

Abstract
The insulin receptor substrate (IRS) proteins serve as essential signaling intermediates for the activation of PI3K by both the insulin-like growth factor 1 receptor (IGF-1R) and its close family member, the insulin receptor (IR). Although IRS-1 and IRS-2 share significant homology, they regulate distinct cellular responses downstream of these receptors and play divergent roles in breast cancer. To investigate the mechanism by which signaling through IRS-1 and IRS-2 results in differential outcomes, we assessed the involvement of the microtubule cytoskeleton in IRS-dependent signaling. Treatment with drugs that either stabilize or disrupt microtubules reveal that an intact microtubule cytoskeleton contributes to IRS-2- but not IRS-1-mediated activation of AKT by IGF-1. Proximal IGF-1R signaling events, including IRS tyrosine phosphorylation and recruitment of PI3K, are not inhibited by microtubule disruption, indicating that IRS-2 requires the microtubule cytoskeleton at the level of downstream effector activation. IRS-2 colocalization with tubulin is enhanced upon Taxol-mediated microtubule stabilization, which, together with the signaling data, suggests that the microtubule cytoskeleton may facilitate access of IRS-2 to downstream effectors such as AKT. Of clinical relevance is that our data reveal that expression of IRS-2 sensitizes breast carcinoma cells to apoptosis in response to treatment with microtubule-disrupting drugs, identifying IRS-2 as a potential biomarker for the response of breast cancer patients to Vinca alkaloid drug treatment.
Keywords: Akt PKB, apoptosis, breast cancer, cell signaling, insulin receptor substrate 1 (IRS-1), microtubule, insulin receptor substrate 2 (IRS-2)
Introduction
Insulin receptor substrate 1 (IRS-1)2 and IRS-2 are cytoplasmic adaptors for the insulin receptor (IR) and insulin-like growth factor 1 receptor (IGF-1R), and they play a major role in determining the cellular response to stimulation of these receptors (1). Notably, the IRS proteins are required for the activation of PI3K downstream of the IR and IGF-1R, which activate AKT and mechanistic target of rapamycin (mTOR) to promote proliferation, survival, motility, protein synthesis, and glucose metabolism (2–5). IRS-1 and IRS-2 are expressed ubiquitously in humans, including in the normal and malignant mammary epithelium (1). Despite their considerable sequence homology, IRS-1 and IRS-2 play divergent roles in breast cancer. In vitro, studies to assess IGF-1-dependent signaling through the IRS proteins in breast carcinoma cells have revealed that IRS-1 primarily regulates proliferation and survival, whereas IRS-2 regulates motility, invasion, and glycolysis (6–10). In vivo, overexpression of either IRS-1 or IRS-2 in the mouse mammary gland promotes mammary tumorigenesis (11). However, metastasis is diminished in the absence of Irs-2 expression and increased in the absence of Irs-1 expression (9, 12).
Differential localization patterns of IRS-1 and IRS-2 in human breast tumors suggest one explanation for their divergent functions in breast cancer (13). In normal breast tissue, ductal carcinoma in situ, and invasive breast tumors, IRS-1 is primarily localized in the nucleus and also diffusely in the cytoplasm, frequently correlating with nuclear expression of estrogen receptor (14–16). Nuclear expression of IRS-1 also correlates with response to tamoxifen in breast cancer patients (17). IRS-1 has been implicated in the regulation of estrogen response genes through its interaction with the estrogen receptor at estrogen response elements in gene promoters (16). The interaction of IRS-1 with β-catenin and its regulation of genes such as c-Myc and Cyclin D1 likely contribute to its role in stimulating proliferation (18, 19). In contrast to IRS-1, IRS-2 is absent from the nucleus in normal breast and breast tumors and is expressed either in the cytoplasm or at the cell membrane (13). Diffuse cytoplasmic IRS-2 staining is associated with better overall survival of breast cancer patients, whereas membrane localization of IRS-2 in breast tumors is associated with decreased overall survival, particularly in patients with progesterone receptor-negative tumors (13).
The localization pattern of IRS-1 and IRS-2 in human tumors suggests that their trafficking to distinct subcellular compartments contributes to their ability to elicit unique signaling responses. However, the mechanism by which the intracellular localization of IRS-1 and IRS-2 is determined is not known. The microtubule cytoskeleton, which plays an important role in intracellular trafficking, has been implicated in insulin-dependent regulation of glucose uptake, and this role for microtubules occurs distal to IRS phosphorylation (20–22). These observations support a role for microtubules in IRS-mediated downstream signaling, potentially through targeting of these adaptor proteins to unique subcellular compartments. We examined the role of the microtubule cytoskeleton in determining differential signaling and functional outcomes for IRS-1 and IRS-2. The data we obtained in this study reveal that IRS-2 co-localizes with microtubules and that the microtubule cytoskeleton contributes to IRS-2- but not IRS-1-dependent signaling to AKT. The differential involvement of the microtubule cytoskeleton in IRS-1- and IRS-2-mediated AKT signaling influences the apoptotic sensitivity of breast carcinoma cells to microtubule-disrupting drugs.
Results
IGF-1R signaling is dependent on the microtubule cytoskeleton
To investigate the role of the microtubule cytoskeleton in IRS-dependent IGF-1R signaling, MDA-MB-231 cells were stimulated with IGF-1 after short-term (30 min to 1 h) treatment with paclitaxel (Taxol) or nocodazole. Taxol, a taxane drug commonly used in cancer treatment, stabilizes microtubules, whereas nocodazole causes depolymerization of the tubulin cytoskeleton (23–25). IRS signaling was measured by assessing the phosphorylation status of AKT, a downstream signaling effector of PI3K, because the IRS proteins are required for the recruitment and activation of PI3K by the IGF-1R (4, 5, 26). Although microtubule stabilization by Taxol treatment did not alter the level of AKT activation (Fig. 1A), disruption of the microtubule cytoskeleton in response to nocodazole treatment significantly reduced phosphorylation of AKT at both threonine 308 and serine 473 (Fig. 1B). In contrast, the activation of ERK1/2 by IGF-1, which can occur independently of the IRS proteins (5), was not decreased by nocodazole treatment (Fig. 1B). IGF-1-dependent AKT activation was also reduced when cells were treated with the Vinca alkaloid drug vinblastine, which also disrupts microtubules and is used clinically in chemotherapy regimens (Fig. 1C) (23). Taken together, these results indicate that an intact microtubule cytoskeleton contributes to IRS-dependent activation of AKT by the IGF-1R.
Figure 1.

