfidation that will not be picked up directly by the MitoA approach, which only responds to free H2S. Therefore it is important in the future to develop related techniques to assess protein persulfidation in parallel to the measurement of free H2S. Finally, it is difficult to relate the MitoN/MitoA ratio to the actual amount of H2S present in vivo.
In summary, we have developed a mass spectrometric probe that will enable for the first time changes in the levels of H2S to be assessed in vivo. Use of this probe will greatly facilitate investigations of the role of H2S in health and disease.
Experimental procedures
Chemical syntheses
MitoA and MitoN were synthesized from triphenylphosphine and the deuterated analogues, d15-MitoA and d15-MitoN, from tri(pentadeuterophenyl)phosphine as shown in supplemental Fig. S1A. The triarylphosphines were each reacted with 1,3-diodopropane to give iodopropyl-TPP salts 1 and d15-1. Displacement of the alkyl iodide by azide gave the azidopropyl-TPP salts 2 and d15-2, and hydrogenation converted these into the amines 3 and d15-3. The latter were coupled with 4-azidobenzoic acid using diisopropylcarbodiimide in acetonitrile (ACN) and the resulting amide precipitated from solution in each case. Ion exchange then gave MitoA and d15-MitoA as their mesylate salts in moderate yield. In a similar way, coupling 4-aminobenzoic acid followed by ion exchange gave MitoN and d15-MitoN as their mesylate salts. 4-Azido-N-(hex-1-yl)benzamide 4 and 4-amino-N-(hex-1-yl)benzamide 5, which are analogues of MitoA and MitoN lacking the TPP targeting group, were prepared for comparison (supplemental Fig. S1B). Further details of the chemical syntheses are given under supplementary materials. Peroxynitrite was prepared according to Refs. 64 and 65. A persulfide analogue of S-nitrosopenicillamine, NAP-SSH, was synthesized as described (52) and was prepared as a 1 mm stock in 10 mm HCl at 4 °C.
Assessment of compound properties
MitoA and MitoN stock solutions (5 mm in ethanol (EtOH)) were stable for at least 6 months at −20 °C. Absorbance spectra were carried out in 1-ml cuvettes in KCl buffer (120 mm KCl, 10 mm Hepes and 1 mm EGTA, pH 7.2 (KOH)) using a Shimadzu UV-2501PC UV-visible spectrophotometer. The molar extinction coefficient for MitoA at 267 nm in KCl buffer was calculated gravimetrically: 20.4 ± 0.1 × 103 m−1 cm−1.
Reversed phase-high performance liquid chromatography (RP-HPLC) was done using a Gilson 321 pump with attached UV-visible 151 system (Gilson). Samples were filtered through a 0.22-μm polyvinylidene difluoride (PVDF) filter (Millex, Millipore), loaded, and run over a C18 column (Jupiter 300 Å, Phenomenex) with a Widepore C18 guard column (Phenomenex). HPLC buffer A (0.1% (v/v) trifluoroacetic acid (TFA) in water) and HPLC buffer B (0.1% (v/v) TFA/acetonitrile) were used with the gradient: 0–2 min, 5% B; 2–17 min, 5–100% B; 17–19 min, 100% B; 19–22 min 100–5% B.
Cell culture
Cells (C2C12 or HCT116 from the American Type Culture Collection) were incubated at 37 °C in a humidified atmosphere of 95% air and 5% CO2 in Dulbecco's modified Eagle's medium (Invitrogen) supplemented with 10% (v/v) fetal bovine serum (FBS) and 100 units/ml of penicillin and 100 μg/ml of streptomycin.
Preparation of H2S solutions
Unless indicated otherwise, standard solutions of H2S were prepared by dissolving NaHS in KCl buffer, the pH was re-adjusted to 7.2 and then the solution was aliquoted into tubes with no head space and kept on ice. The H2S concentration was determined by the methylene blue assay (66).
Mitochondrial preparation and incubation
Liver mitochondria were prepared from female Wistar rats killed by stunning and cervical dislocation, followed by homogenization of the liver and differential centrifugation in ice-cold STE buffer (250 mm sucrose, 10 mm Tris, 1 mm EGTA, pH 7.4). The protein concentration was determined by the biuret assay using bovine serum albumin as a standard (67).
