

Abstract
Isocitrate dehydrogenase 1 (IDH1) catalyzes the reversible NADP+-dependent conversion of isocitrate (ICT) to α-ketoglutarate (αKG) in the cytosol and peroxisomes. Mutations in IDH1 have been implicated in >80% of lower grade gliomas and secondary glioblastomas and primarily affect residue 132, which helps coordinate substrate binding. However, other mutations found in the active site have also been identified in tumors. IDH1 mutations typically result in a loss of catalytic activity, but many also can catalyze a new reaction, the NADPH-dependent reduction of αKG to d-2-hydroxyglutarate (D2HG). D2HG is a proposed oncometabolite that can competitively inhibit αKG-dependent enzymes. Some kinetic parameters have been reported for several IDH1 mutations, and there is evidence that mutant IDH1 enzymes vary widely in their ability to produce D2HG. We report that most IDH1 mutations identified in tumors are severely deficient in catalyzing the normal oxidation reaction, but that D2HG production efficiency varies among mutant enzymes up to ∼640-fold. Common IDH1 mutations have moderate catalytic efficiencies for D2HG production, whereas rarer mutations exhibit either very low or very high efficiencies. We then designed a series of experimental IDH1 mutants to understand the features that support D2HG production. We show that this new catalytic activity observed in tumors is supported by mutations at residue 132 that have a smaller van der Waals volume and are more hydrophobic. We report that one mutation can support both the normal and neomorphic reactions. These studies illuminate catalytic features of mutations found in the majority of patients with lower grade gliomas.
Keywords: brain tumor, enzyme kinetics, enzyme mechanism, enzyme mutation, tumor metabolism
Introduction
Metabolic changes in tumors have been described for nearly a century (1–3), but only relatively recently have enzymes involved in metabolic processes been established as tumor suppressors or oncoproteins. One of the more striking examples of metabolic enzymes playing a role in tumorigenesis includes isocitrate dehydrogenase 1 and 2 (IDH1 and IDH2).3 These homodimeric enzymes are responsible for the reversible NADP+- and Mg2+-dependent conversion of ICT to αKG (Fig. 1A) in the cytosol and peroxisomes (IDH1), or mitochondria (IDH2). IDH3 is responsible for the same reaction within the context of the TCA cycle, although the oxidative decarboxylation catalyzed by this enzyme is non-reversible and NAD+-dependent.
Figure 1.

WT and mutant IDH1 catalytic activities. Shown are the: A, normal oxidative decarboxylation, and B, the neomorphic reduction.
Mutations in IDH1 and IDH2 were identified in glioblastoma multiforme in a large sequencing effort (4), and soon >80% of adult grade II/III gliomas and secondary glioblastomas were found to have IDH1 mutations, commonly R132H or R132C IDH1 (5, 6) (reviewed in Refs. 7–9). Subsequently ∼10–20% of acute myeloid leukemias were shown to have primarily IDH2 mutations, typically R140Q or R172K IDH2 (10). Early mechanisms of tumorigenesis focused on deficient conversion of ICT to αKG (11), suggesting that IDH serves as a tumor suppressor, in part through altering levels of hypoxia-inducible transcription factor-1α (12). However, IDH1 and IDH2 mutations appeared heterozygously in tumors, an unusual feature of a tumor suppressor. In landmark studies (13–15), the most common IDH1 and IDH2 mutations were shown to catalyze a neomorphic reaction: the Mg2+- and NADPH-dependent reduction of αKG to D2HG (Fig. 1B). This suggested IDH1 and IDH2 likely encode for oncoproteins. D2HG is proposed to be an oncometabolite; it competitively inhibits αKG-dependent enzymes including the TET family of 5-methylcytosine hydroxylases and the JmjC family of histone lysine demethylases, resulting in cell de-differentiation (16, 17). Indeed, cancer patients with IDH mutations display hypermethylated phenotypes (18–20) resulting from D2HG-mediated inhibition of histone and DNA demethylation. The proposed oncometabolite D2HG alone can recapitulate tumorigenic phenotypes in cancer models (21, 22), but studies measuring global metabolomics changes between mutant IDH1 expression and D2HG treatment show some differences (23, 24), indicating loss of the normal reaction and/or altered NADPH levels may also play an important role. Development of selected targeted therapy against oncogenic IDH1 and IDH2 mutations is underway and results are promising (8).
A point mutation conferring a new catalytic activity suggests important mechanistic features, and many have used kinetics and structural methods to explore mutant IDH1 and IDH2 activity (13, 25–30). There is evidence that IDH1 mutations produce varying concentrations of D2HG (30), with somewhat subtle but interesting alterations in conformational changes as shown in crystal structures of R132H IDH1 (26, 27, 29). In general, reported kinetic parameters of IDH1 mutants explored to date vary widely, making comparisons difficult. Interestingly, some mutations identified in tumors do not appear to generate D2HG (31, 32), suggesting that loss of the normal reaction itself has important consequences, or perhaps that they are simply passenger mutations.
Here we report a thorough catalytic study of a wide spectrum of IDH1 mutations, including many identified in tumors and several mutants designed to clarify catalytic features. We show that IDH1 mutants vary widely in catalytic efficiency, with more polar and larger residues at position 132 supporting the normal reaction, and more hydrophobic and smaller residues driving the neomorphic reaction. These findings provide significant insight into the types of mutations that may be accommodated at residue 132 for efficient D2HG production. By determining the catalytic features of IDH1 mutations, we reveal features of driver mutations present in the majority of patients with lower grade gliomas and secondary glioblastomas.
Results
Structural modeling and thermal stability of IDH1 mutations
For the mutations explored in this work, only structures of WT and R132H IDH1 in complex with both substrates (ICT and NADP+, or αKG and NADP+) have been reported (13, 27, 29, 33), although a recent high resolution cryo-EM structure shows R132C IDH1 in complex with NADPH (34). To help inform the structural consequences of R132C, R132G, R100Q, A134D, and H133Q IDH1, these mutants were modeled in previously solved structures of R132H IDH1 in complex with αKG, NADP+, and Ca2+ (Protein Data Bank (PDB) 4KZO (27)) and WT IDH1 in complex with ICT, NADP+, and Ca2+ (PDB 1T0L (33)) using the geometry minimization package in Phenix (35). These models were then aligned to the original structures using PyMOL (36) (Fig. 2). In both models, minimal global changes were identified, consistent with previous structural work on R132H IDH1. The presence of the C3 carboxylate in ICT requires some adjustment of the catalytic residue Tyr-139, whereas less movement of this residue is seen in the models containing αKG.
Figure 2.
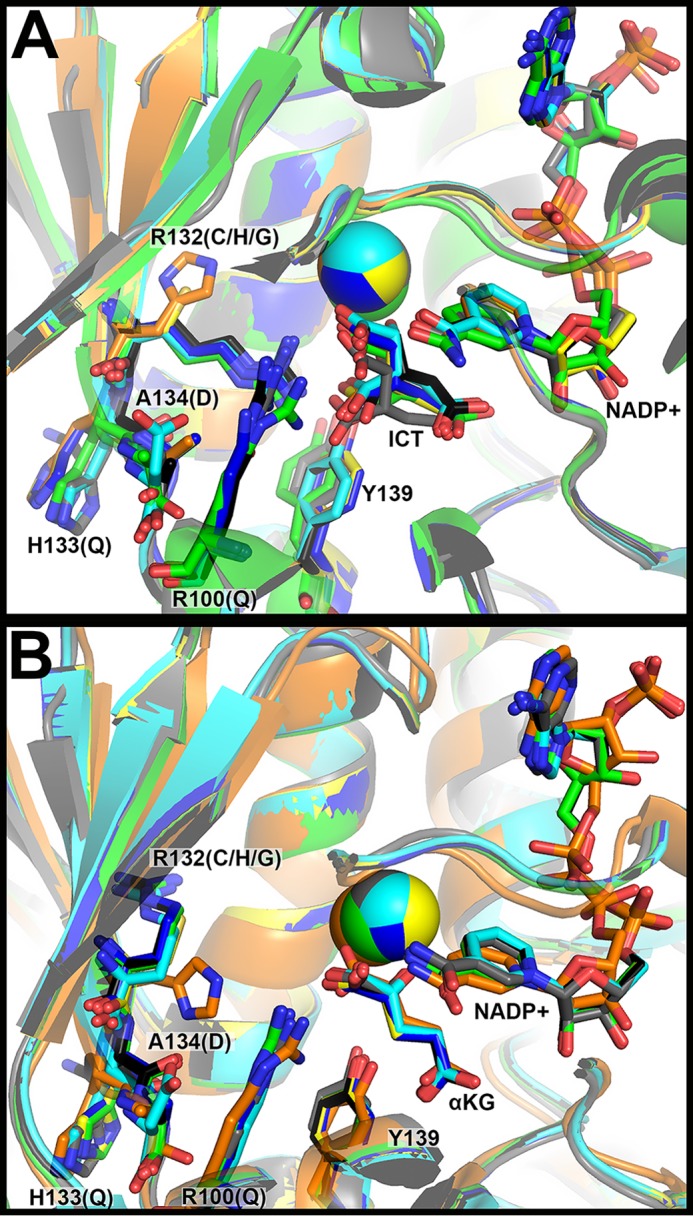
Structural modeling of IDH1 mutations identified in tumors. A, the structure of WT IDH1 complexed with ICT, NADP+, and Ca2+ (PDB 1T0L (33)) and B, R132H IDH1 complexed with αKG, NADP+, and Ca2+ (PDB 4KZO (27)) were used to model additional mutations. In both panels, WT IDH1 is shown in green, A134D in cyan, H133Q in black, R100Q in dark blue, R132H in orange, R132C in yellow, and R132G in gray. Substrates and residues that are mutated are highlighted in stick format, as well as catalytic residue Tyr-139. Ca2+ is shown as a sphere. Ligand restraint generation and optimization of provided cif files were generated using eLBOW in the Phenix software suite (35), and mutations were made using Coot (54). Geometry Minimization (Phenix software suite) (35) was used to regularize geometries of the models, with 500 iterations and 5 macro cycles.