Involvement of the microtubule cytoskeleton in IGF-1-stimulated AKT activation. MDA-MB-231 cells were treated with DMSO, Taxol (20 μm), nocodazole (1 μm), or vinblastine (20 nm) for 30 min and then stimulated with IGF-1 (20 ng/ml) for 5 min. A—C, aliquots of cell extracts containing equivalent amounts of total protein were immunoblotted with antibodies specific for Ser(P)-473AKT (pAKT S473), Thr(P)-308AKT (pAKT T308), or Thr(P)-202/Tyr-204ERK (pERK). The immunoblots were subsequently stripped and reprobed with total AKT and ERK-specific antibodies. D, aliquots of cell extracts containing equivalent amounts of total protein were immunoprecipitated (IP) with antibodies specific for IRS1, IRS2, or IGF-1Rβ subunit and immunoblotted with antibodies specific for phosphotyrosine (pTyr) and the p85 subunit of PI3K (p85). The Tyr(P) immunoblots were subsequently stripped and reprobed with IRS1, IRS2, or IGF-1Rβ-specific antibodies. Total cell extracts were also immunoblotted with antibodies specific for Ser(P)-473AKT and total AKT (whole-cell lysate (WCL), bottom panels). All immunoblots shown are representative of three independent experiments.
To investigate the mechanism by which nocodazole inhibits AKT activation, phosphorylation of the IGF-1R and IRS proteins and IRS/PI3K association were examined. Treatment with nocodazole did not decrease IRS-1 or IRS-2 tyrosine phosphorylation or association with the p85 regulatory subunit of PI3K in response to IGF-1 stimulation, indicating that activation of PI3K upstream of AKT was not inhibited by microtubule disruption (Fig. 1D). Of note, expression of the IGF-1R β subunit and its IGF-1-dependent tyrosine phosphorylation were increased following nocodazole treatment (Fig. 1D). We attribute this increase to the accumulation of the activated receptor at the cell surface or in early endosomes, resulting in sustained expression and activation (27).
IRS-2, but not IRS-1, requires the microtubule cytoskeleton to activate AKT
To determine whether there is a selective role for the microtubule cytoskeleton in IRS-1- or IRS-2-mediated signaling, mammary tumor cells derived from PyMT:WT, PyMT:Irs-1−/−, or PyMT:Irs-2−/− tumors were treated with nocodazole and stimulated with IGF-1 (Fig. 2A). WT cells that express both Irs-1 and Irs-2 demonstrated a 40% reduction in Akt activation after treatment with nocodazole. PyMT:Irs-1−/− cells, which signal exclusively through Irs-2, exhibited a further reduction in IGF-1-dependent Akt activation (∼50%) following treatment with nocodazole. In contrast, Akt activation was equivalent in PyMT:Irs-2−/− cells, which signal exclusively through Irs-1, in response to IGF-1 stimulation following DMSO or nocodazole treatment.
Figure 2.

Selective requirement of the microtubule cytoskeleton for IRS-2-mediated signaling. A, PyMT:WT, PyMT:Irs-1−/− and PyMT:Irs-2−/− cells were treated with DMSO or 20 μm nocodazole (Noc) for 1 h and then stimulated with IGF-1 (10 ng/ml) for 5 min. B, PyMT:Irs-2−/− cells transfected with empty vector (Irs2−/−) or IRS2 (Irs2−/−:IRS2) were treated with DMSO, 1 μm nocodazole, 20 nm vinblastine (Vin), or 20 nm vinorelbine (Vino) for 1 h and then stimulated with IGF-1 (10 ng/ml) for 15 min. C, PyMT:Irs-1−/− cells transfected with empty vector (Irs1−/−) or IRS1 (Irs1−/−:IRS1) were treated with DMSO, 1 μm nocodazole, or 10 μm Taxol for 1 h and then stimulated with IGF-1 (10 ng/ml) for 5 min. D, MDA-MB-231 cells expressing either an shRNA targeting GFP (shGFP) or IRS2 (shIRS2) were treated with DMSO, 1 μm nocodazole, or 20 nm vinblastine for 30 min and then stimulated with IGF-1 (10 ng/ml) for 30 min. E, MDA-MB-231 cells expressing either an shRNA targeting GFP or IRS2 were treated with DMSO or 1 μm nocodazole for 30 min and then stimulated with IGF-1 (10 ng/ml) for the time periods indicated. The data in the graph represent the -fold change in phospho-AKT between DMSO- and nocodazole-treated cells for each cell type. Aliquots of cell extracts containing equivalent amounts of total protein were immunoblotted with antibodies specific for IRS1, IRS2, Ser(P)-473AKT, total AKT, tubulin, or GAPDH. The data shown in the graphs for each immunoblot represent the mean ± S.E. of three independent experiments. *, p ≤ 0.05 relative to shGFP; **, p ≤ 0.01 relative to shGFP.
The role of IRS-2 in the sensitivity of cells to microtubule disruption was explored further using PyMT:Irs-1−/− and PyMT:Irs-2−/− cells that were derived from PyMT:Irs-1fl/fl and PyMT:Irs-2fl/fl cell lines, respectively, after acute adenoviral-Cre infection. PyMT:Irs-2fl/fl cells with or without restored IRS-2 expression were stimulated with IGF-1 after treatment with nocodazole and vinblastine. An additional Vinca alkaloid drug, vinorelbine, which is used to treat breast cancer patients (23, 28), was also assayed (29). As observed previously (Fig. 2A), disruption of microtubules did not decrease Akt activation in cells lacking Irs-2 expression (Irs2−/−), whereas the increase in Akt activation that was observed upon restoration of IRS-2 expression was eliminated by microtubule disruption (Irs2−/−:IRS2) (Fig. 2B). In contrast, the -fold change reduction in Akt activation observed in Irs1−/− cells after nocodazole treatment (50%) was diminished (30%) upon restoration of IRS-1 expression (Irs−/−:IRS1) (Fig. 2C). Taxol treatment did not inhibit Akt activation in mouse mammary tumor cells (Fig. 2C), as was observed for MDA-MB-231 cells (Fig. 1A).
To assess the role of microtubules in IRS-2-mediated signaling in human breast carcinoma cells, IRS2 expression was suppressed by shRNA targeting in MDA-MB-231 cells (Fig. 2D). Total AKT activation in response to IGF-1 stimulation was reduced in shIRS-2 cells compared with shGFP cells (Fig. 2D, second and sixth lanes). Treatment of shGFP cells with either nocodazole or vinblastine significantly reduced AKT activation (Fig. 2D, third and fourth lanes), as was observed for the parental cells (Fig. 1C). In contrast, no significant reduction in AKT phosphorylation occurred in the shIRS2 cells, which signal predominantly through IRS-1, upon treatment with either nocodazole or vinblastine (Fig. 2D, seventh and eighth lanes). The -fold difference in AKT activation observed in shGFP and shIRS2 cells upon microtubule disruption increased with time of stimulation, indicating that microtubules are required to sustain IRS2-dependent signaling (Fig. 2E). Taken together with the mouse cell line data, these results support the hypothesis that an intact microtubule cytoskeleton contributes to IGF-1 signaling through IRS-2 but not IRS-1.