Extraction of MitoA and MitoN
For cells, frozen pellets were thawed, vortexed, and resuspended in 210 μl of 95% acetonitrile (ACN) spiked with internal standard (100 pmol of d15-MitoA and 100 pmol of d15-MitoN). Following protein precipitation (30 min, 4 °C) and centrifugation (16,000 × g, 10 min, room temperature) the supernatant was filtered through a 0.22-μm filter and dried under vacuum (miVac Quattro concentrator; Genevac) and stored at −20 °C. For extraction from tissues, ∼50 mg wet weight tissue was placed in a 2-ml Eppendorf tube containing 210 μl of 95% ACN spiked with internal standards (100 pmol of d15-MitoA and 100 pmol of d15-MitoN). For the blood no further additions were made, but for the tissues we added ∼50 μl of beads (liver, brain, and kidney, 0.5-mm diameter zirconium oxide beads; heart, 0.9–2.0-mm diameter stainless steel beads, both from Next Advance) and homogenized for 3 min using a Bullet Blender (Storm 24; Next Advance) at speed 8. Samples were centrifuged (16,000 × g, 10 min, room temperature), the supernatant was transferred to a new Eppendorf tube. The pellet was re-extracted with 95% ACN, and the supernatants were pooled and incubated for 30 min at 4 °C to precipitate proteins. After centrifugation (16,000 × g, 10 min, room temperature) the supernatant was filtered and dried as above and stored at −20 °C. Prior to analysis the samples were resuspended in 20% ACN, 0.1% FA, centrifuged (16,000 × g, 10 min, room temperature), and transferred to mass spectrometry vials (TrueViewTM LCMS Certified, Waters).
LC-MS/MS analysis
For MS/MS analysis we used a triple-quadrupole mass spectrometer (Waters Xevo TQ-S under positive ion mode: source spray voltage, 3.2 kV; cone voltage, 125 V; ion source temperature, 100 °C; collision energy, 75 V). Nitrogen and argon were used as curtain and collision gas, respectively. MS fragmentation patterns were determined by direct infusion (5 nm at 50 μl/min in 20% ACN, 0.1% FA). For LC-MS/MS analyses the mass spectrometer was connected in series to an I-class Acquity LC system (Waters). Samples were stored in an autosampler at 4 °C and 2-μl samples went into a 15-μl flow-through needle and RP-UPLC at 40 °C using an Acquity UPLC® BEH C18 column (1 × 50 mm, 1.7 μm; Waters) with a Waters UPLC filter (0.2 μm). MS buffers A (95% water, 5% ACN, 0.1% FA) and B (90% ACN, 10% water, 0.1% FA) were infused at 200 μl/min using the following gradient: 0–0.3 min, 5% B; 0.3–3 min, 5–100% B; 3–4 min, 100% B, 4.0–4.10, 100–105% B; 4.10–4.60 min, 5% B. Eluant was diverted to waste from 0 to 1 min and from 4 to 4.6 min. Compounds were detected in multiple reactions monitoring in positive ion mode. Under these conditions MitoA underwent neutral loss of N2 to a nitrene, which was used as the precursor ion. For quantification the following transitions were used: MitoA, 437 > 183; d15-MitoA, 452 > 191; MitoN, 439 > 183; d15-MitoN, 454 > 191. Standard curves with known amounts of MitoA and MitoN were prepared, spiked with IS, and extracted following the protocol outlined above. The peak area of MitoA, MitoN, and IS of samples and standard curves were quantified using the MassLynx 4.1 software.
Liver homogenate incubations
Liver was collected from a female Wistar rat that had been killed by stunning and cervical dislocation, and carried out in accordance with the UK Home Office Guide on Animal Experiments. The liver was transferred to ice-cold STE buffer, cut into small pieces using a razor blade, washed in ice-cold KCl buffer, and homogenized in a Dounce homgenizer, and the protein concentration was determined by biuret assay.