To explore the mechanistic features of these IDH1 mutations identified in tumors, cDNA constructs were generated for heterologous expression and purification in Escherichia coli. WT, R132H, R132C, R132G, R100Q, A134D, and H133Q IDH1 homodimers were expressed and purified to >95% purity (supplemental Fig. S1A). Thermal stability was assessed for each enzyme using circular dichroism to perform thermal shift assays. The melting temperature (Tm) of each of the IDH1 mutants varied little compared with WT IDH1 (supplemental Fig. S2A). R132C IDH1 had the lowest Tm (46.8 °C), but this represents only a 5% change from WT IDH1 (Tm = 49.1 °C). R100Q IDH1 had the highest Tm (51.9 °C), again signifying only a 5% change from WT IDH1.
Efficiency of reactions catalyzed by IDH1 mutants found in tumors
The most common IDH1 mutations found in gliomas are R132H followed by R132C (37). R132G, which has been identified with higher frequency in chondrosarcomas (38, 39), is less frequently seen in gliomas and is a known D2HG producer (30). R100Q IDH1, a long predicted D2HG producer based on the R140Q IDH2 mutation affecting the identical residue, is relatively rare (40). A134D and H133Q IDH1 are rare mutations found in thyroid cancers and are predicted to only be deficient in the normal reaction (31, 40). Steady-state kinetic assays were used to determine the catalytic efficiency of the conversion of ICT to αKG (normal reaction) by monitoring the production of NADPH at A340 nm, or αKG to D2HG (neomorphic reaction) by monitoring the consumption of NADPH, at both 21 and 37 °C. All mutants were deficient in the normal reaction, ranging from a relatively minor 3.5-fold loss of catalytic efficiency (kcat/Km) for H133Q IDH1, whereas the other mutations exhibited more severe ∼300- to 1,340-fold losses in efficiency (Fig. 3, supplemental Fig. S3, Table 1). The observed changes in catalytic efficiency are driven both by decreases in kcat and increases in Km.
Figure 3.

Concentration dependence of the ICT concentration on the observed rate of NADPH production in the normal reaction (37 °C). The determined kobs values were obtained from two different enzyme preparations to ensure reproducibility. The kobs values resulting from each of the two enzyme preparations are distinguished by using either a circle or an × in the plots. The observed rate constants (kobs) were calculated from the linear range of the slopes of plots of concentration versus time using GraphPad Prism software (GraphPad, San Diego, CA). These kobs values were then fit to a hyperbolic equation to generate kcat and Km values, and the standard error listed in Table 1 results from the deviance from these hyperbolic fits is indicated. The determined kobs values were obtained from two different enzyme preparations to ensure reproducibility. Results from assays at 21 °C are shown in supplemental Fig. S3. A, WT IDH1. B, H133Q IDH1. C, A134D IDH1. D, R100Q IDH1. E, R132H IDH1. F, R132C IDH1. G, R132G IDH1.
Table 1.
Kinetic parameters for the normal reaction, conversion of ICT to αKG, catalyzed by IDH1
Values result from fits of kinetic data using two different enzyme preparations. The standard error is determined from the deviance from these hyperbolic fits (Fig. 3, supplemental Fig. S3).
| IDH1 | Relative hydrophobicity of residue 132a | van der Waals volume of side chain at residue 132, Å3b | kcat (37 °C) | Km,ICT (37 °C) | Km,NADP+ (37 °C) | Efficiency, (kcat/Km, ICT, 37 °C) | kcat (21 °C) | Km,ICT (21 °C) | Km,NADP+ (21 °C) | Efficiency, mm−1 s−1 (kcat/Km, ICT, 21 °C) |
|---|---|---|---|---|---|---|---|---|---|---|
| s−1 | mm | mm−1 s−1 | s−1 | mm | mm−1 s−1 | |||||
| WT | −14 (R) | 148 (Arg) | 85 ± 4 | 0.22 ± 0.02 | 0.08 ± 0.03 | 3.9 ± 0.4 × 102 | 11.0 ± 0.4 | 0.015 ± 0.003 | 0.03 ± 0.01 | 7.3 ± 1.3 × 102 |
| H133Q | −14 (Arg) | 148 (Arg) | 45 ± 2 | 0.40 ± 0.08 | 0.16 ± 0.02 | 1.1 ± 0.2 × 102 | 9.4 ± 0.6 | 0.28 ± 0.07 | 0.101 ± 0.008 | 35 ± 9 |
| A134D | −14 (Arg) | 148 (Arg) | 2.3 ± 0.2 | 8 ± 2 | 1.2 ± 0.3 | 0.29 ± 0.08 | 0.200 ± 0.008 | 2.7 ± 0.3 | 0.7 ± 0.1 | 0.074 ± 0.003 |
| R100Q | −14 (Arg) | 148 (Arg) | 5.6 ± 0.4 | 8 ± 2 | 0.18 ± 0.02 | 0.7 ± 0.2 | 1.40 ± 0.06 | 9 ± 1 | 0.070 ± 0.006 | 0.16 ± 0.02 |
| R132H | 8 (His) | 118 (His) | 2.4 ± 0.1 | 4.2 ± 0.6 | 1.6 ± 0.5 | 0.57 ± 0.08 | 0.120 ± 0.006 | 6 ± 1 | 1.0 ± 0.9 | 0.020 ± 0.003 |
| R132C | 49 (Cys) | 86 (Cys) | 4.4 ± 0.1 | 8.2 ± 0.8 | 0.75 ± 0.07 | 0.54 ± 0.05 | 1.61 ± 0.08 | 5.3 ± 0.8 | 0.58 ± 0.08 | 0.30 ± 0.05 |
| R132G | 0 (Gly) | 48 (Gly) | 9.3 ± 0.6 | 7 ± 1 | 0.067 ± 0.007 | 1.3 ± 0.2 | 1.0 ± 0.06 | 3.6 ± 0.6 | 0.14 ± 0.02 | 0.28 ± 0.05 |
Mutants varied widely in their relative catalytic efficiency of D2HG production (Fig. 4, supplemental Fig. S4, Table 2). Only rate-saturating concentrations of substrates generated rates of D2HG production above the signal-to-noise threshold for A134D IDH1 and H133Q IDH1. Thus only upper limits of kcat values are reported as kobs (Fig. 4, B and D) because Km values could not be obtained. R132G IDH1 is the most efficient producer of D2HG (∼125-fold more efficient than WT IDH1), driven primarily by low Km values but also by a high kcat. R132C and R132H IDH1 are ranked next in catalytic efficiency, with a low Km value reported for R132C IDH1 (Table 2). This suggests that production of D2HG in tumors by R132G and R132C IDH1 may be more significant than R132H IDH1 when cytosolic concentration of αKG is considered. This trend is supported by D2HG measurements in glioma tissue (30). Due to its high Km, R100Q IDH1 was one of the least efficient producers of D2HG (Table 2).
Figure 4.

Concentration dependence of αKG concentration on the observed rate of NADPH depletion in the neomorphic reaction (37 °C). The determined kobs values were obtained from two different enzyme preparations to ensure reproducibility. The kobs values resulting from each of the two enzyme preparations are distinguished by using either a circle or an × in the plots. The observed rate constants (kobs) were calculated from the linear range of the slopes of plots of concentration versus time using GraphPad Prism software (GraphPad). These kobs values were then fit to a hyperbolic equation to generate kcat and Km values, and the S.E. results from the deviance from these hyperbolic fits is indicated. Km values and efficiency are in terms of [αKG]. Due to limits of detection, Km values could not be obtained for low efficiency IDH1 enzymes because only saturating kobs rates could be detected. In this case, kobs rates are reported, which approximate kcat rates. Results from assays at 21 °C are shown in supplemental Fig. S4. A, WT IDH1. B, H133Q IDH1. D, R100Q IDH1. E, R132H IDH1. F, R132C IDH1. G, R132G IDH1.
Table 2.
