

Abstract
Zymogen secretory granules in pancreatic acinar cells express two vesicle-associated membrane proteins (VAMP), VAMP2 and -8, each controlling 50% of stimulated secretion. Analysis of secretion kinetics identified a first phase (0–2 min) mediated by VAMP2 and second (2–10 min) and third phases (10–30 min) mediated by VAMP8. Induction of acinar pancreatitis by supramaximal cholecystokinin (CCK-8) stimulation inhibits VAMP8-mediated mid- and late-phase but not VAMP2-mediated early-phase secretion. Elevation of cAMP during supramaximal CCK-8 mitigates third-phase secretory inhibition and acinar damage caused by the accumulation of prematurely activated trypsin. VAMP8−/− acini are resistant to secretory inhibition by supramaximal CCK-8, and despite a 4.5-fold increase in total cellular trypsinogen levels, are fully protected from intracellular trypsin accumulation and acinar damage. VAMP8-mediated secretion is dependent on expression of the early endosomal proteins Rab5, D52, and EEA1. Supramaximal CCK-8 (60 min) caused a 60% reduction in the expression of D52 followed by Rab5 and EEA1 in isolated acini and in in vivo. The loss of D52 occurred as a consequence of its entry into autophagic vacuoles and was blocked by lysosomal cathepsin B and L inhibition. Accordingly, adenoviral overexpression of Rab5 or D52 enhanced secretion in response to supramaximal CCK-8 and prevented accumulation of activated trypsin. These data support that acute inhibition of VAMP8-mediated secretion during pancreatitis triggers intracellular trypsin accumulation and loss of the early endosomal compartment. Maintaining anterograde endosomal trafficking during pancreatitis maintains VAMP8-dependent secretion, thereby preventing accumulation of activated trypsin.
Keywords: autophagy, endosome, pancreas, SNARE proteins, trypsin, trypsinogen
Introduction
In the pancreatic acinar cell, inactive proteolytic digestive enzymes, or zymogens, are sorted into secretory organelles termed zymogen granules (ZGs),3 which fuse with the apical plasma membrane in response to hormonal or neuronal stimulation. We previously described two populations of ZGs based on their differential expression of vesicle-associated membrane proteins (VAMP) VAMP2 and -8, with each population contributing ∼50% of the total secretory response over 30 min (1). Analysis of secretion over time in isolated acini from WT and VAMP8−/− mice demonstrated that VAMP2 mediates an initial (2-min) burst in secretion, and VAMP8 promotes a second peak at 3–5 min that then slowly declines toward basal levels over the following 20–30 min (1).
Acute pancreatitis is an inflammatory disease of the exocrine pancreas thought to be initiated by premature activation of proteolytic zymogens (most notably trypsinogen) as a result of their aberrant mixing with lysosomal hydrolases (most notably cathepsin B (CatB)) in an acidified cellular compartment (2–5). A prominent inflammatory response mediated by nuclear factor NF-κB is also activated, further exacerbating cellular damage and generating systemic complications (6). This mixing phenomenon is proposed to result from abnormalities in zymogen trafficking during onset of the disease. Previous studies reported that VAMP8−/− mice are resistant to experimental pancreatitis induced by either 6 hourly injections of high concentrations of the cholecystokinin (CCK) analog cerulein (CR), or 6-week alcohol feeding followed by 5 hourly injections of the muscarinic agonist carbachol (7, 8). Each study reported reduced serum amylase and improved morphologic parameters of the disease in VAMP8−/− mice. The ethanol feeding study also reported reduced pancreatic and lung inflammation with modestly reduced active trypsin in pancreatic homogenates. Based on the results that acini prepared from VAMP8−/− mice show no basolateral ZG exocytosis when incubated in alcohol as occurs in WT, it was proposed that VAMP8-mediated basolateral exocytosis is the underlying mechanism of alcohol-related acute pancreatitis; however, other parameters of acinar damage were not reported (7, 8).
A common feature of acute experimental pancreatitis is a pronounced inhibition of digestive enzyme secretion. This is believed to be a key pathophysiological mechanism leading to intracellular zymogen activation (9, 10). We recently reported that zymogen activation in response to supramaximal CCK-8, bile salts, or cigarette smoke toxin occurs as a consequence of enhanced EE to LE/lysosome trafficking when acinar secretion is inhibited. Reducing EE to LE trafficking by pharmacological or molecular approaches fully prevented zymogen activation and enhanced acinar basal secretion via an anterograde endosomal pathway (11). Noting that 1) acini prepared from VAMP8−/− mice show significantly elevated basal secretion and a diminished secretory inhibition in response to supramaximal CCK-8, 2) VAMP8-mediated acinar secretion is acutely dependent on endosomal trafficking in WT acini, and 3) VAMP8 is a ubiquitously expressed endosomal SNARE, we investigated the impact of acinar pancreatitis on the VAMP8-secretory pathway and its potential role in intracellular activated zymogen accumulation.
Results
Supramaximal CCK-8 blocks VAMP8-dependent secretion
Low doses of CCK-8 (1–100 pm) and carbachol (1–100 nm) increase digestive enzyme secretion from pancreatic acinar cells, whereas supramaximal concentrations progressively inhibit secretion from a maximal response. Interestingly, the time-course of maximally stimulated secretion from a single group of acinar cells proceeds as an early rapid phase that peaks at 1 min and declines by 2 min and a second longer phase that peaks at 3–4 min and slowly declines to basal levels over the subsequent 20–30 min (1, 12). We recently reported that the early phase is mediated by the ZG SNARE protein VAMP2 and the later phase by VAMP8 (1).
To measure secretion over time from a single population of mouse acinar cells, aliquots of cells and media were sequentially collected at 2-min intervals after stimulation with maximal (10 pm) and supramaximal (10 nm) doses of CCK-8. Analysis of cumulative secretion over 30 min revealed 3 slopes or phases of secretion: a rapid first phase (0–4 min), a moderate second phase (4–10 min), and a slow third phase (10–30 min) (Fig. 1A). The rate of the first phase is equal over the first 2 min for both maximal and supramaximal CCK-8; however, supramaximal CCK-8 reduced the slope of both the second and third phases, ultimately resulting in an ∼50% decrease in total secretion at 30 min (Fig. 1A). Sequential subtraction of the preceding level of secretion from each time point was done to identify the rate of secretion during each 2-min interval. Results indicate that the secretion rate during the first 2 min was equivalent for maximal and supramaximal CCK-8, but a significant reduction in the secretory rate from 2 to 6 min occurred after supramaximal simulation (Fig. 1B). In accordance with our previous study demonstrating that VAMP2 mediates the first and VAMP8 the second phase of secretion, these data suggest that VAMP8-dependent exocytosis is mainly compromised during supramaximal CCK-8 stimulation (1).
Figure 1.

Supramaximal CCK-8 stimulation inhibits the later phases of acinar secretion mediated by VAMP8. Isolated WT mouse pancreatic acini were left untreated (Basal), stimulated with 100 pm CCK-8 (Max), or 10 nm CCK-8 (Supramax) for 30 min. A, acini and media were sequentially removed from the same batch of cells and analyzed for amylase release at indicated time points. Dashed lines indicate the slopes of first- and second-phase secretion. B, the rate of amylase release over each 2-min interval. Dashed lines denote the first (0–2 min) and second (2–10 min) phases of secretion. The third phase of secretion (10–30 min) is not denoted by a dashed line. Note that Supramax CCK-8 greatly diminishes the second and third phases of secretion. C, WT or VAMP8−/− acini were cultured for 4 h with adenovirus (4 × 106 pfu/ml) expressing GFP control or TeTx light chain to cleave VAMP2. D52 and VAMP2 levels were analyzed by immunoblot. D, Max (30 pm) or Supramax (30 nm) CCK-8-stimulated total amylase secretion from GFP- or TeTx-expressing acini (30 min) (data are the mean and S.E. *, p < 0.05 Max to Supramax CCK-8). E, Supramax CCK-8-induced secretory inhibition was calculated as the percent of Max CCK-8 stimulation (data are the mean and S.E. * p < 0.05 to respective GFP control). All experiments were generated from at least three separate acinar preparations, performed in duplicate.