The dependence of IRS2 signaling to Akt on an intact microtubule cytoskeleton indicates a potential interaction of IRS-2 with microtubules. To investigate this possibility, the localization of IRS-1, IRS-2, and tubulin was examined by immunofluorescent staining and confocal imaging. For these experiments, SUM-159 breast carcinoma cells were used because they express both IRS-1 and IRS-2, spread well on coverslips, and retain their spread morphology upon treatment with Taxol and nocodazole, which facilitates detection of co-localization. Both IRS-1 and IRS-2 were expressed in a punctate manner throughout the cytoplasm, with a modest enhancement in the perinuclear region (Fig. 3; left panels). Although no specific pattern of staining was observed for IRS-1, the punctate staining for IRS-2 was more organized, with some apparent alignment along microtubules. To assess further the association of IRS-2 with microtubules, cells were stained after short-term incubation with either Taxol or nocodazole to evaluate their impact on IRS localization. The organized, linear pattern of IRS-2 staining was more apparent upon Taxol stabilization of the microtubules, and a subset of IRS-2 puncta co-localized with tubulin under these conditions (Fig. 3B, center panels). IRS-2 was dispersed throughout the cytoplasm with no tubulin co-localization upon disruption of microtubules by nocodazole (Fig. 3B, right panels). In contrast, IRS-1 staining was not modified in response to Taxol or nocodazole treatment (Fig. 3A).
Figure 3.
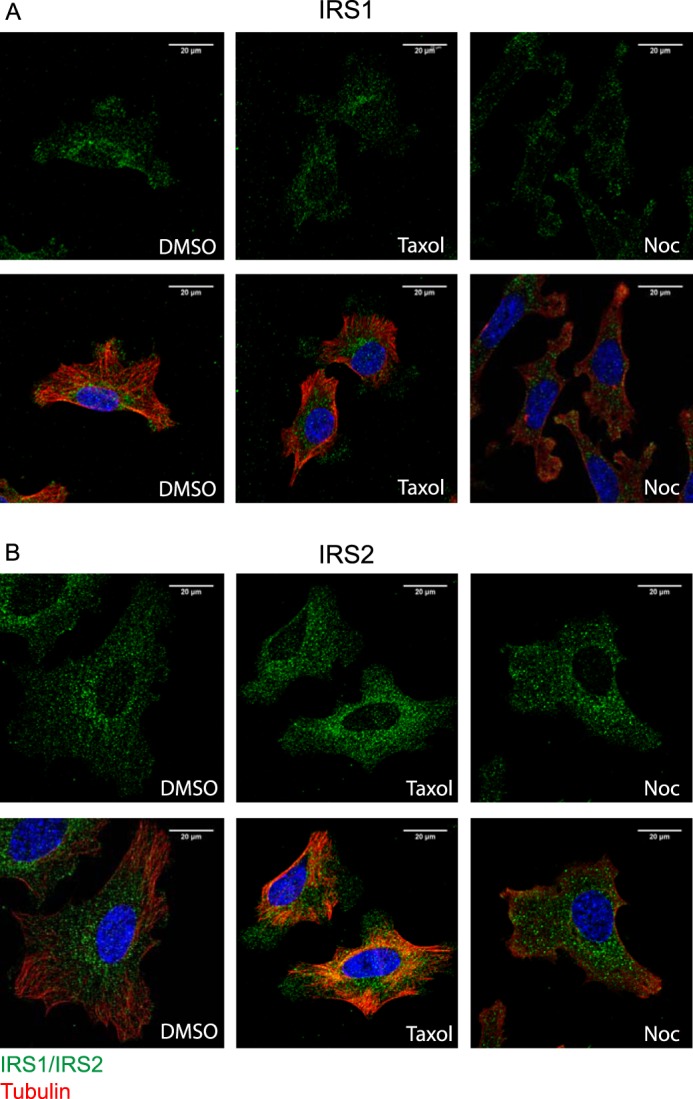
Intracellular localization of IRS proteins and co-localization with microtubules. A and B, SUM159 cells were treated with either DMSO, 20 μm Taxol, or 1 μm nocodazole (Noc) for 1–2 h before fixation. Fixed cells were co-stained for IRS-1 or IRS-2 (green) and tubulin (red). Individual images for IRS-1/IRS-2 and merged IRS/tubulin images are shown (magnification ×63).
IRS-2 determines the cellular response to microtubule disruption
Drugs that target the microtubule cytoskeleton are used clinically for the treatment of cancer (23). To investigate how IRS-2 may impact the response of tumors to microtubule-stabilizing or -disrupting drugs, MDA-MB-231 cells were treated for 48 h with either nocodazole or Taxol and then analyzed for viability by propidium iodide staining and flow cytometry analysis. A significant increase in the sub-G1 population occurred in response to both nocodazole and Taxol treatment in shGFP control cells (Fig. 4A), consistent with an induction of cell death. Compared with shGFP cells, cell death was significantly diminished in shIRS-2 cells in response to nocodazole treatment (Fig. 4A). In contrast, when cells were treated with Taxol, which does not inhibit IRS2-mediated AKT signaling (Figs. 1A and 2C), cell death levels were similar for shGFP and shIRS2 cells. To further investigate the clinical relevance of the IRS2-dependent sensitivity of breast carcinoma cells to microtubule disruption, MDA-MB-231 cells were treated with vinblastine or vinorelbine, both of which are used in the treatment of solid tumors, including breast cancer (23). As was observed for nocodazole treatment, cell death was significantly diminished in shIRS2 cells treated with either vinblastine or vinorelbine compared with shGFP-treated cells (Fig. 4B).
Figure 4.

The Role of IRS-2 in the cellular response to microtubule disruption. A—D, MDA-MB-231 cells were treated with DMSO or the indicated drugs for 48 h. The cells were stained with propidium iodide and analyzed by flow cytometry. Shown are the percentages of cells in the sub-G1 peak (A and B) or cell cycle stages (C and D). The data shown represent the mean of representative experiments performed three times (± S.E.) (nocodazole, vinblastine, and vinorelbine) or twice (± S.D.) (Taxol) independently. *, p ≤ 0.05 relative to shGFP; **, p ≤ 0.01 relative to shGFP.
