

Abstract
Defects in mitochondrial cytochrome c oxidase or respiratory chain complex IV (CIV) assembly are a frequent cause of human mitochondrial disorders. Specifically, mutations in four conserved assembly factors impinging the biogenesis of the mitochondrion-encoded catalytic core subunit 2 (COX2) result in myopathies. These factors afford stability of newly synthesized COX2 (the dystonia-ataxia syndrome protein COX20), a protein with two transmembrane domains, and maturation of its copper center, CuA (cardiomyopathy proteins SCO1, SCO2, and COA6). COX18 is an additional COX2 assembly factor that belongs to the Oxa1 family of membrane protein insertases. Here, we used a gene-editing approach to generate a human COX18 knock-out HEK293T cell line that displays isolated complete CIV deficiency. We demonstrate that COX20 stabilizes COX2 during insertion of its N-proximal transmembrane domain, and subsequently, COX18 transiently interacts with COX2 to promote translocation across the inner membrane of the COX2 C-tail that contains the apo-CuA site. The release of COX18 from this complex coincides with the binding of the SCO1-SCO2-COA6 copper metallation module to COX2-COX20 to finalize COX2 biogenesis. Therefore, COX18 is a new candidate when screening for mitochondrial disorders associated with isolated CIV deficiency.
Keywords: cardiomyopathy, cytochrome c oxidase (complex IV), mitochondrial metabolism, mitochondrial respiratory chain complex, transcription activator-like effector nuclease (TALEN), COX18, COX2, COX20, SCO1, SCO2
Introduction
In nucleated human cells, mitochondria are central organelles that harbor the enzymes required for cellular respiration and aerobic energy production through oxidative phosphorylation (OXPHOS).2 These multimeric complexes are formed by subunits encoded in the nuclear and mitochondrial genomes that are assembled into functional units with the assistance of a myriad of nucleus-encoded ancillary proteins, generally called assembly factors. Mutations in OXPHOS subunits or assembly factors result in failure to fulfill the energy demands of most tissues with a major incidence in those with high energy requirements, such as the central nervous system and skeletal and cardiac muscles (1). Lesions affecting the function of the terminal OXPHOS enzyme, the cytochrome c oxidase or complex IV (CIV), are a frequent cause of mitochondrial disorders. Studies on CIV structure, biogenesis, and function have yielded insight into the molecular basis of these human diseases.
Complex IV is a copper-heme oxidase that couples electron transfer from cytochrome c to oxygen with proton extrusion across the inner membrane to contribute to the proton gradient required for ATP generation. Human CIV is formed by three catalytic core subunits (COX1, -2, and -3) encoded in the mitochondrial genome and 11 additional subunits (COX4, COX5a, COX7a, COX6c, COX7c, COX6b, COX6a, COX7b, COX8, and NDUFA4) encoded in the nuclear genome. CIV biogenesis follows a linear pathway with the different subunits being added in an ordered manner around a seed formed by COX1 (2). COX4 and COX5a are first added to mature COX1 before incorporation of COX2 and most of the remaining structural subunits. Then insertion of COX6a and COX7a/b finalizes assembly of the CIV monomer (2).
COX1 and COX2 contain the CIV redox-active metal centers. Their maturation by incorporation of copper and heme is required for their assembly into the holoenzyme. Despite the linearity of the CIV assembly process, recent data obtained in Saccharomyces cerevisiae has indicated the existence of COX1, COX2, and COX3 stabilization and maturation modules that render the proteins in an assembly-competent state (3). For example, in human mitochondria, a COX1 stabilization module containing the twin CX9C protein CMC1 and assembly factors COA3 (MITRAC12) and COX14 (C12orf62) is formed before the insertion of copper and heme into COX1 and incorporation of COX4 to the growing assembly intermediate (4).
The human COX2 stabilization and maturation module is directly relevant to the present study. COX2 has two transmembrane domains and a long hydrophilic C terminus protruding into the intermembrane space (IMS) containing the dinuclear copper center (CuA). The N-tail of newly synthesized COX2 is translocated across the membrane and then stabilized by interaction with COX20 (5), a transmembrane protein found to be mutated in several cases of dystonia-ataxia syndrome (6). Formation of the CuA center has received major attention because mutations in the SCO1 and SCO2 metallochaperones specific for this function result in severe cardiomyopathies (7–9). Recently, the twin CX9C IMS protein COA6 has been shown to participate in COX2 maturation by interacting with SCO1, SCO2, or both (7, 8, 10). Co-immunoprecipitation (co-IP) assays have detected several COX2 stability and maturation modules containing newly synthesized COX2 and COX20-SCO1-SCO2 (5), COA6-SCO2 (11), or COA6-SCO1 (10). Further research is required to unveil when and how the COX2 C-terminal tail is exported and to clarify how the COX2 module progresses in composition.
All COX2 assembly factors mentioned above are conserved from yeast to human and were first identified in S. cerevisiae. An additional yeast factor, whose human homolog remains largely uncharacterized, is the transmembrane protein COX18, a member of the Oxa1/YidC/Alb3 family of membrane protein insertases (12). S. cerevisiae Cox18 interacts with newly synthesized Cox2 (13) and is required for export of Cox2 C-tail across the inner membrane (12, 13). Whether human COX18 performs the same function remains intriguing because the function of the founder member of the family, OXA1, is not conserved from yeast to human (14).
To characterize the role of human COX18, we used the transcription activator-like effector nuclease (TALEN) technology to knock out COX18 in human embryonic kidney (HEK) 293T cells. We found that COX18 is required for COX2 C terminus translocation across the inner membrane and therefore essential for CIV assembly. We conclude that COX18 is a transient COX2 interactor that connects the COX20-containing COX2 stability module and the SCO1/2-COA6-containing COX2 CuA maturation module during the COX2 biogenetic process. Clinically, it connects two kinds of mitochondrial myopathies: the dystonia-ataxia syndrome due to mutations in COX20 and the cardiomyopathies resulting from mutations in SCO1, SCO2, or COA6.
Results
A COX18 knock-out cell line constructed by TALENs in HEK293T cells displays isolated CIV deficiency
To investigate whether human COX18 is necessary for CIV assembly, we used a TALEN approach to create a stable human COX18 knock-out (KO) line in HEK293T cells. A TALEN pair, designed to bind COX18 gene immediately downstream of the start codon, was co-transfected into HEK293T cells (Fig. 1A). Subsequently, single cells were isolated by fluorescence-activated cell sorting and screened for out-of-frame mutations leading to COX18 protein loss. Because at the time of the screen we did not have an efficient and specific anti-COX18 antibody, we hypothesized that COX18 KO will lead to CIV deficiency and screened for COX2 levels as a surrogate. Among the 43 clones isolated, eight had decreased COX2 steady-state levels (data not shown), five of which were used for DNA extraction, sequenced, and found to carry mutations in COX18 (Table 1).
Figure 1.