Mouse experiments
Wild-type male C57BL/6J mice (8–10 weeks old) were obtained from Charles Rivers Laboratories (UK). Mice were housed under standard laboratory conditions with food and water available ad libitum and experiments were carried out in accordance with the UK Home Office Guide on Animal Experiments and were approved under Project Licenses PPL 70/8238, 70/7963, and PPL 80/2638. To assess MitoA uptake and distribution in vivo, mice were placed in a heating chamber set to 38 °C (Vet-Tech Solutions) for ∼5 min, inserted in a rodent restrainer with free access to the tail, and MitoA (50 nmol) was injected into a lateral tail vein in 100 μl of saline through a 27-gauge needle connected to a 1-ml syringe. Mice were then monitored throughout the incubation period until time points for tissue collection were reached.
For the ex vivo incubation of hearts, mice were anesthetized with isoflurane (Abbott Laboratories, US) and oxygen at 2 liters/min. MitoA (50 nmol) was administered in saline via the inferior vena cava and then the heart was explanted as described (68). In brief, 100 μl of heparin was administered prior to exsanguination by division of the inferior vena cava and aorta. The inferior vena cava, hemiazygous vein, superior vena cava, innominate, left carotid, and left brachiocephalic were ligated prior to division of the descending aorta, pulmonary artery, and pulmonary veins. The heart was perfused with 500 μl of ice-cold Soltran (Baxter Healthcare, UK). During dissection and ligation of vessels the heart was topically cooled with 4 °C saline every 2 min. This process took 12 min and the heart was excised and stored in 10 ml of Belzer UW® Cold Storage Solution, University of Wisconsin solution (Belzer UW® Cold Storage Solution, Bridge to Life Ltd.) on ice for 240 min or snap frozen immediately. Samples were stored at −80 °C until extraction for LC-MS/MS.
In vivo IRI was performed by using an open-chest, in situ mouse (C57BL/6J) heart model in which the LAD is surgically transiently occluded as recently described (69). Mice were anesthetized (pentobarbital sodium; 70 mg/kg; intraperitoneal (i.p.)), intubated endotracheally, and ventilated with 3 cm of H2O positive end-expiratory pressure (110 breaths per min, 125–150 ml tidal volume) using a mouse ventilator (MINIVENT Type 845, Harvard Apparatus, Germany). Body temperature was monitored by rectal probe and maintained at 37 °C via an animal temperature controller (TCAT-2LV, Physitemp, Clifton, NJ). Corneal and withdraw reflexes were checked for anesthesia depth and additional anesthesia was administered throughout the experimental protocol if needed. A left thoracotomy was performed and the heart was exposed via stripping of the pericardium. Regional ischemia was then induced via ligation of the main branch of the LAD. Successful occlusion was confirmed by color change of the anterior wall of the left ventricle and apex from a bright red color typical of a perfused heart to off-white and ischemia was sustained for 30 min, at which point the tissue was reperfused. Reperfusion of the heart was confirmed by a color change of the ischemic zone to red. Control mice were sham treated. MitoA (50 nmol) was administered as a 100-μl bolus via the tail vein at various times during the procedure. Tissue was collected, snap frozen, and stored at −80 °C until analysis.
Author contributions
S. A. characterized MitoA and MitoN. C. B.-G. synthesized MitoA and MitoN. A. L. and S. A. carried out the mass spectrometric analyses. T. R. carried out the mouse LAD experiments. T. A. P. processed samples for mass spectrometry and carried out the ion-selective electrode experiments, J. M. and K. S.-P. carried out the heart storage in transplantation medium experiments, T. K. supervised the mouse LAD experiments. M. R. F. and R. W. carried out the kinetic studies. R. C. H. supervised the chemical synthesis and helped direct the project and write the manuscript. M. P. M. and R. C. H. directed the project and wrote the manuscript, with assistance from all other authors.
Supplementary Material
Acknowledgment
We are grateful to Professor Christopher J. Chang, University of California at Berkeley, for helpful discussions.
This work was supported in part by Medical Research Council UK Grant MC_U105663142, Wellcome Trust Investigator award 110159/Z/15/Z (to M. P. M.), Biotechnology and Biological Sciences Research Council Grant BB/I012826/1, Wellcome Trust Investigator award 110158/Z/15/Z (to R. C. H.), and a Consejo Nacional de Ciencia y Technología studentship (to C. B.-G.). The authors declare that they have no conflicts of interest with the contents of this article.
This article contains supplemental Figs. S1–S5.