Kinetic parameters for the neomorphic reaction, conversion of αKG to D2HG, catalyzed by IDH1
Values result from fits of kinetic data using two different enzyme preparations. The standard error is determined from the deviance from these hyperbolic fits (Fig. 4, supplemental Fig. S4).
| IDH1 | Relative hydrophobicity of residue 132a | van der Waals volume of side chain at residue 132, Å3b | kcat (37 °C) | Km,αKG (37 °C) | Km,NADPH (37 °C) | Efficiency (kcat/Km,αKG, 37 °C) | kcat, s−1 (21 °C) | Km,αKG (21 °C) | Km,NADPH (21 °C) | Efficiency (kcat/Km,αKG, 21 °C) |
|---|---|---|---|---|---|---|---|---|---|---|
| s−1 | mm | mm−1 s−1 | s−1 | mm | mm−1 s−1 | |||||
| WT | −14 (Arg) | 148 (Arg) | 0.019 ± 0.001 | 0.5 ± 0.3 | ≤ 0.010 | 0.04 ± 0.02 | ≤ 0.017 | NDc | ND | ND |
| H133Q | −14 (Arg) | 148 (Arg) | ≤ 0.016 | ND | ND | ND | ≤ 0.034 | ND | ND | ND |
| A134D | −14 (Arg) | 148 (Arg) | ≤ 0.019 | ND | ND | ND | ≤ 0.020 | ND | ND | ND |
| R100Q | −14 (Arg) | 148 (Arg) | 0.34 ± 0.02 | 12 ± 2 | 0.005 ± 0.003 | 0.028 ± 0.005 | 0.128 ± 0.006 | 10 ± 1 | ≤ 0.0025 | 0.013 ± 0.001 |
| R132H | 8 (His) | 118 (His) | 4.2 ± 0.3 | 1.1 ± 0.3 | ≤ 0.025 | 3.8 ± 0.9 | 0.43 ± 0.04 | 1.8 ± 0.6 | ≤ 0.005 | 0.24 ± 0.08 |
| R132C | 49 (Cys) | 86 (Cys) | 1.60 ± 0.07 | 0.36 ± 0.05 | 0.010 ± 0.009 | 4.4 ± 0.6 | 0.84 ± 0.03 | 0.36 ± 0.06 | ≤ 0.025 | 2.3 ± 0.4 |
| R132G | 0 (Gly) | 48 (Gly) | 1.59 ± 0.09 | 0.34 ± 0.08 | <0.025 | 5 ± 1 | 0.45 ± 0.02 | 0.30 ± 0.05 | ≤ 0.025 | 1.5 ± 0.2 |
GC/MS analysis confirms D2HG production by IDH1 tumor mutants
Although the normal reaction is reversible (Fig. 1A), a lower pH and source of CO2 (typically NaHCO3) are required to favor the reverse reaction in vitro, and work by Leonardi et al. (25) have shown that the reverse reaction is deficient in IDH1 mutants. Regardless, we desired to confirm that the less well characterized IDH1 mutants, namely R100Q and R132G, favor D2HG production over ICT when incubated with αKG and NADPH. R132H and R132C IDH1 are well established to produce D2HG (first reported in Ref. 13, but confirmed by many groups). Gas chromatography/mass spectrometry (GC/MS) was used to identify and quantify the amount of D2HG as well as αKG and ICT (not shown) produced in these incubations (Fig. 5) (20). R132G IDH1 gave robust production of D2HG consistent with kinetic data, whereas an incubation with R100Q IDH1 showed D2HG production levels near the lower limit of detection, again consistent with kinetic findings (Figs. 4 and 5). Levels of ICT for both mutants were <0.1 nmol, based on limits of detection. This indicates that NADPH oxidation in the presence of αKG is preferably coupled to D2HG production under these reaction conditions. This also supports previous findings that both mutants generate D2HG in in vitro assays (30). These experiments do not necessarily indicate that the reverse of the normal reaction is ablated, however, as pH < 7 and CO2 are required for this reaction in vitro (25).
Figure 5.
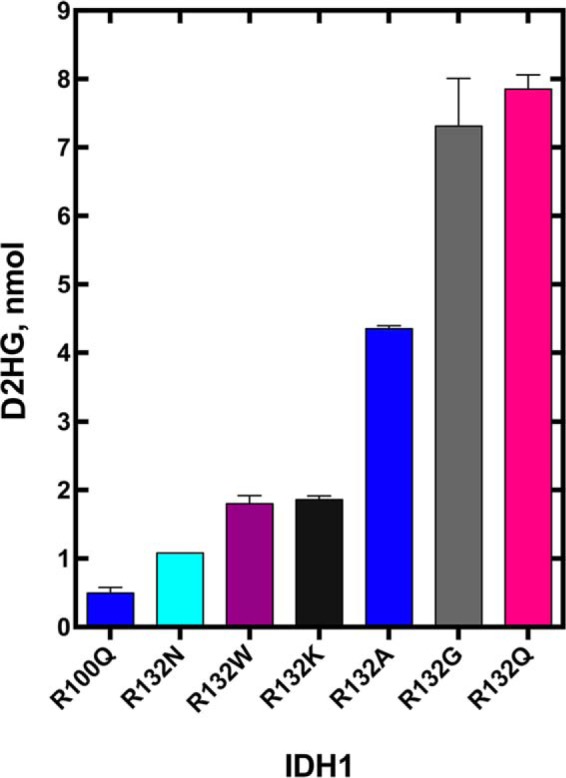
Absolute quantitation of D2HG present in an incubation of IDH1 mutants with αKG and NADPH. Measurements are reported as a calculated mean ± S.E. Only trace amounts of ICT (<0.1 nmol, based on limits of detection) were generated under these experimental conditions, indicating that NADPH oxidation was coupled to D2HG production, rather than ICT production.
Generation of IDH1 mutants engineered to explore mechanistic features of D2HG production
In addition to R132H/R132C/R132G IDH1, several rarer IDH1 mutations in gliomas have been identified, including R132S/R132L/R132V IDH1 (5, 6, 41, 42). In vitro kinetic assays have shown that R132S and R132L IDH1 catalyze production of D2HG at rates similar to the more common R132H and R132C IDH1 mutations (13, 25). Similarly, ectopic expression of R132S and R132L IDH1 in HEK293T cells indicate D2HG production levels are comparable with cell lines expressing R132C and R132H IDH1 (30). R132H/R132C/R132G/R132S/R132L/R132V IDH1 all vary in the degree of hydrophobicity at residue 132, and all have a smaller van der Waals volume than the wild-type arginine. Although these clues illuminate interesting mechanistic characteristics of R132H IDH1, the features that allow IDH1 mutants to generate D2HG with varying catalytic efficiency are not fully clear.
We designed several IDH1 mutations to serve as tools to probe the limits of hydrophobicity (43) and van der Waals volume (44) at residue 132 that support D2HG production. R132A IDH1 is truly an engineered mutation, as to our knowledge it has not been identified in tumors. This residue serves as an example of a more hydrophobic and smaller residue at position 132, similar to R132G IDH1. R132A IDH1 has been shown to be deficient in the normal reaction (29), but its ability to catalyze the neomorphic reaction has not yet been explored. R132N IDH1 also has not been identified in tumors to date. Asparagine has a much smaller van der Waals volume than arginine, although the ranked polarities of these two amino acids are similar. R132Q IDH1 plays an important role in driving chondrosarcomas and a small number of gliomas, and mouse mR132Q IDH1 generates D2HG about 20-fold more efficiently than human R132H IDH1 in vitro (39, 45). This mutation has the most similar ranking in hydrophobicity as compared with WT, but a smaller van der Waals volume. R132K IDH1 is homologous to R172K IDH2, one of the most common D2HG-producing mutations seen in acute myeloid leukemia (11, 41). However, R132K IDH1 has not been reported in tumors, and the activity of this enzyme has not been assessed. R132K IDH1 is most comparable with WT IDH1 when considering both van der Waals volume and polarity ranking of residue 132. Finally, R132W IDH1 is another example of an engineered mutation in that it has not been identified in tumors. It was selected to represent the most extreme case of a large van der Waals volume coupled with high hydrophobicity.
Structural modeling and thermal stability of engineered IDH1 mutations
Because no crystal structures of these mutants are currently available, each was modeled in a previously solved structure of R132H IDH1 in complex with αKG, NADP+, and Ca2+ (PDB code 4KZO (27)) using the geometry minimization package in Phenix (35) followed by alignment in PyMOL (36) (Fig. 6). Few changes are observed globally or within the active site. The size of the amino acid at position 132 does necessitate some local adjustments to avoid steric hindrance, but overall, changes in the models are minimal.
Figure 6.
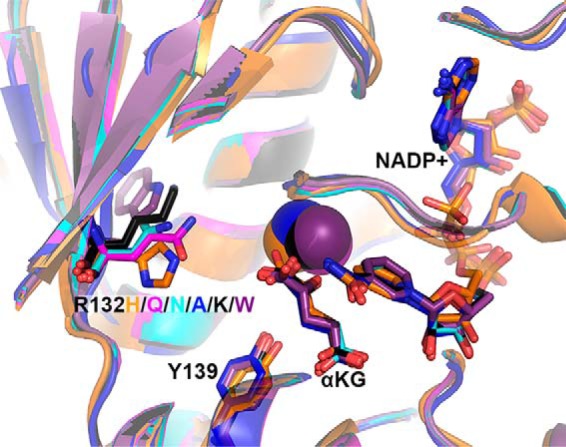
Structural models of experimental IDH1 mutants. The structure of R132H IDH1 complexed with αKG, NADP+, and Ca2+ (PDB 4KZO (27)) was used to model mutations of the tool IDH1 mutations. R132H IDH1 is shown in orange, R132Q in magenta, R132N in cyan, R132A in dark blue, R132K in black, and R132W in purple. Substrates and residues that are mutated are highlighted in stick format, as well as catalytic residue Tyr-139. Ca2+ is shown as a sphere. Ligand restraint generation and optimization of provided cif files were generated using eLBOW in the Phenix software suite (35), and mutations were made using Coot (54). Geometry Minimization (Phenix software suite) (35) was used to regularize geometries of the models, with 500 iterations and 5 macro cycles.
