

Abstract
Members of the IL-12 family perform essential functions in immunoregulation by connecting innate and adaptive immunity and are emerging therapeutic targets. They are unique among other interleukins in forming heterodimers that arise from extensive subunit sharing within the family, leading to the production of at least four functionally distinct heterodimers from only five subunits. This raises important questions about how the assembly of IL-12 family members is regulated and controlled in the cell. Here, using cell-biological approaches, we have dissected basic principles that underlie the biogenesis of the founding member of the family, IL-12. Within the native IL-12 heterodimer, composed of IL-12α and IL-12β, IL-12α possesses three intramolecular and one intermolecular disulfide bridges. We show that, in isolation, IL-12α fails to form its native structure but, instead, misfolds, forming incorrect disulfide bonds. Co-expression of its β subunit inhibits misfolding and thus allows secretion of biologically active heterodimeric IL-12. On the basis of these findings, we identified the disulfide bonds in IL-12α that are critical for assembly-induced secretion and biological activity of IL-12 versus misfolding and degradation of IL-12α. Surprisingly, two of the three disulfide bridges in IL-12α are dispensable for IL-12 secretion, stability, and biological activity. Extending our findings, we show that misfolding also occurs for IL-23α, another IL-12 family protein. Our results indicate that assembly-induced folding is key in IL-12 family biogenesis and secretion. The identification of essential disulfide bonds that underlie this process lays the basis for a simplified yet functional IL-12 cytokine.
Keywords: interleukin, protein assembly, protein folding, protein misfolding, protein secretion, protein turnover
Introduction
For proteins of the secretory pathway, folding and assembly have to be coordinated in the endoplasmic reticulum (ER)3 to guarantee the functionality of, in total, one-third of all human proteins (1). This poses a challenge to the cell, as assembly intermediates may already resemble native states of proteins and are thus difficult to discriminate from mature proteins (2).
Different principles are realized in oligomeric proteins to safeguard assembly. Insights into those have, among others, been obtained for proteins of the immune system, where assembly control is particularly critical in quantitative as well as qualitative terms to maintain immune homeostasis. Accordingly, quality control mechanisms have evolved that specifically fulfil the demands of certain immune proteins. For IgG antibodies, domain folding is coupled to heavy chain (HC)/light chain (LC) interaction and thus allows the ER chaperone machinery to efficiently discriminate native tetrameric IgG (HC2LC2) from immature assembly states (3–5). For the oligomeric IgM, pentamerization of HC2LC2 tetramers also has to be assessed. This is particularly challenging, as HC2LC2 tetramers are already well folded (6, 7). Free cysteines within not yet pentamerized IgM underlie this critical quality control step and allow the recovery of HC2LC2 tetramers from the Golgi via the chaperone ERp44 (8, 9). For oligomeric membrane-embedded B cell receptors, assembly within the membrane can be directly scrutinized (10). In the case of an even more complex membrane protein in the immune system, the αβT cell receptor, multiple checkpoints exist that couple folding, membrane integration, and shielding of retention motifs to scrutinize the assembly of, in total, eight polypeptide chains and thus guarantee correct receptor functionality (11–17).
For one important family of proteins in the immune system, the IL-12 family, which comprises IL-12, IL-23, IL-27, and IL-35 (18) and likely even further members (19), the principles of assembly control are not yet understood but are particularly intriguing: each family member is a heterodimer composed of one α and one β subunit. The α subunits are four-helix bundle proteins; β subunits are composed of fibronectin and Ig domains and are related to IL receptors (20, 21). Furthermore, the four heterodimeric family members are made up of only five shared subunits, three α (IL-12α/p35, IL-23α/p19, and IL-27α/p28), and two β subunits (IL-12β/p40 and Ebi3), adding a layer of combinatorial complexity to assembly regulation and control in the cell, in particular because certain immune cells produce all five subunits simultaneously (see e.g. Ref. 22), and the family members perform highly distinct immunoregulatory roles. IL-12 and IL-23 are mostly proinflammatory cytokines, and targeting IL-12 and IL-23 has emerged as an important therapeutic avenue to treat immune-mediated inflammatory diseases (23). In contrast, IL-27 and IL-35 act immunomodulatory or immunosuppressive, respectively (18, 24).
To dissect the molecular principles of IL-12 family assembly, in this work, we focused on the founding member IL-12/p70 (25–27). IL-12 is produced by antigen-presenting cells and can stimulate T cell differentiation into proinflammatory TH1 cells that secrete IFNγ, which further increases TH1 cell differentiation in a positive feedback loop (28, 29). As such, IL-12 is critical for the eradication of intracellular pathogens and has gained interest in stimulating antitumor responses and regulating autoimmunity (18, 24, 30). IL-12 is a disulfide-bridged heterodimeric glycoprotein composed of IL-12α and IL-12β (21, 27). Both subunits need to be expressed simultaneously to secrete bioactive IL-12 (26, 31). Of note, isolated IL-12β can be secreted as a monomer or homodimer (27, 32, 33), whereas isolated IL-12α is retained in cells, and its secretion depends on IL-12β expression (34, 35). Why IL-12α as opposed to IL-12β is retained and how IL-12β induces secretion of IL-12α, and thus biologically active IL-12, has remained unclear but is critical to understand IL-12 family-mediated immune reactions as well as principles governing cellular protein assembly control.