As we previously reported, 4-h adenoviral expression of tetanus toxin light chain in acinar cells cleaves >80% of VAMP2 but has no effect on VAMP8, which has no tetanus toxin cleavage site (Fig. 1C) (1). Analysis of stimulated secretion, calculated by subtracting basal from total secretion, demonstrates that loss of VAMP2 reduces maximal CCK-stimulated secretion by 50% in WT acini, whereas loss of VAMP2 in VAMP8−/− acini results in an almost complete loss of secretion (Fig. 1D). The residual secretion remaining in tetanus toxin (TeTx)-expressing VAMP8−/− cells is likely due to incomplete VAMP2 cleavage. Supramaximal CCK-8 inhibited secretion (ratio of supramaximal to maximal secretion) by 55% in GFP-control acini and 82% after VAMP2 cleavage when VAMP8 is the primary ZG SNARE (Fig. 1E). In contrast, in VAMP8−/− acini, where VAMP2 is the primary ZG SNARE, supramaximal stimulation reduced secretion by only 22%, supporting that VAMP8-mediated secretion is mainly inhibited by acinar hyperstimulation.
Elevated cAMP enhances VAMP8-dependent secretion during supramaximal CCK stimulation
Elevated cAMP in response to the hormone secretin or the cell-permeant cAMP analog 8-(4-chlorophenylthio) adenosine 3′,5′-cyclic monophosphate (CPT-cAMP), each, produces a small increase in amylase secretion; however, when added in combination with CCK-8, they significantly potentiate secretion (13). We previously reported that VAMP8−/− acini have increased basal secretion that is inhibited by elevated cAMP (1); this is best detected by analyzing stimulated secretion where basal is subtracted from total secretion (compare WT and VAMP8−/− in Fig. 2, A and B). In WT acini, secretin and CPT-cAMP enhanced maximal CCK-8 secretion by 173 and 200% of maximal CCK-8 alone, respectively (Fig. 2A). Supramaximal CCK-8-stimulated secretion was increased from 2.2 to 3.2 units/μg of DNA in the presence of secretin but was not statistically significant. However, supramaximal CCK-8-stimulated secretion was significantly enhanced by 232%, in response to CPT-cAMP fully blocking the supramaximal CCK-induced secretory inhibition. In contrast to WT acini, no significant potentiation was detected in VAMP8−/− acini in response to maximal or supramaximal CCK-8 (Fig. 2B).
Figure 2.

Elevated cAMP differentially augments early and late phases of secretion in response to maximal or supramaximal CCK-8. A and B, amylase activity released from WT (A) or VAMP8−/− acini (B) left untreated (basal) or stimulated with 10 nm secretin (Sec), 100 μm CPT-cAMP, Max CCK-8 (10 pm for WT, 30 pm for VAMP8−/−), Supramax CCK-8 (10 nm for WT, 30 nm for VAMP8−/−), or combinations for 30 min. Data are stimulated secretion calculated by subtracting basal from total secretion. Note that elevated cAMP fully prevented the secretory inhibition caused by supramax CCK-8 in WT acini but had no effect in VAMP8−/− acini. C and D, WT pancreatic acini were left untreated (Basal), stimulated with 100 μm CPT-cAMP, 100 pm CCK-8 (Max), 10 nm CCK-8 (Supramax), or combinations of CPT-cAMP and CCK-8 for 30 min. Acini and media were sequentially removed from the same batch of cells and analyzed for amylase release at the indicated time points. E and F, the rate of amylase release over each 2-min interval was calculated from panels A and C. Note that elevated cAMP augments mainly the first and third phases of secretion with Max CCK-8 but only the third phase of secretion with Supramax CCK-8 (data are the mean and S.E. *, p < 0.05). All experiments were generated from at least three separate acinar preparations, performed in duplicate.
Elevated cAMP acts on third-phase secretion to prevent supramaximal CCK-mediated secretory inhibition
As treatment of WT acini with CPT-cAMP and supramaximal CCK-8 fully prevents secretory inhibition (Fig. 2A), we examined the time course of secretion for this response (Fig. 2C). CPT-cAMP alone had no effects during the first 10 min and slightly elevated secretion at times >15 min. When combined with maximal CCK-8, CPT-cAMP clearly elevated third-phase (>10 min) secretion (Fig. 2C). Analysis of the secretory rate over the first 10 min (Fig. 2D) and calculation of the area under the curve (Table 1) indicated both first and second-phase secretion were significantly increased compared with maximal CCK-8 alone. Unexpectedly, the reduction in the second phase caused by supramaximal CCK-8 remained impeded in the presence of CPT-cAMP; however, third-phase secretion was markedly elevated, restoring total secretion over 30 min to levels seen for maximal CCK-8 alone (Fig. 2E). The lack of effect of CPT-cAMP on the second phase was further identified by analyzing the secretory rate over the first 10 min of secretion (Fig. 2F and Table 1). Thus, although elevated cAMP acts on VAMP8-dependent secretion, it does not restore the loss of VAMP8-mediated second-phase secretion during supramaximal CCK-8 but rather acts on the late third phase (>10 min). Moreover, VAMP2-mediated first-phase secretion is not compromised by supramaximal CCK-8 and, strikingly, is potentiated by elevated cAMP in response to maximal but not supramaximal CCK-8.
Table 1.
Area under the curve for cAMP effect on CCK-8 stimulation (0–10 min)
| Time | Max CCK | Max CCK + cAMP |
|---|---|---|
| 0–10 min | 23.6 ± 1.0 | 36.4 ± 6.2a |
| 0–4 min | 11.7 ± 0.5 | 18.7 ± 3.0a |
| 6–10 min | 11.9 ± 0.5 | 17.7 ± 3.2a |
| Time | SupraMax CCK | SupraMax CCK + cAMP |
|---|---|---|
| 0–10 min | 14.0 ± 1.3 | 16.4 ± 0.8 |
| 0–4 min | 8.6 ± 0.8 | 9.9 ± 0.5 |
| 6–10 min | 5.5 ± 0.5 | 6.5 ± 0.3 |
a p < 0.02.
VAMP8−/− prevents intracellular accumulation of activated zymogens in response to supramaximal CCK-8
VAMP8−/− mice are resistant to cerulein or alcohol-induced experimental pancreatitis in vivo; however, as VAMP8 is ubiquitously expressed and has been shown to mediate secretion in various immune cells (14–17), the acinar cell-specific effects in the whole animal knock-out are not clear. Isolated acini are free from the inflammatory component of the disease, thus allowing investigation of the earliest cellular events leading to acinar damage. VAMP8−/− acini contain ∼4-fold more total trypsin activity (activated by enterokinase addition to lysates) than WT when normalized to cellular DNA (Fig. 3A). WT acini show an ∼10-fold induction in intracellular trypsin accumulation after supramaximal stimulation (Fig. 3B). Strikingly, VAMP8−/− show no increase in intracellular trypsin activity in response to supramaximal CCK-8. This is evident when normalizing to total cellular DNA or total intracellular trypsin activity, indicating basal levels of trypsin activity are extremely low in VAMP8−/− acini (Fig. 3C). Finally, as support that intracellular trypsin mediates cellular damage during pancreatitis, VAMP8−/− acini show no increase in necrosis as measured by lactate dehydrogenase (LDH) release, compared with WT acini, which have a 6-fold increase (Fig. 3D). These results suggest that VAMP8-regulated membrane trafficking is essential to accumulate activated zymogens during the onset of acute pancreatitis.
Figure 3.