All five IDH1 mutants were successfully heterologously expressed and purified to >95% purity (supplemental Fig. S1B). Thermal stability was assessed using circular dichroism in thermal shift assays. Again, minimal changes were observed in Tm (supplemental Fig. S2B). R132K IDH1 had the highest Tm (49.8 °C), which varied from WT IDH1 by only 2%.
Kinetic analysis of engineered IDH1 mutants
The catalytic efficiency of the normal reaction was measured for all mutants, and efficiencies were plotted against relative hydrophobicity according to Monera et al. (43) (Fig. 7A), and against van der Waals volume (44) (Fig. 7B). All mutants were significantly deficient in converting ICT to αKG, driven both by a decrease in kcat as well as an increase in Km (Table 3, supplemental Fig. S5). Two IDH1 mutations maintained moderate oxidative decarboxylation activity; R132Q and R132K IDH1 had 33- and 56-fold losses of αKG production efficiency relative to WT IDH1, respectively. All other mutations had ≥220-fold decreases in catalytic efficiency.
Figure 7.

Comparisons of catalytic efficiency by IDH1 with mutations at residue 132. The observed rate constants (kobs) were calculated from the linear range of the slopes of plots of concentration versus time, and then fit to a hyperbolic equation to generate kcat and Km values. All experiments were performed at 37 °C. These catalytic parameters result from fits of kinetic data resulting from two different enzyme preparations to ensure reproducibility. A, relative catalytic efficiencies (kcat/Km) of the conversion of ICT to αKG using Km values for ICT are plotted against relative hydrophobicity (43). B, relative catalytic efficiencies (kcat/Km) of the conversion of ICT to αKG using Km values for ICT are plotted against van der Waals volume (44). C, relative catalytic efficiencies (kcat/Km) of the conversion of αKG to D2HG using Km values for αKG are plotted against relative hydrophobicity (43). D, relative catalytic efficiencies (kcat/Km) of the conversion of αKG to D2HG using Km values for αKG are plotted against van der Waals volume (44).
Table 3.
Kinetic parameters for the normal reaction, conversion of ICT to αKG, catalyzed by IDH1
Values result from fits of kinetic data using two different enzyme preparations. The standard error is determined from the deviance from these hyperbolic fits (supplemental Fig. S5).
| IDH1 | Relative hydrophobicity of residue 132a | van der Waals volume of side chain at residue 132, Å3 b | kcat (37 °C) | Km,ICT (37 °C) | Efficiency (kcat/Km,ICT, 37 °C) |
|---|---|---|---|---|---|
| s−1 | mm | mm−1s−1 | |||
| R132W | 97 (Trp) | 163 (Trp) | 1.21 ± 0.08 | 3.6 ± 0.6 | 0.34 ± 0.06 |
| R132A | 41 (Ala) | 67 (Ala) | 10.4 ± 0.2 | 5.7 ± 0.4 | 1.8 ± 0.1 |
| R132Q | −10 (Gln) | 114 (Gln) | 9.2 ± 0.3 | 0.8 ± 0.2 | 12 ± 3 |
| R132K | −23 (Lys) | 135 (Lys) | 7.2 ± 0.4 | 1.1 ± 0.2 | 7 ± 1 |
| R132N | −28 (Asn) | 96 (Asn) | 0.047 ± 0.001 | 1.5 ± 0.1 | 0.031 ± 0.008 |
IDH1 mutants were also incubated with αKG and NADPH to measure presumptive D2HG production efficiency (Fig. 7, Table 4, supplemental Fig. S6). R132Q IDH1 was the most efficient D2HG producer of the mutants explored in this work, with 4-fold higher efficiency than the next most efficient mutant, R132G IDH1. There was a notable decrease in efficiency in all other mutants, with R132A IDH1 having similar catalytic efficiencies as R132G/R132C/R132H IDH1. R132N/R132K/R132W and WT IDH1 were all very poor at producing D2HG. The severely deficient catalytic efficiency seen in R132N IDH1 was primarily driven by a very high Km value (Table 4). This suggests that like R100Q IDH1, D2HG production by R132N IDH1 may not be physiologically relevant when the cytosolic concentration of αKG is considered. Relative efficiencies of the other mutants were driven both by changes in kcat and Km, with a low Km value driving R132A IDH1 production (Table 4).
Table 4.
Kinetic parameters for the neomorphic reaction, conversion of αKG to D2HG, catalyzed by IDH1
Values result from fits of kinetic data using two different enzyme preparations. The standard error is determined from the deviance from these hyperbolic fits (supplemental Fig. S6).
| IDH1 | Relative hydrophobicity of residue 132a | van der Waals volume of side chain at residue 132, Å3 b | kcat (37 °C) | Km,αKG (37 °C) | Efficiency (kcat/Km,αKG, 37 °C) |
|---|---|---|---|---|---|
| s−1 | mm | mm−1s−1 | |||
| R132W | 97 (Trp) | 163 (Trp) | 0.54 ± 0.01 | 0.82 ± 0.08 | 0.659 ± 0.007 |
| R132A | 41 (Ala) | 67 (Ala) | 0.37 ± 0.01 | 0.11 ± 0.02 | 3.4 ± 0.6 |
| R132Q | −10 (Gln) | 114 (Gln) | 4.7 ± 0.2 | 0.26 ± 0.04 | 18 ± 3 |
| R132K | −23 (Lys) | 135 (Lys) | 0.57 ± 0.02 | 0.61 ± 0.07 | 0.9 ± 0.1 |
| R132N | −28 (Asn) | 96 (Asn) | 0.79 ± 0.06 | 10 ± 2 | 0.08 ± 0.02 |
GC/MS analysis confirms D2HG production by engineered IDH1 mutants
To confirm that an incubation of the engineered IDH1 mutants with αKG and NADPH favors D2HG production rather than the reverse of the normal reaction (i.e. ICT production), GC/MS was used to quantify levels of D2HG, ICT, and αKG of the engineered IDH1 mutants. Measured amounts of D2HG were as expected under the incubation lengths at experimentally measured kinetic efficiency. R132Q and R132G IDH1 generated the highest levels of D2HG (Fig. 5), followed by R132A IDH1. Again, levels of ICT were difficult to measure due to their very low concentrations (<0.1 nmol, based on limits of detection). This suggests that NADPH oxidation is coupled primarily to D2HG production, rather than ICT, under these experimental conditions.
Discussion
Here we report the first in-depth, simultaneous catalytic characterization of 11 IDH1 mutations and WT IDH1, including mutations identified in tumors (R132H/R132C/R132G/R132Q, R100Q, A134D, and H133Q IDH1) and additional mutations (R132A/R132K/R132N/R132W IDH1) designed to measure the effects of hydrophobicity (43) and van der Waals volume (44) at residue 132 on catalytic efficiency. To date, a relatively wide range of catalytic rates have been reported for WT and mutant IDH1. For WT IDH1, kcat values for the normal reaction typically range from ∼9 to 12 s−1 (26, 27, 29) at ambient temperature. This is in good agreement with our findings (Table 1), although there are reports of much higher rates (13, 15). Km values for both ICT and NADP+ typically range from 5 to 65 μm at ambient temperature (13, 15, 25–27, 29), again consistent with our values (Table 1). Plasma levels of αKG have been reported to be ∼23 μm (46), which is in line with measured Km values.
H133Q IDH1 displays a relatively minor change in catalytic efficiency for αKG production compared with WT IDH1, and D2HG production was extremely slow, indicating that this may be a passenger mutation (Tables 1 and 2). R100Q IDH1 shows drastic increases in Km values both for ICT and αKG, resulting in significant decreases in catalytic efficiency for both reactions studied (Tables 1 and 2). This was surprising because the homologous mutation in IDH2, R140Q, is the most common IDH mutation found in AML (47). However, kinetic characterizations of IDH2 mutants are limited (28, 48). Kinetic and structural comparisons for R100Q IDH1 and R140Q IDH2 will be important for characterizing any mechanistic differences between these homologous mutations. Currently, crystal structures of R140Q IDH2 are limited to complexes with inhibitors (49). R100Q IDH1 has been characterized as a D2HG-producer, but our data suggests this mutant does so only weakly.
Similarly, R132K IDH1 is homologous to R172K IDH2, the second most common IDH2 mutation identified in AML (50). Thus the relative catalytic inefficiency of D2HG production by R132K IDH1 (Table 4) was also surprising. Kinetic and structural analysis of R132K IDH1 and R172K IDH2 will also be critical for understanding any functional differences between these homologous mutations. Nearly negligible in vitro D2HG production by R132K and R100Q IDH1 may explain why these mutations are rare (or not identified) in gliomas, despite being frequently observed as R172K and R140Q IDH2 in AML.