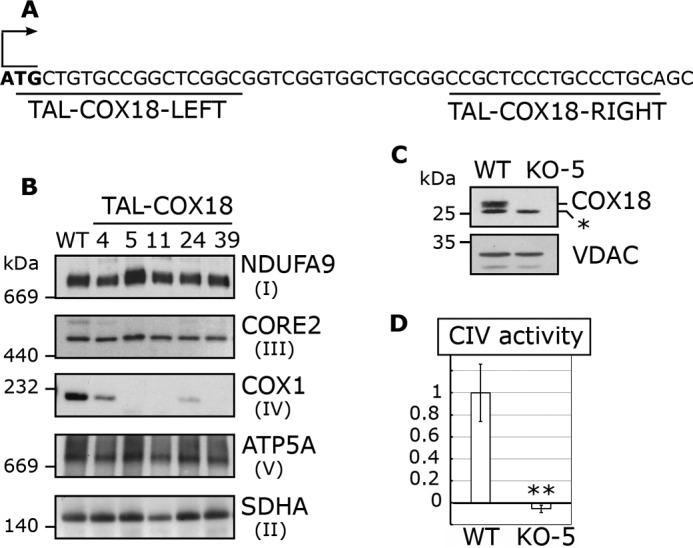
TALEN-generated HEK293T KO-COX18 clones display isolated CIV deficiency. A, schematic representation of COX18 gene downstream of the start codon and the sequence recognition sites of the TALEN pair used. B, steady-state levels of OXPHOS complexes analyzed by BN-PAGE and immunoblotting with the indicated antibodies. SDHA (complex II subunit) served as a loading control. C, immunoblotting analysis of COX18 abundance in mitochondria isolated from HEK293T (WT) and HEK293T KO-COX18 clone 5 cell lines. *, unspecific band. VDAC was used as a loading control. D, cytochrome c oxidase activity measured in KO-COX18 clone 5 cells normalized by citrate synthase activity and expressed as percentage of the WT. The bars represent average and the error bars represent S. D. of three repetitions. ** denotes p < 0.001.
Table 1.
COX18 alleles in TAL-COX18 clones
The DNA numbering refers to the coding sequence (c.), and the protein (p.) number refers to the predicted full polypeptide (39). C, compound; Mut, mutant; Hetero, heterozygous; Homo, homozygous; del, deletion; +, position in introns; ins, insertion; fs, frameshift.
| TAL clones | COX18 genotype | DNA | Protein |
|---|---|---|---|
| TAL1-4 | Mut. Homo | c.25_46del | p.9_16del |
| TAL1-5 | Mut. Homo | c.20_39del | p.Gly7fs |
| TAL1-11 | Mut. Homo | c.34_49del | p.Pro13fs |
| TAL1-24 | C. Hetero | c.10_45del/c.17_24del | p.4_15del/p.Gly6fs |
| TAL1-39 | C. Hetero | c.17_42del/c.24_25insGT | p.Gly6fs/ p.Trp9fs |
BN-PAGE followed by immunoblotting analysis of lauryl maltoside extracts showed that the five cell lines present a drastic decrease in CIV abundance without detectable alterations in the other OXPHOS complexes (Fig. 1B). The residual level of CIV detected in each cell line correlated with their COX18 genotype. Two clones (clones 4 and 24) carry in-frame small deletions in COX18 N terminus where the predicted mitochondrial targeting sequence is located and retain some residual CIV (Fig. 1B and Table 1), indicating that, although the mutations probably affect COX18 import efficiency, they allow for enough functional COX18 within mitochondria to account for the residual CIV detected (Fig. 1B). Three other clones genetically predicted to be complete knockouts (clones 5, 11, and 39) have no detectable CIV, which allowed us to conclude that COX18 is essential for CIV stability or assembly in human cells. When an appropriate anti-COX18 became available, we confirmed that the randomly selected TAL clone 5 has no residual COX18 (Fig. 1C) or residual CIV activity (Fig. 1D) and henceforth refer to this clone as KO-COX18 for all subsequent analyses.
COX18-FLAG is a mitochondrial transmembrane protein and complements the CIV deficiency of KO-COX18
To confirm the COX18 role in CIV biogenesis and eliminate the possibility of off-target effects during COX18 gene editing, we complemented the KO-COX18 line with COX18-FLAG cloned in the pIRESpuro2 plasmid under the control of a CMV promoter. In stable WT or KO lines expressing COX18-FLAG, the protein level was severalfold higher than the WT COX18 endogenous level (Fig. 2A).
Figure 2.
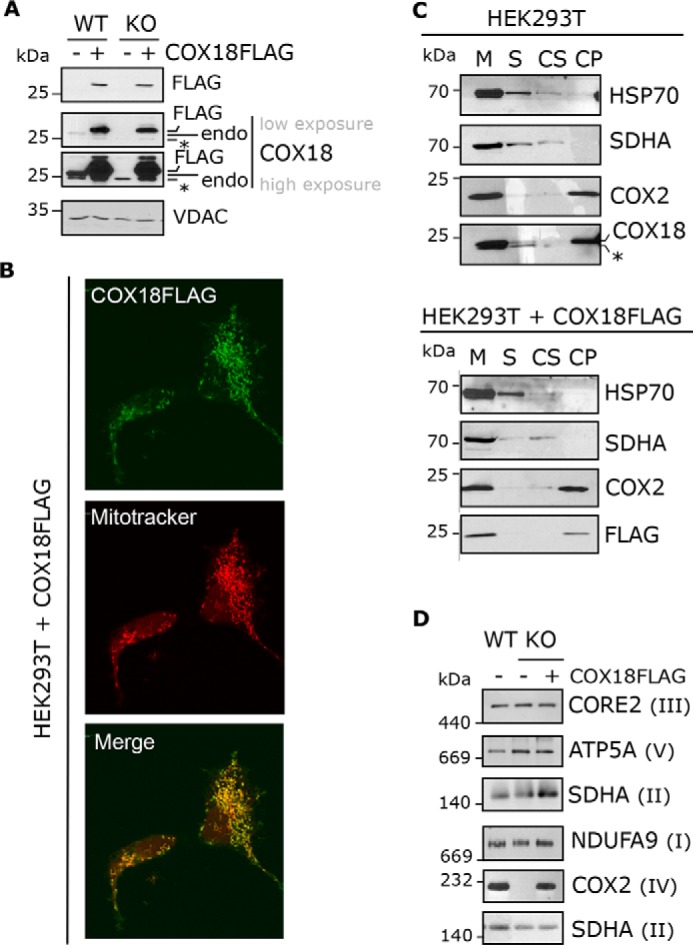
COX18-FLAG is a transmembrane mitochondrial protein and complements the absence of COX18. A, immunoblotting analysis of COX18-FLAG and COX18 steady-state levels in HEK293T and KO-COX18 cells stably transfected with an empty (−) or COX18-FLAG (+) vector. *, unspecific band. B, immunofluorescence analysis of HEK293T stably expressing COX18-FLAG using an anti-FLAG antibody and a secondary antibody conjugated to Alexa Fluor 488. Mitochondria (M) from HEK293T or Š and membrane-bound fractions were separated by centrifugation. The pellet, containing membrane-bound proteins, was then extracted. The fractions were analyzed by immunoblotting using antibodies against COX18 or the controls COX2 (membrane protein), SDHA (loosely bound to membrane), and HSP70 (soluble protein). D, BN-PAGE and immunoblotting analyses of OXPHOS complexes extracted with lauryl maltoside in HEK293T or KO-COX18 stably transfected with an empty (−) or COX18-FLAG (+) plasmid. endo, endogenous.