VAMP8−/− acini are fully protected from supramaximal CCK-induced intracellular trypsin accumulation and cell damage. A, total cellular trypsin activity in acinar lysates after enterokinase cleavage of trypsinogen was normalized to total DNA in WT and VAMP8−/− acini. B and C, WT or VAMP8−/− acini were left untreated (Basal) or stimulated with Max CCK-8 (10 pm in WT, 30 pm in VAMP8−/−) or Supramax CCK-8 (10 nm in WT, 30 nm in VAMP8−/−) for 30 min. Intracellular trypsin activity was measured and normalized to total DNA in panel B or to total cellular trypsin in panel C. D, WT or VAMP8−/− acini were treated as above for 3 h, and lactate dehydrogenase was released to the medium was calculated as a percentage of total cellular levels. Note that VAMP8−/− acini are fully protected from Supramax CCK-8 induced damage (data are the mean and S.E. *, p < 0.05). All experiments were generated from at least three separate acinar preparations, performed in duplicate.
Because VAMP8 is involved in endosome and lysosome fusion reactions, the possibility that chronically impaired lysosome formation prevented intracellular zymogen activation in VAMP8−/− acini was evaluated by analysis of CatB maturation (Fig. 4, A and B). In comparison to WT, VAMP8−/− acini had greatly reduced levels of the proenzyme and single chain forms of the enzyme, whereas the mature double-chain form was highly elevated. Consistent with immunoblotting, CatB activity, which in WT acini is increased by supramaximal CCK-8 stimulation, was significantly elevated under basal conditions but not further increased by CCK-8 (Fig. 4C). As CatB activation requires a low pH environment typical of lysosomes and intracellular trypsinogen activation likewise occurs in an acidic compartment, we utilized the acidic dye Lysotracker Red to identify acidic organelles in acinar cells (Fig. 4D). Quantification of punctate structures <1 μm diameter amounted to ∼97% of total in WT acini, but was significantly reduced in VAMP8−/− acini (Fig. 4E). Rather, Lysotracker-positive punctate structures greater than 3 and 4 μm in diameter were increased by ∼4- and 10-fold, respectively, in VAMP8−/− acini (Fig. 4F). The enlarged acidic organelles support a role for VAMP8 in lysosome formation but also indicate that lysosome function remains intact in VAMP8−/− acini. Thus, the absence of intracellular trypsin accumulation with supramaximal CCK-8 may likely be due to a lack of zymogen and lysosome mixing rather than a direct inhibition of trypsinogen activation.
Figure 4.

VAMP8−/− have enhanced lysosomal CatB activity. CatB maturation was determined by immunoblotting (A) and quantified by densitometry (B) in WT or VAMP8−/− acinar lysates. Pro, proenzyme. C, isolated WT or VAMP8−/− acini were left untreated or stimulated with 10 nm CCK-8 for 60 min. Intracellular CatB activity was measured using a fluorogenic substrate. D, WT or VAMP8−/− acini were incubated with 50 nm Lysotracker Red DND-99 for 45 min and imaged by confocal microscopy (scale bars, 5 μm). E, Lysotracker Red punctate structures <1 μm were quantified by ImageJ. F, Lysotracker Red punctate structures binned to the indicated sizes for VAMP8 were quantified as -fold WT (data are the mean and S.E. *, p < 0.05). All experiments were generated from at least three separate acinar preparations, performed in duplicate.
Supramaximal CCK-8-stimulation reduces endosomal regulatory protein expression
We recently reported that expression of the endosomal protein Rab5 and its binding proteins D52 and EEA1 are essential to maintain VAMP8-dependent secretory activity in acini (1). Suspension culture of acini (16 h) causes a >90% reduction in Rab5, D52, and EEA1, resulting in a complete loss of VAMP8-dependent secretion that is restored by rescuing Rab5/D52/EEA1 expression (1). Based on our findings that VAMP8-mediated second and third phases of secretion are greatly attenuated by supramaximal CCK-8 (Fig. 1), we examined its effects on endosomal protein expression. Supramaximal CCK-8 (100 nm) over 60 min in rat acini caused a >60% reduction in D52, Rab5, and EEA1 (Fig. 5 A). Likewise, analysis of D52/Rab5/EEA1 after 1 h of supramaximal CR stimulation in vivo in the rat caused a 30–40% reduction in D52 and EEA1 with the reduction in Rab5 not significant (Fig. 5B). The transcription factor spliced XBP1, which is reported to be reduced in pancreatitis (18), decreased an average of 75% but was not significant. In contrast to protein, mRNA levels of these proteins remained stable (Fig. 5C). More aggressive pancreatitis by 4 hourly injections of CR in rat caused a marked reduction in all 3 proteins, similar to what is seen in rat acinar cells (Fig. 5D). In mice, which are more resistant to CR-pancreatitis, 7 hourly injections of CR or 72-h choline-deficient ethionine-supplemented feeding similarly resulted in reductions in Rab5/D52/EEA1 protein levels (Fig. 5E). These data suggest that the loss of VAMP8-dependent secretion during experimental pancreatitis is concordant with a progressive loss of endosomal regulatory proteins necessary to maintain the VAMP8-dependent secretory pathway.
Figure 5.
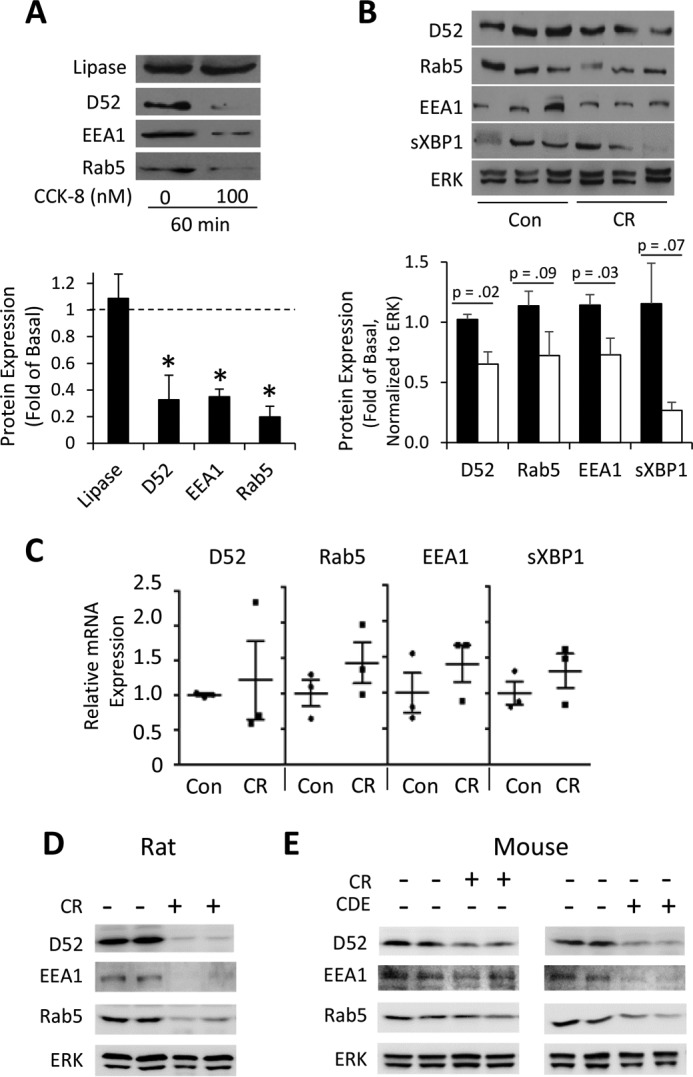
Endosomal proteins are lost during in vivo and in vitro pancreatitis. Isolated rat pancreatic acini were left untreated or stimulated with supramax CCK-8 (100 nm) for 60 min. A, whole cell lysates were immunoblotted for pancreatic lipase and endosomal proteins (D52, EEA1, Rab5). Immunoblots were quantified and plotted as the -fold of untreated levels indicated by the dashed line (data are the mean and S.E. *, p < 0.05). All experiments were generated from at least three separate acinar preparations, performed in duplicate. B and C, pancreatitis was induced in rats by a single IV injection of supramximal CR (50 μg/kg of body weight) or saline control. 1-h post injection the indicated protein (B) and mRNA (C) levels were analyzed in pancreatic homogenates. Data are the mean and S.E., n = 3–4 animals per group. D, endosomal protein (D52, EEA1, Rab5) expression was analyzed after induction of in vivo pancreatitis in mice given 7 hourly IP injections of CR or after 72 h feeding of a choline-deficient/ethionine-supplemented diet (CDE). E, endosomal protein (D52, EEA1, Rab5) expression after induction of in vivo pancreatitis in rats given 4 hourly i.p. injections of CR. Note that after 1-h CR pancreatitis in vivo (panel B), although all endosomal proteins were reduced, only D52 and EEA1 were significant (p < 0.05); however, at longer periods in both rat and mouse (panels D and E, respectively) all endosomal proteins are markedly reduced.
