

Abstract
Enveloped viruses infect host cells by fusing their membranes with those of the host cell, a process mediated by viral glycoproteins upon binding to cognate host receptors or entering into acidic intracellular compartments. Whereas the effect of receptor density on viral infection has been well studied, the role of cell type-specific factors/processes, such as pH regulation, has not been characterized in sufficient detail. Here, we examined the effects of cell-extrinsic factors (buffer environment) and cell-intrinsic factors (interferon-inducible transmembrane proteins, IFITMs), on the pH regulation in early endosomes and on the efficiency of acid-dependent fusion of the avian sarcoma and leukosis virus (ASLV), with endosomes. First, we found that a modest elevation of external pH can raise the pH in early endosomes in a cell type-dependent manner and thereby delay the acid-induced fusion of endocytosed ASLV. Second, we observed a cell type-dependent delay between the low pH-dependent and temperature-dependent steps of viral fusion, consistent with the delayed enlargement of the fusion pore. Third, ectopic expression of IFITMs, known to potently block influenza virus fusion with late compartments, was found to only partially inhibit ASLV fusion with early endosomes. Interestingly, IFITM expression promoted virus uptake and the acidification of endosomal compartments, resulting in an accelerated fusion rate when driven by the glycosylphosphatidylinositol-anchored, but not by the transmembrane isoform of the ASLV receptor. Collectively, these results highlight the role of cell-extrinsic and cell-intrinsic factors in regulating the efficiency and kinetics of virus entry and fusion with target cells.
Keywords: endocytosis, fusion protein, pH regulation, viral protein, virus entry
Introduction
Infectivity of enveloped viruses often varies depending on the cell type, even for cells expressing comparable levels of cognate receptors. This is largely due to the varied efficiency of multiple steps of entry leading to productive infection. Whereas multiple host factors are involved in late post-fusion steps of virus entry (1–9), the effects of intrinsic, cell type-dependent factors and extrinsic factors on viral fusion are poorly characterized. After the initial interaction of viruses with cellular receptors or attachment factors, low endosomal pH is required to trigger fusion-inducing conformational changes in most viral proteins (reviewed in Refs. 10 and 11). The low pH requirement of virus entry could also stem from the need for optimal milieu (e.g. endosomal protease activity) for priming the viral glycoproteins for the fusion reaction (10). It is thus likely that cell type-dependent regulation of endosomal pH modulates the efficiency and kinetics of virus fusion. To date, however, only a few studies have directly examined the link between the pH in virus-carrying endosomes and the efficiency/kinetics of subsequent viral fusion (12–15).
Endosome-resident lipids and proteins have been implicated in the completion of virus fusion and/or the nucleocapsid release into the cytoplasm (8, 16–22). Cells can also express restriction factors that interfere with the viral fusion step. Recent studies have shown that expression of interferon-inducible transmembrane proteins (IFITMs)3 inhibits fusion of unrelated enveloped viruses in vitro and in vivo (23–25). Using single virus imaging, we have shown that human IFITM3 blocks the formation of small fusion pores without inhibiting the influenza virus hemifusion with endosomes (26). Different models for the IFITMs' antiviral activity have been proposed, including the “stiffening” of cell membranes that disfavors viral fusion (24). However, direct evidence supporting the current models is lacking.
The avian sarcoma and leukosis virus (ASLV) is an excellent model for studies of viral entry, as its envelope glycoprotein (Env) requires two consecutive triggers, binding to cognate receptors and acidic pH, to mediate membrane fusion (27–29). Subtype A ASLV undergoes robust fusion with cells expressing either of the two naturally occurring isoforms of the TVA receptor: the glycosylphosphatidylinositol (GPI)-anchored TVA800 and the transmembrane TVA950 proteins. These alternative receptor isoforms direct the virus entry through distinct endocytic pathways (13, 30, 31). These features make ASLV amenable to biophysical studies of virus entry at a single particle level. We have previously investigated the entry/fusion mechanism of ASLV by single virus imaging and delineated the relationship between endosomal pH and the probability and kinetics of ASLV fusion (13, 14, 32). We have also shown that ASLV fuses with early or maturing endosomes, depending on the receptor isoform (13).
Here, we examined the progression of ASLV entry through the sequential steps: endocytosis, acid-dependent conformational changes in Env, and completion of the fusion process, using both bulk and single virus imaging assays. These experiments revealed the existence of a cell type- and external buffer-dependent delay in low pH-induced steps of ASLV fusion relative to endocytosis. This delay was largely due to the pH regulation in early endosomes of A549-derived cells, whereas no significant delay was detected in CV-1-derived cells. We further found that ectopic expression of the virus restriction factor IFITM3 in either cell type inhibited ASLV fusion, albeit less potently than the influenza virus fusion. Strikingly, IFITM3 accelerated endocytosis and acidification of ASLV-carrying vesicles in A549 cells expressing TVA800, but not TVA950. Our results show the importance of endosomal pH regulation, which is influenced by both cell-extrinsic and -intrinsic factors, for entry/fusion of ASLV and likely other viruses that undergo acid-induced fusion. We also present evidence supporting the notion that cell-specific factors other than endosomal pH regulate the kinetics of ASLV fusion.
Results
Analysis of ASLV entry and fusion kinetics
ASLV fusion progresses through two major consecutive steps: receptor-mediated priming of Env followed by low pH-dependent refolding of the viral protein into the final 6-helix bundle structure that drives membrane fusion (27–29, 33–35). As we and others have previously demonstrated, the progression of virus-cell fusion through distinct intermediate steps can be measured by adding specific inhibitors of these steps at varied time points post-infection (e.g. Refs. 31, 36, and 37). The ASLV Env-derived R99 peptide blocks fusion by preventing the formation of the final helical bundle structure (28, 35). Because the membrane-impermeable R99 peptide targets only surface-accessible virions, the addition of this peptide after varied times of virus-cell incubation at 37 °C permits the measurement of the rate of receptor-mediated endocytosis that culminates in fusion (31). Furthermore, the time-of-addition experiments using NH4Cl or bafilomycin A1, agents that raise endosomal pH and interfere with the low pH-dependent steps of ASLV fusion, report the kinetics of completion of acid-dependent steps of this process. Finally, one can take advantage of the steep temperature-dependence of viral fusion (38–42). Several studies, including ours, have shown that low pH initiates refolding of pH-dependent viral fusion proteins, such as influenza and ASLV, whereas the late steps of fusion, the formation and dilation of a fusion pore, can proceed at neutral pH and physiological temperature (43–46). Lowering the temperature at distinct times post-infection will block the late steps of fusion, most likely the fusion pore opening and/or dilation (47). Thus, the comparison of the ASLV escape kinetics from R99, NH4Cl, and the temperature block (TB) reports the progression of ASLV fusion through receptor-mediated endocytosis followed by acid- and temperature-dependent steps, respectively (32, 37).
Temperature-dependent steps of ASLV fusion with CV-1 cells are delayed relative to acid-dependent steps
We applied the above time-of-addition strategy to examine the progression of ASLV pseudoviral particle (ASLVpp) fusion with CV-1-derived cells expressing either the full-length (TVA950) or the GPI-anchored (TVA800) TVA receptor (48, 49). Virus-cell fusion was measured using the β-lactamase (BlaM) assay that detects the delivery of virus-incorporated BlaM-Vpr chimeric protein into the cytoplasm (50). In both cell lines, the acid-dependent steps of fusion (escape from NH4Cl) were completed shortly after virus uptake (escape from R99), whereas resistance to low temperature was acquired after a pronounced delay of about 10 min following the completion of acid-dependent steps (Fig. 1, A and B). Delayed escape from TB is unlikely to result from temperature-dependent post-fusion processes, because the activity of BlaM released into the cytoplasm appears to be independent of viral core uncoating (36, 51). In addition, the BlaM signal reaches a maximum shortly after the synchronized ASLV fusion with endosomes of CV-1 cells is stopped by chilling the samples (32) (see also the fusion kinetics with A549 cells below), further supporting the notion that post-fusion steps do not contribute to the BlaM signal.
Figure 1.