Immunofluorescence microscopy experiments localized COX18-FLAG to mitochondria (Fig. 2B). As expected, endogenous COX18 and COX18-FLAG could not be extracted with alkaline carbonate, corroborating that they are transmembrane proteins (Fig. 2C). Also, expression of COX18-FLAG in the KO-COX18 cell line restored CIV steady-state levels (Fig. 2D), indicating that the tagged protein is functional.
Supercomplexes I–III are stable in KO-COX18 cell line
To analyze how the absence of COX18 affects the formation and stability of mitochondrial respiratory supercomplexes, we used BN-PAGE and immunoblotting to characterize digitonin-extracted WT and KO-COX18 cell extracts. In KO-COX18, the respirasome (I + III2 + IVn) and all other CIV-containing supercomplexes were absent (Fig. 3A), whereas III2 and I + III2 accumulated normally. A larger I + III-containing supercomplex is detected in KO-COX18 cells and could correspond to some aberrant In + IIIn. These data confirm that the absence of COX18 results in an isolated complete CIV deficiency.
Figure 3.

KO-COX18 cells accumulate CIV subcomplexes containing COX1, COX4, and COX5a. A, BN-PAGE analysis of respiratory chain supercomplexes in HEK293T and KO-COX18 cells extracted with digitonin. The left panel is an in-gel complex I activity staining, and the other three panels show immunoblots probed with the indicated antibodies. B, immunoblotting analysis of CIV subunit steady-state levels in mitochondria isolated from HEK293T and KO-COX18 stably expressing an empty or COX18-FLAG vector. Two exposure times are shown for COX2, COX3, and COX6b. C, BN-PAGE and immunoblot analyses of CIV subcomplexes (sub-CIV), highlighting the accumulation of early assembly intermediates. SDHA was used as a loading control.
CIV subcomplexes containing COX1, COX4, and COX5A accumulate in the absence of COX18
To discern the CIV assembly step that is blocked in KO-COX18 cells, we analyzed the accumulation of CIV subunits and assembly subcomplexes. Whereas COX1, COX4, and COX5a remain stable in KO-COX18 cells, COX2, COX3, and COX6b were barely detected (Fig. 3B). BN-PAGE analysis showed that the absence of COX18 results in the accumulation of several CIV subcomplexes (Fig. 3C), which contain the subunits COX1 and COX4 (Fig. 3C). These assembly intermediates are similar to those previously described in cell lines defective in COX2 biogenesis, like mutants of COX20 (5), SCO1 (15), or SCO2 (16), pointing toward a role of COX18 in the biogenesis of COX2.
COX2 is normally synthesized but unstable in the KO-COX18 cell line
To understand the molecular function of COX18, we analyzed COX2 synthesis by mitochondrial ribosomes. Following a 30-min pulse with [35S]methionine, all mitochondrial DNA-encoded polypeptides were synthesized in KO-COX18 as in WT cells (Fig. 4A). However, after a 5-h chase, only the signals of COX2 and at a much lesser extent COX3 were markedly attenuated in KO-COX18 compared with WT cells (Fig. 4A). A chase time course showed that after a 15-min chase newly synthesized COX2 is quite stable in the absence of COX18, but its degradation can already be observed after 1 h (Fig. 4B). Hence, human COX18 is not required for COX2 synthesis but essential for its post-translational stability.
Figure 4.

COX2 is synthesized but unstable in KO-COX18 cells. A and B, mitochondrial translation products were pulsed-labeled with [35S]methionine for 30 min after inhibition of the cytosolic translation with emetine. HEK293T and KO-COX18 cells stably transfected with empty (−) or COX18-FLAG (+) plasmids were washed and incubated with fresh media for chase periods of 5 h (A) or 15 or 60 min (B). In B, the digitalized signals from X-ray films were quantified using the ImageJ program and expressed as percentage of the signal for each newly synthesized polypeptide at time 0. The bars represent the average, and the error bars represent S.D. of three repetitions. Cyt, cytochrome.
COX2 co-immunoprecipitates with COX18
The requirement of COX18 to stabilize newly synthesized COX2 suggests a potential interaction between these two proteins. To test this possibility, we used the KO-COX18 line stably expressing COX18-FLAG to perform an IP experiment with FLAG-conjugated beads. This experiment yielded a weak but reproducible specific enrichment of COX2, illustrating a probable transient interaction of COX18 with COX2 during its maturation (Fig. 5A).
Figure 5.
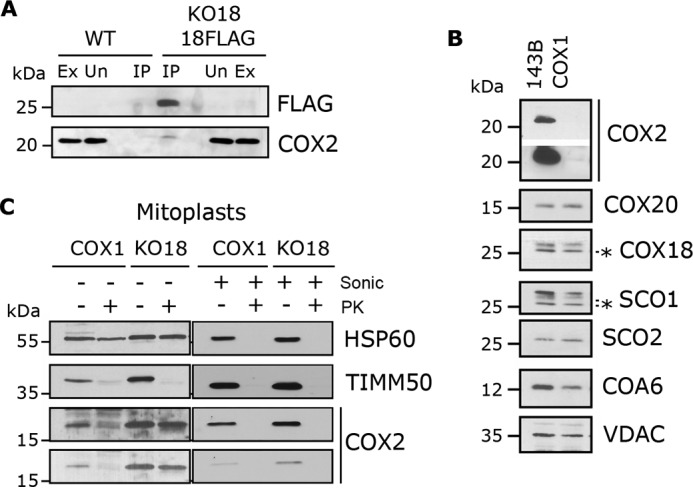
COX18 translocates the COX2 C terminus across the inner membrane following COX2 stabilization by COX20. A, mitochondria from HEK293T (WT) or KO-COX18 + COX18-FLAG were extracted and absorbed onto FLAG-conjugated beads. The eluate was analyzed by immunoblotting. B, steady-state-level analysis of the indicated proteins in a homoplasmic COX1 mutant cybrid cell line compared with the control 143B. Two exposure times are presented for COX2. C, mitoplasts and sonicated (Sonic) mitochondria from homoplasmic COX1 mutant cybrids and KO-COX18 were incubated in hypotonic buffer supplemented or not with proteinase K (PK). HSP60 is a matrix protein, and TIMM50 an inner transmembrane protein. Ex, extract; Un, unbound; IP, immunoprecipitate. IP corresponds to 40-fold the amount of extract loaded. The asterisk indicates antibody cross reacting bands.