Here we show that, in isolation, IL-12α fails to fold properly, forming non-native disulfide bonds. Misfolding is inhibited by IL-12β, giving rise to the native IL-12 heterodimer. Furthermore, we identify which of the seven cysteine residues in IL-12α contribute to folding and heterodimerization versus misfolding and degradation. Last, we extend our insights to another IL-12 family member, IL-23, which possesses a different α subunit than IL-12 (IL-23α/p19) but shares IL-12β (36). This study thus establishes assembly-induced folding as a general mechanism in IL-12 family biogenesis.
Results
IL-12β releases IL-12α from ER retention
The secretion of IL-12 depends on the expression of both subunits of the IL-12 heterodimer (Fig. 1A), IL-12α and IL-12β (26, 31). It has been shown that IL-12α is retained in the cell in isolation, whereas isolated IL-12β is readily secreted, and its expression is limiting for the secretion of heterodimeric IL-12 (34). In agreement with these findings, our data show that human IL-12α is retained in 293T cells, an established model cell line for studying IL-12 family assembly (34, 36–38), and its secretion is induced by human IL-12β (Fig. 1B). In contrast, IL-12β alone is readily secreted (Fig. 1C). Although IL-12 is a covalent heterodimer (Fig. 1A), mutation of the cysteine residues in IL-12α and IL-12β that form the interchain disulfide bond in the heterodimer to Ser (IL-12αC96S and IL-12βC199S, respectively) neither significantly affected IL-12β-induced secretion of IL-12α (Fig. 1B) (21, 39) nor secretion of IL-12βC199S alone (Fig. 1C).
Figure 1.

IL-12α secretion depends on IL-12β co-expression. A, IL-12 structure. IL-12 is composed of two covalently linked subunits (IL-12α and IL-12β, structure based on PDB code 3HMX). The IL-12α subunit is depicted in gray and the IL-12β subunit in blue. The intermolecular disulfide bond between IL-12α Cys-96 and IL-12β Cys-199 is shown in a yellow CPK representation. B, IL-12α secretion analyzed by immunoblotting. IL-12α is retained in the cell when expressed in isolation (L, lysate), and co-expression of IL-12β induces its secretion (M, medium) independent of the presence or absence of the cysteine residues that form the IL-12α-IL-12β disulfide bridge. 1% lysate or medium was applied to the gel and blotted with the indicated antibodies. Hsc70 served as a loading control. MW, molecular weight. C, IL-12β secretion analyzed by immunoblotting. The same as in B, except isolated IL-12β and its C199S mutant were analyzed. D, IL-12α secretion and glycan modification by metabolic labeling. Cells were transfected with IL-12α or empty pSVL vector (mock) and metabolically labeled for 1 h before they were chased for the indicated times. Immunoprecipitations (IP) from cell lysates or media were performed with IL-12α antibodies and samples were treated with Endo H (H) or PNGase F (F) where indicated. E, IL-12β secretion and glycan modification by metabolic labeling. The same as in D, except IL-12β was analyzed. F, IL-12 secretion and glycan modification upon co-expression of IL-12α and IL-12β by metabolic labeling. The same as in D, except IL-12α was co-transfected with IL-12β.
To define the site of intracellular retention of IL-12α and thus assembly with IL-12β, we performed metabolic labeling experiments coupled to immunoprecipitation and enzymatic deglycosylation. In these experiments, the entire pool of IL-12α expressed in isolation was Endo H-sensitive, indicative of its retention in the ER (Fig. 1D). IL-12β became partially Endo H-resistant upon secretion but remained PNGase F-sensitive, as expected for a protein transported through the Golgi (Fig. 1E). In agreement with previous studies (33, 34, 40) and our immunoblotting data (Fig. 1B), co-expression of IL-12β induced secretion of IL-12α concomitant with it becoming Endo H-resistant, as expected for bona fide secretion (Fig. 1F). Of note, upon co-expression of both subunits, IL-12α could be co-immunoprecipitated with IL-12β in both its Endo H-sensitive as well as Endo H-resistant forms from cell lysates (Fig. 1F). Extending earlier studies (33, 34, 40), our data show that IL-12 assembly occurs in the ER and that passage time through the Golgi is long enough to also allow co-immunoprecipitation of Endo H-resistant IL-12α with IL-12β.