A similar resistance to cell death upon treatment with nocodazole was observed for PyMT:Irs2−/− cells compared with parental cells (PyMT:Irs2fl/fl) or Irs2−/− cells in which WT Irs-2 expression was restored (Irs2−/−:IRS2) (Fig. 5A). In contrast, restoration of IRS1 expression to PyMT:Irs1−/− cells reduced cell death in response to nocodazole treatment (Fig. 5B). Similar to MDA-MB-231 cells, cell death in response to Taxol treatment was not dependent upon Irs expression in PyMT mammary tumor cells (Fig. 5, A and B).
Figure 5.

The differential impact of Irs1 and Irs2 on the cellular response to microtubule disruption. A—G, PyMT cells were treated with DMSO or the indicated drugs for 48 h. The cells were stained with propidium iodide and analyzed by flow cytometry. Shown are the percentages of cells in the sub-G1 peak (A and B) or cell cycle stages (C–G). The data shown represent the mean ± S.E. of representative experiments performed three times or twice (Taxol;Irs2 cells) independently. 2fl/fl, PyMT:Irs2fl/fl cells; 2−/−, PyMT;Irs2−/− cells; 2−/−:IRS2, PyMT:Irs2−/−:IRS2 cells; 1−/−, PyMT;Irs1−/− cells; 1−/−:IRS1, PyMT:Irs1−/−:IRS1 cells. *, p ≤ 0.05 relative to Irsfl/fl; **, p ≤ 0.001 relative to Irsfl/fl.
As has been reported previously, cells undergo a G2/M arrest in response to microtubule disruption or stabilization (30). The cell cycle profiles of cells treated with nocodazole or Taxol were analyzed to determine whether IRS2 expression influences the cell cycle response to microtubule-targeting drugs. MDA-MB-231:shGFP cells exhibited an increase in G2/M arrest when treated with nocodazole (Fig. 4C). In contrast, a significantly higher percentage of MDA-MB-231:shIRS2 cells remained in G1/G0 upon disruption of microtubules (Fig. 4D). This increase in G1/G0 cells was also observed in PyMT:Irs2−/− cells (Fig. 5D), and rescue of IRS-2 expression restored the G2/M arrest profile to that of the parental PyMT:Irs2fl/fl cells (Fig. 5, C and E). In contrast, PyMT:Irs1−/− cells exhibited G2/M arrest, and restoration of IRS-1 expression promoted an increase in G1/G0 cells (Fig. 5, F and G), similar to the profile of the IRS2-deficient cells that signal only through IRS1. All cells treated with Taxol exhibited a similar G2/M arrest profile (Figs. 4 and 5). These data indicate that breast carcinoma cells that express and signal through IRS-2 are more sensitive to drugs that depolymerize microtubules and respond by undergoing cell cycle arrest and increased cell death, whereas the Taxol response is not determined by IRS-2 expression.
Given that AKT signaling is selectively resistant to microtubule disruption in cells deficient for IRS2 expression, we hypothesized that the resistance of these cells to nocodazole-mediated cell death was the result of sustained AKT signaling in these cells. To test this hypothesis, cells were treated with nocodazole for 48 h in the presence or absence of an AKT-specific inhibitor, MK2206. Treatment of MDA-MB-231 shGFP and shIRS2 cells with MK2206 alone did not alter the percentage of sub-G1 cells (Fig. 6A) or cell cycle profile (Fig. 6, B and C), as has been reported previously for parental MDA-MB-231 cells at the concentration of inhibitor used in these assays (28). This concentration of MK2206 was, however, sufficient to inhibit AKT activation (Fig. 6D). Combined treatment of the MDA-MB-231:shGFP cells with MK2206 and nocodazole did not increase cell death (Fig. 6A) or alter cell cycle progression (Fig. 6B). In contrast, addition of MK2206 to the nocodazole-treated MDA-MB-231:shIRS2 cells increased the percentage of sub-G1 cells to that observed for shGFP cells treated with nocodazole alone (Fig. 6A). Combined treatment with MK2206 and nocodazole also promoted G2/M arrest in these cells, restoring the cell cycle profile to that of the nocodazole-treated, IRS2-expressing cells (Fig. 6C).
Figure 6.

The role of AKT in the IRS-2-dependent response to microtubule disruption. MDA-MB-231 cells were treated with DMSO or the indicated drugs for 48 h. A—C, cells were stained with propidium iodide and analyzed by flow cytometry. Shown are the percentages of cells in the sub-G1 peak (A) or cell cycle stages (B and C). The data shown represent the mean ± S.E. of representative experiments performed three times independently. *, p ≤ 0.05 relative to shGFP. D, aliquots of cell extracts containing equivalent amounts of total protein were immunoblotted with antibodies specific for IRS1, IRS2, Ser(P)-473AKT, total AKT, or tubulin. The data shown in the bottom panel represent the mean ± S.E. of three independent experiments. Noc, nocodazole. *, p ≤ 0.05 relative to DMSO; **, p ≤ 0.001 relative to DMSO.
To investigate the mechanism of cell death in response to microtubule disruption, cell extracts from MDA-MB-231 cells treated with nocodazole for 48 h in the presence or absence of MK2206 were immunoblotted for cleaved caspase 3. Caspase 3 cleavage increased significantly upon treatment of shGFP cells with nocodazole, confirming that these cells undergo apoptotic cell death (Fig. 7A) (31). Cleaved caspase 3 levels were significantly lower in shIRS2 nocodazole-treated cells. However, combined treatment of shIRS2 cells with both nocodazole and MK2206 increased caspase 3 cleavage, supporting a role for AKT signaling in the enhanced viability of these cells. Analysis of upstream apoptotic effectors identified the BCL2 homology 3 (BH3)-only protein BIM as a potential regulator of caspase 3 activation in response to microtubule disruption (31). Specifically, the BIM-EL and BIM-L isoforms of BIM were expressed at elevated levels in shGFP cells compared with shIRS2 cells, and expression increased upon co-treatment with MK2206 in shIRS2 cells (Fig. 7B).
Figure 7.

Involvement of the apoptotic effector BIM in the response to microtubule disruption. A and B, MDA-MB-231 cells were treated with DMSO or the indicated drugs for 48 h. Aliquots of cell extracts containing equivalent amounts of total protein were immunoblotted with antibodies specific for cleaved caspase 3 (A) or BIM (B) and tubulin. The data shown in the bottom panels represent the mean ± S.E. of three independent experiments. *, p ≤ 0.05 relative to DMSO; **, p ≤ 0.01 relative to DMSO; #, p ≤ 0.05 relative to shGFP-Nocodazole; ##, p ≤ 0.01 relative to shGFP-Nocodazole. Noc, nocodazole.