COX18 is essential for the translocation of COX2 C terminus across the inner membrane
To test whether human COX18 plays a role in the translocation of the hydrophilic COX2 C terminus across the inner membrane like its yeast counterpart, we used a homoplasmic COX1 mutant cybrid cell line (17) where COX1 is absent, thus preventing any CIV assembly. In the COX1 cybrid line, all other mtDNA-encoded proteins, including COX2, are synthesized normally (4, 17), but COX2 is quickly degraded as it cannot join the CIV assembly line. The absence of COX1 does not strongly affect the stability of any COX2-specific assembly factor even if COA6 and SCO1 are slightly decreased (Fig. 5B). In S. cerevisiae cox1 mutants, COX2 has been shown to contain copper (11), which could also be true in the human COX1 mutant cybrids, thus providing an opportunity to analyze COX2 translocation by protease-protection assays. In support of this possibility, COX2 is proteinase K-sensitive in mitoplasts prepared by hypotonic swelling of mitochondria isolated from COX1 cybrid cells (Fig. 5C). On the contrary, the small fraction of COX2 still detected in KO-COX18 mitoplasts, which corresponds to newly synthesized COX2, is protected from degradation by proteinase K (Fig. 5C). This is not due to COX2 misfolding and adopting a protease-insensitive conformation in the absence of COX18 because COX2 was fully digested in a sample treated with brief sonication to expose all proteins to the protease (Fig. 5C). Therefore, human COX18 is essential for the translocation of the COX2 C terminus, which roughly represents 50% of the protein, across the inner membrane where its CuA site will be populated.
COX18 cooperates with COX20, COA6, SCO1, and SCO2 within the Cox2 maturation module
To decipher whether COX18 is part of the COX2 stabilization and maturation modules and at which stage the COX2 C-tail is exported, we performed several IP experiments. When COX18-FLAG was immunoprecipitated using mitochondrial extracts incubated with anti-FLAG-conjugated beads, we detected in the eluate COX20 and a small fraction of SCO1 but not SCO2 or COA6 (Fig. 6A). Instead, using a previously constructed KO-COX20 + COX20-FLAG cell line (5), COX20-FLAG co-immunoprecipitated with COX2, SCO1, and SCO2 (Fig. 6B) as reported (5) and with COX18 and COA6 (Fig. 6B). The network of interactions between COX20 and other COX2 assembly factors depends on the presence of newly synthesized COX2 because they are largely disrupted by doxycycline-mediated inhibition of COX2 synthesis (Fig. 6B).
Figure 6.

COX18 cooperates with COX20, COA6, SCO1, and SCO2 to mature COX2. A, mitochondria from HEK293T (WT) or KO-COX18 + COX18-FLAG were extracted and absorbed onto FLAG-conjugated beads. Data from three independent experiments are presented. B, HEK293T and KO-COX20 + COX20-FLAG cells were incubated for 24 h in the presence or absence of doxycycline. Mitochondria were then isolated, solubilized, and incubated with anti FLAG-conjugated beads. C, mitochondria from HEK293T and KO-COX18 stably expressing an empty or COX20-FLAG vector were solubilized and incubated with FLAG-conjugated beads. In each panel, the eluates were separated by SDS-PAGE and analyzed by immunoblotting. Two exposures are presented for COX18 and COX20. Ex, extract; Un, unbound; IP, immunoprecipitate; endo, endogenous. IP corresponds to 40-fold (in A) or 20-fold (in B and C) the amount of extract loaded. The asterisk indicates antibody cross reacting bands.
Our results highlight the cooperation of at least five assembly factors to channel COX2 through synthesis, membrane insertion, stabilization, and CuA site formation. The question remains of whether the assembly factors interact with COX2 and among them in a transient and ordered fashion. Our data suggest that COX2 maturation could be as a minimum a two-step process, the first step involving COX20-COX18-SCO1 and the second step following COX18 release with at least COX20-SCO2-COA6 and probably SCO1.
In the absence of COX18, COX20 still interacts with COX2 but not with SCO1, SCO2, or COA6
To further set apart the different steps of COX2 maturation, we analyzed the COX20 protein-interacting network in KO-COX18 cells. When COX18 is absent, COX20-FLAG does not co-precipitate any longer with SCO1, SCO2, or COA6. However, it still binds some COX2 albeit less efficiently than in the control line (Fig. 6C). These data indicate that COX18 acts transiently in COX2 biogenesis following the binding of COX20 to newly synthesized COX2 and before the action of the SCO proteins in CuA maturation.
Discussion
The TALEN-mediated generation of a KO-COX18 HEK293T cell line has provided a tool for the identification of human COX18 as the essential mitochondrial COX2 C terminus insertase and brought light to the COX2 biogenesis process. Our data support a model for COX2 maturation (presented in Fig. 6) that has at least four distinct steps. 1) COX20 is the first chaperone that binds and stabilizes newly synthesized COX2, either during or following membrane insertion of COX2 N terminus. COX20 remains bound to COX2 though all its maturation process. 2) COX18 is subsequently recruited to promote the membrane insertion of the second transmembrane domain of COX2, thus translocating its C terminus across the inner membrane. 3) The incorporation of SCO1 most probably coincides with the release of COX18 from the complex because a tiny fraction of SCO1 was already detected in the COX20-COX2-COX18 complex. 4) The copper insertion module formed by SCO1, SCO2, and COA6 joins to complete COX2 maturation by copper insertion into the CuA site (Fig. 7).
Figure 7.

COX2 maturation model. The schematic depicts the role of COX18 as a transient interactor of newly synthesized COX2 to promote the translocation of its C-tail (Ct) across the inner mitochondrial membrane.
COX18 belongs to the evolutionarily conserved family of Oxa1 proteins and is found in mitochondria from fungi to mammals (12, 18). Other members of the family include mammalian mitochondrial OXA1L, yeast mitochondrial Oxa1, chloroplast Alb3, and bacterial YidC, all of which are involved in the insertion of several hydrophobic proteins into phospholipid bilayers (19). Remarkably, Cox2 is the only known substrate of Cox18 in S. cerevisiae. Cox18 is required to export the large hydrophilic Cox2 C terminus but not the Cox2 N-tail across the inner membrane (12, 13). In this study, we have shown that human COX18 fulfills the same molecular function as its yeast counterpart and is, therefore, essential for CIV assembly. The Oxa1/YicC/Alb3 protein family members have significant similarity in their secondary structure and topology, all with at least five transmembrane domains, but the primary sequences even of the core domains display only a few invariant amino acid residues and low similarity. Oxa1 members have an extra C terminus region that binds to the ribosome (20), whereas bacterial YidC proteins contain an additional N-terminal transmembrane domain required for membrane targeting (21). Several studies have tested the functional overlapping between Oxa1/YicC and Cox18 in different organisms, particularly the fungi S. cerevisiae, Schizosaccharomyces pombe, and Neurospora crassa and the bacterium Escherichia coli (18, 22–24). Mild to extremely weak heterologous complementation by overexpression of chimeric proteins has been observed among members of this family (18, 22–24). For example, YidC can partially complement an S. cerevisiae cox18 deletion strain (23), and a fusion of the extra N terminus transmembrane domain of YidC to ScCox18 or ScOxa1 can partially complement the absence of YidC in E. coli (24). An overexpressed chimera of N-terminal ScCox18 fused to HsCOX18 could also complement, albeit extremely weakly, a truncation of ScCOX18 or SpCOX18 (22). These data highlight the substrate recognition specificity for members of this protein family and explain why, after the independent evolution of COX2 and COX18 in yeast and human mitochondria, and despite their conserved functions HsCOX18 could not restore the respiratory deficiency of a ScCOX18 deletion mutant strain (22) or ScCOX18 overexpression could not complement the CIV assembly defect of our human KO-COX18 cell line (not shown).