As noted, Km values for D2HG production for some IDH1 mutants are higher than physiologically relevant αKG concentrations. However, IDH1 mutations are found heterozygously in tumors, and a caveat to this work in that mutant IDH1 homodimers were studied. For IDH1 mutant/WT heterodimers, local concentration of αKG may be much higher due to production of this metabolite at the WT IDH1 monomer, particularly if substrate channeling occurs. Furthermore, Km values may be lower overall due to favorable substrate binding at the WT monomer of the heterodimer. Ward et al. (28) have shown that D2HG production by IDH1 mutations in cells is increased if WT IDH1 activity is retained. However, in cases of very high Km values such as those observed for R100Q IDH1, it is possible that the reaction measured is not physiologically relevant, even in heterodimeric form. Future work includes exploring these mutants as heterodimers using two unique tags and sequential affinity chromatography as described by others (25, 26), although this method does not necessarily preclude the possibility of interconversion of heterodimers and homodimers.
Importantly, Pusch and colleagues (30) have quantified D2HG production in cells and in tumor tissue by R132G/R132C/R132H IDH1. In both cases, the highest levels of D2HG in tumors were associated with R132G IDH1 mutations, followed by R132C IDH1 and R132H IDH1 (30). R100Q IDH1 had very low D2HG production in cells (30). These trends are recapitulated well in our in vitro studies (Table 2). The in vitro enzyme assays by Pusch et al. (30) yielded the highest Km,αKG values for R100Q IDH1, followed by R132H, R132G, and R132C IDH1. Again, our findings well support these trends, and our kcat values allow the first comparisons of these mutations in catalytic efficiency (Table 2). Others have also performed kinetic assays with R132C, R132H, and/or R132G IDH1, although kcat and Km parameters have only been reported for R132C and R132H IDH1. Catalytic efficiencies of D2HG production for R132H IDH1 homodimers range from ∼0.47 to 0.7 mm−1 s−1 (with one report of >1,000 mm−1 s−1), and an efficiency of 8.2 mm−1 s−1 for R132C IDH1, although a much faster kcat and higher Km were reported (13, 15, 27, 29). Although efficiencies were not measured, more D2HG was produced by R132C and R132G IDH1 than by R132H IDH1 (13, 27, 29, 51). Overall, of the kinetic parameters characterizing a limited number of IDH1 mutations, our findings are generally in agreement with reported values.
Pusch et al. (30) also find that D2HG production levels are inversely related to glioma mutation frequency. Our findings support this observation; the relatively rare R132G and R132Q IDH1 mutants were much more efficient producers of D2HG than the common R132C and R132H IDH1 mutants. However, R132C and R132G are more common than R132H IDH1 in chondrosarcomas and intrahepatic cholangiocarcinomas (38, 39). We also show that nearly negligible producers of D2HG (H133Q, A134D, and R100Q IDH1) are also rare in tumors.
To date, only structures of human R132H and WT IDH1 in complex with both substrates have been solved (13, 27, 29, 33), so computational models of the mutations explored in this work were generated. There are significant limitations in the information that can be inferred from our structural models, which in general we find to be somewhat inconclusive. Six possible hydrogen bonds (defined as interatomic distances ranging from 2.5 to 3.2 Å) are seen between αKG and non-solvent atoms for WT and R100Q IDH1, whereas 7 and 8 hydrogen bonds can be observed for H133Q and R132C IDH1, and A134D, R132H, and R132G IDH1, respectively. Thus to some degree, more hydrogen bonds are observed for the more efficient D2HG producers. However, there are definite exceptions to this rule, such as R132C and A134D IDH1. As less obvious patterns were observed among normal reaction efficiency among the mutants, it is difficult to infer trends. Twelve possible hydrogen bonds were observed between ICT and non-solvent atoms for WT IDH1, followed by 11 for H133Q IDH1, 10 for R132G IDH1, 9 for A134D IDH1, 8 for R100Q IDH1, 7 for R132C IDH1, and 6 for R132H IDH1. In general, we find these mutations affect Km values for ICT and αKG more than Km values for NADP+ and NADPH. This is not surprising as most of the mutations explored in this work are associated with coordination of ICT or αKG rather than NADP+ or NADPH. Crystal structures of these mutants will help clarify the mechanisms of this observation.
Models of the mutations designed to test attributes of the amino acid at position 132 (R132Q/R132N/R132A/R132K/R132W IDH1) reveal that there is space for a larger residue at position 132 when oriented away from αKG. This indicates that a mutation like R132W IDH1 could still support substrate binding. Indeed, inefficient yet measurable D2HG production was observed for this mutant. In general, no notable changes were identified in αKG binding or orientation of the catalytic residue Tyr-139. In this model, 7 possible hydrogen bonds are observed between αKG and non-solvent atoms for R132H IDH1, 5 possible hydrogen bonds are observed for R132Q, R132K, R132W, and R132N IDH1, and 4 possible hydrogen bonds are observed for R132A IDH1. Obtaining crystal structures for the mutants explored in this work will be required to successfully define structure/function relationships among the IDH1 mutants.
The Arg-132 IDH1 mutations showed a trend with respect to hydrophobicity for the normal reaction (Fig. 7A). Amino acid residues lysine, arginine, and glutamine rank very closely in relative hydrophobicity (values of −23, −14, and −10 for Lys, Arg, and Gln, respectively (43)), and mutants containing these residues at position 132 had the most efficient rates of ICT oxidative decarboxylation (Tables 1 and 3). This maintained catalytic efficiency is also likely driven by similar van der Waals volume and shape of these residues. Inconsistent with these trends, however, is R132N IDH1. With a hydrophobicity value of −28 (43) as well as similar albeit smaller van der Waals volume, this mutant is surprisingly inefficient. This inefficiency is driven by a very slow kcat, which is the slowest measurable rate in this work. In contrast, R132N IDH1 had a Km similar to the other designed mutants explored. It is possible this mutation causes a shift of the catalytic residue Tyr-139, or simply is unable to orient ICT adequately. A structure of this mutant will be important to clarify this inefficiency, and moderate Km values suggest a ternary complex could theoretically be obtained. Without exception, rates of the normal reaction drop drastically with residues more hydrophobic than R132Q IDH1 (Fig. 7A). All of these mutations, except R132H and R132W IDH1, also have a smaller van der Waals volume than lysine, arginine, and glutamine. This implies amino acid size also is important for supporting ICT oxidative decarboxylation. However, employing volume as a surrogate parameter for size discounts the overall shape of the residue. For example, despite having similar volumes, the ring structure of the histidine and the long side chain of lysine are quite different in shape. Overall, polar and larger (and perhaps particularly longer) side chains, such as lysine and glutamine support the normal reaction. This is unsurprising due to similarity in structural features of arginine, found in WT IDH1.
Trends with respect to the neomorphic reaction are less clear, but may be driven more by relative hydrophobicity than van der Waals volume (Fig. 7, C and D). The more hydrophobic residues, short of tryptophan, are associated with more efficient rates of D2HG production, although the abrupt change in efficiency between R132Q and WT IDH1 is surprising. Additionally, more efficient rates of D2HG production are seen with smaller van der Waals volumes. However, notable exceptions include R132N, R132Q, and R132H IDH1. R132N IDH1 is drastically inefficient, with the high Km value resulting in poor catalytic efficiency of this mutant. R132Q and R132H IDH1 are efficient, but have larger van der Waals volumes than most of the efficient D2HG producers. Overall, converse to trends seen for the normal reaction, relatively hydrophobic and small residues at position 132 favor the neomorphic reaction. Expansion of this work to include additional mutants will be valuable.
R132Q IDH1 is the only mutant studied in this work that was relatively efficient at both αKG and D2HG production, driven primarily by fast kcat rates in both cases (Tables 3 and 4). It will be interesting to determine whether conversion of αKG to ICT (reversible normal reaction) is also relatively efficient under conditions of lower pH and a source of CO2, and how catalytic rates compare between R132Q IDH1 mutant homodimers and WT/R132Q IDH1 heterodimers. It is not yet clear if R132Q IDH1 is a driver of tumorigenesis, but mouse models indicate that expression of this mutation is sufficient to cause enchondromatosis (39).
Overall, our results represent the first large-scale kinetic analysis of a wide variety of IDH1 mutations. These mutations include several identified in tumors and others we designed to clarify catalytic features. We show that most IDH1 mutations are uniformly deficient in the conversion of ICT to αKG, and show wide variation in the ability to convert αKG to D2HG, varying up to 640-fold in catalytic efficiency. This work provides critical guidance in clarifying driver versus passenger IDH1 mutations, and mutations with oncogenic versus tumor suppressive properties.
Experimental procedures
Materials
Dithiothreitol, isopropyl 1-thio-β-d-galactopyranoside, Triton X-100, α-ketoglutarate acid sodium salt, dl-isocitric acid trisodium salt hydrate, and MgCl2 were purchased from Fisher Scientific (Hampton, NH). β-Mercaptoethanol was purchased from MP Biomedicals (Santa Ana, CA). β-Nicotinamide adenine dinucleotide phosphate-reduced tetrasodium salt and β-nicotinamide adenine dinucleotide phosphate disodium salt were purchased from EMD Millipore (Darmstadt, Germany). cOmplete protease inhibitor mixture tablets were purchased from Roche Applied Science.