Discussion
In this study, we demonstrate a differential involvement of the microtubule cytoskeleton in IRS-dependent activation of AKT. AKT activation in response to IGF-1 stimulation is maintained when the microtubule cytoskeleton is disrupted in cells that signal only through IRS-1. In contrast, microtubule disruption significantly diminishes AKT activation when the IGF-1R signals through IRS-2. Proximal IGF-1R signaling events, including receptor tyrosine phosphorylation, IRS tyrosine phosphorylation, and recruitment of PI3K, are not inhibited by microtubule disruption, indicating that IRS-2 requires the microtubule cytoskeleton at the level of downstream effector activation. The co-localization of IRS-2, but not IRS-1, with tubulin is enhanced upon Taxol-mediated microtubule stabilization, which, in concert with the signaling data, suggests an interaction of IRS-2 with the microtubule cytoskeleton that may facilitate its access to effectors such as AKT. Functionally, IRS-2 sensitizes breast carcinoma cells to apoptotic cell death in response to treatment with microtubule-disrupting drugs through a mechanism involving the inhibition of AKT signaling and regulation of the BH3-only apoptotic activator BIM. Our data identify IRS-2 as a potential biomarker for the response of breast cancer patients to Vinca alkaloid drug treatment.
The IRS proteins function as signaling intermediates for both the IGF-1R and IR. Previous studies have investigated the importance of the microtubule cytoskeleton in signaling through the IR in insulin-responsive cell types such as adipocytes and muscle (20, 21). Similar to our findings with IGF-1R signaling, proximal IR signaling events are not impacted by microtubule disruption, whereas distal events such as GLUT4 translocation to the plasma membrane are inhibited (20). The impact of microtubule disruption on AKT activation in response to insulin stimulation is cell type-dependent. Insulin-induced AKT activation was modestly reduced in 3T3-L1 adipocytes, maintained in CHO cells that express IR and IRS-1 (CHO.IR.IRS-1), and inhibited in skeletal muscle cells upon treatment with microtubule-disrupting drugs (21, 32). Importantly, the involvement of either IRS-1 or IRS-2 was not investigated in these different cell models. The differential expression and activation of IRS-1 and IRS-2 in each cell type may explain the differential responses of these cells to microtubule disruption.
Our demonstration that IRS-1 and IRS-2 differ in their dependence upon an intact microtubule cytoskeleton for their downstream signaling adds to the mechanistic understanding of how signaling by these homologous proteins regulates distinct functional outcomes. IRS-1 and IRS-2 are both capable of recruiting PI3K and activating AKT, but the functional response to this activation is quite different for each adaptor protein (1). Stimulation of cells with insulin or IGF-1 promotes proliferation when IRS-1 is the dominant signaling adaptor (8, 33). Although a direct role for AKT in this response has not been demonstrated, it is dependent upon activation of PI3K (33). In contrast, stimulation of breast carcinoma cells that express IRS-2 as the dominant signaling adaptor respond by increasing migration/invasion and glycolytic metabolism (7–10). IRS-2-dependent activation of PI3K is also required for these cellular responses (5). With regard to metabolism, the ability of IRS-2 to selectively regulate distinct downstream AKT effectors contributes to this differential outcome. IRS-2-dependent activation of AKT results in the phosphorylation and inactivation of GSK3, and this inactivation is required for IRS-2-mediated regulation of glucose uptake (5). In this study, expression of the apoptotic activator BIM increased in response to disruption of IRS-2-dependent AKT activation, a mechanism that may involve the selective regulation of another AKT effector pathway. Specifically, BIM expression is positively regulated by FOXO transcription factors, which are inactivated by AKT phosphorylation (34–36). IRS-2 has been shown previously to mediate insulin regulation of Foxo1 activity and Bim expression in neonatal mouse hepatocytes (37).
Our data support the hypothesis that the ability of IRS-2 to interact with and potentially traffic along microtubules may determine its access to distinct subsets of effectors to elicit unique functional responses. Of note, a selective role for IRS-1- and IRS-2 dependent signaling in skeletal muscle has been reported that involves differential AKT isoform activation. Insulin-stimulated myoblast differentiation and glucose metabolism are regulated by IRS-1/AKT2 signaling, whereas signaling through IRS-2/AKT1 controls lipid metabolism (38). The possibility that AKT isoforms are selectively sensitive to microtubule disruption could contribute to the differential sensitivity of IRS-1 and IRS-2 to microtubule loss.
The results of this study are consistent with the work of other groups that suggests a central role for AKT in the tumor cell response to microtubule-disrupting drugs (39–41). Specifically, AKT promotes the phosphorylation of microtubule-binding proteins that stabilize microtubules and, in doing so, increases the resistance of tumor cells to drugs that function by disrupting the microtubule cytoskeleton. Our data now reveal that the mechanism by which tumor cells regulate AKT activity will also influence the response to these drugs. Our results have implications for the use of microtubule-disrupting drugs, such as vinblastine and vinorelbine, for the treatment of breast cancer. The differential responses of IRS-1- and IRS-2-dependent signaling to these chemotherapeutic drugs raise the possibility that IRS-2 may influence how breast tumors respond to Vinca alkaloid drug treatment. We reported previously that expression of IRS-2 at the cell membrane is associated with a statistically significant decrease in overall survival in breast cancer patients (13). We hypothesize that IRS-2 at the cell membrane is indicative of active signaling, and patients who have tumors with this staining pattern may be more sensitive to microtubule-disrupting drugs than patients without active IRS-2 signaling. Moreover, breast tumors with low IRS2 expression/function could be responsive to combination therapies that pair an AKT inhibitor with a Vinca alkaloid drug. Therefore, IRS-2 could be used as a biomarker to identify patients for targeted treatment with these drugs.
Experimental procedures
Cell lines, shRNA, and transfection
The MDA-MB-231 cell line was obtained from the ATCC Cell Biology Collection. SUM-159 cells were a kind gift from Art Mercurio (UMass Medical School, Worcester, MA). WT, Irs-1−/−, and Irs-2−/− mammary tumor cell lines were established from MMTV-PyV-MT-derived tumors as described previously (9). Irs-1fl/fl and Irs-2fl/fl mammary tumor cells were isolated from female FVB MMTV-PyMT::Irs-1fl/fl and MMTV-PyMT::Irs-2fl/fl mice, respectively, and Irs−/− cells were generated by infection with adenoviral Cre-recombinase as described previously (5). Lentiviral vectors containing shRNAs targeting GFP and human IRS-2 were obtained from Open Biosystems (Huntsville, AL). MDA-MB-231 cells were infected with the virus, and stably expressing cells were selected by the addition of 2 μg/ml puromycin. IRS expression was restored in Irs−/− mammary tumor cells by transfection with HA-tagged human IRS-1 or IRS-2 (kindly provided by Adrian Lee, University of Pittsburgh, Pittsburgh, PA) and selection in G418 (0.5 mg/ml) (11).