Despite the recent work devoted to solving the COX2 biogenesis pathway in human mitochondria and the important information the current work provides (see a model in Fig. 7), several questions remain unanswered. First, it is not clear how COX2 mRNA translation is activated and regulated. In S. cerevisiae mitochondrial mRNAs have long 5′-UTRs where specific translation activators, such as Pet111 for COX2 mRNA, will bind (25). These activators are not conserved in human mitochondria, whose mRNAs have extremely short 5′-UTRs, and therefore it is unclear how COX2 mRNA translation initiates. Recent data, however, have suggested that COX2 mRNA translation could be regulated by downstream COX2 maturation steps. COX2 synthesis has been reported to be decreased in fibroblasts from patients with mutations in SCO2 but not in SCO1 (26). However, it is not affected in HEK293T cells knocked out for either COX20 (5) or COX18 (this work) or knocked down for SCO2 (27). Cell type-specific effects could account for the conflicting reports. Also, some deviation in experimental details, such as the use of long mitochondrial translation pulses that could reflect degradation of newly synthesized proteins or the preincubation of cell cultures in media supplemented with chloramphenicol with the objective of enhancing overall mitochondrial protein synthesis upon washing, could be behind the reported differences.
Second, it remains obscure how the COX2 N-tail is inserted into the inner membrane. Although it is assumed that it occurs co-translationally, the insertase and mechanism involved remain to be deciphered. In yeast, the process depends on Oxa1 (28), but there is no evidence that OXA1L performs the same function in human mitochondria. In fact, knockdown of OXA1L in HEK293 cells affects the biogenesis of the F1F0-ATP synthase and complex I without altering the abundance of complex III or IV (14), and OXA1L was not enriched in COX20-FLAG IPs (5). Our data shows that COX18 is not involved in the process. It is plausible that COX20, an integral inner membrane protein whose hydrophilic domains are located in the IMS, could guide the co-translational insertion of COX2 N-proximal transmembrane domain into the membrane given that, in human COX2, the soluble N-tail is shorter and, contrary to ScCox2, does not require processing.
Third, although our studies have clearly shown that in the absence of COX18 no other protein can at least partially taker over its function, the possibility exists that additional proteins could cooperate with COX18. This is at least the case in S. cerevisiae where the fungi-specific Mss2 and Pnt1 have been identified as COX18 interactors and are required for full COX2 C-tail export across the inner membrane (12, 13, 29).
And fourth, whereas it is well established that the metallochaperones SCO1 and SCO2 are essential for CuA formation by performing non-overlapping functions (26, 30), their actual roles might need further clarification. According to Leary et al. (26), based on studies in cultured human cells, SCO2 and then SCO1 successively transfer a copper ion to COX2, and subsequently SCO2 acts as a thiol-disulfide oxidoreductase for SCO1. However, a recent in vitro biochemical study (31) reported that SCO1 is a metallochaperone that selectively transfers Cu(I) ions based on loop recognition, whereas SCO2 is a copper-dependent thiol reductase of the CuA cysteine ligands in COX2. Hence, in this scenario, the reduction of the apo-CuA disulfide by SCO2 Cu(I) is carried out by the metal and not by the thiols. Finally, the possible role of COA6 in COX2 metallation also requires clarification because functional interactions with either SCO2 (11) or SCO1 (10) have been reported. Our study suggests that SCO1 could interact with COX2 prior to the action of SCO2-COA6, perhaps to stabilize the protein upon release of COX18 from the COX2 N-tail translocation complex.
In addition to its biological significance, the COX2 biogenetic pathway proteins are at the frontline of biomedical relevance because mutations in most of its components have been associated with mitochondrial encephalomyopathies and cardiomyopathies (6–9, 32). Mutations in COX18, the single component not yet directly connected to human disorders, are expected to cause isolated CIV deficiency associated with similar disease scenarios.
Experimental procedures
Human cell lines, transfection, and culture conditions
HEK293T embryonic kidney cells (CRL-3216) and 143B osteosarcoma cells (CRL-8303) were obtained from ATCC. COX1 mutant cybrid cells carrying a homoplasmic G6930A mitochondrial mutation that leads to a truncated version of COX1 missing 33% of its sequence was reported before (17). KO-COX20 and a stable KO-COX20 + COX20-FLAG cell lines were previously constructed in HEK293T using the TALEN technology (5). Cells were grown in high-glucose DMEM with 110 mg/liter sodium pyruvate, 50 mg/liter uridine, and 10% FBS at 37 °C under 5% CO2. Cells were transfected with plasmids mixed with Lipofectamine 2000 (Invitrogen) in Opti-MEM I (Gibco). Stable lines were established by transfection of HEK293T or KO-COX18 cell lines with pIRESpuro2 empty vector or pIRESpuro2 containing COX18-FLAG or COX20-FLAG (5). Two days after transfection, the media were supplemented with 2.5 μg/ml puromycin, and drug selection was maintained for at least 1 month.
Plasmids
TALEN plasmids were obtained from Cellectis (France). The left TALEN was designed to bind the TGCTGTGCCGGCTCGGC DNA sequence, and the right TALEN was designed to bind to the CCGCTCCCTGCCCTGC sequence. We used a TALEN approach (33, 34) to create a stable human COX18 KO line in HEK293T cells as we previously reported for the generation of a COX20 KO cell line. A TALEN pair was designed to bind COX18 gene immediately downstream of the start codon. In this approach, upon dimerization, the nuclease introduces a DNA break that, when repaired by endogenous non-homologous recombination, occasionally results in small deletions or insertions into the genome.
HsCOX18-FLAG was amplified by PCR from HEK293T cDNA using primers 5′-ccggGCTAGCatgctgtgccggctcggcgg-3′ and 5′-gcgcGGATCCttacttgtcgtcatcgtctttgtagtcTTTTCTTGAAATGAACTTGGT-3′. The COX18 gene was cloned in pIRESpuro2 plasmid using NheI and BamHI sites.