Plasmid mutagenesis
All IDH1 constructs are in a pET-28b vector containing an N-terminal hexahistidine tag. Site-directed mutagenesis (Kapa Biosciences, Wilmington, MA) was used to generate R132C (forward primer, 5′-GGTTAAACCGATCATTATTGGTTGCCATGCCTATGGTGATCAGTATC, reverse primer, 5′-GATACTGATCACCATAGGCATGGCAACCAATAATGATCGGTTTAACC); R100Q (forward primer, 5′-GCCCGAATGGCACCATTCAGAATATTCTGGG, reverse primer, 5′-CCCAGAATATTCTGAATGGTGCCATTCGGGC); A134D (forward primer, 5′-GGTCGTCATGATTATGGTGATCAGTATCG, reverse primer, 5′-CGATACTGATCACCATAATCATGACGACC); H133Q (forward primer, 5′-AACCGATCATTATTGGTCGTCAGGCCTATGGTGATC, reverse primer, 5′-GATCACCATAGGCCTGACGACCAATAATGATCGGTT); R132G (forward primer, 5′-AACCGATCATTATTGGTGGTCATGCCTATGGTGATC, reverse primer, 5′-GATCACCATAGGCATGACCACCAATAATGATCGGTT); R132A (forward primer, 5′-GGTTAAACCGATCATTATTGGTGCGCATGCCTATGGTGATCAGTATC, reverse primer, 5′-GATACTGATCACCATAGGCATGCGCACCAATAATGATCGGTTTAACC); R132Q (forward primer, 5′-GTTAAACCGATCATTATTGGTCAGCATGCCTATGGTGATCAGTATC, reverse primer, 5′-GATACTGATCACCATAGGCATGCTGACCAATAATGATCGGTTTAAC); R132K (forward primer, 5′-GGGTTAAACCGATCATTATTGGTAAACATGCCTATGGTGATCAGTATCG, reverse primer, 5′-CGATACTGATCACCATAGGCATGTTTACCAATAATGATCGGTTTAACCC); R132W (forward primer, 5′-GGTTAAACCGATCATTATTGGTTGGCATGCCTATGGTGATCAGTATC, reverse primer, 5′-GATACTGATCACCATAGGCATGCCAACCAATAATGATCGGTTTAACC); and R132N (forward primer, 5′-CGATACTGATCACCATAGGCATGGTTACCAATAATGATCGGTTTAACCC, reverse primer, 5′-GGGTTAAACCGATCATTATTGGTAACCATGCCTATGGTGATCAGTATCG). All constructs were sequenced to confirm accuracy.
Protein purification
The following conditions were used for both WT and mutant IDH1 homodimer expression and purification. BL21 gold (DE3) cells transformed with the IDH1 construct were incubated in 0.5–2 liters of terrific broth supplemented with 30 μg/ml of kanamycin at 37 °C, 200 rpm until an A600 of 1.0–1.2 was reached. The temperature was reduced to ambient and expression was induced with 1 mm isopropyl 1-thio-β-d-galactopyranoside (final concentration) followed by ∼18–20-h incubation at ambient temperature at 130 rpm. Cell pellets were harvested and resuspended in lysis buffer (20 mm Tris, pH 7.5, at 4 °C, 500 mm NaCl, 0.1% Triton X-100, and a protease inhibitor tablet). Following cell lysis via sonication, lysate was collected after separation via centrifugation at 12,000 rpm for 1 h. The lysate was loaded on to a pre-equilibrated nickel-nitrilotriacetic acid nickel affinity column (Qiagen, Valencia, CA), washed with Buffer A (50 mm Tris, pH 7.5, at 4 °C, 500 mm NaCl, and 5 mm β-mercaptoethanol), and eluted in bulk with Buffer B (50 mm Tris, pH 7.5, at 4 °C, 500 mm NaCl, 500 mm imidazole, pH 7.5, and 5 mm β-mercaptoethanol). The elution was concentrated and dialyzed in storage buffer (50 mm Tris, pH 7.5, at 4 °C, 100 mm NaCl, 20% glycerol, and 1 mm dithiothreitol). SDS-PAGE (stain-free 4–12% gels, Bio-Rad Laboratories) was used to confirm >95% protein purity. Purified IDH1 was concentrated, the concentration was determined using a calculated molar extinction coefficient of 64,143 cm−1 m−1 at 280 nm, flash frozen in liquid nitrogen, and stored at −80 °C. We noted that enzyme preparations of IDH1 lost activity following ≤2 month storage at −80 °C, so all protein was discarded within 2 months of freezing or if activity was noted to decline, whichever occurred first.
Steady-state activity assays
Assays were performed at 21 and/or 37 °C, and two protein preparations were used to obtain rates in all cases. The reactions were performed at 21 °C to allow comparison of values to published results, and reactions at 37 °C were to determine more physiologically relevant rates. Unless otherwise noted, an 8452 diode array spectrophotometer was used (OLIS, Atlanta, GA) for enzymatic assays. Enzyme concentrations were optimized to ensure signal was sufficient and that enzyme concentrations were within the linear range. For conversion of ICT to αKG, a cuvette containing IDH1 assay buffer (50 mm Tris, pH 7.5, at room temperature or 37 °C, assay depending, 150 mm NaCl, 10 mm MgCl2, 1 mm dithiothreitol), and IDH1 (100 nm for H133Q, R100Q, R132C, R132G, R132A, and R132Q IDH1, and 500 nm for A134D, R132H, R132W, R132K, and R132N IDH1) were preincubated for 3 min at the assay temperature. Reactions were initiated by adding NADP+ and ICT, with either saturating NADP+ and titrating ICT to determine Km,ICT, or saturating ICT and titrating NADP+ to determine Km,NADP+. The change in absorbance due to NADPH formation was monitored at 340 nm. For the conversion of αKG to D2HG, a cuvette containing IDH1 assay buffer and IDH1 (500 nm for all mutants, except R132G and R132A IDH1, which were at a concentration of 200 nm) were preincubated for 3 min at the assay temperature. Reactions were initiated by adding NADPH and αKG, with either saturating NADPH and titrating αKG to determine Km,αKG, or saturating αKG and titrating NADPH to determine Km,NADPH. The pH of αKG was adjusted to 7.0 before use. The change in absorbance due to NADPH consumption was monitored at 340 nm. For both reactions, the slope of the linear range of the incubations were calculated and converted to nanomolar NADPH using the molar extinction coefficient for NADPH of 6.22 cm−1 mm−1 to obtain kobs (i.e. nm NADPH/nm enzyme s−1 at each concentration of substrate). Results were fit to hyperbolic plots in GraphPad Prism (GraphPad Software, La Jolla, CA) to estimate kcat and Km mean values ± S.E.
For the WT IDH1 normal oxidation reaction, an RSM-1000 stopped-flow spectrophotometer (OLIS) operated in the absorbance mode was used to increase the number of points within the time frame assayed. Here, 30 nm WT IDH1 (final concentration) was used and measurements were performed in replicates of 3–5, and the other parameters were the same. A DM45 spectrofluoromer (OLIS) was used for NADPH titrations for R100Q, R132H, and R132C IDH1 (50 nm enzyme), and NADP+ titrations for WT IDH1 (3 nm enzyme) after first generating a standard curve for [NADP(H)].
GC/MS analysis
A reaction with 19 μm IDH1 (R100Q, R132G, R132A, R132K, R132N, R132Q, or R132W) was incubated with IDH1 assay buffer, and the reaction was initiated with 240 μm NADPH and 240 μm αKG and monitored at 340 nm. After 100 s, an aliquot of the reaction was removed, quenched with 0.13 m EDTA (final concentration), and this was lyophilized to dryness. These extracts were then redissolved in 50% methanol containing l-norvaline to serve as an internal standard, and then re-dried in a MiVac (SP Scientific, Warminster, PA). A standard 1 mm solution of l-norvaline, αKG, d-2-HG, and dl-ICT was prepared. Standards and incubations were derivatized with O-isobutylhydroxylamine hydrochloride and N-tert-butyldimethylsilyltrifluoroacetamide and analyzed using GC/MS as described in Ref. 52. Measurements were performed in duplicate.
Thermal stability using circular dichroism
IDH1 was diluted to ∼5 μm in a buffer containing 10 mm potassium phosphate at pH 7.5 and 100 mm NaCl. The thermal melt experiment was initiated at 5 °C and the temperature was increased to 70 °C in 1° increments. The IDH1 secondary structure, which is rich in α helices, was monitored via the 222 nm peak, which corresponds to α helicity. Analysis was performed using the Igor software package (WaveMetrics) and was analyzed with a Santoro-Bolen fit (53).
Structural modeling of IDH1 mutations
PDB code 4KZO (27) was used as the model for an IDH1 structure containing αKG and NADP+, and code 1T0L (33) was used for an IDH1 structure containing ICT and NADP+. Ligand-restraint generation and optimization of provided cif files were generated using eLBOW in the Phenix software suite (35), and mutations were made in the PDB file using Coot (54). Geometry Minimization in the Phenix software suite (35) was used to regularize geometries (bond lengths, nonbonded distances, bond angles, dihedral angles, chirality, planarity, and parallelity) of the structural models of IDH1 mutants in complex with the ligands, with 500 maximum iterations and 5 macrocycles.