Misfolding of isolated IL-12α is inhibited by IL-12β
To assess the molecular basis for ER retention of IL-12α, we analyzed its redox status in the cell as an indicator of its folding state (41). IL-12α possesses seven cysteines, six of which form three intramolecular disulfide bonds in the native state, and one forms an intermolecular disulfide bridge with IL-12β (21). Analysis of the redox status of IL-12α by non-reducing SDS-PAGE coupled to immunoblotting revealed multiple redox species for IL-12α expressed in isolation that collapsed into a single species upon treatment of cells with the reducing agent DTT (Fig. 2A). These species included one with a higher electrophoretic mobility than the reduced protein, indicating disulfide bridge formation, a species with the same mobility as the reduced protein, and a prominent species at approximately the size of an IL-12α dimer as well as several higher-molecular-weight forms (Fig. 2A). Under the IL-12α/IL-12β expression ratio of our experiments, co-expression of IL-12β led to secretion of the covalent IL-12α-IL-12β heterodimer concomitant with the disappearance of all intracellular redox species except for the heterodimer (Fig. 2A). Of note, when IL-12αC96S was analyzed analogously, a very similar mispairing behavior of its disulfide bonds was observed (Fig. 2B). This argues that the multiple redox species of IL-12α are not caused by its Cys-96 residue, which is unpaired in the absence of IL-12β. Similar to the WT pair, IL-12βC199S induced secretion of IL-12αC96S, which, when secreted, migrated with an electrophoretic mobility in between the species observed in the lysate (Fig. 2B). This suggests the formation of intrachain disulfide bonds and further modification of the N-linked glycans in the Golgi in IL-12αC96S, as was expected to occur upon its secretion induced by IL-12βC199S.
Figure 2.

Misfolding of isolated IL-12α is inhibited by IL-12β. A, IL-12α redox status in the cell. The oxidation state of IL-12α was analyzed by non-reducing SDS-PAGE. Where indicated, samples were treated with β-mercaptoethanol (β-Me) after cell lysis to provide a standard for completely reduced protein, or cells were treated with DTT for 1 h before lysis to reduce disulfide bonds in cells. Alternatively, IL-12β was co-expressed to analyze its effect on the redox status of IL-12α. 293T cells were (co-)transfected with the indicated IL-12α/β subunits, and 2% lysate (L) or medium (M) was applied to the gel and blotted with the indicated antibodies. IL-12 heterodimers are indicated by an asterisk. MW, molecular weight. B, IL-12αC96S redox status in the cell. The same as in A, except IL-12αC96S was analyzed in the absence or presence of IL-12βC199S as indicated. Secreted IL-12αC96S is indicated by an asterisk. C, IL-12α oligomerization in the cell. Possible IL-12α dimerization was assessed by co-expression of untagged and HA-tagged IL-12α subunits. IL-12α untagged/HA-tagged-only samples served as size standard in comparison with the co-transfected sample. 1% lysate was analyzed by non-reducing SDS-PAGE (8% gel) and blotted with the indicated antibodies.
The different high-molecular-weight redox species observed for IL-12α could either be due to covalent IL-12α oligomerization, interaction with other cysteine-containing proteins, e.g. ER oxidoreductases, or both. To discriminate between these scenarios for the major IL-12α species of ∼50–60 kDa (Fig. 2, A and B), we co-expressed HA-tagged and untagged IL-12α. If the α subunit was able to form homodimers, then a mixed complex of untagged and HA-tagged α subunits with a size in between the homogenous complexes would be expected. Indeed, in these experiments, three bands of the expected sizes of the possible IL-12α dimers were present (Fig. 2C). Thus, in absence of the IL-12β subunit, IL-12α is prone to form disulfide-bridged homodimers.
To assess whether our findings were IL-12α-specific or more general within the IL-12 family, we analogously analyzed the redox status of isolated IL-23α. Like IL-12α, IL-23α also pairs with IL-12β, but to form another IL-12 family member, IL-23 (36). Similar to IL-12α, we observed covalent misfolding for isolated IL-23α, and, interestingly, even comparable redox species were present (supplemental Fig. 1).