Antibodies
The following commercial antibodies were obtained as indicated: FLAG, VDAC, HSP70, SCO2, COX20, COX5a, COA6, and COX18 from Sigma-Aldrich and COX1, COX2, COX3, COX4–1, COX6b, ATP5α, NDUFA9, CORE2, and SDHA from Abcam. SCO1 antibody was purified from immunized rabbit (5). Secondary antibodies coupled to horseradish peroxidase were obtained from Santa Cruz Biotechnology. Secondary mouse TrueBlot (Rockland) was used for immunoblotting after immunoprecipitation to eliminate interference of the heavy and light chains of the immunoprecipitation antibody.
HEK293T DNA extraction
Cells from a confluent HEK293T culture grown in one 1.9-cm2 well (24-well plate) were trypsinized and washed in PBS. Cells were resuspended in 500 μl of RSB buffer (10 mm Tris-HCl, pH 7.5, 10 mm NaCl, 25 mm EDTA, pH8, 1% SDS) with 25 μl of 20 mg/ml proteinase K (final 1 mg/ml) and incubated for 2 h at 55 °C. Then 0.1 μg/μl RNase was added, and the extract was incubated at 37 °C for 15 min. Following an extraction with 500 μl of phenol/chloroform/isoamyl (125:25:1), the mixture was vortexed and centrifuged for 5 min at 12,000 × g. The upper phase was collected and precipitated in 0.2 m NaCl (final concentration) and 2 volumes of ethanol. DNA was pelleted at 13,000 rpm for 15 min and washed in 75% ethanol, dried, and resuspended in 25 μl of water.
Whole-cell extracts and isolation of mitochondria
Cells were solubilized in radioimmune precipitation assay buffer (25 mm Tris-HCl, pH 7.6, 150 mm NaCl, 1% Nonidet P-40, 1% sodium deoxycholate, 0.1% SDS) with 1 mm PMSF and 1× mammalian protease inhibitor mixture (Sigma) and centrifuged 5 min at 10,000 × g at 4 °C to obtain whole-cell extracts. Mitochondria from human cells were isolated as described previously (5, 35).
BN-PAGE analysis
Cells were extracted as already described (5) either with digitonin for supercomplex analysis or lauryl maltoside for individual complex analysis.
Immunoblotting
Folin phenol reagent was used to measured protein concentration (36). 20–250 μg of mitochondrial proteins or 30–60 μg of whole-cell protein extracts were separated by SDS-PAGE in the Laemmli buffer system (37). After transfer, nitrocellulose membranes were probed with antibodies followed by a second reaction with anti-mouse or anti-rabbit IgG conjugated to horseradish peroxidase. Chemiluminescence was used for the final detection.
Determination of cytochrome c oxidase and citrate synthase activities
The activities of complex IV and the tricarboxylic acid cycle enzyme citrate synthase were determined spectrophotometrically in frozen-thawed cells as described previously (38).
Mitochondrial protein synthesis
Six-well plates were coated for 1 h at room temperature with 0.1 mg/ml collagen. Cells were grown until 80% confluence in DMEM with 110 mg/liter sodium pyruvate, 50 mg/liter uridine, and 10% FBS under 5% CO2 at 37 °C. Cells were washed twice in PBS and incubated for 20 min in DMEM devoid of methionine. Then the media were supplemented with 100 μg/ml emetine for 10 min to inhibit cytoplasmic protein synthesis followed by addition of 10 μl of [35S]methionine and incubation for 30-min pulse time. After incubation, cells were washed in PBS and incubated for an additional 5 min with complete medium for pulse samples. When a chase was performed, the cells were washed twice in PBS and incubated for the appropriate chase time in complete medium. Cells were then harvested by trypsinization, and whole-cell extracts were prepared by solubilization in radioimmune precipitation assay buffer. Following quantification by the procedure of Lowry et al. (36), 100 μg were loaded on a 17.5% SDS-polyacrylamide running gel with a 5% stacking gel. After running, the gel was transferred to a nitrocellulose membrane and exposed to an X-ray film.
Mitochondrial protein solubility assay
200 μg of mitochondria in 200 μl of STE buffer (0.6 m sorbitol, 20 mm Tris, pH 7.5, 1 mm EDTA, 1 mm PMSF) were sonicated for 3 s at intensity 2 using a Virtis Virsonic 100 ultrasonic cell disrupter and then centrifuged at 35,000 × g for 15 min at 4 °C. The supernatant containing the soluble proteins was removed. The pellet was then resuspended in 100 μl of 200 mm Na2CO3, pH 11.5, to extract extrinsic proteins, incubated for 30 min on ice, and centrifuged at 35,000 × g for 15 min at 4 °C. The pellet was extracted a second time in the same conditions. The 200 μl of supernatant after alkaline carbonate extraction contains the extrinsic proteins loosely associated to membranes. The pellet, containing intrinsic transmembrane proteins, was resuspended in 200 μl of STE. Equivalent samples from the different fractions were analyzed by immunoblotting.
Proteinase K protection assay
To prepare mitoplasts by hypotonic swelling of mitochondria, 250 μg were washed once and then resuspended in 200 μl of 20 mm Hepes, pH 7.5. In a parallel control sample, mitochondria were disrupted by brief sonication. The samples were incubated for 50 min at 4 °C with 2 μl of proteinase K at 0.2 μg/ml, and the reaction was stopped with 5 μl of 0.2 m PMSF. Mitoplasts were centrifuged at 15,000 × g for 15 min at 4 °C, and the resulting pellet was resuspended in 80 μl of 2× Laemmli buffer. Samples of sonicated mitochondria were also supplemented with Laemmli buffer. All samples were separated by SDS-PAGE and analyzed by immunoblotting.
Immunoprecipitation
Mitochondria were isolated, and 600 μg were extracted in 390 μl of 20 mm Tris, pH 7.4, 50 mm KCl, 0.4% lauryl maltoside, 1 mm PMSF with 4 μl of a protease inhibitor mixture (Sigma, P8340). After 30-min centrifugation at 22,000 × g, the extract was incubated with 50 μl of anti-FLAG-conjugated agarose or magnetic beads (Sigma M2 or Clontech) previously washed in 50 mm KCl, 20 mm Tris, pH 7.4. Following 4-h incubation at 4 °C on an orbital shaker, the unbound material was recovered, and the beads were washed 10 times in 500 μl of 20 mm Tris, pH7.5, 50 mm KCl, 0.02% lauryl maltoside. The beads were then supplemented with 50 μl of 2× Laemmli buffer and boiled 5 min to release the bound material. Representative amounts of all fractions were loaded on an SDS-polyacrylamide gel in the following proportions: extract, 1×; unbound material, 1×; immunoprecipitate, 20× for COX20-FLAG IP and extract, 1×; unbound material, 1×; immunoprecipitate, 50× for COX18-FLAG IP.