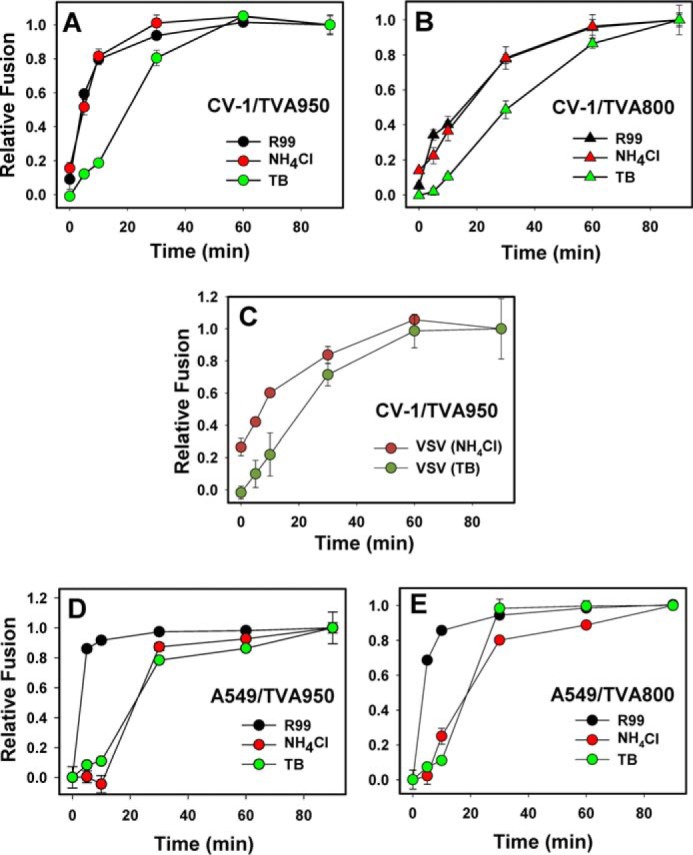
Progression of the ASLV and VSV pseudovirus entry and fusion with CV-1- and A549-derived cells. A and B, ASLV pseudovirus fusion with CV-1/TVA950 (A) and CV-1/TVA800 (B) cells was measured using the BlaM assay, as described under “Experimental procedures.” Fusion inhibitors (R99, NH4Cl, or low temperature (TB)) were applied at the indicated times after virus entry/fusion was initiated by raising the temperature to 37 °C. C, VSV pseudovirus fusion with CV-1/TVA950 cells experiments were carried out, as described above. D and E, ASLV pseudovirus fusion with A549 cells expressing either TVA950 (D) or TVA800 (E). Data are mean ± S.E. from 3 independent triplicate experiments.
Comparable kinetic differentials between the virus escape from NH4Cl and cold were observed upon fusion of particles pseudotyped with ASLV Env and the vesicular stomatitis virus (VSV) G glycoprotein, which mediates acid-dependent fusion with early endosomes (16, 17, 52, 53) (Fig. 1C). The similarity in the lag-time between escapes from NH4Cl and TB for the different viral fusion glycoproteins in CV-1-derived cells may reflect the slow enlargement of fusion pores, which is a temperature-dependent process (54), or the delayed opening of a fusion pore after the low pH trigger.
Acid- and temperature-dependent steps of ASLV fusion with A549 cells are delayed relative to virus uptake
To determine whether the lag between the acid- and temperature-dependent steps of ASLV fusion with CV-1-derived cells is observed in other cell types, we examined the virus entry kinetics in the human alveolar epithelial cell line A549 engineered to express the cognate TVA receptor isoforms. The ASLV fusion kinetics with CV-1 and A549 cells was strikingly different. First, A549/TVA800 cells internalized ASLV faster than CV-1/TVA800 cells, suggesting marked differences in the rate of endocytosis of GPI-anchored proteins (Fig. 1E). However, in contrast to the CV-1 cells, in which the acid-dependent steps were completed shortly after endocytosis, a profound lag was detected in the time of R99 and NH4Cl addition experiments in A549-derived cells, irrespective of the TVA isoform (Fig. 1, D and E). In fact, the kinetics of ASLV escape from NH4Cl in A549/TVA800 and A549/TVA950 cells overlapped with the TB kinetics. The delayed completion of the low pH-dependent stage(s) of viral fusion after receptor-mediated endocytosis may be due to a slow acidification of endocytic vesicles carrying the virus or, alternatively, to delayed fusion following the acidification of endosomal lumen. To discern between these possibilities, we measured the time course of bulk virus endocytosis and delivery into mildly acidic compartments, using the imaging approach developed in our laboratory (31). Pseudoviruses bearing the HIV-1 Gag-mCherry chimera were co-labeled with ecliptic pHluorin (EcpH, maximum emission ∼508 nm (55)) fused to the transmembrane domain of ICAM-1 (Fig. 2A). The pH-sensitive EcpH fluorescence (pKa ∼7.0) is strongly quenched at mildly acidic pH (31) and therefore serves as an all-or-none indicator of virus entry into acidic compartments (Fig. 2A). Here, the fluorescence of acid-resistant intraviral mCherry (pKa<4.5 (56)) fused to the Gag polyprotein serves as a reference signal for the visualization of endosomal acidification.
Figure 2.

Kinetics of ASLV fusion and measurement of endosomal pH. A, illustration of the virus labeling strategy with Gag-mCherry (red) and EcpH-ICAM (green) to assess the pH drop in virus-carrying endosomes (top) and images of CV-1 cells before (0 h) and after (1 h) internalization of labeled viruses (bottom). Virus entry into acidic endosomes is manifested in disappearance of the EcpH signal and accumulation of Gag-mCherry in the perinuclear areas. B, kinetics of ASLV fusion with A549/TVA950 cells in DMEM measured by the BlaM assay and EcpH quenching measured in parallel imaging experiments. C, images of Gag-mCherry/EcpH-ICAM co-labeled ASLV particles internalized by A549/TVA950 cells at different pH. Viruses were pre-bound to cells in the cold and allowed to enter by incubation at 37 °C for 15 min. Cells were then placed in buffers of the indicated acidity supplemented with monensin and nigericin to equilibrate the external and endosomal pH (see “Experimental procedures” for details). The EcpH signal is virtually lost in the background cell fluorescence at pH ≤ 6.2. A triangle shows the expected fluorescence ratio in DMEM equilibrated with air (pH ∼ 7.9). D, calibration of the mean ratio of EcpH and mCherry signals from intracellular compartments as a function of endosomal pH (as illustrated in panel C). Data are mean ratios ± S.E. from at least 4 image fields acquired for each pH value. The light pink and blue colored regions represent the pH range conducive for ASLV fusion and the background EcpH/mCherry ratio, respectively. E and F, kinetics of ASLV fusion with A549/TVA950 cells in LIB (E) or DMEM buffered with HEPES at pH 7.4 (F), as measured by the BlaM assay. EcpH quenching in panel E was measured in parallel imaging experiments. Data are mean ± S.E. from 3 (panels B and E) and 2 (panel F) independent triplicate experiments.
Analysis of the ratio of integrated green and red signals from cell-bound particles over time showed that ASLV entry into acidic endosomes in CV-1-derived cells occurred with the rate that was close to the kinetics of virus escape from R99 and NH4Cl (Ref. 31 and data not shown). This is as expected for viruses undergoing acid-dependent fusion shortly after entering into acidic endosomes. In sharp contrast, the EcpH quenching kinetics in A549/TVA950 cells overlapped with the rate of virus uptake, but was much faster than the virus escape from NH4Cl (Fig. 2B). This striking phenotype implies that internalized ASLV quickly enters into mildly acidic compartments, but completes acid-dependent steps of fusion after 10–20 min, which is at least an order of magnitude slower than the low pH-triggered ASLV fusion with CV-1-derived cells (14).