Author contributions
D. A. M. and A. J. G. performed most of the kinetic assays, and E. R. G., S. L. A., M. A. K., and P. M. also contributed kinetic assays. E. R. G. and P. M. prepared a cDNA construct, and D. A. M. and E. R. G. performed circular dichroism experiments. D. A. S. performed the GC/MS experiments. A. H. aided in protein purifications and kinetic assays. C. D. S. conceived the idea for the project, prepared cDNA constructs, performed the modeling, and wrote the paper. All authors contributed to data analysis and manuscript editing, and approved the final version of the manuscript.
Supplementary Material
Acknowledgments
We thank Charles Rock (St. Jude's Hospital) for providing the WT and R132H IDH1 clones, and Brian Maniaci, Melissa Lokensgard, Peter Suon, and Dr. John Love (SDSU) for assistance with circular dichroism measurements. Finally, we thank Dr. Tom Huxford for critical reading of this manuscript.
This work was supported by National Institute of Health Grants K99 CA187594 (to C. D. S.), R00 CA187594 (to C. D. S.), 5T34 GM008303 (to E. R. G. and M. A. K.), and P30 CA030199 (to D. A. S.), a Summer Undergraduate Research Program Grant from San Diego State University (to M. A. K.), and San Diego State University startup funds (to C. D. S.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains supplemental Figs. S1–S6.
- IDH
- isocitrate dehydrogenase
- ICT
- isocitrate
- αKG
- α-ketoglutarate
- D2HG
- d-2-hydroxyglutarate
- AML
- acute myeloid leukemia.
References
- 1. Warburg O. (1925) Über den Stoffwechsel der Carcinomzelle. Klin. Wochenschr. 4, 534–536 [Google Scholar]
- 2. Warburg O. (1956) Origin of cancer cells. Oncologia 9, 75–83 [PubMed] [Google Scholar]
- 3. Racker E. (1972) Bioenergetics and the problem of tumor growth. Am. Sci. 60, 56–63 [PubMed] [Google Scholar]
- 4. Parsons D. W., Jones S., Zhang X., Lin J. C., Leary R. J., Angenendt P., Mankoo P., Carter H., Siu I. M., Gallia G. L., Olivi A., McLendon R., Rasheed B. A., Keir S., Nikolskaya T., et al. (2008) An integrated genomic analysis of human glioblastoma multiforme. Science 321, 1807–1812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Balss J., Meyer J., Mueller W., Korshunov A., Hartmann C., and von Deimling A. (2008) Analysis of the IDH1 codon 132 mutation in brain tumors. Acta Neuropathol. 116, 597–602 [DOI] [PubMed] [Google Scholar]
- 6. Bleeker F. E., Lamba S., Leenstra S., Troost D., Hulsebos T., Vandertop W. P., Frattini M., Molinari F., Knowles M., Cerrato A., Rodolfo M., Scarpa A., Felicioni L., Buttitta F., Malatesta S., Marchetti A., and Bardelli A. (2009) IDH1 mutations at residue p.R132 (IDH1(R132)) occur frequently in high-grade gliomas but not in other solid tumors. Hum. Mutat. 30, 7–11 [DOI] [PubMed] [Google Scholar]
- 7. Losman J. A., and Kaelin W. G. Jr. (2013) What a difference a hydroxyl makes: mutant IDH, (R)-2-hydroxyglutarate, and cancer. Genes Dev. 27, 836–852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Dang L., Yen K., and Attar E. C. (2016) IDH mutations in cancer and progress toward development of targeted therapeutics. Ann. Oncol. 27, 599–608 [DOI] [PubMed] [Google Scholar]
- 9. Dang L., Jin S., and Su S. M. (2010) IDH mutations in glioma and acute myeloid leukemia. Trends Mol. Med. 16, 387–397 [DOI] [PubMed] [Google Scholar]
- 10. Mardis E. R., Ding L., Dooling D. J., Larson D. E., McLellan M. D., Chen K., Koboldt D. C., Fulton R. S., Delehaunty K. D., McGrath S. D., Fulton L. A., Locke D. P., Magrini V. J., Abbott R. M., Vickery T. L., et al. (2009) Recurring mutations found by sequencing an acute myeloid leukemia genome. N. Engl. J. Med. 361, 1058–1066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Yan H., Parsons D. W., Jin G., McLendon R., Rasheed B. A., Yuan W., Kos I., Batinic-Haberle I., Jones S., Riggins G. J., Friedman H., Friedman A., Reardon D., Herndon J., Kinzler K. W., et al. (2009) IDH1 and IDH2 mutations in gliomas. N. Engl. J. Med. 360, 765–773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zhao S., Lin Y., Xu W., Jiang W., Zha Z., Wang P., Yu W., Li Z., Gong L., Peng Y., Ding J., Lei Q., Guan K. L., and Xiong Y. (2009) Glioma-derived mutations in IDH1 dominantly inhibit IDH1 catalytic activity and induce HIF-1α. Science 324, 261–265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Dang L., White D. W., Gross S., Bennett B. D., Bittinger M. A., Driggers E. M., Fantin V. R., Jang H. G., Jin S., Keenan M. C., Marks K. M., Prins R. M., Ward P. S., Yen K. E., Liau L. M., et al. (2009) Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 462, 739–744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ward P. S., Patel J., Wise D. R., Abdel-Wahab O., Bennett B. D., Coller H. A., Cross J. R., Fantin V. R., Hedvat C. V., Perl A. E., Rabinowitz J. D., Carroll M., Su S. M., Sharp K. A., Levine R. L., and Thompson C. B. (2010) The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting alpha-ketoglutarate to 2-hydroxyglutarate. Cancer Cell 17, 225–234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gross S., Cairns R. A., Minden M. D., Driggers E. M., Bittinger M. A., Jang H. G., Sasaki M., Jin S., Schenkein D. P., Su S. M., Dang L., Fantin V. R., and Mak T. W. (2010) Cancer-associated metabolite 2-hydroxyglutarate accumulates in acute myelogenous leukemia with isocitrate dehydrogenase 1 and 2 mutations. J. Exp. Med. 207, 339–344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Xu W., Yang H., Liu Y., Yang Y., Wang P., Kim S. H., Ito S., Yang C., Wang P., Xiao M. T., Liu L. X., Jiang W. Q., Liu J., Zhang J. Y., Wang B., et al. (2011) Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of α-ketoglutarate-dependent dioxygenases. Cancer Cell 19, 17–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chowdhury R., Yeoh K. K., Tian Y. M., Hillringhaus L., Bagg E. A., Rose N. R., Leung I. K., Li X. S., Woon E. C., Yang M., McDonough M. A., King O. N., Clifton I. J., Klose R. J., Claridge T. D., et al. (2011) The oncometabolite 2-hydroxyglutarate inhibits histone lysine demethylases. EMBO Rep. 12, 463–469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lu C., Ward P. S., Kapoor G. S., Rohle D., Turcan S., Abdel-Wahab O., Edwards C. R., Khanin R., Figueroa M. E., Melnick A., Wellen K. E., O'Rourke D. M., Berger S. L., Chan T. A., Levine R. L., et al. (2012) IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature 483, 474–478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Turcan S., Rohle D., Goenka A., Walsh L. A., Fang F., Yilmaz E., Campos C., Fabius A. W., Lu C., Ward P. S., Thompson C. B., Kaufman A., Guryanova O., Levine R., Heguy A., et al. (2012) IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype. Nature 483, 479–483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Figueroa M. E., Abdel-Wahab O., Lu C., Ward P. S., Patel J., Shih A., Li Y., Bhagwat N., Vasanthakumar A., Fernandez H. F., Tallman M. S., Sun Z., Wolniak K., Peeters J. K., et al. (2010) Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell 18, 553–567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Losman J. A., Looper R. E., Koivunen P., Lee S., Schneider R. K., McMahon C., Cowley G. S., Root D. E., Ebert B. L., and Kaelin W. G. Jr. (2013) (R)-2-Hydroxyglutarate is sufficient to promote leukemogenesis and its effects are reversible. Science 339, 1621–1625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ma S., Jiang B., Deng W., Gu Z. K., Wu F. Z., Li T., Xia Y., Yang H., Ye D., Xiong Y., and Guan K. L. (2015) d-2-Hydroxyglutarate is essential for maintaining oncogenic property of mutant IDH-containing cancer cells but dispensable for cell growth. Oncotarget 6, 8606–8620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Reitman Z. J., Jin G., Karoly E. D., Spasojevic I., Yang J., Kinzler K. W., He Y., Bigner D. D., Vogelstein B., and Yan H. (2011) Profiling the effects of isocitrate dehydrogenase 1 and 2 mutations on the cellular metabolome. Proc. Natl. Acad. Sci. U.S.A. 108, 3270–3275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Oizel K., Gratas C., Nadaradjane A., Oliver L., Vallette F. M., and Pecqueur C. (2015) d-2-Hydroxyglutarate does not mimic all the IDH mutation effects, in particular the reduced etoposide-triggered apoptosis mediated by an alteration in mitochondrial NADH. Cell Death Dis. 6, e1704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Leonardi R., Subramanian C., Jackowski S., and Rock C. O. (2012) Cancer-associated isocitrate dehydrogenase mutations inactivate NADPH-dependent reductive carboxylation. J. Biol. Chem. 287, 14615–14620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Pietrak B., Zhao H., Qi H., Quinn C., Gao E., Boyer J. G., Concha N., Brown K., Duraiswami C., Wooster R., Sweitzer S., and Schwartz B. (2011) A tale of two subunits: how the neomorphic R132H IDH1 mutation enhances production of αHG. Biochemistry 50, 4804–4812 [DOI] [PubMed] [Google Scholar]