Cathepsin inhibition prevents the loss of D52 expression during acinar pancreatitis
Similar to in vivo studies in mice where 7 hourly CR injections are necessary to produce significant decreases in endosomal proteins, supramaximal CCK-8 for 60 min in mouse acini reduced D52 by ∼50% (Fig. 6, A and B), with Rab5 and EEA1 remaining relatively stable (not shown). D52 contains putative PEST sequences in both the amino and carboxyl termini, suggesting it may be susceptible to ubquitination and proteasomal degradation. Unexpectedly, treatment of mouse acini with the proteasome inhibitor MG-132 alone markedly reduced D52 expression to the same extent as CCK-8 treatment (Fig. 6A). Moreover, when added together, these agents fully depleted cellular D52 levels. Proteasomal inhibition by MG-132 is known to induce a large compensatory increase in cellular autophagy, suggesting D52 degradation might result from this process (19). Noting that the LAMP endolysosomal proteins are degraded during acute pancreatitis by CatB, acini were pretreated (30 min) with the CatB and CatL inhibitor Ca-074Me, resulting in a complete inhibition of D52 degradation in response to supramaximal CCK-8 (Fig. 6B). Acinar cells undergo a high rate of autophagic flux that is further enhanced by both secretory and supramaximal stimulation (20). We, therefore, examined the colocalization of D52 with the lysosomal marker LAMP2 and autophagy protein LC3, which when recruited from the cytosol to the growing phagophore by lipidation appears as cytoplasmic puncta indicative of autophagosmes (Fig. 6C). As previously seen for LAMP1 (21), D52 partially colocalizes with LAMP2 under basal conditions when sparse LC3 puncta are present. Exposure to maximal and supramaximal concentrations of CCK-8 caused a large increase in LC-3 puncta, indicating enhanced autophagic flux. Quantification of D52/LC3/LAMP2 puncta revealed a >2-fold increase with secretory stimulation and 3-fold after supramaximal CCK-8, supporting that a portion of cellular D52 is targeted to the autophagic pathway during stimulation (Fig. 6D). These results were confirmed by immunoelectron microscopy demonstrating D52 immunoreactivity within autophagic vacuoles in acinar cells (Fig. 6E).
Figure 6.

D52 is present in autophagic vacuoles, and its degradation is blocked by cathepsin B/L inhibition. A, mouse pancreatic acinar cells were pretreated for 30 min with the proteasome inhibitor MG132 (25 μm) or DMSO vehicle (0.1%) before 1 h of supramaximal CCK-8 stimulation (100 nm). Note that D52 levels are significantly reduced by proteasome inhibition alone and the addition of CCK-8 fully depleting cellular D52. B, mouse pancreatic acinar cells were pretreated 30 min with the cathepsin B and L inhibitor Ca-074ME (ME; 50 μm) or DMSO vehicle (0.1%) before 1 h of supramaximal CCK-8 stimulation (100 nm). Note that the depletion of D52 levels by supramaximal CCK-8 is blocked by cathepsin B/L inhibition. C and D, D52 was colocalized with lysosomal membrane protein LAMP2 and LC3 in sections of pancreatic lobules that were treated with maximal (30 pm) or supramaximal (100 nm) CCK-8 for 30 min. Data in D quantifies the number of white puncta representing all three fluorophores under each condition. Note that acinar stimulation rapidly enhances LC3 puncta, which partially colocalizes with D52. Data in A–D are the mean and S.E. (*, p < 0.05) from at least three separate preparations). E, immunoelectron microscopy of D52 in sections of pancreatic lobules treated with 100 pm CCK (2 min). Note that D52 (yellow arrows) is present in autophagic vacuoles (arrowheads) consistent with its colocalization with LAMP2 and LC3 in panel C. A single representative image seen in three independent cell preparations is shown.
Enhancement of anterograde endosomal trafficking prevents intracellular trypsinogen activation
Based on the current evidence that VAMP8-dependent secretion is lost early during CCK-pancreatitis, we hypothesized that enhancing secretion from the VAMP8-secretory pathway by overexpression of D52 or Rab5 would reduce intracellular trypsin accumulation. Suspension culture of acinar cells for 16 h causes reduced secretory activity, a loss of Rab5 and D52 expression, and no supramaximal CCK-stimulated trypsinogen activation. Additionally, maintaining either D52 or Rab5 by adenoviral expression will maintain the expression of the other protein (21). On the other hand, mouse acini cultured on collagen-coated plates in the presence of the isobutyl methyl xanthine and 10% FBS retain secretory activity, trypsinogen activation, and D52/Rab5 expression for 24 h (11, 20, 22). Unlike suspension culture, adenoviral overexpression of HA-tagged D52 or canine-Rab5 in mouse acini maintained on collagen did not alter the expression of the other protein (Fig. 7A). HA-D52 and canine-Rab5 overexpression resulted in an approximate 235% increase in basal secretion and a 150% increase in supramaximal CCK-8-stimulated secretion (Fig. 7B). Correspondingly, overexpression of HA-D52 had no effect on basal activity but reduced CCK-8-stimulated intracellular trypsin activity by 50% compared with CCK-8 effects in GFP expressing control cells (Fig. 7C). Similarly, canine-Rab5 overexpression reduced basal activity by 50% and fully inhibited CCK-8-induced trypsin accumulation compared with stimulated control cells. Taken together with our previous work demonstrating that D52 and Rab5-mediated endosomal trafficking is required for secretion from the VAMP8-pathway, these results support that inhibition of the VAMP8-secretory pathway plays a pivotal role in promoting active zymogen accumulation early during acute pancreatitis.
Figure 7.

Enhanced endosomal trafficking prevents zymogen activation. WT mouse acini were cultured on collagen-coated plates in the presence of 10% FBS, 0.5 mm isobutylmethylxanthine, and 106 pfu/ml adenovirus encoding GFP control, HA-D52, or canine Rab5 for 24 h. A, D52 and Rab5 levels as determined by immunoblot. B, amylase release after basal and supramaximal (100 nm) CCK-8 (30 min) stimulation. C, intracellular trypsin activity after basal and supramaximal CCK-8 stimulation (data are the mean and S.E. *, p < 0.05). All experiments were generated from at least three separate acinar preparations, performed in duplicate.
Discussion
Our previous work described two functional populations of ZGs in pancreatic acinar cells based on the expression of VAMP2 and VAMP8, which control the early (0–2 min) and late phases of secretion (>2 min), respectively (1) (Fig. 8). Here we subdivided the late-phase into second-phase (2–10 min) and third-phase (>10 min) secretion (1, 23). Elevated cAMP augments all phases of maximally stimulated secretion. With supramaximal stimulation, VAMP8-mediated second- and third-phase secretion is compromised, and inhibition of the third phase is alleviated by elevated cAMP. We previously reported the early endosomal proteins Rab5 and D52 are necessary for VAMP8-dependent ZG secretion and also promote basal secretion from an endolysosome-like secretory compartment termed the constitutive-like pathway that arises from removal of membrane and digestive enzymes from maturing ZGs (1). Inhibition of VAMP8-dependent secretion during pancreatitis leads to an accumulation of intracellular trypsinogen in the constitutive-like pathway that traffics from EE to late endosomes/lysosomes and becomes activated. Interestingly the fusion of late endosomes (Fig. 8) and autophagic vacuoles (not shown in Fig. 8) with lysosomes entails VAMP8-dependent fusion events. In VAMP8−/− acini, which show no intracellular trypsin activity during pancreatitis, it appears the lysosomal compartment forms likely as a consequence of CatB trafficking from the Golgi to the LE/lysosome by a mannose 6-phosphate receptor pathway.