- SQR
- sulfide:quinone oxidoreductase
- PTM
- posttranslational modification
- NAP
- N-acetylpenicillamine
- IS
- internal standards
- TPP
- triphenylphosphonium
- LAD model
- left anterior descending model
- ACN
- acetonitrile
- FA
- formic acid.
References
- 1. Abe K., and Kimura H. (1996) The possible role of hydrogen sulfide as an endogenous neuromodulator. J. Neurosci. 16, 1066–1071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Wang X., Wang Q., Guo W., and Zhu Y. Z. (2011) Hydrogen sulfide attenuates cardiac dysfunction in a rat model of heart failure: a mechanism through cardiac mitochondrial protection. Biosci. Rep. 31, 87–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Li L., Hsu A., and Moore P. K. (2009) Actions and interactions of nitric oxide, carbon monoxide and hydrogen sulphide in the cardiovascular system and in inflammation: a tale of three gases! Pharmacol. Ther. 123, 386–400 [DOI] [PubMed] [Google Scholar]
- 4. Li L., Bhatia M., Zhu Y. Z., Zhu Y. C., Ramnath R. D., Wang Z. J., Anuar F. B., Whiteman M., Salto-Tellez M., and Moore P. K. (2005) Hydrogen sulfide is a novel mediator of lipopolysaccharide-induced inflammation in the mouse. FASEB J. 19, 1196–1198 [DOI] [PubMed] [Google Scholar]
- 5. Whiteman M., and Winyard P. G. (2011) Hydrogen sulfide and inflammation: the good, the bad, the ugly and the promising. Expert Rev. Clin. Pharmacol. 4, 13–32 [DOI] [PubMed] [Google Scholar]
- 6. Szabo C., Ransy C., Módis K., Andriamihaja M., Murghes B., Coletta C., Olah G., Yanagi K., and Bouillaud F. (2014) Regulation of mitochondrial bioenergetic function by hydrogen sulfide: part I. biochemical and physiological mechanisms. Br. J. Pharmacol. 171, 2099–2122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kimura H., Nagai Y., Umemura K., and Kimura Y. (2005) Physiological roles of hydrogen sulfide: synaptic modulation, neuroprotection, and smooth muscle relaxation. Antioxid. Redox Signal. 7, 795–803 [DOI] [PubMed] [Google Scholar]
- 8. Yang G., Wu L., Jiang B., Yang W., Qi J., Cao K., Meng Q., Mustafa A. K., Mu W., Zhang S., Snyder S. H., and Wang R. (2008) H2S as a physiologic vasorelaxant: hypertension in mice with deletion of cystathionine γ-lyase. Science 322, 587–590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Calvert J. W., Coetzee W. A., and Lefer D. J. (2010) Novel insights into hydrogen sulfide: mediated cytoprotection. Antioxid. Redox Signal. 12, 1203–1217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Calvert J. W., Jha S., Gundewar S., Elrod J. W., Ramachandran A., Pattillo C. B., Kevil C. G., and Lefer D. J. (2009) Hydrogen sulfide mediates cardioprotection through Nrf2 signaling. Circ. Res. 105, 365–374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kondo K., Bhushan S., King A. L., Prabhu S. D., Hamid T., Koenig S., Murohara T., Predmore B. L., Gojon G. Sr., Gojon G. Jr., Wang R., Karusula N., Nicholson C. K., Calvert J. W., and Lefer D. J. (2013) H2S protects against pressure overload-induced heart failure via upregulation of endothelial nitric oxide synthase. Circulation 127, 1116–1127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. King A. L., and Lefer D. J. (2011) Cytoprotective actions of hydrogen sulfide in ischaemia-reperfusion injury. Exp. Physiol. 96, 840–846 [DOI] [PubMed] [Google Scholar]
- 13. Johansen D., Ytrehus K., and Baxter G. F. (2006) Exogenous hydrogen sulfide (H2S) protects against regional myocardial ischemia-reperfusion injury: evidence for a role of K ATP channels. Basic Res. Cardiol. 101, 53–60 [DOI] [PubMed] [Google Scholar]