Immunoblotting and immunoprecipitation
Cells were either serum-starved overnight (MDA-MB-231) or for 4 h (PyMT cells) in serum-free medium. Drugs were added to the medium prior to stimulation with IGF-1. MDA-MB-231 cells were treated with nocodazole or vinblastine for 30 min and Taxol for 2 h. The mouse tumor cell lines were treated with nocodazole, vinblastine, or vinorelbine for 1 h. The concentrations (see figure legends) and time periods of incubation were determined to stabilize or disrupt the microtubule cytoskeleton, as assessed by immunofluorescence staining for tubulin. For microtubule-altering drugs, paclitaxel (T7402) was obtained from Sigma-Aldrich (St. Louis, MO), and nocodazole (S2775), vinblastine (S1248), and vinorelbine (S4269) were obtained from Selleckchem (Houston, TX). MK2206 (S1078) was obtained from Selleckchem. Cells were stimulated for 5–30 min with human recombinant IGF-1 (R&D Systems, Minneapolis, MN) prior to extraction.
For total cell extract immunoblots, cells were solubilized at 4 °C in radioimmune precipitation assay lysis buffer (25 mm Tris (pH 8.0), 0.1% sodium dodecyl sulfate, 1% sodium deoxycholate, 1% Nonidet P-40, 150 mm sodium chloride, 10 mm sodium fluoride, and 1 mm sodium orthovanadate) containing protease inhibitors (Roche). Cell extracts containing equivalent amounts of protein were resolved by SDS-PAGE and transferred to nitrocellulose membranes. The membranes were blocked for 1 h with a 50 mm Tris buffer (pH 7.5) containing 0.15 m NaCl, 0.05% Tween 20, and 5% (w/v) dry milk or 5% BSA, incubated overnight at 4 °C in the same buffer containing primary antibodies, and then incubated for 1 h in 5% blocking buffer with milk containing peroxidase-conjugated secondary antibodies. Proteins were detected by enhanced chemiluminescence (Bio-Rad, Hercules, CA) using a ChemiDoc XRS+ (Bio-Rad) with Image Lab software. Only signals within a linear range were used for quantitation, and signals were normalized to total protein and/or housekeeping genes. The following antibodies were used for immunoblotting: IRS-1 (human: C20, Santa Cruz Biotechnology, Santa Cruz, CA; mouse: Bethyl Custom Immunochemistry Services), IRS-2 (4502, Cell Signaling Technology, Danvers, MA), p85 (05-212, Millipore, Billerica, MA), IGF-1Rβ (3025, Cell Signaling Technology), phospho-AKT Ser-473 (9271 and 4060, Cell Signaling Technology), phospho-AKT Thr-308 (2965, Cell Signaling Technology), AKT (sc-8312, Santa Cruz Biotechnology; 9272, Cell Signaling Technology), α-tubulin (T5168, Sigma-Aldrich), phospho-ERK (9106, Cell Signaling Technology), ERK (9102, Cell Signaling Technology), GAPDH (A300-642A, Bethyl), cleaved caspase 3 (9664, Cell Signaling Technology), BIM (2933, Cell Signaling Technology), peroxidase-conjugated goat anti-rabbit IgG (111-035-144, Jackson ImmunoResearch Laboratories, Inc., West Grove, PA), and peroxidase-conjugated goat anti-mouse IgG (711-035-151, Jackson ImmunoResearch Laboratories, Inc.).
For immunoprecipitations, cells were extracted using a 20 mm Tris (pH 7.4) buffer containing 1% Nonidet P-40, 0.137 m NaCl, 10% glycerol, 10 mm sodium fluoride, 1 mm sodium orthovanadate, and protease inhibitors (Roche). Aliquots of cell extracts containing equivalent amounts of protein were precleared for 30 min with protein A-Sepharose beads and then incubated overnight at 4 °C with antibodies and protein A-Sepharose beads (Amersham Biosciences, Piscataway, NJ) with constant agitation. The beads were washed three times in extraction buffer. Laemmli sample buffer was added to the samples, and immune complexes were resolved by SDS-PAGE, transferred to nitrocellulose membranes, and immunoblotted as described above. The following antibodies were used for immunoprecipitation: IRS-1 (C20, Santa Cruz Biotechnology), IRS-2 (Bethyl Custom Immunochemistry Services), IGF-1R (3025, Cell Signaling Technology), rabbit IgG (sc-2027, Santa Cruz Biotechnology), and mouse IgG2b (ab18421, Abcam).
Immunofluorescence microscopy
Subconfluent, adherent cells plated on glass coverslips were treated with nocodazole (1 μm) for 1 h or Taxol (20 μm) for 2 h. Cells were washed three times with Dulbecco's PBS and fixed in 3.8% paraformaldehyde in Dulbecco's PBS with 0.5% Tween (PBST) for 1 h. Permeabilized cells were blocked for 1 h using 3% BSA in PBST. Primary antibodies diluted in blocking buffer were added to cells and incubated at room temperature for 1 h. Secondary antibodies were diluted in the same buffer, and cells were incubated at room temperature for an additional 30 min. Cells were washed three times with PBST after each antibody incubation. Coverslips were then mounted on glass slides using Prolong Gold containing DAPI (Cell Signaling Technology), and the slides were viewed by confocal microscopy (Zeiss LSM700, ×63 oil immersion objective). All images were adjusted equally for brightness/contrast using ImageJ software. Antibodies used for immunofluorescence included IRS-1 (H165, Santa Cruz Biotechnology), IRS-2 (NB110-57138, Novus, Littleton, CO), α-tubulin (T5168, Sigma-Aldrich), Alexa Fluor 488 donkey anti-rabbit IgG (A-21206, Thermo Fisher Scientific, Waltham, MA), and Alexa Fluor 568 donkey anti-mouse IgG (A10037, Thermo Fisher Scientific). The specificity of the IRS-1 and IRS-2 antibody staining was validated by staining PyMT:Irs1−/−,Irs2−/− double-null cells that were transfected with either empty vector, IRS-1, or IRS-2 (supplemental Fig. 1).
Cell cycle analysis
MDA-MB-231 cells were treated with nocodazole (1 μm), vinblastine (20 nm), vinorelbine (20 nm), Taxol (10 μm), or MK2206 (250 nm) for 48 h. The mouse PyMT mammary tumor cell lines were treated with nocodazole (1 μm) or Taxol (1 or 10 μm) for 48 h. Adherent cells were collected by trypsinization after treatment and combined with non-adherent cells from the culture medium for cell cycle analysis. After centrifugation, the cell pellet was washed once in cold PBS, and the cells were then fixed in 70% methanol and stored overnight at −20 °C. The fixed cells were washed once in PBS and then resuspended in PBS containing 0.1% Triton X-100, 0.1 mm EDTA, 0.05 mg/ml RNase A (50 units/mg), and 50 μg/ml propidium iodide. The cells were analyzed by flow cytometry using a BD Biosciences FACSCalibur after 1-h incubation at room temperature.