Figure 8.
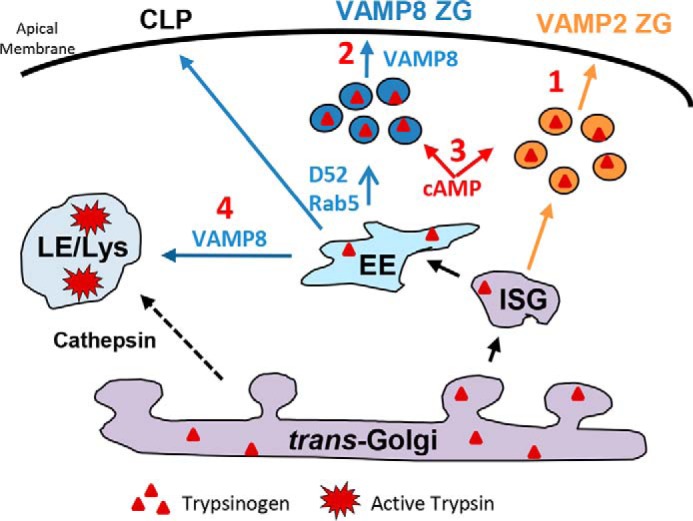
Loss of VAMP8-dependent secretion leads to intracellular trypsinogen activation. Immature secretory granules (ISG) give rise to VAMP2- and VAMP8-contianing ZGs. Under normal conditions acinar stimulation is characterized by VAMP2-mediated ZG exocytosis at <2 min (1) and VAMP8-dependent ZG exocytosis at later times (2). Elevated cAMP augments early and late phases of maximally stimulated secretion. With supramaximal stimulation, VAMP8-mediated late-phase secretion is compromised, and this inhibition is alleviated by elevated cAMP (3). The early endosomal proteins, Rab5 and D52, are necessary for VAMP8-dependent secretion and also promote basal secretion from an endolysosome-like secretory compartment termed the constitutive-like pathway (CLP) (1, 21). Inhibition of VAMP8-dependent secretion leads to an accumulation of intracellular trypsinogen that traffics from early (EE) to the late endosome (LE)/lysosome (Lys), also involving VAMP8-dependent fusion events. Cathepsin B is directly trafficked from the Golgi to the LE/lysosome by a mannose 6-phosphate receptor pathway where it mixes with trypsinogen to produce active trypsin leading to the pathogenesis of pancreatitis.
Evidence that elevated cAMP markedly augments all phases of secretion in response to maximal CCK-8 but only impacts late-phase secretion in response to supramaximal CCK-8 suggests that regulatory proteins responsive to cAMP may differentially impact the VAMP2- and VAMP8-mediated pathways (Fig. 8). The effects of cAMP in acini have been tied to both the activation of PKA and exchange protein directly activated by cAMP (EPAC) (24); however, use of PKA inhibitors or EPAC-specific cAMP analogs have indicated a large degree of overlap of these pathways with regard to augmentation of supramaximal CCK- or carbachol-stimulated secretion and intracellular accumulation of activated zymogens (25). PKA activation was also reported to specifically modulate inositol triphosphate receptor-mediated Ca2+ release to the cytosol (26). Other studies indicate that both PKA and EPAC accelerate basal to apical Ca2+ waves in response to carbachol, and these effects are mediated by the ryanodine receptor (27). Of particular interest, elevated cAMP actually enhances levels of intracellular zymogen activity, but the cells remain healthy due to the eventual secretion of these zymogens (13). This suggests that zymogen mixing with lysosomes occurs in the presence of cAMP, but by preventing secretory inhibition, cAMP circumvents the retention of active zymogens. Our current results demonstrating that VAMP8−/− acini have enlarged acidic compartments consistent with lysosomes together with significantly enhanced levels of activated CatB suggest that lysosomal function is intact. Whether zymogen activation occurs before secretion is not certain; however, unlike the cAMP response, where intracellular active zymogens are initially retained at 30 min, we detected no intracellular trypsin activity, suggesting that CatB and zymogen mixing does not occur in VAMP8−/− acini.
Secretory inhibition caused by supramaximal CCK-8 compromised mainly the VAMP8-mediated second (2–10 min) and third phases (>10 min) of secretion, which we recently reported is dependent on the expression of the EE proteins Rab5 and D52 (Fig. 8) (1). Interestingly, VAMP8−/− acini have a >80% reduction in Rab5 and D52 levels and undergo a redistribution of other endosomal proteins including Ti-VAMP7, VAMP4, and LAMP1 to purified ZG fractions (1). Consequently, VAMP8−/− acini have enhanced basal secretion, which when analyzing total secretion (basal plus stimulated values) prevents the secretory inhibition caused by supramaximal CCK-8. The current results that VAMP8−/− acini are fully resistant to supramaximal CCK-induced intracellular trypsin accumulation closely parallel our work demonstrating that inhibition of EE to LE trafficking by genetic or pharmacological manipulations likewise elevates basal secretion and protects against intracellular trypsin accumulation (11). Thus the current results demonstrating that overexpression of D52 or Rab5 elevates basal and supramaximal CCK-8-stimulated secretion and protects against intracellular trypsin accumulation are clearly in line with a pivotal role for anterograde endosomal trafficking in preventing the accumulation of activated zymogens.
Although the loss of Rab5 and D52 during acute pancreatitis implicate an acute dysregulation of the VAMP8-secretory pathway in the pathobiology of acute pancreatitis, the time course for degradation (60 min) is not early enough to account for the rapid loss of VAMP8-dependent secretion caused by supramaximal CCK-8 that occurs during the second (4–10 min) and third phases (>10 min) of secretion. Regulated exocytosis proceeds through a sequence of tethering, docking, priming, and fusion of secretory granules with the plasma membrane (28, 29). Padfield and Panesar (12) using α-hemolysin-permeabilized acini to control intracellular Ca2+ and ATP levels, demonstrated that early-phase secretion in acini, triggered by elevating cellular Ca2+, is ATP-independent and involves ZGs that are docked and primed at the plasma membrane, whereas late-phase secretion requires ATP-dependent priming of ZGs. A number of studies support that the ATP-dependent priming step for exocytosis represents synthesis of phosphatidylinositol 4,5-bisphosphate (PIP2) in nanodomains on the inner leaflet of the plasma membrane that are essential for SNARE complex formation and membrane fusion (28–30). Intriguingly, PIP2 synthesis is regulated by phosphatidylinositol 4-phosphate 5-kinase, which undergoes rapid plasma membrane recycling in an ADP-ribosylation factor-6-dependent endosomal pathway that when impaired, inhibits exocytosis (31). Although synthesis of PIP2 nanodomains appears to be an essential component of secretion in neural and endocrine cells, it is largely unexplored in acinar cells and could provide valuable insight into the mechanism of the rapid secretory inhibition seen during acute pancreatitis.
In addition to the early retention of activated zymogens during acute pancreatitis, acinar cells also undergo enhancement of autophagic flux that is eventually inhibited by a loss of lysosomal function, resulting in the accumulation of autophagic vacuoles containing ZGs and various organelles (20). The mechanism of lysosomal dysfunction has been tied to the loss of lysosomal membrane proteins, LAMP1 and -2 (32, 33). Accordingly, pancreas-specific deletion of LAMP2 in rodent pancreas leads to spontaneous pancreatitis (33). Consistent with our current results in isolated acini and in vivo showing an approximate 50% decrease in D52 expression at 1 h, partial LAMP1 and -2 degradation was also recently reported in 1-h supramaximally stimulated pancreatic lobules (33). Demonstration that a portion of cellular D52 enters autophagic vacuoles and cathepsin B/L inhibition blocks D52 degradation indicates that endosomal dysfunction during acute pancreatitis is tied to the autophagic pathway, a critical component of the pathogenic response leading to pancreatitis. Results that the decline in Rab5 and EEA1 lag behind D52 during pancreatitis in mouse suggest an alternative mechanism that will require further investigation.