Initial acidification of endosomes in A549 cells may not be sufficient to effectively trigger ASLV fusion
Given the relatively high pH threshold for ASLV fusion (14, 46, 57), we have originally assumed that quenching the EcpH probe reports entry into early acidic compartments that support viral fusion (31). To test this notion, we estimated the pH that effectively quenches the EcpH signal of single virions. ASLV Env-pseudotyped HIV-1 particles co-labeled with EcpH-ICAM/Gag-mCherry were allowed to enter A549/TVA950 cells for 15 min, during which time, nearly all pseudoviruses were internalized by cells (Fig. 2B). Cells were then exposed to buffers adjusted to pH between 4.7 and 7.7, which were supplemented with monensin/nigericin to clamp the endosomal pH at a desired level (58), and imaged. Whereas the viral particles in endosomes exhibited strong EcpH fluorescence at neutral pH, a nearly complete quenching of the EcpH signal was observed at pH 6.3 and below (Fig. 2C). Measurements of the EcpH/mCherry ratio as a function of endosomal pH, using the above buffers, yielded the calibration curve that enabled the assessment of endosomal pH in live cell experiments (Fig. 2D). Our estimation of early endosomal pH in A549 cells that effectively quenched EcpH suggested that the pH may not initially drop below 6.2–6.3, which is not sufficiently acidic to trigger optimal ASLV fusion (14, 46). In other words, the observed delay of ASLV fusion with A549 cells (as measured by the time of NH4Cl addition, Figs. 1, D and E, and 2B) may reflect the need for virus trafficking from early, marginally acidic endosomes to late, more acidic compartments prior to fusion. We thus predicted that the initial pH drop upon entry into CV-1 cells, which support ASLV fusion with early endosomes (14), is more robust than in A549 cells.
We hypothesized that the delayed ASLV fusion with A549 cells, but not CV-1 cells, results from the use of a CO2-dependent medium (DMEM), which becomes mildly alkaline (pH 7.8–8.0) under ambient conditions. Indeed, when the BlaM fusion and parallel EcpH quenching experiments were conducted in a HEPES-containing live cell imaging buffer (LIB), ASLV uptake (escape from R99 and EcpH quenching), and virus escape from NH4Cl occurred very quickly and with virtually superimposable kinetics (Fig. 2E). It thus appears that the use of CO2-dependent DMEM for time-of-addition experiments, which require transient exposure to air (see “Experimental procedures”), raises the extracellular and early endosomal pH in A549 cells. Accordingly, the lag between ASLV uptake and escape from NH4Cl in A549 cells disappeared when DMEM was buffered with HEPES (Fig. 2F).
By contrast, CV-1 cells appear to more robustly control the initial pH drop in early endosomes down to the level that effectively triggers ASLV fusion. Another critical distinction between ASLV fusion with CV-1 and A549 cells is that, in the latter cells, escape from the TB was not delayed relative to escape from NH4Cl (Figs. 1 and 2). The lag between the pH- and temperature-dependent steps of the ASLV (and VSV) fusion observed for CV-1 cells (Fig. 1, A–C) was virtually undetectable in A549 cells. The overlapping NH4Cl and temperature-block kinetics in A549 cells argue against the requirement for maintaining physiological temperature after fusion for the BlaM activity. In other words, delayed escape from the cold block observed in CV-1 cells is not due to post-fusion temperature-dependent processes that are required for the BlaM activity.
Acidification of early endosomes in A549 cells is delayed in mildly alkaline medium
To test the notion that the initial pH drop in A549 cells may not trigger ASLV fusion, we evaluated the dynamics of bulk endosomal pH in CV-1 and A549 cells, using a mixture of fluorescent transferrins as a marker for early endosomes. Cells were incubated with a mixture of two transferrin (Trf) preparations, one labeled with a pH-sensitive FITC dye and the other labeled with a pH-insensitive Alexa Fluor 594 dye (AF594). Uptake of this Trf mixture into acidic endosomes leads to the reduction in the FITC fluorescence (pKa ∼ 6.5), whereas the AF594 signal remains relatively constant (supplemental Fig. S1). A calibration curve relating the fluorescence ratio to endosomal pH was obtained by allowing Trf endocytosis for 15 min and imposing a known pH value onto endosomal compartments, using a membrane-permeable monensin/nigericin buffer system described above (Fig. 2 and supplemental Fig. S2). The obtained linear pH dependence of the FITC/AF594 ratio, with >3-fold change between 4.5 and 8.0 (Fig. 3A), enables the measurements of endosomal pH in a physiologically relevant range.
Figure 3.

Effect of extracellular buffers on endosomal pH and on ASLV fusion. A, calibration of the pH in early endosomes by measuring the ratio of fluorescence signals from a mixture of internalized transferrin labeled with FITC (pH sensitive) or with AF594, as described under “Experimental procedures.” Briefly, cells were incubated with transferrin mixture for 15 min at 37 °C and exposed to buffers of different acidity supplemented with monensin and nigericin. The resulting changes in transferrin fluorescence are illustrated in supplemental Fig. S2. Data are mean ratios ± S.E. from at least 4 image fields acquired for each pH value. B, the dynamics of endosomal pH in A549/TVA950 and CV-1/TVA950 cells bathed in DMEM versus LIB measured by FITC/AF594 transferrin fluorescence ratio, as also illustrated in supplemental Fig. S1. Data are mean ratios ± S.E. from 4 or more image fields acquired for each pH value. C, the efficiency of ASLV fusion with A549 and CV-1 cells expressing TVA950 in different extracellular buffers (DMEM and LIB). The total number of co-labeled particles analyzed for each condition (2–3 independent experiments) is shown in the graph. D, the kinetics of single ASLV (co-labeled with YFP-Vpr and Gag-imCherry) fusion with TVA950-expressing A549 and CV-1 cells bathed in DMEM or LIB. Note that, despite different fusion efficiencies (panel C), a comparable number of events were annotated and plotted to ensure an appropriate sample size for all conditions. **, p < 0.01; ***, p < 0.001.
We were thus able to evaluate the drop in endosomal pH in CV-1 and A549 cells shortly after the initiation of Trf uptake by shifting from cold to 37 °C. As predicted, early endosomes of CV-1 cells acidified to pH < 6.0 within a few minutes, irrespective of the buffer used (Fig. 3B). In comparison, the overall endosomal pH in A549 cells bathed in DMEM was considerably less acidic than in LIB (Fig. 3B). Within 20 min at 37 °C, the endosomal pH in A549 cells bathed in LIB approached 6.0, which is sufficient to trigger effective ASLV fusion (14). By contrast, the average endosomal pH in DMEM remained close to neutral. Moreover, we reproducibly observed the pH to “overshoot” (become slightly alkaline) at about 5 min after initiating endocytosis in DMEM (Fig. 3B), likely due to Trf recycling from early endosomes.
Together, the results shown in Fig. 3 confirm that A549 and CV-1 cells differentially regulate endosomal pH. Under identical conditions, Trf-carrying endosomes in A549 cells bathed in DMEM were on average considerably less acidic than in CV-1 cells. The differences in the kinetics of endosomal pH drop in CV-1 and A549 cells may be related to the regulation of cargo uptake and recycling pathways. Whereas Trf remain largely peripheral within 2–5 min after initiation of uptake by A549 cells, this marker quickly accumulated in perinuclear recycling compartments of CV-1 cells (supplemental Fig. S1). These findings support the notion that weak initial acidification of endosomes of A549 cells bathed in DMEM delays ASLV fusion.
The extent and rate of ASLV fusion correlate with the pH drop in early endosomes
Poor initial acidification of endosomes of A549 cells bathed in DMEM may necessitate ASLV trafficking to more acidic late compartments to undergo fusion. To measure the extent and kinetics of ASLV fusion in these cells, we used ASLVpp co-labeled with YFP-Vpr (viral core marker) and Gag-imCherry (fluid-phase marker). In keeping with the less efficient acidification profiles in DMEM (Fig. 3B), single ASLV fusion was reduced 2–3-fold in DMEM compared with LIB in both cell lines (Fig. 3C). As expected for a marginal initial acidification in A549 cells bathed in DMEM, these cells were the least conducive to single ASLV fusion, with only ∼7% of double-labeled particles releasing the mCherry marker into the cytoplasm. The fact that ASLV fusion occurred at all under these conditions may be indicative of preferential virus sorting into somewhat more acidic endosomes than those carrying Trf (Fig. 3B), in agreement with the previous study (12).