- 14. Wen Y. D., Wang H., Kho S. H., Rinkiko S., Sheng X., Shen H. M., and Zhu Y. Z. (2013) Hydrogen sulfide protects HUVECs against hydrogen peroxide induced mitochondrial dysfunction and oxidative stress. PloS One 8, e53147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Elrod J. W., Calvert J. W., Morrison J., Doeller J. E., Kraus D. W., Tao L., Jiao X., Scalia R., Kiss L., Szabo C., Kimura H., Chow C. W., and Lefer D. J. (2007) Hydrogen sulfide attenuates myocardial ischemia-reperfusion injury by preservation of mitochondrial function. Proc. Natl. Acad. Sci. U.S.A. 104, 15560–15565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kang J., Li Z., Organ C. L., Park C. M., Yang C. T., Pacheco A., Wang D., Lefer D. J., and Xian M. (2016) pH-controlled hydrogen sulfide release for myocardial ischemia-reperfusion injury. J. Am. Chem. Soc. 138, 6336–6339 [DOI] [PubMed] [Google Scholar]
- 17. Blackstone E., Morrison M., and Roth M. B. (2005) H2S induces a suspended animation-like state in mice. Science 308, 518. [DOI] [PubMed] [Google Scholar]
- 18. Derwall M., Francis R. C., Kida K., Bougaki M., Crimi E., Adrie C., Zapol W. M., and Ichinose F. (2011) Administration of hydrogen sulfide via extracorporeal membrane lung ventilation in sheep with partial cardiopulmonary bypass perfusion: a proof of concept study on metabolic and vasomotor effects. Crit. Care 15, R51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Drabek T., Kochanek P. M., Stezoski J., Wu X., Bayir H., Morhard R. C., Stezoski S. W., and Tisherman S. A. (2011) Intravenous hydrogen sulfide does not induce hypothermia or improve survival from hemorrhagic shock in pigs. Shock 35, 67–73 [DOI] [PubMed] [Google Scholar]
- 20. Kabil O., and Banerjee R. (2014) Enzymology of H2S biogenesis, decay and signaling. Antioxid. Redox Signal. 20, 770–782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Libiad M., Yadav P. K., Vitvitsky V., Martinov M., and Banerjee R. (2014) Organization of the human mitochondrial hydrogen sulfide oxidation pathway. J. Biol. Chem. 289, 30901–30910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lagoutte E., Mimoun S., Andriamihaja M., Chaumontet C., Blachier F., and Bouillaud F. (2010) Oxidation of hydrogen sulfide remains a priority in mammalian cells and causes reverse electron transfer in colonocytes. Biochim. Biophys. Acta 1797, 1500–1511 [DOI] [PubMed] [Google Scholar]
- 23. Mishanina T. V., Libiad M., and Banerjee R. (2015) Biogenesis of reactive sulfur species for signaling by hydrogen sulfide oxidation pathways. Nat. Chem. Biol. 11, 457–464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wedmann R., Onderka C., Wei S., Szijártó I. A., Miljkovic J. L., Mitrovic A., Lange M., Savitsky S., Yadav P. K., Torregrossa R., Harrer E. G., Harrer T., Ishii I., Gollasch M., Wood M. E., et al. (2016) Improved tag-switch method reveals that thioredoxin acts as depersulfidase and controls the intracellular levels of protein persulfidation. Chem. Sci. 7, 3414–3426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Nicholls P., Marshall D. C., Cooper C. E., and Wilson M. T. (2013) Sulfide inhibition of and metabolism by cytochrome c oxidase. Biochem. Soc. Trans. 41, 1312–1316 [DOI] [PubMed] [Google Scholar]
- 26. Filipovic M. R., Miljkovic J. Lj., Nauser T., Royzen M., Klos K., Shubina T., Koppenol W. H., Lippard S. J., and Ivanović-Burmazović I. (2012) Chemical characterization of the smallest S-nitrosothiol, HSNO: cellular cross-talk of H2S and S-nitrosothiols. J. Am. Chem. Soc. 134, 12016–12027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Miljkovic J. Lj, Kenkel I., Ivanović-Burmazović I., and Filipovic M. R. (2013) Generation of HNO and HSNO from nitrite by heme-iron-catalyzed metabolism with H2S. Angew. Chem. Int. Ed. Engl. 52, 12061–12064 [DOI] [PubMed] [Google Scholar]