Mitochondrial immunocytochemistry
HEK293T cells were plated on collagen-coated (0.1 mg/ml collagen for 1 h at room temperature) coverslips and grown until 50% confluence. Cells were stained for 30 min with 50 nm MitoTracker Red (Molecular Probes), fixed with 2% paraformaldehyde for 20 min, and treated with cold methanol for 5 min before a 2-h incubation with an anti-FLAG primary antibody (Sigma) at 1:500 in PBS with 2% BSA. Anti-mouse secondary antibody conjugated to Alexa Fluor 488 (Invitrogen) was used for immunofluorescence detection. Images were obtained on a Leica SP5 confocal microscope (DMI6000 stand). The following parameters were used for the detection: for Alexa Fluor 488, argon ion laser; photomultiplier tube gain, 600; and 510–540-nm emission band; for MitoTracker Red, 561-nm laser; photomultiplier tube gain, 600; and 580–630-nm emission band.
Statistical analysis
All the experiments have been repeated at least in triplicate. Enzymatic activity data are presented as average ± S.D. of the percentage of control. Values were analyzed for statistical significance by Student's t test. A p value <0.05 (*) was considered significant.
Author contributions
M. B. and A. B. designed the experiments. M. B. performed the experiments. M. B. and A. B. wrote the manuscript.
Acknowledgments
We thank Dr. G. Manfredi for providing cell lines. We thank Dr. Peter Rehling and Dr. Sven Dennerlein for useful discussions.
This work was supported by NIGMS, National Institutes of Health Grants GM071775, GM105781, GM112179, and R35GM118141 (to A. B.), Muscular Dystrophy Association Grant 381828 (to A. B.), and an American Heart Association fellowship (to M. B.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- OXPHOS
- oxidative phosphorylation
- CIV
- complex IV
- COX
- cytochrome c oxidase
- TALEN
- transcription activator-like effector nuclease
- BN
- blue native
- IMS
- intermembrane space
- IP
- immunoprecipitation
- Sc
- S. cerevisiae
- Hs
- Homo sapiens
- Sp
- S. pombe
- SDHA
- succinate dehydrogenase subunit A
- VDAC
- voltage-dependent anion channel.
References
- 1. Fontanesi F., and Barrientos A. (2013) Mitochondrial cytochrome c oxidase assembly in health and in human diseases, in Mitochondrial Disorders Caused by Nuclear Genes. Part 3 (Wong L. E., ed) pp. 239–259, Springer Science, New York [Google Scholar]
- 2. Nijtmans L. G., Taanman J. W., Muijsers A. O., Speijer D., and Van den Bogert C. (1998) Assembly of cytochrome-c oxidase in cultured human cells. Eur. J. Biochem. 254, 389–394 [DOI] [PubMed] [Google Scholar]
- 3. McStay G. P., Su C. H., and Tzagoloff A. (2013) Modular assembly of yeast cytochrome oxidase. Mol. Biol. Cell 24, 440–452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bourens M., and Barrientos A. (2017) A CMC1-knockout reveals translation-independent control of human mitochondrial complex IV biogenesis. EMBO Rep. 18, 477–494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bourens M., Boulet A., Leary S. C., and Barrientos A. (2014) Human COX20 cooperates with SCO1 and SCO2 to mature COX2 and promote the assembly of cytochrome c oxidase. Hum. Mol. Genet. 23, 2901–2913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Szklarczyk R., Wanschers B. F., Nijtmans L. G., Rodenburg R. J., Zschocke J., Dikow N., van den Brand M. A., Hendriks-Franssen M. G., Gilissen C., Veltman J. A., Nooteboom M., Koopman W. J., Willems P. H., Smeitink J. A., Huynen M. A., et al. (2013) A mutation in the FAM36A gene, the human ortholog of COX20, impairs cytochrome c oxidase assembly and is associated with ataxia and muscle hypotonia. Hum. Mol. Genet. 22, 656–667 [DOI] [PubMed] [Google Scholar]
- 7. Ghosh A., Trivedi P. P., Timbalia S. A., Griffin A. T., Rahn J. J., Chan S. S., and Gohil V. M. (2014) Copper supplementation restores cytochrome c oxidase assembly defect in a mitochondrial disease model of COA6 deficiency. Hum. Mol. Genet. 23, 3596–3606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Baertling F., van den Brand M. A., Hertecant J. L., Al-Shamsi A., van den Heuvel L. P., Distelmaier F., Mayatepek E., Smeitink J. A., Nijtmans L. G., and Rodenburg R. J. (2015) Mutations in COA6 cause cytochrome c oxidase deficiency and neonatal hypertrophic cardiomyopathy. Hum. Mutat. 36, 34–38 [DOI] [PubMed] [Google Scholar]
- 9. Papadopoulou L. C., Sue C. M., Davidson M. M., Tanji K., Nishino I., Sadlock J. E., Krishna S., Walker W., Selby J., Glerum D. M., Coster R. V., Lyon G., Scalais E., Lebel R., Kaplan P., et al. (1999) Fatal infantile cardioencephalomyopathy with COX deficiency and mutations in SCO2, a COX assembly gene. Nat. Genet. 23, 333–337 [DOI] [PubMed] [Google Scholar]
- 10. Stroud D. A., Maher M. J., Lindau C., Vögtle F. N., Frazier A. E., Surgenor E., Mountford H., Singh A. P., Bonas M., Oeljeklaus S., Warscheid B., Meisinger C., Thorburn D. R., and Ryan M. T. (2015) COA6 is a mitochondrial complex IV assembly factor critical for biogenesis of mtDNA-encoded COX2. Hum. Mol. Genet. 24, 5404–5415 [DOI] [PubMed] [Google Scholar]
- 11. Pacheu-Grau D., Bareth B., Dudek J., Juris L., Vögtle F. N., Wissel M., Leary S. C., Dennerlein S., Rehling P., and Deckers M. (2015) Cooperation between COA6 and SCO2 in COX2 maturation during cytochrome c oxidase assembly links two mitochondrial cardiomyopathies. Cell Metab. 21, 823–833 [DOI] [PubMed] [Google Scholar]
- 12. Saracco S. A., and Fox T. D. (2002) Cox18p is required for export of the mitochondrially encoded Saccharomyces cerevisiae Cox2p C-tail and interacts with Pnt1p and Mss2p in the inner membrane. Mol. Biol. Cell 13, 1122–1131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Fiumera H. L., Broadley S. A., and Fox T. D. (2007) Translocation of mitochondrially synthesized Cox2 domains from the matrix to the intermembrane space. Mol. Cell. Biol. 27, 4664–4673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Stiburek L., Fornuskova D., Wenchich L., Pejznochova M., Hansikova H., and Zeman J. (2007) Knockdown of human Oxa1l impairs the biogenesis of F1Fo-ATP synthase and NADH:ubiquinone oxidoreductase. J. Mol. Biol. 374, 506–516 [DOI] [PubMed] [Google Scholar]
- 15. Williams S. L., Valnot I., Rustin P., and Taanman J. W. (2004) Cytochrome c oxidase subassemblies in fibroblast cultures from patients carrying mutations in COX10, SCO1, or SURF1. J. Biol. Chem. 279, 7462–7469 [DOI] [PubMed] [Google Scholar]