The kinetics of single ASLV fusion also correlated with the early pH dynamics in cells. For both CV-1 and A549 cells, viral fusion in LIB was significantly faster than in DMEM (Fig. 3D). Interestingly, A549 cells seemed to support faster kinetics of fusion than CV-1 cells for a comparable or even less pronounced endosomal acidification rate. Although the initial endosome acidification was less rapid in A549 cells, as compared with CV-1 cells in LIB (Fig. 3B), the kinetics of ASLV fusion was considerably faster with the former cells (Fig. 3D). Furthermore, despite the virtual lack of acidification of Trf-carrying endosomes in A549 cells in DMEM (Fig. 3B), the fusion kinetics overlapped with that for CV-1 cells bathed in the same medium (Fig. 3D). This finding implies that, in addition to the endosome acidity, additional cell type-specific factors control the rate of ASLV fusion (see “Discussion”).
Note that we observed no significant reduction of the extents of ASLV fusion with A549 compared with CV-1 cells in DMEM, using the bulk BlaM assay (data not shown). The difference in the results of BlaM and single virus assays is likely due to an inherently shorter window of observation in the imaging experiments (20–30 min), as compared with a 90-min long BlaM experiment. The shorter observation window for single particle imaging is due in part to YFP-Vpr quenching in acidic endosomes and to particle crowding in the perinuclear area (13, 59). These events complicate the reliable detection of late fusion events and thus truncate the distribution of single virus fusion events. This should exaggerate the difference between ASLV fusion in DMEM versus LIB in which the fusion is considerably faster.
ASLV fusion is inhibited in cells expressing IFITM3
Expression of IFITM3, an interferon-inducible transmembrane protein, blocks fusion of a number of unrelated viruses entering cells by different endocytic routes (23–25, 60, 61). Viruses entering from late endosomes (e.g. influenza virus) are generally more susceptible to IFITM3 inhibition than those fusing with early endosomes (e.g. VSV) (24, 25), in line with the reported IFITM3 accumulation in late endosomes (62, 63). We have previously shown that ASLV tends to fuse with early endosomes of CV-1 cells expressing TVA950, whereas TVA800-mediated fusion occurs primarily in maturing endosomes (13, 14). We thus sought to determine whether ASLV fusion was sensitive to IFITM3 and whether the potency of this restriction factor is modulated by TVA800- and TVA950-mediated virus entry from distinct compartments.
The effect of IFITM3 expression on ASLV fusion with A549/TVA800 and A549/TVA950 cells was examined using the BlaM assay. In both cell lines expressing comparable levels of IFITM3 (Fig. 4A), ASLV fusion was inhibited to a similar extent (∼50%) compared with the respective control cells lacking IFITM3 (Fig. 4B). IFITM3 also caused a comparable reduction in ASLV fusion in CV-1 cells expressing either receptor isoform (Fig. 4, C and D). Thus, the efficiency of IFITM3-mediated restriction appears independent of the ASLV receptor isoform. The IFITM3 effect on ASLV and VSV fusion was similar in magnitude. By contrast, and in agreement with the previous studies (23, 26, 62, 63), IFITM3 potently inhibited fusion of pseudoviruses bearing the influenza A virus HA/NA glycoproteins in both cell lines (Fig. 4B).
Figure 4.

Ectopic expression and virus restriction activity of IFITM3. A, immunofluorescence staining for IFITM3 expression in A549/TVA800 and A549/TVA950 cells transduced with the IFITM3-encoding vector or an empty vector. B, effect of IFITM3 expression on ASLV, VSV, and influenza A virus (IAV) pseudovirus fusion with A549/TVA950 and A549/TVA800 cells was measured by the BlaM assay. Data are mean ± S.E. from 3 independent triplicate experiments. C, immunostaining of CV-1/TVA800 cells ectopically expressing or lacking IFITM3 with anti-IFITM3 serum. D, effect of IFITM3 expression on ASLV and VSV pseudovirus fusion with CV-1/TVA950 and CV-1/TVA800 cells. Data are mean ± S.E. from 3 independent triplicate experiments. **, p < 0.01; ***, p < 0.001.
We further assessed the effect of IFITM3 expression on ASLV fusion with A549 cells by single particle tracking. Consistent with the BlaM results, IFITM3 inhibited the content release from single ASLV pseudoparticles in cells expressing TVA800 or TVA950 (Fig. 5A). Our results imply that the IFITM3 potency against ASLV is largely independent of the TVA receptor isoform.
Figure 5.
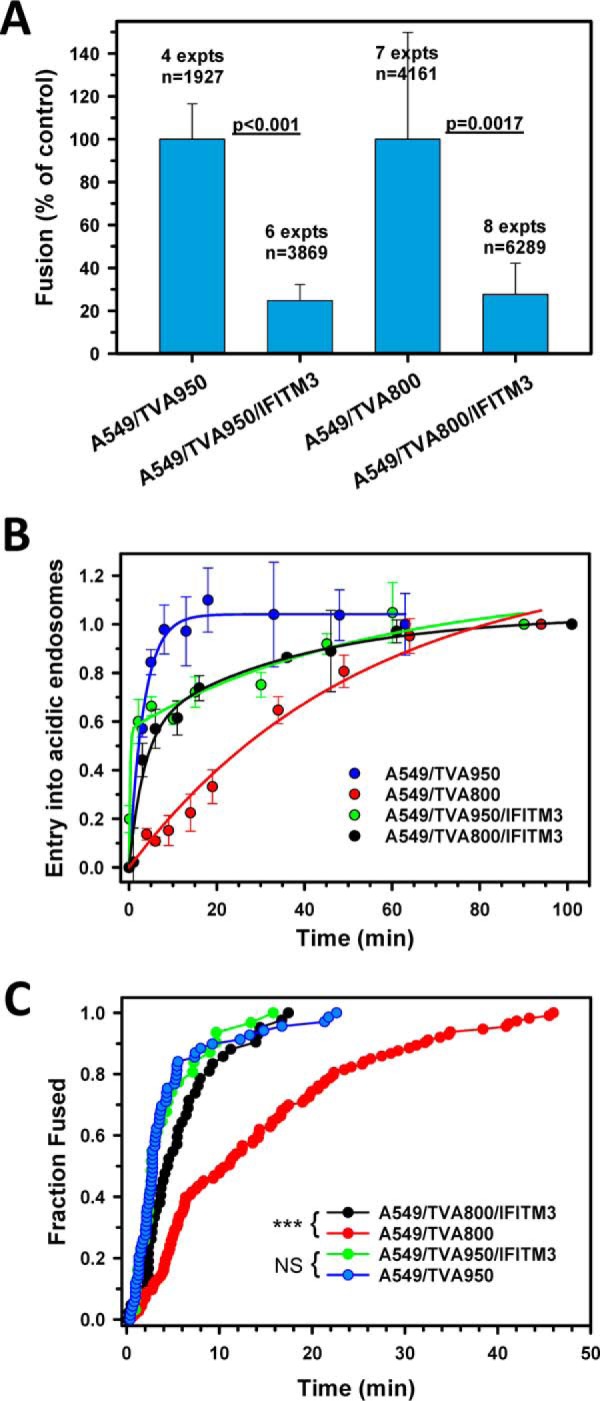
The effect of IFITM3 on extent and kinetics of single ASLV uptake and fusion with A549 cells expressing alternative TVA receptor isoforms. A, percent of double-labeled ASLV pseudoviruses that fused with the indicated cell lines expressing or lacking IFITM3 bathed in LIB. Data are mean ± S.E. from 4 to 8 independent experiments. B, kinetics of ASLV uptake and entry into acidic endosomes in A549/TVA800 and A549/TVA950 cells expressing or lacking IFITM3, as measured by EcpH quenching (see Fig. 2 and “Experimental procedures”). Data are mean ± S.E. from 3 independent experiments. Solid lines are curve fits using a single- or double-exponential raise model. C, the kinetics of single ASLV (co-labeled with YFP-Vpr and Gag-imCherry) fusion with A549/TVA950 and A549/TVA800 cells expressing or lacking IFITM3. Measurements were done in LIB. Despite different fusion efficiencies, a comparable number of events were annotated and plotted to ensure an appropriate sample size for all conditions. Note that a roughly equal number of data points was analyzed for each condition. ***, p < 0.001; NS, not significant.