- 27. Rendina A. R., Pietrak B., Smallwood A., Zhao H., Qi H., Quinn C., Adams N. D., Concha N., Duraiswami C., Thrall S. H., Sweitzer S., and Schwartz B. (2013) Mutant IDH1 enhances the production of 2-hydroxyglutarate due to its kinetic mechanism. Biochemistry 52, 4563–4577 [DOI] [PubMed] [Google Scholar]
- 28. Ward P. S., Lu C., Cross J. R., Abdel-Wahab O., Levine R. L., Schwartz G. K., and Thompson C. B. (2013) The potential for isocitrate dehydrogenase mutations to produce 2-hydroxyglutarate depends on allele specificity and subcellular compartmentalization. J. Biol. Chem. 288, 3804–3815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Yang B., Zhong C., Peng Y., Lai Z., and Ding J. (2010) Molecular mechanisms of “off-on switch” of activities of human IDH1 by tumor-associated mutation R132H. Cell Res. 20, 1188–1200 [DOI] [PubMed] [Google Scholar]
- 30. Pusch S., Schweizer L., Beck A. C., Lehmler J. M., Weissert S., Balss J., Miller A. K., and von Deimling A. (2014) d-2-Hydroxyglutarate producing neo-enzymatic activity inversely correlates with frequency of the type of isocitrate dehydrogenase 1 mutations found in glioma. Acta Neuropathol. Commun. 2, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hemerly J. P., Bastos A. U., and Cerutti J. M. (2010) Identification of several novel non-p.R132 IDH1 variants in thyroid carcinomas. Eur. J. Endocrinol. 163, 747–755 [DOI] [PubMed] [Google Scholar]
- 32. Zhang C., Moore L. M., Li X., Yung W. K., and Zhang W. (2013) IDH1/2 mutations target a key hallmark of cancer by deregulating cellular metabolism in glioma. Neuro Oncol. 15, 1114–1126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Xu X., Zhao J., Xu Z., Peng B., Huang Q., Arnold E., and Ding J. (2004) Structures of human cytosolic NADP-dependent isocitrate dehydrogenase reveal a novel self-regulatory mechanism of activity. J. Biol. Chem. 279, 33946–33957 [DOI] [PubMed] [Google Scholar]
- 34. Merk A., Bartesaghi A., Banerjee S., Falconieri V., Rao P., Davis M. I., Pragani R., Boxer M. B., Earl L. A., Milne J. L., and Subramaniam S. (2016) Breaking Cryo-EM resolution barriers to facilitate drug discovery. Cell 165, 1698–1707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., et al. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Schrodinger L. (2010) The PyMOL Molecular Graphics System, version 1.5.0.4, Schroedinger, LLC, New York [Google Scholar]
- 37. Ichimura K., Pearson D. M., Kocialkowski S., Bäcklund L. M., Chan R., Jones D. T., and Collins V. P. (2009) IDH1 mutations are present in the majority of common adult gliomas but rare in primary glioblastomas. Neuro Oncol. 11, 341–347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Grassian A. R., Pagliarini R., and Chiang D. Y. (2014) Mutations of isocitrate dehydrogenase 1 and 2 in intrahepatic cholangiocarcinoma. Curr. Opin. Gastroenterol. 30, 295–302 [DOI] [PubMed] [Google Scholar]
- 39. Hirata M., Sasaki M., Cairns R. A., Inoue S., Puviindran V., Li W. Y., Snow B. E., Jones L. D., Wei Q., Sato S., Tang Y. J., Nadesan P., Rockel J., Whetstone H., Poon R., et al. (2015) Mutant IDH is sufficient to initiate enchondromatosis in mice. Proc. Natl. Acad. Sci. U.S.A. 112, 2829–2834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ward P. S., Cross J. R., Lu C., Weigert O., Abel-Wahab O., Levine R. L., Weinstock D. M., Sharp K. A., and Thompson C. B. (2012) Identification of additional IDH mutations associated with oncometabolite R(−)-2-hydroxyglutarate production. Oncogene 31, 2491–2498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hartmann C., Meyer J., Balss J., Capper D., Mueller W., Christians A., Felsberg J., Wolter M., Mawrin C., Wick W., Weller M., Herold-Mende C., Unterberg A., Jeuken J. W., Wesseling P., et al. (2009) Type and frequency of IDH1 and IDH2 mutations are related to astrocytic and oligodendroglial differentiation and age: a study of 1,010 diffuse gliomas. Acta Neuropathol. 118, 469–474 [DOI] [PubMed] [Google Scholar]
- 42. Agarwal S., Sharma M. C., Jha P., Pathak P., Suri V., Sarkar C., Chosdol K., Suri A., Kale S. S., Mahapatra A. K., and Jha P. (2013) Comparative study of IDH1 mutations in gliomas by immunohistochemistry and DNA sequencing. Neuro Oncol. 15, 718–726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Monera O. D., Sereda T. J., Zhou N. E., Kay C. M., and Hodges R. S. (1995) Relationship of sidechain hydrophobicity and α-helical propensity on the stability of the single-stranded amphipathic α-helix. J. Pept. Sci. 1, 319–329 [DOI] [PubMed] [Google Scholar]
- 44. Richards F. M. (1974) The interpretation of protein structures: total volume, group volume distributions and packing density. J. Mol. Biol. 82, 1–14 [DOI] [PubMed] [Google Scholar]
- 45. Liu A., Hou C., Chen H., Zong X., and Zong P. (2016) Genetics and epigenetics of glioblastoma: applications and overall incidence of IDH1 mutation. Front. Oncol. 6, 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Halámková L., Mailloux S., Halámek J., Cooper A. J., and Katz E. (2012) Enzymatic analysis of α-ketoglutaramate: a biomarker for hyperammonemia. Talanta 100, 7–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Marcucci G., Maharry K., Wu Y. Z., Radmacher M. D., Mrózek K., Margeson D., Holland K. B., Whitman S. P., Becker H., Schwind S., Metzeler K. H., Powell B. L., Carter T. H., Kolitz J. E., Wetzler M., et al. (2010) IDH1 and IDH2 gene mutations identify novel molecular subsets within de novo cytogenetically normal acute myeloid leukemia: a Cancer and Leukemia Group B study. J. Clin. Oncol. 28, 2348–2355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. DiNardo C. D., Propert K. J., Loren A. W., Paietta E., Sun Z., Levine R. L., Straley K. S., Yen K., Patel J. P., Agresta S., Abdel-Wahab O., Perl A. E., Litzow M. R., Rowe J. M., Lazarus H. M., et al. (2013) Serum 2-hydroxyglutarate levels predict isocitrate dehydrogenase mutations and clinical outcome in acute myeloid leukemia. Blood 121, 4917–4924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Wang F., Travins J., DeLaBarre B., Penard-Lacronique V., Schalm S., Hansen E., Straley K., Kernytsky A., Liu W., Gliser C., Yang H., Gross S., Artin E., Saada V., Mylonas E., et al. (2013) Targeted inhibition of mutant IDH2 in leukemia cells induces cellular differentiation. Science 340, 622–626 [DOI] [PubMed] [Google Scholar]
- 50. Koszarska M., Bors A., Feczko A., Meggyesi N., Batai A., Csomor J., Adam E., Kozma A., Orban T. I., Lovas N., Sipos A., Karaszi E., Dolgos J., Fekete S., Reichardt J., et al. (2013) Type and location of isocitrate dehydrogenase mutations influence clinical characteristics and disease outcome of acute myeloid leukemia. Leuk. Lymphoma 54, 1028–1035 [DOI] [PubMed] [Google Scholar]
- 51. Jin G., Reitman Z. J., Spasojevic I., Batinic-Haberle I., Yang J., Schmidt-Kittler O., Bigner D. D., and Yan H. (2011) 2-Hydroxyglutarate production, but not dominant negative function, is conferred by glioma-derived NADP-dependent isocitrate dehydrogenase mutations. PLoS One 6, e16812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Ratnikov B., Aza-Blanc P., Ronai Z. A., Smith J. W., Osterman A. L., and Scott D. A. (2015) Glutamate and asparagine cataplerosis underlie glutamine addiction in melanoma. Oncotarget 6, 7379–7389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Santoro M. M., and Bolen D. W. (1988) Unfolding free energy changes determined by the linear extrapolation method: 1. unfolding of phenylmethanesulfonyl α-chymotrypsin using different denaturants. Biochemistry 27, 8063–8068 [DOI] [PubMed] [Google Scholar]
- 54. Emsley P., and Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.