The different intra- and interchain disulfide bonds of IL-12α vary in their impact on IL-12 folding, assembly, and secretion
Our data reveal that isolated IL-12 family α subunits populate non-native redox species in the cell whose formation is suppressed by IL-12β. To assess the impact of the different cysteine residues in IL-12α on this behavior, we individually replaced each of the disulfide bond-forming cysteine pairs in IL-12α (SS1–3) with serines (Fig. 3A), denoted as ΔSS1/2/3. Our data show that all IL-12α mutants individually lacking one disulfide bridge had a qualitatively comparable redox pattern to WT IL-12α, including monomers, dimers, and larger species (Fig. 3B, left panel). However, the relative amount of the species varied significantly for the different mutants in comparison with the WT. To assess the extent of covalent misfolding of the different disulfide mutants, we quantitatively compared the percentage of high-molecular-weight (HMW) and low-molecular-weight (LMW) species, defined as species migrating above or below 35 kDa, respectively (Fig. 3B, left panel). Remarkably, all disulfide mutants showed significantly less HMW species than the WT, with mutants ΔSS1 and ΔSS2 having a particularly strong impact (Fig. 3B, right panel). All mutants were still retained in the cell when expressed in isolation and thus unable to pass ER quality control (Fig. 3C). Upon IL-12β co-expression, IL-12α ΔSS1 and ΔSS2 were secreted to a very similar extent as IL-12α WT, whereas IL-12α ΔSS3 showed no secretion upon co-expression of IL-12β (Fig. 3C). Furthermore, all secretion-competent IL-12α disulfide mutants were still able to form the heterodimeric IL-12 complex inside the cell, albeit with an apparently slightly reduced efficiency, as judged by the presence of different IL-12α redox species even in the presence of IL-12β (supplemental Fig. 2). To further analyze the secretion behavior of our mutants, we co-transfected them with IL-12βC199S, which cannot form the intermolecular disulfide bridge, resulting in a non-covalent IL-12 complex. IL-12α ΔSS1 and ΔSS2 were still secreted with IL-12βC199S but much less efficiently than the IL-12α WT/IL-12βC199S pair (Fig. 3D), revealing an interplay between the different disulfide bonds. Again, no secretion of IL-12α ΔSS3 could be detected (Fig. 3D), which is in agreement with the observed redox species (supplemental Fig. 2).
Figure 3.
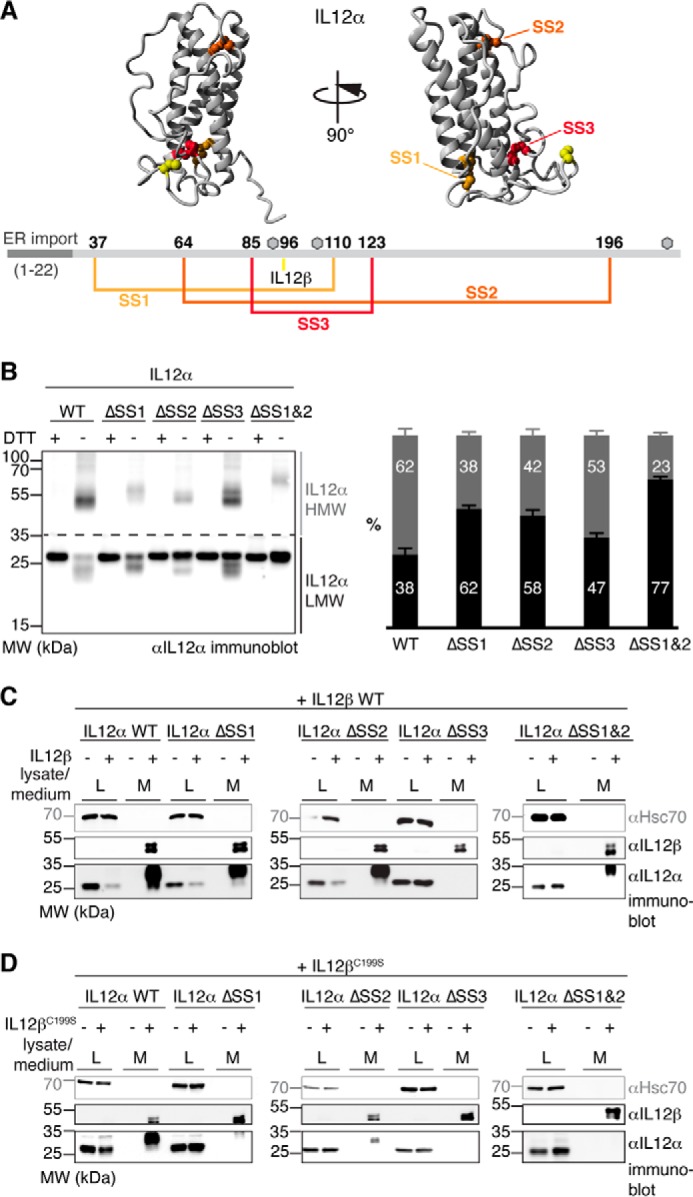
Disulfide bridges affect IL-12α misfolding and secretion differently. A, disulfide bonds within IL-12α. Cysteines forming intra- or intermolecular disulfide bridges are highlighted in IL-12α (structure based on PDB code 3HMX), numbered, and depicted in the corresponding colors in IL-12α. Gray hexagons indicate predicted glycosylation sites. B, impact of intramolecular disulfide bridges on the IL-12α redox status. IL-12α subunits individually lacking one disulfide bridge (ΔSS1–3, respectively; cysteine pairs were mutated to Ser) or a combination of two mutants (ΔSS1&2) were analyzed by non-reducing SDS-PAGE (left panel). 293T cells were transfected with the different IL-12α subunits and treated with DTT where indicated, and 2% lysate (L) was applied to the gel and blotted with IL-12α antibody. A quantitative analysis (right panel) indicates the percentage of HMW and LMW IL-12α species (n = 4 ± S.E.). LMW species (black) were defined as smaller than 35 kDa and HMW species (gray) as larger than 35 kDa. MW, molecular weight. C, secretion behavior of IL-12α disulfide bridge mutants. 293T cells were transfected with the indicated constructs, and 2% lysate or medium (M) was applied to the gel and blotted with the indicated antibodies. Hsc70 served as a loading control. D, secretion behavior of IL-12α disulfide bridge mutants. The same as in C, except secretion of IL-12α constructs in the presence of IL-12βC199S was analyzed.