- 16. Leary S. C., Kaufman B. A., Pellecchia G., Guercin G. H., Mattman A., Jaksch M., and Shoubridge E. A. (2004) Human SCO1 and SCO2 have independent, cooperative functions in copper delivery to cytochrome c oxidase. Hum. Mol. Genet. 13, 1839–1848 [DOI] [PubMed] [Google Scholar]
- 17. D'Aurelio M., Pallotti F., Barrientos A., Gajewski C. D., Kwong J. Q., Bruno C., Beal M. F., and Manfredi G. (2001) In vivo regulation of oxidative phosphorylation in cells harboring a stop-codon mutation in mitochondrial DNA-encoded cytochrome c oxidase subunit I. J. Biol. Chem. 276, 46925–46932 [DOI] [PubMed] [Google Scholar]
- 18. Funes S., Nargang F. E., Neupert W., and Herrmann J. M. (2004) The Oxa2 protein of Neurospora crassa plays a critical role in the biogenesis of cytochrome oxidase and defines a ubiquitous subbranch of the Oxa1/YidC/Alb3 protein family. Mol. Biol. Cell 15, 1853–1861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bonnefoy N., Fiumera H. L., Dujardin G., and Fox T. D. (2009) Roles of Oxa1-related inner-membrane translocases in assembly of respiratory chain complexes. Biochim. Biophys. Acta 1793, 60–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Haque M. E., Spremulli L. L., and Fecko C. J. (2010) Identification of protein-protein and protein-ribosome interacting regions of the C-terminal tail of human mitochondrial inner membrane protein Oxa1L. J. Biol. Chem. 285, 34991–34998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Urbanus M. L., Fröderberg L., Drew D., Björk P., de Gier J. W., Brunner J., Oudega B., and Luirink J. (2002) Targeting, insertion, and localization of Escherichia coli YidC. J. Biol. Chem. 277, 12718–12723 [DOI] [PubMed] [Google Scholar]
- 22. Gaisne M., and Bonnefoy N. (2006) The COX18 gene, involved in mitochondrial biogenesis, is functionally conserved and tightly regulated in humans and fission yeast. FEMS Yeast Res. 6, 869–882 [DOI] [PubMed] [Google Scholar]
- 23. Preuss M., Ott M., Funes S., Luirink J., and Herrmann J. M. (2005) Evolution of mitochondrial Oxa proteins from bacterial YidC. Inherited and acquired functions of a conserved protein insertion machinery. J. Biol. Chem. 280, 13004–13011 [DOI] [PubMed] [Google Scholar]
- 24. van Bloois E., Koningstein G., Bauerschmitt H., Herrmann J. M., and Luirink J. (2007) Saccharomyces cerevisiae Cox18 complements the essential Sec-independent function of Escherichia coli YidC. FEBS J. 274, 5704–5713 [DOI] [PubMed] [Google Scholar]
- 25. Poutre C. G., and Fox T. D. (1987) PET111, a Saccharomyces cerevisiae nuclear gene required for translation of the mitochondrial mRNA encoding cytochrome c oxidase subunit II. Genetics 115, 637–647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Leary S. C., Sasarman F., Nishimura T., and Shoubridge E. A. (2009) Human SCO2 is required for the synthesis of CO II and as a thiol-disulphide oxidoreductase for SCO1. Hum. Mol. Genet. 18, 2230–2240 [DOI] [PubMed] [Google Scholar]
- 27. Richter-Dennerlein R., Oeljeklaus S., Lorenzi I., Ronsör C., Bareth B., Schendzielorz A. B., Wang C., Warscheid B., Rehling P., and Dennerlein S. (2016) Mitochondrial protein synthesis adapts to influx of nuclear-encoded protein. Cell 167, 471.e10–483.e10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hell K., Herrmann J., Pratje E., Neupert W., and Stuart R. A. (1997) Oxa1p mediates the export of the N- and C-termini of pCoxII from the mitochondrial matrix to the intermembrane space. FEBS Lett. 418, 367–370 [DOI] [PubMed] [Google Scholar]
- 29. Broadley S. A., Demlow C. M., and Fox T. D. (2001) Peripheral mitochondrial inner membrane protein, Mss2p, required for export of the mitochondrially coded Cox2p C tail in Saccharomyces cerevisiae. Mol. Cell. Biol. 21, 7663–7672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Horng Y. C., Leary S. C., Cobine P. A., Young F. B., George G. N., Shoubridge E. A., and Winge D. R. (2005) Human Sco1 and Sco2 function as copper-binding proteins. J. Biol. Chem. 280, 34113–34122 [DOI] [PubMed] [Google Scholar]
- 31. Morgada M. N., Abriata L. A., Cefaro C., Gajda K., Banci L., and Vila A. J. (2015) Loop recognition and copper-mediated disulfide reduction underpin metal site assembly of CuA in human cytochrome oxidase. Proc. Natl. Acad. Sci. U.S.A. 112, 11771–11776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Valnot I., Osmond S., Gigarel N., Mehaye B., Amiel J., Cormier-Daire V., Munnich A., Bonnefont J. P., Rustin P., and Rötig A. (2000) Mutations of the SCO1 gene in mitochondrial cytochrome c oxidase deficiency with neonatal-onset hepatic failure and encephalopathy. Am. J. Hum. Genet. 67, 1104–1109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Li T., Huang S., Zhao X., Wright D. A., Carpenter S., Spalding M. H., Weeks D. P., and Yang B. (2011) Modularly assembled designer TAL effector nucleases for targeted gene knockout and gene replacement in eukaryotes. Nucleic Acids Res. 39, 6315–6325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Christian M., Cermak T., Doyle E. L., Schmidt C., Zhang F., Hummel A., Bogdanove A. J., and Voytas D. F. (2010) Targeting DNA double-strand breaks with TAL effector nucleases. Genetics 186, 757–761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Fernández-Vizarra E., Ferrín G., Pérez-Martos A., Fernández-Silva P., Zeviani M., and Enríquez J. A. (2010) Isolation of mitochondria for biogenetical studies: an update. Mitochondrion 10, 253–262 [DOI] [PubMed] [Google Scholar]
- 36. Lowry O. H., Rosebrough N. J., Farr A. L., and Randall R. J. (1951) Protein measurement with the Folin phenol reagent. J. Biol. Chem. 193, 265–275 [PubMed] [Google Scholar]
- 37. Laemmli U. K. (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227, 680–685 [DOI] [PubMed] [Google Scholar]
- 38. Barrientos A., Fontanesi F., and Díaz F. (2009) Evaluation of the mitochondrial respiratory chain and oxidative phosphorylation system using polarography and spectrophotometric enzyme assays. Curr. Protoc. Hum. Genet. Chapter 19, Unit19.3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. den Dunnen J. T., and Antonarakis S. E. (2000) Mutation nomenclature extensions and suggestions to describe complex mutations: a discussion. Hum. Mutat. 15, 7–12 [DOI] [PubMed] [Google Scholar]