We propose that secretory inhibition early during acute pancreatitis 1) prevents endolysosomal secretion from the constitutive-like secretory pathway, 2) inhibits the maturation and secretion from the VAMP8-ZG secretory pathway, and 3) by default, diverts EE trafficking of zymogens toward the late endosome and lysosome thereby promoting premature zymogen activation and accumulation in the cell (Fig. 8). At later times (≥1 h), Rab5/D52/EEA1 degradation maintains impairment of the VAMP8-secretory pathway, which together with the loss of lysosomal membrane proteins and the consequent inhibition of autophagic flux greatly exacerbates disease progression The significantly elevated basal secretion seen in VAMP8−/− acini, caused by a marked enhancement and redistribution of regulatory proteins including Ti-VAMP7 and Rab11a during development (1), maintains anterograde secretory flux thereby mitigating zymogen activation. Clearly, additional studies aimed at further understanding the relationship of endosomal trafficking to the high rate of autophagic flux in acini should provide valuable insight into the pathobiology of acute pancreatitis.
Experimental procedures
Antibodies
Anti-D52 antibodies were previously described (34). Antibodies for VAMP2 (104 211) and VAMP8 (104 303) were from Synaptic Systems. Anti-EEA1 (610456) and anti-canine Rab5 (610725) were from BD Biosciences. Anti-hemagglutinin mouse monoclonal antibody was from Cell Signaling Technology. Anti-Rab5 (ab18211) and anti-CatB (ab33538) recognizing mature and immature forms was purchased from Abcam. Anti-LAMP2 (OAAB06711) was from Avia. Anti-LC3 α/β (SC-16756) was from Santa Cruz. Alexa-conjugated secondary antibodies were from Life Technologies. Ultra Small ImmunoGold secondary was from Aurion. Peroxidase-conjugated secondary antibodies were from GE Healthcare. All antibodies were characterized before immunoblotting studies by serial dilutions to determine optimal conditions and negative controls to ensure specificity.
Other reagents
Phadebas Amylase Assay kit was from Magle Life Sciences, CCK-8 from Research Plus, cerulein was from Sigma, Dulbecco's minimal essential medium (DMEM), essential amino acids, fetal bovine serum, penicillin and streptomycin, Image-iT® FX Signal Enhancer, and Prolong gold antifade reagent with 4,6-diamidino-2-phenylindole (DAPI) were all from Invitrogen. Bovine serum albumin, CPT-cAMP, and a protease inhibitor mixture containing AEBSF (4-(2-aminoethyl)benzenesulfonyl fluoride), aprotinin, EDTA, leupeptin, and E64 were from Calbiochem. Protein determination reagent and nonfat dry milk were from Bio-Rad. SuperSignal West Femto Chemiluminescent Substrate was from Thermo Scientific. All other reagents were purchased from Sigma. HA-D52, canine-Rab5, TeTx, and GFP adenovirus were previously described (11, 21, 35). Harvested adenovirus from AD-293 was purified and concentrated by cesium chloride centrifugation. Adenovirus titer was determined by plaque assay using agarose overlay on AD-293 cells.
Culture of pancreatic acini
The University of Wisconsin Committee on Use and Care of Animals approved all studies involving animals. Pancreatic acini were isolated from male Harlan Sprague-Dawley rats and C57BL/6 mice by collagenase digestion as previously described (36). Culturing of acini was performed as described (21). Where indicated, mouse acini were cultured for 24 h on collagen-coated plates as previously described (20).
Animal models of pancreatitis
The experimental protocol for 1 h pancreatitis in Fig. 5, B and C, was approved by the animal research committee of the University of Wisconsin in accordance with the National Institutes of Health guidelines. Male rats (∼150 g) were anesthetized with 1.5–2% isoflurane and given a single IV injection of 50 μg/kg body weight CR or saline vehicle (100 μl) as control into the jugular vein. After 1 h the pancreas was removed for analysis. The experimental protocols for Fig. 5, D and E, were approved by the animal research committee of the Veterans Affairs Greater Los Angeles Healthcare System, in accordance with the National Institutes of Health guidelines. Acute cerulein pancreatitis was induced as described elsewhere (20, 33) in male Sprague-Dawley rats and in C57BL/6 mice that received hourly intraperitoneal injections of 50 μg/kg cerulein or physiologic saline. Rats were euthanized 4 h and mice 7 h after the first injection. Choline-deficient, ethionine-supplemented pancreatitis was induced in young (∼5 weeks old) female mice fed either a choline-deficient diet or control diet. Both the choline-deficient and control diets were obtained from Harlan Teklad (Madison, WI) and were provided fresh to the animals every 12 h in 3-g aliquots. At each feeding, the choline-deficient diet was supplemented with 0.5% ethionine. Mice were sacrificed 72 h after the initiation of the diet.
Assays: amylase secretion
For secretion of amylase, acinar cells were maintained in salt-balanced HEPES buffer containing the indicated agents at 37 °C for 30 min before measuring amylase in the medium as previously described (21). The cumulative time course of amylase secretion was conducted by incubating acini in 50 ml of HEPES buffer and removing 1 ml of acini and media every 2 min. To calculate the rate of release per 2-min intervals, sequential subtraction from the previous time point was performed.
Intracellular protease activity
Analysis of intracellular protease activity was conducted on acinar lysates from cells treated as control or supramaximally stimulated with CCK-8. Trypsin and CatB activities were measured fluorometrically using a method described previously (37, 38).
Immunoblotting
Immunoblotting of acinar lysates was conducted as described (39, 40).
LDH release
Acini were left untreated or stimulated with 10 nm CCK-8 for 3 h. Supernatant was collected from the final 3 h of culture and analyzed for LDH activity using a previously described procedure (13). All experiments were performed in duplicate from at least three independent cell preparations.
Quantitative PCR
Total RNA was extracted and purified with the RNeasy Plus Mini Kit (Qiagen, 74134). Total RNA was used for cDNA synthesis (iScriptTM cDNA synthesis kit, Bio-Rad). Real time quantitative PCR was performed with the KAPA SYBR® FAST qPCR kit (KAPA Biosystems, KK4611) and analyzed with the Roche LightCycler® 480. Relative expression levels were calculated using the 2−ΔΔCt method using cyclophilin A as the internal control. The primer pairs utilized are provided in Table 2.
Table 2.
Primers utilized for quantitative real time PCR
| Gene | Forward | Reverse |
|---|---|---|
| Cyclophilin A | CCAAGACTGAGTGGCTGGAT | TCCATGGCTTCCACAATGCT |
| Tpd52 | GCCATCACCTGGCATGGATT | CGCTCGGAGAGAGGTAGAGA |
| EEA1 | AACTGAGCTGCTTCAGAGACC | CCGAGTTAGCGTGTTCTGACT |
| Rab5a | AGCTGGCCAAGAACGGTATC | CACAACTATGGCGGCTTGTG |
| sXbp1 | TGCTGAGTCCGCAGCAGGTGC | GCTTGGCTGATGAGGTCCCC |
Lysotracker Red DND-99 labeling
Freshly prepared acini were attached to poly-l-lysine-coated coverslips for 15 min at 37 °C before 45 min of 50 nm Lysotracker Red DND-99 incubation. Acini were fixed 12 min in 4% formaldehyde at room temperature and mounted with Prolong anti-fade Gold plus DAPI. Images were captured at room temperature using a Nikon A1R-Si+ confocal microscope, a Nikon AP0 Apo ×100 oil objective with a numerical aperture of 1.49, and a Nikon DS-Qi2 camera with a work station running NIS-Elements software. WT and VAMP8−/− acini representing 4–15 cells/acini from 3 independent preparations were used for Lysotracker Red punctuate structure quantification, images were binarized by threshold function, and circular objects were detected by particle analysis function in ImageJ software. The number of punctuate structures was variable among acini, but the size distribution was highly consistent as a percent of total puncta.