Author contributions
J. M. M., J. L. C., and L. M. S. conceived the project and wrote the manuscript. J. M. M. designed, performed, and analyzed the experiments shown in Figs. 2 and 4–7. J. L. C. designed, performed, and analyzed the experiments shown in Figs. 1 and 2. A. J. P. performed and analyzed the experiments shown in Figs. 5 and 6. J. J. performed and analyzed the experiments shown in Figs. 2 and 3. All authors analyzed the results and approved the final version of the manuscript.
Supplementary Material
Acknowledgment
We thank Art Mercurio for helpful comments on the manuscript.
This work was supported by National Institutes of Health Grant CA142782 (to L. M. S.), National Institutes of Health F31 Predoctoral Fellowship CA180706 (to J. M. M.), and Department of Defense Breast Cancer Predoctoral Fellowship W81XWH-10-1-0038 (to J. L. C.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains supplemental Fig. 1.
- IRS
- insulin receptor substrate
- IR
- insulin receptor
- IGF
- insulin-like growth factor.
References
- 1. Mardilovich K., Pankratz S. L., and Shaw L. M. (2009) Expression and function of the insulin receptor substrate proteins in cancer. Cell Commun. Signal. 7, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Myers M. G. Jr, Backer J. M., Sun X. J., Shoelson S., Hu P., Schlessinger J., Yoakim M., Schaffhausen B., and White M. F. (1992) IRS-1 activates phosphatidylinositol 3′-kinase by associating with src homology 2 domains of p85. Proc. Natl. Acad. Sci. U.S.A. 89, 10350–10354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hadari Y. R., Tzahar E., Nadiv O., Rothenberg P., Roberts C. T. Jr, LeRoith D., Yarden Y., and Zick Y. (1992) Insulin and insulinomimetic agents induce activation of phosphatidylinositol 3′-kinase upon its association with pp185 (IRS-1) in intact rat livers. J. Biol. Chem. 267, 17483–17486 [PubMed] [Google Scholar]
- 4. Myers M. G. Jr, Sun X. J., Cheatham B., Jachna B. R., Glasheen E. M., Backer J. M., and White M. F. (1993) IRS-1 is a common element in insulin and insulin-like growth factor-I signaling to the phosphatidylinositol 3′-kinase. Endocrinology 132, 1421–1430 [DOI] [PubMed] [Google Scholar]
- 5. Landis J., and Shaw L. M. (2014) Insulin receptor substrate 2-mediated phosphatidylinositol 3-kinase signaling selectively inhibits glycogen synthase kinase 3β to regulate aerobic glycolysis. J. Biol. Chem. 289, 18603–18613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Jackson J. G., and Yee D. (1999) IRS-1 expression and activation are not sufficient to activate downstream pathways and enable IGF-I growth response in estrogen receptor negative breast cancer cells. Growth Horm. IGF Res. 9, 280–289 [DOI] [PubMed] [Google Scholar]
- 7. Jackson J. G., Zhang X., Yoneda T., and Yee D. (2001) Regulation of breast cancer cell motility by insulin receptor substrate-2 (IRS-2) in metastatic variants of human breast cancer cell lines. Oncogene 20, 7318–7325 [DOI] [PubMed] [Google Scholar]
- 8. Byron S. A., Horwitz K. B., Richer J. K., Lange C. A., Zhang X., and Yee D. (2006) Insulin receptor substrates mediate distinct biological responses to insulin-like growth factor receptor activation in breast cancer cells. Br. J. Cancer 95, 1220–1228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Nagle J. A., Ma Z., Byrne M. A., White M. F., and Shaw L. M. (2004) Involvement of insulin receptor substrate 2 in mammary tumor metastasis. Mol. Cell Biol. 24, 9726–9735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Pankratz S. L., Tan E. Y., Fine Y., Mercurio A. M., and Shaw L. M. (2009) Insulin receptor substrate-2 regulates aerobic glycolysis in mouse mammary tumor cells via glucose transporter 1. J. Biol. Chem. 284, 2031–2037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Dearth R. K., Cui X., Kim H. J., Kuiatse I., Lawrence N. A., Zhang X., Divisova J., Britton O. L., Mohsin S., Allred D. C., Hadsell D. L., and Lee A. V. (2006) Mammary tumorigenesis and metastasis caused by overexpression of insulin receptor substrate 1 (IRS-1) or IRS-2. Mol. Cell Biol. 26, 9302–9314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ma Z., Gibson S. L., Byrne M. A., Zhang J., White M. F., and Shaw L. M. (2006) Suppression of insulin receptor substrate 1 (IRS-1) promotes mammary tumor metastasis. Mol. Cell Biol. 26, 9338–9351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Clark J. L., Dresser K., Hsieh C. C., Sabel M., Kleer C. G., Khan A., and Shaw L. M. (2011) Membrane localization of insulin receptor substrate-2 (IRS-2) is associated with decreased overall survival in breast cancer. Breast Cancer Res. Treat. 130, 759–772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Koda M., Sulkowska M., Kanczuga-Koda L., and Sulkowski S. (2005) Expression of insulin receptor substrate 1 in primary breast cancer and lymph node metastases. J. Clin. Pathol. 58, 645–649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sisci D., Morelli C., Garofalo C., Romeo F., Morabito L., Casaburi F., Middea E., Cascio S., Brunelli E., Andò S., and Surmacz E. (2007) Expression of nuclear insulin receptor substrate 1 (IRS-1) in breast cancer. J. Clin. Pathol. 60, 633–641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Morelli C., Garofalo C., Sisci D., del Rincon S., Cascio S., Tu X., Vecchione A., Sauter E. R., Miller W. H. Jr, and Surmacz E. (2004) Nuclear insulin receptor substrate 1 interacts with estrogen receptor α at ERE promoters. Oncogene 23, 7517–7526 [DOI] [PubMed] [Google Scholar]
- 17. Migliaccio I., Wu M. F., Gutierrez C., Malorni L., Mohsin S. K., Allred D. C., Hilsenbeck S. G., Osborne C. K., Weiss H., and Lee A. V. (2010) Nuclear IRS-1 predicts tamoxifen response in patients with early breast cancer. Breast Cancer Res. Treat. 123, 651–660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Chen J., Wu A., Sun H., Drakas R., Garofalo C., Cascio S., Surmacz E., and Baserga R. (2005) Functional significance of type 1 insulin-like growth factor-mediated nuclear translocation of the insulin receptor substrate-1 and β-catenin. J. Biol. Chem. 280, 29912–29920 [DOI] [PubMed] [Google Scholar]