- 28. Murphy M. P. (2012) Mitochondrial thiols in antioxidant protection and redox signaling: distinct roles for glutathionylation and other thiol modifications. Antioxid. Redox Signal. 16, 476–495 [DOI] [PubMed] [Google Scholar]
- 29. Kamoun P., Belardinelli M. C., Chabli A., Lallouchi K., and Chadefaux-Vekemans B. (2003) Endogenous hydrogen sulfide overproduction in Down syndrome. Am. J. Med. Genet. A 116A, 310–311 [DOI] [PubMed] [Google Scholar]
- 30. Manna P., Gungor N., McVie R., and Jain S. K. (2014) Decreased cystathionine-γ-lyase (CSE) activity in livers of type 1 diabetic rats and peripheral blood mononuclear cells (PBMC) of type 1 diabetic patients. J. Biol. Chem. 289, 11767–11778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Tiranti V., Viscomi C., Hildebrandt T., Di Meo I., Mineri R., Tiveron C., Levitt M. D., Prelle A., Fagiolari G., Rimoldi M., and Zeviani M. (2009) Loss of ETHE1, a mitochondrial dioxygenase, causes fatal sulfide toxicity in ethylmalonic encephalopathy. Nat. Med. 15, 200–205 [DOI] [PubMed] [Google Scholar]
- 32. Yu F., Li P., Song P., Wang B., Zhao J., and Han K. (2012) An ICT-based strategy to a colorimetric and ratiometric fluorescence probe for hydrogen sulfide in living cells. Chem. Commun. (Camb.) 48, 2852–2854 [DOI] [PubMed] [Google Scholar]
- 33. Lin V. S., Lippert A. R., and Chang C. J. (2013) Cell-trappable fluorescent probes for endogenous hydrogen sulfide signaling and imaging H2O2-dependent H2S production. Proc. Natl. Acad. Sci. U.S.A. 110, 7131–7135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lord S. J., Conley N. R., Lee H. L., Samuel R., Liu N., Twieg R. J., and Moerner W. E. (2008) A photoactivatable push-pull fluorophore for single-molecule imaging in live cells. J. Am. Chem. Soc. 130, 9204–9205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Shimamoto K., and Hanaoka K. (2015) Fluorescent probes for hydrogen sulfide (H2S) and sulfane sulfur and their applications to biological studies. Nitric Oxide 46, 72–79 [DOI] [PubMed] [Google Scholar]
- 36. Lin V. S., Chen W., Xian M., and Chang C. J. (2015) Chemical probes for molecular imaging and detection of hydrogen sulfide and reactive sulfur species in biological systems. Chem. Soc. Rev. 44, 4596–4618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Henthorn H. A., and Pluth M. D. (2015) Mechanistic insights into the H2S-mediated reduction of aryl azides commonly used in H2S Detection. J. Am. Chem. Soc. 137, 15330–15336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Chen W., Pacheco A., Takano Y., Day J. J., Hanaoka K., and Xian M. (2016) A single fluorescent probe to visualize hydrogen sulfide and hydrogen polysulfides with different fluorescence signals. Angew. Chem. Int. Ed. Engl. 55, 9993–9996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hartle M. D., Hansen R. J., Tresca B. W., Prakel S. S., Zakharov L. N., Haley M. M., Pluth M. D., and Johnson D. W. (2016) A synthetic supramolecular receptor for the hydrosulfide anion. Angew. Chem. Int. Ed. Engl. 55, 11480–11484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hartle M. D., Prell J. S., and Pluth M. D. (2016) Spectroscopic investigations into the binding of hydrogen sulfide to synthetic picket-fence porphyrins. Dalton Trans. 45, 4843–4853 [DOI] [PubMed] [Google Scholar]
- 41. Dufton N., Natividad J., Verdu E. F., and Wallace J. L. (2012) Hydrogen sulfide and resolution of acute inflammation: a comparative study utilizing a novel fluorescent probe. Sci. Rep. 2, 499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Qian Y., Zhang L., Ding S., Deng X., He C., Zheng X. E., Zhu H.-L., and Zhao J. (2012) A fluorescent probe for rapid detection of hydrogen sulfide in blood plasma and brain tissues in mice. Chem. Sci. 3, 2920–2923 [Google Scholar]