Immunofluorescence and immunoelectron microscopy
Immunofluorescence microscopy was conducted on cryostat sections of paraformaldehyde-fixed pancreatic lobules as previously detailed (21). Immunofluorescent dots were quantified per cell from at least three separate tissue preparations. For immunoelectron microscopy, acinar cells were treated as indicated with CCK-8 and then transferred to microcentrifuge tubes for all processing. Briefly, cells were fixed for 1 h at room temperature in 4% formaldehyde and 0.25% glutaraldehyde. Cells were then treated with 1 m glycine, pH 7.4, for 30 min at room temperature for aldehyde inactivation. Cells were then permeabilized and blocked in sync with PBS containing 0.05% Triton X-100, 4.0% bovine serum albumin, 4.0% goat serum, and 0.1% cold-water fish skin gelatin for 30 min at room temperature with gentle agitation. Cells were then resuspended in acetylated BSA/PBS with TPD-52 primary antibody and placed on a wheel at 4 °C overnight. The next day cells were incubated overnight at 4 °C in acetylated BSA/PBS with Ultra small gold-conjugated secondary antibody. Silver secondary was enhanced using Aurion R-Gent silver-enhancing kit, and the reaction was terminated with 0.3 m sodium thiosulfate. Cells were post-fixed in 2.5% glutaraldehyde, 0.5% osmium tetroxide, and dehydrated in an ethanol gradient followed by propylene oxide and then incubated overnight in equal volumes of 1:1 EM bed 812-Spurs low-viscosity resin and propylene oxide. After the tissues were sectioned, they were placed on copper grids and stained with Reynolds lead citrate and uranyl acetate. Sections were evaluated with a Philips CM 120 electron microscope. Captured images were converted to TIFF files and edited for publication in Adobe Photoshop.
Statistics
All data are the mean and S.E. of at least three separate tissue preparations performed in duplicate or triplicate. p values were calculated using analysis of variance (ANOVA) and post-hoc paired Student's t test.
Author contributions
G. E. G., S. W. M., J. A. B., and A. S. G. contributed to the study concept and design. S. W. M., E. K. J., C. L. H., D. D. H. T., M. M. C., and O. A. M. acquired the data. S. W. M., E. K. J., and G. E. G. drafted the manuscript.
This work was supported, in whole or in part, by National Institutes of Health Grant RO1DK07088 (to G. E. G.). This work was also supported by USDA/HATCH Grants WIS01583 (to G. E. G.) and National Institutes of Health Grants PO1DK098108 (to G. E. G. and A. S. G.) and T32DK007665 (to S. W. M and E. K. J.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- ZG
- zymogen granule
- VAMP
- vesicle-associated membrane protein
- CatB
- cathepsin B
- CR
- cerulein
- CCK-8
- cholecystokinin-8
- EE
- early endosome
- LE
- late endosome
- EEA1
- early endosomal antigen 1
- LDH
- lactate dehydrogenase
- D52
- tumor protein D52
- PIP2
- phosphatidylinositol 4,5-bisphosphate
- CPT-cAMP
- 8-(4-chlorophenylthio) adenosine 3′,5′-cyclic monophosphate
- TeTx
- tetanus toxin
- EPAC
- exchange protein directly activated by cAMP.
References
- 1. Messenger S. W., Falkowski M. A., Thomas D. D., Jones E. K., Hong W., Gaisano H. Y., Giasano H. Y., Boulis N. M., and Groblewski G. E. (2014) Vesicle-associated membrane protein 8 (VAMP8)-mediated zymogen granule exocytosis is dependent on endosomal trafficking via the constitutive-like secretory pathway. J. Biol. Chem. 289, 28040–28053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lerch M. M., Halangk W., and Krüger B. (2000) The role of cysteine proteases in intracellular pancreatic serine protease activation. Adv. Exp. Med. Biol. 477, 403–411 [DOI] [PubMed] [Google Scholar]
- 3. Halangk W., Lerch M. M., Brandt-Nedelev B., Roth W., Ruthenbuerger M., Reinheckel T., Domschke W., Lippert H., Peters C., and Deussing J. (2000) Role of cathepsin B in intracellular trypsinogen activation and the onset of acute pancreatitis. J. Clin. Invest. 106, 773–781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Grady T., Mah'Moud M., Otani T., Rhee S., Lerch M. M., and Gorelick F. S. (1998) Zymogen proteolysis within the pancreatic acinar cell is associated with cellular injury. Am. J. Physiol. 275, G1010–G1017 [DOI] [PubMed] [Google Scholar]
- 5. Otani T., Chepilko S. M., Grendell J. H., and Gorelick F. S. (1998) Codistribution of TAP and the granule membrane protein GRAMP-92 in rat caerulein-induced pancreatitis. Am. J. Physiol. 275, G999–G1009 [DOI] [PubMed] [Google Scholar]
- 6. Gukovsky I., Gukovskaya A. S., Blinman T. A., Zaninovic V., and Pandol S. J. (1998) Early NF-κB activation is associated with hormone-induced pancreatitis. Am. J. Physiol. 275, G1402–G1414 [DOI] [PubMed] [Google Scholar]
- 7. Cosen-Binker L. I., Binker M. G., Wang C.-C., Hong W., and Gaisano H. Y. (2008) VAMP8 is the v-SNARE that mediates basolateral exocytosis in a mouse model of alcoholic pancreatitis. J. Clin. Invest. 118, 2535–2551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Dolai S., Liang T., Lam P. P., Fernandez N. A., Chidambaram S., and Gaisano H. Y. (2012) Effects of ethanol metabolites on exocytosis of pancreatic acinar cells in rats. Gastroenterology 143, 832–843 [DOI] [PubMed] [Google Scholar]
- 9. Frossard J.-L., Steer M. L., and Pastor C. M. (2008) Acute pancreatitis. Lancet 371, 143–152 [DOI] [PubMed] [Google Scholar]
- 10. Saluja A. K., Lerch M. M., Phillips P. A., and Dudeja V. (2007) Why does pancreatic overstimulation cause pancreatitis? Annu. Rev. Physiol. 69, 249–269 [DOI] [PubMed] [Google Scholar]
- 11. Messenger S. W., Thomas D. D., Cooley M. M., Jones E. K., Falkowski M. A., August B. K., Fernandez L. A., Gorelick F. S., and Groblewski G. E. (2015) Early to late endosome trafficking controls secretion and zymogen activation in rodent and human pancreatic acinar cells. Cell. Mol. Gastroenterol. Hepatol. 1, 695–709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Padfield P. J., and Panesar N. (1995) Ca2+-dependent amylase secretion from SLO-permeabilized rat pancreatic acini requires diffusible cytosolic proteins. Am. J. Physiol. 269, G647–G652 [DOI] [PubMed] [Google Scholar]
- 13. Chaudhuri A., Kolodecik T. R., and Gorelick F. S. (2005) Effects of increased intracellular cAMP on carbachol-stimulated zymogen activation, secretion, and injury in the pancreatic acinar cell. Am. J. Physiol. Gastrointest. Liver Physiol. 288, G235–G243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Loo L. S., Hwang L.-A., Ong Y. M., Tay H. S., Wang C.-C., and Hong W. (2009) A role for endobrevin/VAMP8 in CTL lytic granule exocytosis. Eur. J. Immunol. 39, 3520–3528 [DOI] [PubMed] [Google Scholar]