IFITM3 selectively accelerates ASLV entry and fusion in A549 cells expressing TVA800, but not TVA950
Next, the kinetics of virus uptake into acidic compartments was measured using the EcpH-ICAM/Gag-mCherry-labeled reporter viruses, as above (Fig. 2). Unlike the control A549/TVA950 cells, in which the quick ASLV uptake could be described by a single exponent, the kinetics of virus internalization by IFITM3 expressing cells was biphasic, with approximately half of the viruses losing the EcpH signal at a slower rate (Fig. 5B). Strikingly, IFITM3 expression had an opposite effect on the normally much slower ASLV uptake by A549/TVA800 cells (see Ref. 31 and Fig. 5B). The slow, single exponential kinetics of virus uptake by these cells was markedly accelerated and became biphasic (double-exponential) upon IFITM3 expression. Interestingly, the time course of EcpH quenching in IFITM3-expressing cells was virtually identical, irrespective of the TVA isoform (Fig. 5B). The faster rate of virus delivery into acidic compartments of IFITM3+ A549/TVA800 cells may reflect the accelerated endocytosis and/or accelerated acidification of early carrier vesicles.
In accordance with the kinetics of ASLV uptake into acidic endosomes, the rates of fusion of particles co-labeled with YFP-Vpr and Gag-imCherry were close in cells expressing TVA950, irrespective of the presence of IFITM3, as well as for IFITM3-expressing A549/TVA800 cells (Fig. 5C). By comparison, and in agreement with the delayed uptake/acidification kinetics (Fig. 5B), single ASLV fusion proceeded slower with the control cells expressing TVA800 (Fig. 5C). Thus, IFITM3 expression does not significantly delay ASLV fusion following the acidification of endosomal lumen, but it reduces the probability of the viral content release compared with control cells. The proportionality of the rates of EcpH quenching and viral fusion also suggests that the initial acidification of virus-carrying compartments is sufficiently robust to induce ASLV fusion with cells bathed in LIB. In other words, in contrast to DMEM, which appears to raise the pH in early endosomes of A549 cells, IFITM3 expression appears to actually promote the initial acidification and, therefore, does not interfere with the triggering of ASLV Env.
Discussion
Our results show that both cell-extrinsic (buffer) and -intrinsic (IFITM3) factors can regulate the acidification of early endosomes in a cell type-specific manner and thus modulate the extent and the rate of ASLV fusion. Whereas CV-1 and HeLa (data not shown) cells were less sensitive to the variation of extracellular pH, endosome acidification was delayed in A549 cells bathed in a mildly alkaline DMEM. This finding highlights the need to tightly control the extracellular pH, especially when studying processes that are sensitive to endosomal pH, such as acid-mediated virus fusion. As a result of insufficient acidification of early endosomes, ASLV fusion was delayed relative to the virus uptake in A549 cells bathed in mildly alkaline medium. It is thus prudent to supplement CO2-dependent media with HEPES or other buffers to prevent pH elevation during experiments that require moving the cells in and out of a CO2 incubator.
We found that IFITM3 expression affected ASLV endocytosis and likely the rate of endosome acidification. Interestingly, IFITM3 slightly delayed ASLV uptake by TVA950 expressing cells, whereas markedly accelerating endocytosis/entry into acidic compartments of TVA800-expressing cells (Fig. 5, B and C). This observation is consistent with the reported role of IFITM3 in regulation of clathrin-mediated endocytosis and endosomal pH through stabilizing the vATPase complex (64). The increased acidity of early endosomes in A549/TVA800 cells expressing IFITM3 could be the reason we did not see a stronger inhibition of ASLV fusion with these cells versus A549/TVA950 cells. Based on our previous finding that ASLV fuses with early endosomes of TVA950-expressing cells and with intermediate/maturing endosomes in TVA800-expressing CV-1 cells (13), it was reasonable to expect that IFITM3 would more potently inhibit TVA800-driven ASLV fusion. However, the increased acidity of early endosomes in IFITM3-expressing TVA800 cells may favor the ASLV fusion with early endosomes that are less enriched in IFITM3 than intermediate compartments, which normally support ASLV fusion mediated by TVA800.
The finding that IFITM3 expression partially inhibits ASLV fusion (Figs. 4 and 5), is consistent with the previous reports that viruses entering from late endosomes (e.g. influenza virus) are generally more sensitive to IFITM3 than those entering from early endosomes (e.g. VSV) (24, 25). Our results thus support the notion that the antiviral potency of IFITM3 is largely determined by its subcellular localization.
Besides the endosomal pH regulation, additional cell-specific factors/processes appear to modulate the extent and kinetics of ASLV fusion. Under identical conditions, and for similar average endosomal pH, ASLV fuses faster with A549 cells than with CV-1 cells (Fig. 3D). Note that this difference does not appear to be due to the higher endogenous level of IFITM3 in CV-1 cells, as judged by immunostaining (Fig. 4, A and C). We also observed a large delay between temperature-dependent and low pH-dependent steps of ASLV fusion in CV-1, but not in A549 cells (Fig. 1). This delay suggests slow completion of the fusion process (e.g. slow pore dilation) after progressing through low pH-dependent steps in CV-1 cells. This result thus supports a role of cell type-specific, late endosome-resident factors in completion of ASLV fusion. Interestingly, it has been reported that Dengue virus hemifuses with early endosomes, but undergoes full fusion after entering late endosomes enriched in an unusual anionic lipid, lysobisphosphatidic acid (17). Future studies will help elucidate the host-dependence factors that modulate fusion of ASLV and other viruses.
Experimental procedures
Cell lines and reagents
Human embryonic kidney HEK293T/17, human alveolar adenocarcinoma A549, and African green monkey kidney CV-1 cells were obtained from ATCC (Manassas, VA). CV-1/TVA800, CV-1/TVA950, A549/TVA800, A549/TVA950, and TZM-bl/TVA950 cells expressing high levels of alternative TVA receptors have been described previously (31, 32, 59). The A549/TVA and CV-1/TVA cells stably expressing IFITM3 were obtained by transducing with VSV-G-pseudotyped viruses encoding wild-type IFITM3 or with the vector pQCXIP (Clontech) and selecting with 0.5 and 8 μg/ml of puromycin (InvivoGen, San Diego, CA), respectively. Unless otherwise indicated, the cells were cultured in Dulbecco's modified Eagle's medium (DMEM, Mediatech, Manassas, VA) supplemented with 10% fetal bovine serum (FBS), heat inactivated (Sigma), and 100 units/ml of penicillin-streptomycin (Gemini Bio-Products, Sacramento, CA). The growth medium for HEK293T/17 cells was supplemented with 0.5 mg/ml of G418 sulfate (Mediatech). The ASLV-A Env-derived R99 peptide (∼95% purity by HPLC) was synthesized by Macromolecular Resources (Fort Collins, CO). Live Cell Imaging Buffer (LIB), FluoroBriteTM DMEM, transferrin-FITC, transferrin-Alexa Fluor 594, goat anti-rabbit Alexa Fluor 568, and CCF4-AM substrate were purchased from Life Technologies. Rabbit anti-IFITM3 serum (against the IFITM3 N terminus) was from Abgent (San Diego, CA). HEPES stock solution was from HyClone (Logan, Utah). Calf skin collagen and nigericin were from Sigma. Monensin sodium salt was from Calbiochem.
Plasmids and virus production
The expression vectors for the ASLV subtype A Env lacking the cytoplasmic domain, HIV-1 backbone pR8ΔEnv, pMM310 (encoding for BlaM-Vpr), pcRev, HIV-1-Gag-imCherryΔEnv, psPAX2-Gag-imCherry, ecliptic pHlourin-ICAM-1 (EcpH-ICAM), pMDG (encoding for VSV-G), and pCAGGS vectors encoding influenza H1N1 WSN HA and NA were described previously (26, 36, 59, 65, 66). The YFP-Vpr plasmid was a gift from Dr. T. Hope (Northwestern University). The expression vector encoding human IFITM3 was a gift from Dr. A. L. Brass (University of Massachusetts).