Because our data revealed that disulfide bridges 1 and 2 in IL-12α were dispensable for secretion of a covalent IL-12α-IL-12β heterodimer (Fig. 3C), in the next step we combined these deletions (ΔSS1&2), giving rise to an IL-12α mutant with only three cysteine residues. This mutant populated the least amount of HMW species of all mutants tested (Fig. 3B). Of note, it was the only mutant that showed exclusively one monomeric species, migrating the same as the reduced protein (Fig. 3B, left panel). Furthermore, secretion of IL-12α ΔSS1&2 could still be induced by IL-12β WT but not by IL-12βC199S (Fig. 3, C and D). Secretion of IL-12α ΔSS1&2 in the presence of IL-12β, however, appeared to be somewhat less efficient than for the individual deletions (Fig. 3C and supplemental Fig. 2).
A limited role of the IL-12α cysteine residues in its degradation
The reduction of disulfide bonds can be rate-limiting in ER-associated protein degradation (ERAD) (42). Because we had observed covalent misfolding for IL-12α, we assessed the half-life of our IL-12α disulfide mutants by cycloheximide (CHX) chase experiments. For WT IL-12α, we observed a half-life of ∼1.6 h (Fig. 4A), in agreement with a previous study that reported a half-life of ∼2 h (34). Unexpectedly, we observed no significant differences in the half-lives of all IL-12α disulfide mutants in comparison with WT IL-12α (Fig. 4A). Taken together, these data suggested an overall limited role of disulfide bridges on the stability of unassembled IL-12α in the cell, and we only observed slightly different degradation kinetics of IL-12α HMW species versus LMW species (supplemental Fig. 3). Nevertheless, IL-12α HMW species most likely need to be reduced prior to degradation. Thus, to further understand the biological fate of misassembled IL-12 α subunits, we assessed whether the BiP co-chaperone ERdj5 (43) was involved in their degradation. For the truncated α1-antitrypsin mutant NHK, which aberrantly forms disulfide-bridged dimers, ERdj5 accelerates its degradation (42). Interestingly, overexpression of ERdj5 shifted the HMW/LMW ratio of IL-12α toward the monomeric species, arguing that ERdj5 can indeed reduce covalent IL-12α assemblies (Fig. 4B). Degradation of IL-12α in the presence of ERdj5, however, was not accelerated but, rather, appeared slightly decelerated, indicating that reducing the disulfide bridges in misassembled IL-12α is not rate-limiting for its degradation (Fig. 4C).
Figure 4.