- 15. He J., Johnson J. L., Monfregola J., Ramadass M., Pestonjamasp K., Napolitano G., Zhang J., and Catz S. D. (2016) Munc13–4 interacts with syntaxin 7, regulates late endosomal maturation, endosomal signaling and TLR9-initiated cellular responses. Mol. Biol. Cell 27, 572–587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Marshall M. R., Pattu V., Halimani M., Maier-Peuschel M., Müller M.-L., Becherer U., Hong W., Hoth M., Tschernig T., Bryceson Y. T., and Rettig J. (2015) VAMP8-dependent fusion of recycling endosomes with the plasma membrane facilitates T lymphocyte cytotoxicity. J. Cell Biol. 210, 135–151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Paumet F., Le Mao J., Martin S., Galli T., David B., Blank U., and Roa M. (2000) Soluble NSF attachment protein receptors (SNAREs) in RBL-2H3 mast cells: functional role of syntaxin 4 in exocytosis and identification of a vesicle-associated membrane protein 8-containing secretory compartment. J. Immunol. 164, 5850–5857 [DOI] [PubMed] [Google Scholar]
- 18. Lugea A., Tischler D., Nguyen J., Gong J., Gukovsky I., French S. W., Gorelick F. S., and Pandol S. J. (2011) Adaptive unfolded protein response attenuates alcohol-induced pancreatic damage. Gastroenterology 140, 987–997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ding W.-X., Ni H.-M., Gao W., Yoshimori T., Stolz D. B., Ron D., and Yin X.-M. (2007) Linking of autophagy to ubiquitin-proteasome system is important for the regulation of endoplasmic reticulum stress and cell viability. Am. J. Pathol. 171, 513–524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Mareninova O. A., Hermann K., French S. W., O'Konski M. S., Pandol S. J., Webster P., Erickson A. H., Katunuma N., Gorelick F. S., Gukovsky I., and Gukovskaya A. S. (2009) Impaired autophagic flux mediates acinar cell vacuole formation and trypsinogen activation in rodent models of acute pancreatitis. J. Clin. Invest. 119, 3340–3355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Messenger S. W., Thomas D. D., Falkowski M. A., Byrne J. A., Gorelick F. S., and Groblewski G. E. (2013) Tumor protein D52 controls trafficking of an apical endolysosomal secretory pathway in pancreatic acinar cells. Am. J. Physiol. Gastrointest. Liver Physiol. 305, G439–G452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sphyris N., Logsdon C. D., and Harrison D. J. (2005) Improved retention of zymogen granules in cultured murine pancreatic acinar cells and induction of acinar-ductal transdifferentiation in vitro. Pancreas 30, 148–157 [DOI] [PubMed] [Google Scholar]
- 23. Weng N., Thomas D. D., and Groblewski G. E. (2007) Pancreatic acinar cells express vesicle-associated membrane protein 2- and 8-specific populations of zymogen granules with distinct and overlapping roles in secretion. J. Biol. Chem. 282, 9635–9645 [DOI] [PubMed] [Google Scholar]
- 24. Sabbatini M. E., Chen X., Ernst S. A., and Williams J. A. (2008) Rap1 activation plays a regulatory role in pancreatic amylase secretion. J. Biol. Chem. 283, 23884–23894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chaudhuri A., Husain S. Z., Kolodecik T. R., Grant W. M., and Gorelick F. S. (2007) Cyclic AMP-dependent protein kinase and Epac mediate cyclic AMP responses in pancreatic acini. Am. J. Physiol. Gastrointest. Liver Physiol. 292, G1403–G1410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Giovannucci D. R., Groblewski G. E., Sneyd J., and Yule D. I. (2000) Targeted phosphorylation of inositol 1,4,5-trisphosphate receptors selectively inhibits localized Ca2+ release and shapes oscillatory Ca2+ signals. J. Biol. Chem. 275, 33704–33711 [DOI] [PubMed] [Google Scholar]
- 27. Shah A. U., Grant W. M., Latif S. U., Mannan Z. M., Park A. J., and Husain S. Z. (2008) Cyclic AMP accelerates calcium waves in pancreatic acinar cells. Am. J. Physiol. Gastrointest. Liver Physiol. 294, G1328–G1334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. James D. J., and Martin T. F. J. (2013) CAPS and Munc13: CATCHRs that SNARE vesicles. Front. Endocrinol. (Lausanne) 4, 187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Messenger S. W., Falkowski M. A., and Groblewski G. E. (2014) Ca2+-regulated secretory granule exocytosis in pancreatic and parotid acinar cells. Cell Calcium 55, 369–375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Martin T. F. (2015) PI(4,5)P2-binding effector proteins for vesicle exocytosis. Biochim. Biophys. Acta 1851, 785–793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Aikawa Y., and Martin T. F. (2003) ARF6 regulates a plasma membrane pool of phosphatidylinositol(4,5)bisphosphate required for regulated exocytosis. J. Cell Biol. 162, 647–659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Fortunato F., Bürgers H., Bergmann F., Rieger P., Büchler M. W., Kroemer G., and Werner J. (2009) Impaired autolysosome formation correlates with Lamp-2 depletion: role of apoptosis, autophagy, and necrosis in pancreatitis. Gastroenterology 137, 350–360 [DOI] [PubMed] [Google Scholar]
- 33. Mareninova O. A., Sendler M., Malla S. R., Yakubov I., French S. W., Tokhtaeva E., Vagin O., Oorschot V., Lüllmann-Rauch R., Blanz J., Dawson D., Klumperman J., Lerch M. M., Mayerle J., Gukovsky I., and Gukovskaya A. S. (2015) Lysosome associated membrane proteins maintain pancreatic acinar cell homeostasis: LAMP-2-deficient mice develop pancreatitis. Cell. Mol. Gastroenterol. Hepatol. 1, 678–694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Groblewski G. E., Yoshida M., Yao H., Williams J. A., and Ernst S. A. (1999) Immunolocalization of CRHSP28 in exocrine digestive glands and gastrointestinal tissues of the rat. Am. J. Physiol. 276, G219–G226 [DOI] [PubMed] [Google Scholar]
- 35. Teng Q., Tanase D., Tanase D. K., Liu J. K., Garrity-Moses M. E., Baker K. B., and Boulis N. M. (2005) Adenoviral clostridial light chain gene-based synaptic inhibition through neuronal synaptobrevin elimination. Gene Ther. 12, 108–119 [DOI] [PubMed] [Google Scholar]
- 36. Thomas D. D., Taft W. B., Kaspar K. M., and Groblewski G. E. (2001) CRHSP-28 regulates Ca2+-stimulated secretion in permeabilized acinar cells. J. Biol. Chem. 276, 28866–28872 [DOI] [PubMed] [Google Scholar]
- 37. Kawabata S., Miura T., Morita T., Kato H., Fujikawa K., Iwanaga S., Takada K., Kimura T., and Sakakibara S. (1988) Highly sensitive peptide-4-methylcoumaryl-7-amide substrates for blood-clotting proteases and trypsin. Eur. J. Biochem. 172, 17–25 [DOI] [PubMed] [Google Scholar]
- 38. Mach L., Mort J. S., and Glössl J. (1994) Maturation of human procathepsin B: proenzyme activation and proteolytic processing of the precursor to the mature proteinase in vitro are primarily unimolecular processes. J. Biol. Chem. 269, 13030–13035 [PubMed] [Google Scholar]
- 39. Thomas D. D., Kaspar K. M., Taft W. B., Weng N., Rodenkirch L. A., and Groblewski G. E. (2002) Identification of annexin VI as a Ca2+-sensitive CRHSP-28-binding protein in pancreatic acinar cells. J. Biol. Chem. 277, 35496–35502 [DOI] [PubMed] [Google Scholar]
- 40. Kaspar K. M., Thomas D. D., Taft W. B., Takeshita E., Weng N., and Groblewski G. E. (2003) CaM kinase II regulation of CRHSP-28 phosphorylation in cultured mucosal T84 cells. Am. J. Physiol. Gastrointest. Liver Physiol. 285, G1300–G1309 [DOI] [PubMed] [Google Scholar]