Fluorescent pseudoviruses were produced by transfecting HEK293T/17 cells using JetPrime reagent (Polyplus Transfection, Illkirch-Graffenstaden, France). Cells grown in a 10-cm dish were transfected with 1 μg of pR8ΔEnv, 2 μg of HIV-1-Gag-imCherryΔEnv or psPAX2-Gag-imCherry, 1 μg of pcRev, 2 μg of glycoprotein envelope expression vector, and either 3 μg of EcpH-ICAM or 2 μg of YFP-Vpr. To produce pseudoviruses bearing β-lactamase-Vpr (BlaM-Vpr), cells were transfected with 3 μg of pR8ΔEnv, 2 μg of BlaM-Vpr, 1 μg of pcRev, and 2 μg of envelope glycoprotein expression vector. The virus-containing medium was collected at 48 h post-transfection, passed through a 0.45-μm filter, aliquoted, frozen, and stored at −80 °C. The infectious titer was determined by a β-Gal assay in TZM-bl/TVA cells, as described (36).
β-Lactamase (BlaM) virus-cell fusion assay
The virus-cell fusion measured by the BlaM assay was carried out as described previously (36, 59, 66). Briefly, the target cells were cultured in 96-well strip plate (Corning, Costar). Pseudoviruses bearing the BlaM-Vpr (m.o.i. = 1, unless indicated otherwise) were bound to target cells by centrifugation at 1550 × g, 4 °C, for 30 min. Unbound virus was washed off using cold PBS. Fusion was initiated by shifting to 37 °C and stopped after the indicated times, either by adding fusion inhibitors or by placing cells on ice (referred to as TB). Cells were then loaded with fluorescent CCF4-AM β-lactamase substrate and incubated overnight at 11 °C. Intracellular β-lactamase activity (ratio of coumarin to fluorescein fluorescence) was measured using the Synergy HT fluorescence microplate reader (Bio-Tek, Germany).
Single-virus imaging
Single-virus fusion assays were performed by imaging fluorescently labeled ASLVpp entry in CV-1 and A549 cells expressing the TVA950 or TVA800 receptor isoform, based on previous protocols (59, 65). Briefly, cells were grown on collagen-coated number 1.5 glass-bottomed Petri dishes (MatTek, MA) up to 90% confluent in FluoroBrite DMEM with 10% FBS. Prior to the imaging assay, cells were incubated on ice for 5 min, washed with cold PBS, inoculated with ∼105 infectious units of chilled YFP-Vpr- and Gag-imCherry-labeled ASLVpp, and spun at 1500 × g for 20 min at 4 °C. After spinoculation, the cells were washed with cold PBS. Synchronous viral entry was initiated by the addition of appropriate warm buffer, with the dish placed on the objective inside a pre-warmed incubation chamber on a Zeiss LSM 780 confocal microscope (Carl Zeiss, Germany) at 37 °C. Images were collected every 5–6 s, a typical experiment lasted ∼45 min, after which time, no additional viral fusion events were observed.
Endosomal pH measurements
Transferrin uptake experiments were performed on cells grown to 90% confluence in FluoroBrite DMEM with 10% FBS. Cells were washed twice with 1 ml of warm buffer and incubated for 30 min at 37 °C in an incubator in LIB without serum containing 2 μg/ml of Hoechst-33342 to stain nuclei. The samples were placed on ice, washed with cold LIB without serum, and incubated for 10 min at 4 °C in LIB without serum containing 100 μg/ml of each labeled transferrin. Medium with labeled transferrins was removed, washed with cold buffer, and the cells were imaged at the indicated times after raising the temperature on the microscope by the addition of warm LIB. In separate experiments in 8-well chambered slides (Nunc Lab-Tek, ThermoScientific), a calibration curve was generated to determine the ratiometric fluorescence response of FITC/AF594 to pH for endosomal pH measurements. A similar protocol was followed for endocytosis of the mixture of transferrins in cells for 15 min at 37 °C, followed by treatment with 20 μm monensin and 10 μm nigericin in citrate-phosphate buffers of varying pH for 10 min to equilibrate external and intracellular pH. The cells were imaged for calibration under identical conditions and imaging parameters as the transferrin uptake assay in untreated cells. Ratiometric measurements were based on intensity thresholding and identification of integrated FITC and AF594 signals in Volocity (PerkinElmer Life Sciences) (58). Estimation of EcpH/mCherry ratio in endosomes followed a similar protocol, where a chilled viral suspension of ASLVpp bearing the fluorophores EcpH-ICAM and Gag-mCherry was spun onto cells at 4 °C, followed by entry at 37 °C for 10 min, and finally incubation with monensin and nigericin containing citrate-phosphate buffer at the appropriate pH.
Immunofluorescence assay
IFITM3-expressing cells were grown to 90% confluence on collagen-coated glass coverslips in full growth medium, fixed with 2% paraformaldehyde, permeabilized with 125 μg/ml of digitonin, blocked with PBS, 10% FBS, and sequentially incubated with primary rabbit anti-IFITM3 antibody and goat anti-rabbit Alexa Fluor 568 secondary antibody for 1 h each. Images were collected on a Zeiss LSM 780 laser scanning microscope using a 40×/1.2 NA water immersion objective.
Statistical analysis
Data were analyzed using the Student's t test or non-parametric Mann-Whitney test, as appropriate (* denotes p < 0.05; **, p < 0.01; ***, p < 0.001, n.s., not significant).
Author contributions
T. M. D. and M. M. designed, performed, and analyzed the experiments and co-wrote the paper. C. M. performed the experiments and edited the manuscript. G. B. M. conceived and coordinated the study, analyzed the results, and wrote the paper. All authors reviewed the results and approved the final version of the manuscript.
Supplementary Material
Acknowledgments
We are grateful to the National Institutes of Health AIDS Research and Reference Reagent Program for reagents. We also thank the members of the Melikyan laboratory for critical reading of the manuscript.
This work was supported, in whole or in part, by National Institutes of Health Grant R01 AI053668 (to G. B. M.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains supplemental Figs. S1 and S2.
- IFITM
- interferon-inducible transmembrane protein
- ASLV
- avian sarcoma and leukosis virus
- BlaM
- β-lactamase
- EcpH
- ecliptic pHluorin
- LIB
- live cell imaging buffer
- TB
- temperature block
- Trf
- transferrin
- TVA
- tumor virus A receptor
- Vpr
- viral protein R of HIV
- VSV
- vesicular stomatitis virus
- Env
- envelope glycoprotein
- GPI
- glycosylphosphatidylinositol
- ASLVpp
- ASLV pseudoviral particle.