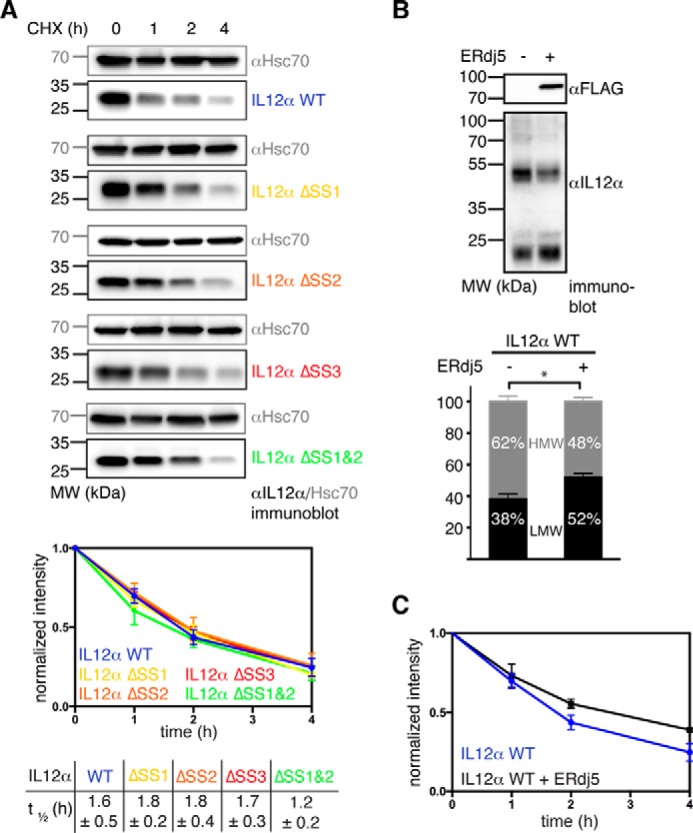
Influence of intramolecular disulfide bridges and ERdj5 on IL-12α degradation. A, measurement of IL-12α turnover by CHX chase assays. 293T cells were transfected with the indicated IL-12α subunits and incubated with CHX for up to 4 h. Cell lysates were analyzed by immunoblotting with the indicated antibodies. Hsc70 served as a loading control (top panel). The anti-IL-12α immunoblot signal was normalized to the signal present at the beginning of the chase for the respective IL-12α constructs (bottom panel, n = 4 ± S.E.). Half-lives from exponential fits of the curves (± S.D.) are shown below the graph. MW, molecular weight. B, influence of ERdj5 overexpression on the IL-12α redox state. The amount of HMW and LMW IL-12α species in the absence or presence of ERdj5 overexpression was analyzed (n = 3 ± S.E.; *, p < 0.05). Expression of FLAG-tagged ERdj5 was verified by immunoblotting. C, influence of ERdj5 overexpression on IL-12α degradation. IL-12α turnover by CHX chase assays in the absence (data taken from A) or in the presence of ERdj5 overexpression (n = 3 ± S.E.).
A simplified, biologically active IL-12
Because our data revealed that two of three disulfide bridges in IL-12α are dispensable for secretion and did not change the rate of IL-12α degradation, we next investigated their impact on the stability and biological activity of IL-12. Toward this end, we incubated supernatants of 293T cells co-transfected with IL-12β and either IL-12α WT, ΔSS1, ΔSS2, or ΔSS1&2 for extended times at 37 °C (IL-12α ΔSS3 was excluded because it was not secreted with IL-12β). None of the constructs showed detectable aggregation within 48 h, and all revealed a stability comparable with WT IL-12 (Fig. 5A).
Figure 5.
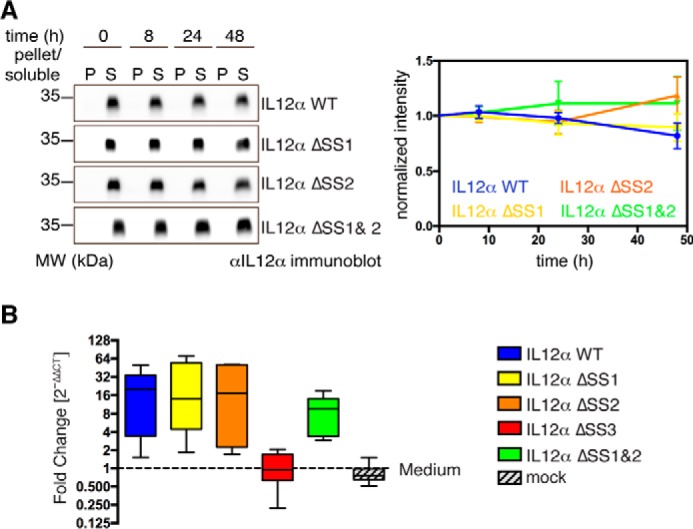
Influence of intramolecular disulfide bridges on IL-12α extracellular stability and biological activity. A, for stability tests, supernatants of 293T cells transfected with the indicated IL-12α constructs and IL-12β were incubated at 37 °C for the indicated times. 2% pellet (P) and soluble (S) material were analyzed by SDS-PAGE and blotted with IL-12α antibody (left panel). The anti-IL-12α immunoblot signal for soluble protein was normalized to the signal at 0-h incubation for the respective IL-12α constructs (right panel, n = 4 ± S.E.). MW, molecular weight. B, biological functionality of different IL-12 disulfide mutants. To assess the biological activity of IL-12 complexes containing different IL-12α mutant subunits, hPBMCs (from six blood donors) were stimulated with supernatants from 293T cells expressing IL-12β and one of the different IL-12α constructs as indicated. The relative expression of IFNγ was determined by quantitative PCR and normalized to IFNγ expression in hPBMCs in the presence of medium only.
Next, to dissect the impact of the different cysteine deletions in IL-12α on IL-12 activity, we used supernatants of 293T cells co-transfected with IL-12β WT and all different IL-12α mutants and assessed their effect on IFNγ induction in human peripheral blood mononuclear cells (hPBMCs). For all mutants that were secreted in the presence of IL-12 (Fig. 3C), we also observed induction of IFNγ in hPBMCs (Fig. 5B), indicating that most cysteines within IL-12α are not essential for IL-12 activity.