- 43. Smith R. A., Hartley R. C., Cochemé H. M., and Murphy M. P. (2012) Mitochondrial pharmacology. Trends Pharmacol. Sci. 33, 341–352 [DOI] [PubMed] [Google Scholar]
- 44. Smith R. A., Porteous C. M., Gane A. M., and Murphy M. P. (2003) Delivery of bioactive molecules to mitochondria in vivo. Proc. Natl. Acad. Sci. U.S.A. 100, 5407–5412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Montoya L. A., and Pluth M. D. (2012) Selective turn-on fluorescent probes for imaging hydrogen sulfide in living cells. Chem. Commun. (Camb.) 48, 4767–4769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lippert A. R., New E. J., and Chang C. J. (2011) Reaction-based fluorescent probes for selective imaging of hydrogen sulfide in living cells. J. Am. Chem. Soc. 133, 10078–10080 [DOI] [PubMed] [Google Scholar]
- 47. Cochemé H. M., Quin C., McQuaker S. J., Cabreiro F., Logan A., Prime T. A., Abakumova I., Patel J. V., Fearnley I. M., James A. M., Porteous C. M., Smith R. A., Saeed S., Carré J. E., Singer M., et al. (2011) Measurement of H2O2 within living Drosophila during aging using a ratiometric mass spectrometry probe targeted to the mitochondrial matrix. Cell Metab. 13, 340–350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Cochemé H. M., Logan A., Prime T. A., Abakumova I., Quin C., McQuaker S. J., Patel J. V., Fearnley I. M., James A. M., Porteous C. M., Smith R. A., Hartley R. C., Partridge L., and Murphy M. P. (2012) Using the mitochondria-targeted ratiometric mass spectrometry probe MitoB to measure H2O2 in living Drosophila. Nat. Protoc. 7, 946–958 [DOI] [PubMed] [Google Scholar]
- 49. Logan A., Cochemé H. M., Li Pun P. B., Apostolova N., Smith R. A., Larsen L., Larsen D. S., James A. M., Fearnley I. M., Rogatti S., Prime T. A., Finichiu P. G., Dare A., Chouchani E. T., Pell V. R., et al. (2014) Using exomarkers to assess mitochondrial reactive species in vivo. Biochim. Biophys. Acta 1840, 923–930 [DOI] [PubMed] [Google Scholar]
- 50. Pun P. B., Logan A., Darley-Usmar V., Chacko B., Johnson M. S., Huang G. W., Rogatti S., Prime T. A., Methner C., Krieg T., Fearnley I. M., Larsen L., Larsen D. S., Menger K. E., Collins Y., et al. (2014) A mitochondria-targeted mass spectrometry probe to detect glyoxals: implications for diabetes. Free Radic. Biol. Med. 67, 437–450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Ono H., and Ito A. (1984) Transport of the precursor for sulfite oxidase into intermembrane space of liver mitochondria: characterization of import and processing activities. J. Biochem. 95, 345–352 [DOI] [PubMed] [Google Scholar]
- 52. Artaud I., and Galardon E. (2014) A persulfide analogue of the nitrosothiol SNAP: formation, characterization and reactivity. ChemBioChem 15, 2361–2364 [DOI] [PubMed] [Google Scholar]
- 53. Yadav P. K., Martinov M., Vitvitsky V., Seravalli J., Wedmann R., Filipovic M. R., and Banerjee R. (2016) Biosynthesis and reactivity of cysteine persulfides in signaling. J. Am. Chem. Soc. 138, 289–299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Asin-Cayuela J., Manas A. R., James A. M., Smith R. A., and Murphy M. P. (2004) Fine-tuning the hydrophobicity of a mitochondria-targeted antioxidant. FEBS Lett. 571, 9–16 [DOI] [PubMed] [Google Scholar]