References
- 1. Banerjee I., Miyake Y., Nobs S. P., Schneider C., Horvath P., Kopf M., Matthias P., Helenius A., and Yamauchi Y. (2014) Influenza A virus uses the aggresome processing machinery for host cell entry. Science 346, 473–477 [DOI] [PubMed] [Google Scholar]
- 2. He J., Sun E., Bujny M. V., Kim D., Davidson M. W., and Zhuang X. (2013) Dual function of CD81 in influenza virus uncoating and budding. PLoS Pathog. 9, e1003701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Karlas A., Machuy N., Shin Y., Pleissner K. P., Artarini A., Heuer D., Becker D., Khalil H., Ogilvie L. A., Hess S., Mäurer A. P., Müller E., Wolff T., Rudel T., and Meyer T. F. (2010) Genome-wide RNAi screen identifies human host factors crucial for influenza virus replication. Nature 463, 818–822 [DOI] [PubMed] [Google Scholar]
- 4. Stauffer S., Feng Y., Nebioglu F., Heilig R., Picotti P., and Helenius A. (2014) Stepwise priming by acidic pH and a high K+ concentration is required for efficient uncoating of influenza A virus cores after penetration. J. Virol. 88, 13029–13046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lee A. S., Burdeinick-Kerr R., and Whelan S. P. (2014) A genome-wide small interfering RNA screen identifies host factors required for vesicular stomatitis virus infection. J. Virol. 88, 8355–8360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Marceau C. D., Puschnik A. S., Majzoub K., Ooi Y. S., Brewer S. M., Fuchs G., Swaminathan K., Mata M. A., Elias J. E., Sarnow P., and Carette J. E. (2016) Genetic dissection of Flaviviridae host factors through genome-scale CRISPR screens. Nature 535, 159–163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Savidis G., McDougall W. M., Meraner P., Perreira J. M., Portmann J. M., Trincucci G., John S. P., Aker A. M., Renzette N., Robbins D. R., Guo Z., Green S., Kowalik T. F., and Brass A. L. (2016) Identification of zika virus and dengue virus dependency factors using functional genomics. Cell Rep. 16, 232–246 [DOI] [PubMed] [Google Scholar]
- 8. Brass A. L., Dykxhoorn D. M., Benita Y., Yan N., Engelman A., Xavier R. J., Lieberman J., and Elledge S. J. (2008) Identification of host proteins required for HIV infection through a functional genomic screen. Science 319, 921–926 [DOI] [PubMed] [Google Scholar]
- 9. König R., Zhou Y., Elleder D., Diamond T. L., Bonamy G. M., Irelan J. T., Chiang C. Y., Tu B. P., De Jesus P. D., Lilley C. E., Seidel S., Opaluch A. M., Caldwell J. S., Weitzman M. D., Kuhen K. L., et al. (2008) Global analysis of host-pathogen interactions that regulate early-stage HIV-1 replication. Cell 135, 49–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. White J. M., and Whittaker G. R. (2016) Fusion of enveloped viruses in endosomes. Traffic 17, 593–614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Smith A. E., and Helenius A. (2004) How viruses enter animal cells. Science 304, 237–242 [DOI] [PubMed] [Google Scholar]
- 12. Lakadamyali M., Rust M. J., and Zhuang X. (2006) Ligands for clathrin-mediated endocytosis are differentially sorted into distinct populations of early endosomes. Cell 124, 997–1009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Padilla-Parra S., Marin M., Kondo N., and Melikyan G. B. (2014) Pinpointing retrovirus entry sites in cells expressing alternatively spliced receptor isoforms by single virus imaging. Retrovirology 11, 47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Padilla-Parra S., Matos P. M., Kondo N., Marin M., Santos N. C., and Melikyan G. B. (2012) Quantitative imaging of endosome acidification and single retrovirus fusion with distinct pools of early endosomes. Proc. Natl. Acad. Sci. U.S.A. 109, 17627–17632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kielian M. C., Marsh M., and Helenius A. (1986) Kinetics of endosome acidification detected by mutant and wild-type Semliki Forest virus. EMBO J. 5, 3103–3109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Le Blanc I., Luyet P. P., Pons V., Ferguson C., Emans N., Petiot A., Mayran N., Demaurex N., Fauré J., Sadoul R., Parton R. G., and Gruenberg J. (2005) Endosome-to-cytosol transport of viral nucleocapsids. Nat. Cell. Biol. 7, 653–664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zaitseva E., Yang S.-T., Melikov K., Pourmal S., and Chernomordik L. V. (2010) Dengue virus ensures its fusion in late endosomes using compartment-specific lipids. PLoS Pathog. 6, e1001131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Pirooz S. D., He S., Zhang T., Zhang X., Zhao Z., Oh S., O'Connell D., Khalilzadeh P., Amini-Bavil-Olyaee S., Farzan M., and Liang C. (2014) UVRAG is required for virus entry through combinatorial interaction with the class C-Vps complex and SNAREs. Proc. Natl. Acad. Sci. U.S.A. 111, 2716–2721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Dubé M., Etienne L., Fels M., and Kielian M. (2016) Calcium-dependent rubella virus fusion occurs in early endosomes. J. Virol. 90, 6303–6313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Nour A. M., Li Y., Wolenski J., and Modis Y. (2013) Viral membrane fusion and nucleocapsid delivery into the cytoplasm are distinct events in some flaviviruses. PLoS Pathog. 9, e1003585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ooi Y. S., Stiles K. M., Liu C. Y., Taylor G. M., and Kielian M. (2013) Genome-wide RNAi screen identifies novel host proteins required for alphavirus entry. PLoS Pathog. 9, e1003835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Perreira J. M., Aker A. M., Savidis G., Chin C. R., McDougall W. M., Portmann J. M., Meraner P., Smith M. C., Rahman M., Baker R. E., Gauthier A., Franti M., and Brass A. L. (2015) RNASEK is a V-ATPase-associated factor required for endocytosis and the replication of rhinovirus, influenza A virus, and dengue virus. Cell Rep. 12, 850–863 [DOI] [PubMed] [Google Scholar]
- 23. Brass A. L., Huang I. C., Benita Y., John S. P., Krishnan M. N., Feeley E. M., Ryan B. J., Weyer J. L., van der Weyden L., Fikrig E., Adams D. J., Xavier R. J., Farzan M., and Elledge S. J. (2009) The IFITM proteins mediate cellular resistance to influenza A H1N1 virus, West Nile virus, and dengue virus. Cell 139, 1243–1254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Perreira J. M., Chin C. R., Feeley E. M., and Brass A. L. (2013) IFITMs restrict the replication of multiple pathogenic viruses. J. Mol. Biol. 425, 4937–4955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bailey C. C., Zhong G., Huang I. C., and Farzan M. (2014) IFITM-family proteins: the cell's first line of antiviral defense. Annu. Rev. Virol. 1, 261–283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Desai T. M., Marin M., Chin C. R., Savidis G., Brass A. L., and Melikyan G. B. (2014) IFITM3 restricts influenza A virus entry by blocking the formation of fusion pores following virus-endosome hemifusion. PLoS Pathog. 10, e1004048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Mothes W., Boerger A. L., Narayan S., Cunningham J. M., and Young J. A. (2000) Retroviral entry mediated by receptor priming and low pH triggering of an envelope glycoprotein. Cell 103, 679–689 [DOI] [PubMed] [Google Scholar]
- 28. Markosyan R. M., Bates P., Cohen F. S., and Melikyan G. B. (2004) A study of low pH-induced refolding of Env of avian sarcoma and leukosis virus into a six-helix bundle. Biophys. J. 87, 3291–3298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Barnard R. J., Narayan S., Dornadula G., Miller M. D., and Young J. A. (2004) Low pH is required for avian sarcoma and leukosis virus Env-dependent viral penetration into the cytosol and not for viral uncoating. J. Virol. 78, 10433–10441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Narayan S., Barnard R. J., and Young J. A. (2003) Two retroviral entry pathways distinguished by lipid raft association of the viral receptor and differences in viral infectivity. J. Virol. 77, 1977–1983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Jha N. K., Latinovic O., Martin E., Novitskiy G., Marin M., Miyauchi K., Naughton J., Young J. A., and Melikyan G. B. (2011) Imaging single retrovirus entry through alternative receptor isoforms and intermediates of virus-endosome fusion. PLoS Pathog. 7, e1001260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Padilla-Parra S., Marin M., Kondo N., and Melikyan G. B. (2012) Synchronized retrovirus fusion in cells expressing alternative receptor isoforms releases the viral core into distinct sub-cellular compartments. PLoS Pathog. 