Discussion
Since its discovery, IL-12 has gained particular interest because of its heterodimeric nature (25–27). Early on, it had been speculated that its β subunit could potentially act as a scaffold for more than one co-subunit because IL-12β could not only be secreted in complex with IL-12α but also alone (27, 31). This idea was proven correct when IL-23 was discovered (36) and, more recently, has come into focus again when mass spectrometric studies revealed several IL-12β-interacting proteins in mouse plasma (44). This interaction promiscuity is a general theme within the IL-12 family, where at least four heterodimers (IL-12, IL-23, IL-27, and IL-35) are made up by only five subunits (18, 19). Because IL-12α as well as IL-23α secretion are dependent on IL-12β (26, 31, 34, 36), and secreted IL-12β can compete with IL-12 signaling (32, 45), important questions arise about how IL-12β can induce the secretion of multiple α subunits and how this process, and thus downstream immune reactions, are regulated and controlled in the ER. The IL-12 family thus extends assembly-induced secretion as a principle in ER quality control (1, 46) by important questions.
Our data show that assembly-induced folding of IL-12α is key: isolated IL-12α misfolds and forms non-native disulfide bonds. IL-12β inhibits these side reactions in the ER and induces formation of native IL-12. It is noteworthy that isolated IL-12α predominantly forms covalent homodimers. Such a well defined interaction suggests a certain degree of structure in isolated IL-12α. In other proteins of the immune system, where assembly-induced folding underlies ER quality control, no such well defined non-native covalent assembly states have been observed (5, 11, 47), but, interestingly, we also observed a similar behavior for IL-23α.
In more general terms, the formation of non-native disulfide bonds is frequently an initial step in oxidative protein folding, and it is often assumed that folding drives correct disulfide bond formation (48–50). Because IL-12α depends on IL-12β for proper folding, oxidation can be separated from folding. It remains to be seen whether IL-12β can rescue IL-12α from misfolding, as has been observed for the αβTCR upon assembly in vitro (11), or whether assembly has to occur early on to inhibit otherwise irreversible IL-12α misfolding. In addition to their effects on folding, non-native disulfide bonds can decelerate degradation by ERAD. We find that overexpression of ERdj5, an ER oxidoreductase involved in ERAD (42) but also in disulfide isomerization during productive folding (51), reduces the amount of non-native covalently assembled IL-12α but does not accelerate IL-12α degradation. In combination with our findings that the half-life of the different IL-12α disulfide mutants was unaltered, this argues that ERAD factors downstream of ERdj5/IL-12α reduction are likely rate-limiting for its degradation.
Native IL-12α possesses three intramolecular and one intermolecular disulfide bridge connecting it with IL-12β (21). Although the intermolecular disulfide bridge is dispensable for the secretion of bioactive IL-12 (21, 39), our data show that an interplay exists between the various disulfide bridges within IL-12α and the IL-12 heterodimer. Surprisingly, two of the three disulfide bridges within IL-12α can be deleted while still allowing secretion of IL-12, but only if these IL-12α mutants can form covalent dimers with IL-12β. If not, then IL-12β-induced secretion of IL-12α lacking any one of its disulfide bridges is drastically reduced, arguing for a mutual stabilization by intra- and intermolecular disulfide bridges in IL-12. Previous studies had shown that tunicamycin, which globally inhibits N-linked glycosylation, significantly reduces IL-12 secretion (33). Often an interplay between glycosylation and oxidative folding exists, e.g. by chaperone recruitment to or exclusion from certain sites within a polypeptide and mediated by Calnexin/Calreticulin-recruited protein disulfide isomerases (1, 52). Abolishment of IL-12 glycosylation by tunicamycin treatment may thus also impact correct oxidative folding of IL-12α, which we now report to be important in IL-12 biogenesis. However, our findings also show that a simplified IL-12 heterodimer is possible. Deleting two of the three disulfide bridges in IL-12α did still allow for secretion in the presence of IL-12β. Furthermore, these deletions did not appear to significantly reduce the stability of IL-12, whereas biological activity was maintained, and covalent misfolding within cells was reduced. Based on our data, we cannot exclude, however, that biological activity under limiting concentrations of IL-12 was reduced or stability under more drastic conditions or in more complex physiological environments was compromised. Nevertheless, the combination of these beneficial traits may render this simplified IL-12α an interesting molecule, as IL-12 has regained interest in immunotherapy (53, 54). Of note, this simplified IL-12 also shows that population of a more compact IL-12α species, which argues for disulfide bond formation within the subunit (see Fig. 3B), is not a prerequisite for assembly with IL-12β and IL-12 secretion. This highlights intriguing questions regarding the structural biology of IL-12 cytokines: what extent of structure within IL-12α and IL-23α is necessary for specific recognition by IL-12β to form the similar but structurally distinct IL-12 or IL-23 heterodimers (21, 55)? And how, being incompletely structured, does IL-12α specifically recognize even a second interaction partner, Ebi3, to give rise to IL-35 (38, 56)? The answers to these questions, building on our findings (Fig. 6), will depend on more detailed in vitro studies as well as insights into which ER chaperones regulate and control these processes (57) to reveal how folding and assembly of IL-12 family members is orchestrated in immune cells to shape immune responses.