- 55. Dare A. J., Bolton E. A., Pettigrew G. J., Bradley J. A., Saeb-Parsy K., and Murphy M. P. (2015) Protection against renal ischemia-reperfusion injury in vivo by the mitochondria targeted antioxidant MitoQ. Redox. Biol. 5, 163–168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Dare A. J., Logan A., Prime T. A., Rogatti S., Goddard M., Bolton E. M., Bradley J. A., Pettigrew G. J., Murphy M. P., and Saeb-Parsy K. (2015) The mitochondria-targeted anti-oxidant MitoQ decreases ischemia-reperfusion injury in a murine syngeneic heart transplant model. J. Heart Lung Transplant. 34, 1471–1480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Barca E., Kleiner G., Tang G., Ziosi M., Tadesse S., Masliah E., Louis E. D., Faust P., Kang U. J., Torres J., Cortes E. P., Vonsattel J. P., Kuo S. H., and Quinzii C. M. (2016) Decreased coenzyme Q10 levels in multiple system atrophy cerebellum. J. Neuropathol. Exp. Neurol. 75, 663–672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Yadav P. K., Yamada K., Chiku T., Koutmos M., and Banerjee R. (2013) Structure and kinetic analysis of H2S production by human mercaptopyruvate sulfurtransferase. J. Biol. Chem. 288, 20002–20013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Porteous C. M., Logan A., Evans C., Ledgerwood E. C., Menon D. K., Aigbirhio F., Smith R. A., and Murphy M. P. (2010) Rapid uptake of lipophilic triphenylphosphonium cations by mitochondria in vivo following intravenous injection: implications for mitochondria-specific therapies and probes. Biochim. Biophys. Acta 1800, 1009–1017 [DOI] [PubMed] [Google Scholar]
- 60. Porteous C. M., Menon D. K., Aigbirhio F. I., Smith R. A., and Murphy M. P. (2013) P-glycoprotein (Mdr1a/1b) and breast cancer resistance protein (Bcrp) decrease the uptake of hydrophobic alkyl triphenylphosphonium cations by the brain. Biochim. Biophys. Acta 1830, 3458–3465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Denekamp C., Pocsfalvi G., and Claeys M. (1999) Charge-remote and charge-proximate fragmentations in deuterium-labeled n-hexyadecyltriphenylphosphonium cations. Int. J. Mass Spectrom. 188, 163–175 [Google Scholar]
- 62. Chouchani E. T., Pell V. R., Gaude E., Aksentijević D., Sundier S. Y., Robb E. L., Logan A., Nadtochiy S. M., Ord E. N., Smith A. C., Eyassu F., Shirley R., Hu C. H., Dare A. J., James A. M., et al. (2014) Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature 515, 431–435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Chouchani E. T., Pell V. R., James A. M., Work L. M., Saeb-Parsy K., Frezza C., Krieg T., and Murphy M. P. (2016) A unifying mechanism for mitochondrial superoxide production during ischemia-reperfusion injury. Cell Metab. 23, 254–263 [DOI] [PubMed] [Google Scholar]
- 64. Beckman J. S., Beckman T. W., Chen J., Marshall P. A., and Freeman B. A. (1990) Apparent hydroxyl radical production by peroxynitrite: implications for endothelial injury from nitric oxide and superoxide. Proc. Natl. Acad. Sci. U.S.A. 87, 1620–1624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Hughes M. N., and Nicklin H. G. (1968) The chemistry of pernitrites: part 1. kinetics of decomposition of pernitrous acid. J. Am. Chem. Soc. A 1968, 450–452 [Google Scholar]
- 66. Cline J. D. (1969) Spectrophotometric determination of hydrogen sulfide in natural waters. Limnology Oceanography 14, 454–458 [Google Scholar]
- 67. Gornall A. G., Bardawill C. J., and David M. M. (1949) Determination of serum protein by means of the biuret reaction. J. Biol. Chem. 177, 751–766 [PubMed] [Google Scholar]
- 68. Corry R. J., Winn H. J., and Russell P. S. (1973) Heart transplantation in congenic strains of mice. Transplant. Proc. 5, 733–735 [PubMed] [Google Scholar]
- 69. Schmidt B., Metzner A., Chun K. R., Leftheriotis D., Yoshiga Y., Fuernkranz A., Neven K., Tilz R. R., Wissner E., Ouyang F., and Kuck K. H. (2010) Feasibility of circumferential pulmonary vein isolation using a novel endoscopic ablation system. Circ. Arrhythm. Electrophysiol. 3, 481–488 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.