- 19. Wu A., Chen J., and Baserga R. (2008) Nuclear insulin receptor substrate-1 activates promoters of cell cycle progression genes. Oncogene 27, 397–403 [DOI] [PubMed] [Google Scholar]
- 20. Olson A. L., Trumbly A. R., and Gibson G. V. (2001) Insulin-mediated GLUT4 translocation is dependent on the microtubule network. J. Biol. Chem. 276, 10706–10714 [DOI] [PubMed] [Google Scholar]
- 21. Liu L. Z., Cheung S. C., Lan L. L., Ho S. K., Chan J. C., and Tong P. C. (2013) Microtubule network is required for insulin-induced signal transduction and actin remodeling. Mol. Cell. Endocrinol. 365, 64–74 [DOI] [PubMed] [Google Scholar]
- 22. Granger E., McNee G., Allan V., and Woodman P. (2014) The role of the cytoskeleton and molecular motors in endosomal dynamics. Semin. Cell Dev. Biol. 31, 20–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Dumontet C., and Jordan M. A. (2010) Microtubule-binding agents: a dynamic field of cancer therapeutics. Nat. Rev. Drug Discov. 9, 790–803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Schiff P. B., Fant J., and Horwitz S. B. (1979) Promotion of microtubule assembly in vitro by Taxol. Nature 277, 665–667 [DOI] [PubMed] [Google Scholar]
- 25. Samson F., Donoso J. A., Heller-Bettinger I., Watson D., and Himes R. H. (1979) Nocodazole action on tubulin assembly, axonal ultrastructure and fast axoplasmic transport. J. Pharmacol. Exp. Ther. 208, 411–417 [PubMed] [Google Scholar]
- 26. Franke T. F., Kaplan D. R., Cantley L. C., and Toker A. (1997) Direct regulation of the Akt proto-oncogene product by phosphatidylinositol-3,4-bisphosphate. Science 275, 665–668 [DOI] [PubMed] [Google Scholar]
- 27. Rohatgi R. A., Janusis J., Leonard D., Bellvé K. D., Fogarty K. E., Baehrecke E. H., Corvera S., and Shaw L. M. (2015) Beclin 1 regulates growth factor receptor signaling in breast cancer. Oncogene 34, 5352–5362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sangai T., Akcakanat A., Chen H., Tarco E., Wu Y., Do K. A., Miller T. W., Arteaga C. L., Mills G. B., Gonzalez-Angulo A. M., and Meric-Bernstam F. (2012) Biomarkers of response to Akt inhibitor MK-2206 in breast cancer. Clin. Cancer Res. 18, 5816–5828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Aapro M., and Finek J. (2012) Oral vinorelbine in metastatic breast cancer: a review of current clinical trial results. Cancer Treat. Rev. 38, 120–126 [DOI] [PubMed] [Google Scholar]
- 30. Tishler R. B., Lamppu D. M., Park S., and Price B. D. (1995) Microtubule-active drugs Taxol, vinblastine, and nocodazole increase the levels of transcriptionally active p53. Cancer Res. 55, 6021–6025 [PubMed] [Google Scholar]
- 31. Taylor R. C., Cullen S. P., and Martin S. J. (2008) Apoptosis: controlled demolition at the cellular level. Nat. Rev. Mol. Cell Biol. 9, 231–241 [DOI] [PubMed] [Google Scholar]
- 32. Thomas E. C., Zhe Y., Molero J. C., Schmitz-Peiffer C., Ramm G., James D. E., and Whitehead J. P. (2006) The subcellular fractionation properties and function of insulin receptor substrate-1 (IRS-1) are independent of cytoskeletal integrity. Int. J. Biochem. Cell Biol. 38, 1686–1699 [DOI] [PubMed] [Google Scholar]
- 33. Myers M. G. Jr, Zhang Y., Aldaz G. A., Grammer T., Glasheen E. M., Yenush L., Wang L. M., Sun X. J., Blenis J., Pierce J. H., and White M. F. (1996) YMXM motifs and signaling by an insulin receptor substrate 1 molecule without tyrosine phosphorylation sites. Mol. Cell Biol. 16, 4147–4155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Linseman D. A., Phelps R. A., Bouchard R. J., Le S. S., Laessig T. A., McClure M. L., and Heidenreich K. A. (2002) Insulin-like growth factor-I blocks Bcl-2 interacting mediator of cell death (Bim) induction and intrinsic death signaling in cerebellar granule neurons. J. Neurosci. 22, 9287–9297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Urbich C., Knau A., Fichtlscherer S., Walter D. H., Brühl T., Potente M., Hofmann W. K., de Vos S., Zeiher A. M., and Dimmeler S. (2005) FOXO-dependent expression of the proapoptotic protein Bim: pivotal role for apoptosis signaling in endothelial progenitor cells. FASEB J. 19, 974–976 [DOI] [PubMed] [Google Scholar]
- 36. De Bruyne E., Bos T. J., Schuit F., Van Valckenborgh E., Menu E., Thorrez L., Atadja P., Jernberg-Wiklund H., and Vanderkerken K. (2010) IGF-1 suppresses Bim expression in multiple myeloma via epigenetic and posttranslational mechanisms. Blood 115, 2430–2440 [DOI] [PubMed] [Google Scholar]
- 37. Valverde A. M., Fabregat I., Burks D. J., White M. F., and Benito M. (2004) IRS-2 mediates the antiapoptotic effect of insulin in neonatal hepatocytes. Hepatology 40, 1285–1294 [DOI] [PubMed] [Google Scholar]
- 38. Huang C., Thirone A. C., Huang X., and Klip A. (2005) Differential contribution of insulin receptor substrates 1 versus 2 to insulin signaling and glucose uptake in l6 myotubes. J. Biol. Chem. 280, 19426–19435 [DOI] [PubMed] [Google Scholar]
- 39. Gagnon V., Van Themsche C., Turner S., Leblanc V., and Asselin E. (2008) Akt and XIAP regulate the sensitivity of human uterine cancer cells to cisplatin, doxorubicin and Taxol. Apoptosis 13, 259–271 [DOI] [PubMed] [Google Scholar]
- 40. Yang S. X., Costantino J. P., Kim C., Mamounas E. P., Nguyen D., Jeong J. H., Wolmark N., Kidwell K., Paik S., and Swain S. M. (2010) Akt phosphorylation at Ser473 predicts benefit of paclitaxel chemotherapy in node-positive breast cancer. J. Clin. Oncol. 28, 2974–2981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Page C., Lin H. J., Jin Y., Castle V. P., Nunez G., Huang M., and Lin J. (2000) Overexpression of Akt/AKT can modulate chemotherapy-induced apoptosis. Anticancer Res. 20, 407–416 [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.