8, e1002694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Melikyan G. B., Barnard R. J., Abrahamyan L. G., Mothes W., and Young J. A. (2005) Imaging individual retroviral fusion events: from hemifusion to pore formation and growth. Proc. Natl. Acad. Sci. U.S.A. 102, 8728–8733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Smith J. G., Mothes W., Blacklow S. C., and Cunningham J. M. (2004) The mature avian leukosis virus subgroup A envelope glycoprotein is metastable, and refolding induced by the synergistic effects of receptor binding and low pH is coupled to infection. J. Virol. 78, 1403–1410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Netter R. C., Amberg S. M., Balliet J. W., Biscone M. J., Vermeulen A., Earp L. J., White J. M., and Bates P. (2004) Heptad repeat 2-based peptides inhibit avian sarcoma and leukosis virus subgroup a infection and identify a fusion intermediate. J. Virol. 78, 13430–13439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Miyauchi K., Kim Y., Latinovic O., Morozov V., and Melikyan G. B. (2009) HIV enters cells via endocytosis and dynamin-dependent fusion with endosomes. Cell 137, 433–444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Miyauchi K., Kozlov M. M., and Melikyan G. B. (2009) Early steps of HIV-1 fusion define the sensitivity to inhibitory peptides that block 6-helix bundle formation. PLoS Pathog. 5, e1000585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Melikyan G. B., Markosyan R. M., Hemmati H., Delmedico M. K., Lambert D. M., and Cohen F. S. (2000) Evidence that the transition of HIV-1 gp41 into a six-helix bundle, not the bundle configuration, induces membrane fusion. J. Cell Biol. 151, 413–423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Chernomordik L. V., Leikina E., Frolov V., Bronk P., and Zimmerberg J. (1997) An early stage of membrane fusion mediated by the low pH conformation of influenza hemagglutinin depends upon membrane lipids. J. Cell Biol. 136, 81–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Gallo S. A., Clore G. M., Louis J. M., Bewley C. A., and Blumenthal R. (2004) Temperature-dependent intermediates in HIV-1 envelope glycoprotein-mediated fusion revealed by inhibitors that target N- and C-terminal helical regions of HIV-1 gp41. Biochemistry 43, 8230–8233 [DOI] [PubMed] [Google Scholar]
- 41. Pak C. C., Krumbiegel M., and Blumenthal R. (1994) Intermediates in influenza virus PR/8 haemagglutinin-induced membrane fusion. J. Gen. Virol. 75, 395–399 [DOI] [PubMed] [Google Scholar]
- 42. Stegmann T., White J. M., and Helenius A. (1990) Intermediates in influenza induced membrane fusion. EMBO J. 9, 4231–4241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Chernomordik L. V., Frolov V. A., Leikina E., Bronk P., and Zimmerberg J. (1998) The pathway of membrane fusion catalyzed by influenza hemagglutinin: restriction of lipids, hemifusion, and lipidic fusion pore formation. J. Cell Biol. 140, 1369–1382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Leikina E., Ramos C., Markovic I., Zimmerberg J., and Chernomordik L. V. (2002) Reversible stages of the low-pH-triggered conformational change in influenza virus hemagglutinin. EMBO J. 21, 5701–5710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Markosyan R. M., Melikyan G. B., and Cohen F. S. (2001) Evolution of intermediates of influenza virus hemagglutinin-mediated fusion revealed by kinetic measurements of pore formation. Biophys. J. 80, 812–821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Melikyan G. B., Barnard R. J., Markosyan R. M., Young J. A., and Cohen F. S. (2004) Low pH is required for avian sarcoma and leukosis virus Env-induced hemifusion and fusion pore formation but not for pore growth. J. Virol. 78, 3753–3762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Markosyan R. M., Cohen F. S., and Melikyan G. B. (2003) HIV-1 envelope proteins complete their folding into six-helix bundles immediately after fusion pore formation. Mol. Biol. Cell 14, 926–938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Bates P., Young J. A., and Varmus H. E. (1993) A receptor for subgroup A Rous sarcoma virus is related to the low density lipoprotein receptor. Cell 74, 1043–1051 [DOI] [PubMed] [Google Scholar]
- 49. Elleder D., Melder D. C., Trejbalova K., Svoboda J., and Federspiel M. J. (2004) Two different molecular defects in the Tva receptor gene explain the resistance of two tvar lines of chickens to infection by subgroup A avian sarcoma and leukosis viruses. J. Virol. 78, 13489–13500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Cavrois M., De Noronha C., and Greene W. C. (2002) A sensitive and specific enzyme-based assay detecting HIV-1 virion fusion in primary T lymphocytes. Nat. Biotechnol. 20, 1151–1154 [DOI] [PubMed] [Google Scholar]
- 51. Cavrois M., Neidleman J., Yonemoto W., Fenard D., and Greene W. C. (2004) HIV-1 virion fusion assay: uncoating not required and no effect of Nef on fusion. Virology 328, 36–44 [DOI] [PubMed] [Google Scholar]
- 52. Pasqual G., Rojek J. M., Masin M., Chatton J. Y., and Kunz S. (2011) Old world arenaviruses enter the host cell via the multivesicular body and depend on the endosomal sorting complex required for transport. PLoS Pathog. 7, e1002232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Sieczkarski S. B., and Whittaker G. R. (2003) Differential requirements of Rab5 and Rab7 for endocytosis of influenza and other enveloped viruses. Traffic 4, 333–343 [DOI] [PubMed] [Google Scholar]
- 54. Cohen F. S., and Melikyan G. B. (2004) The energetics of membrane fusion from binding, through hemifusion, pore formation, and pore enlargement. J. Membr. Biol. 199, 1–14 [DOI] [PubMed] [Google Scholar]
- 55. Miesenböck G., De Angelis D. A., and Rothman J. E. (1998) Visualizing secretion and synaptic transmission with pH-sensitive green fluorescent proteins. Nature 394, 192–195 [DOI] [PubMed] [Google Scholar]
- 56. Shaner N. C., Steinbach P. A., and Tsien R. Y. (2005) A guide to choosing fluorescent proteins. Nat. Methods 2, 905–909 [DOI] [PubMed] [Google Scholar]
- 57. Delos S. E., La B., Gilmartin A., and White J. M. (2010) Studies of the “chain reversal regions” of the avian sarcoma/leukosis virus (ASLV) and ebolavirus fusion proteins: analogous residues are important, and a His residue unique to EnvA affects the pH dependence of ASLV entry. J. Virol. 84, 5687–5694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Altan N., Chen Y., Schindler M., and Simon S. M. (1998) Defective acidification in human breast tumor cells and implications for chemotherapy. J. Exp. Med. 187, 1583–1598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Desai T. M., Marin M., Sood C., Shi J., Nawaz F., Aiken C., and Melikyan G. B. (2015) Fluorescent protein-tagged Vpr dissociates from HIV-1 core after viral fusion and rapidly enters the cell nucleus. Retrovirology 12, 88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Diamond M. S., and Farzan M. (2013) The broad-spectrum antiviral functions of IFIT and IFITM proteins. Nat. Rev. Immunol. 13, 46–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Li K., Markosyan R. M., Zheng Y. M., Golfetto O., Bungart B., Li M., Ding S., He Y., Liang C., Lee J. C., Gratton E., Cohen F. S., and Liu S. L. (2013) IFITM proteins restrict viral membrane hemifusion. PLoS Pathog. 9, e1003124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Feeley E. M., Sims J. S., John S. P., Chin C. R., Pertel T., Chen L. M., Gaiha G. D., Ryan B. J., Donis R. O., Elledge S. J., and Brass A. L. (2011) IFITM3 inhibits influenza A virus infection by preventing cytosolic entry. PLoS Pathog. 7, e1002337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Amini-Bavil-Olyaee S., Choi Y. J., Lee J. H., Shi M., Huang I. C., Farzan M., and Jung J. U. (2013) The antiviral effector IFITM3 disrupts intracellular cholesterol homeostasis to block viral entry. Cell Host Microbe 13, 452–464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Wee Y. S., Roundy K. M., Weis J. J., and Weis J. H. (2012) Interferon-inducible transmembrane proteins of the innate immune response act as membrane organizers by influencing clathrin and v-ATPase localization and function. Innate Immun. 18, 834–845 [DOI] [PubMed] [Google Scholar]
- 65. Padilla-Parra S., Marin M., Gahlaut N., Suter R., Kondo N., and Melikyan G. B. (2013) Fusion of mature HIV-1 particles leads to complete release of a Gag-GFP-based content marker and raises the intraviral pH. PloS One 8, e71002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. de la Vega M., Marin M., Kondo N., Miyauchi K., Kim Y., Epand R. F., Epand R. M., and Melikyan G. B. (2011) Inhibition of HIV-1 endocytosis allows lipid mixing at the plasma membrane, but not complete fusion. Retrovirology 8, 99. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.