Figure 6.
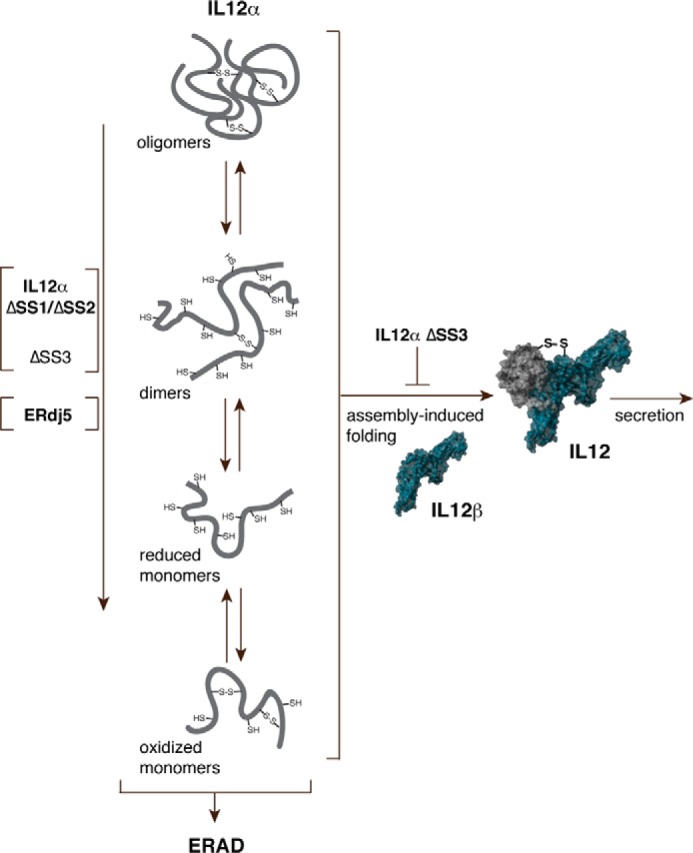
A model for IL-12 biogenesis. In isolation, IL-12α covalently misfolds, including a prominent homodimeric species. Assembly with IL-12β inhibits misfolding and allows secretion of bioactive IL-12. Disulfide bridge 3 within IL-12α is essential for IL-12β-induced secretion, whereas disulfide bridges 1 and 2 are dispensable. The deletion of any disulfide bridge as well as ERdj5 overexpression leads to a reduced amount of covalently misfolded IL-12α, with a particularly strong effect of disulfide bridges 1 and 2 on misfolding. Misfolded IL-12α is ultimately targeted for ERAD if no IL-12β is present.
Experimental procedures
Constructs
Human interleukin cDNAs were obtained from Origene (Rockville) and cloned into the pSVL vector (Amersham Biosciences) for mammalian expression. FLAG-tagged murine ERdj5 (42) was a kind gift from Kenji Inaba (Tohoku University). Mutants were generated by site-directed mutagenesis. All constructs were sequenced.
Cell culture and transient transfections
293T cells were grown in DMEM containing L-Ala-L-Gln (AQmedia, Sigma-Aldrich) supplemented with 10% (v/v) fetal bovine serum (Biochrom) at 37 °C and 5% CO2. The medium was complemented with a 1% (v/v) antibiotic-antimycotic solution (25 μg/ml amphotericin B, 10 mg/ml streptomycin, and 10,000 units of penicillin; Sigma-Aldrich). Transient transfections were carried out for 24 h in either p35 or p60 poly d-lysine-coated dishes (BD Biosciences) using GeneCellin (BioCellChallenge) according to the protocol of the manufacturer. In p35 dishes, 1 μg of IL-12α DNA and 2 μg of IL-12β DNA were always (co-)transfected, and in p60 dishes, 2 μg of IL-12α DNA and 4 μg of IL-12β DNA were always (co-)transfected unless stated otherwise. For ERdj5 experiments, 1 μg of IL-12α DNA was co-transfected with 1 μg of ERdj5 DNA.
Immunoblotting experiments
For secretion experiments by immunoblotting, cells were transfected for 8 h in p35 dishes, washed twice with PBS, and then supplemented with 0.5 ml of fresh medium for another 16 h. For CHX chase assays, cells were treated with 50 μg/ml CHX (Sigma-Aldrich) for the times indicated in the figures before lysis. Prior to lysis, when indicated, cells were treated with 10 mm DTT or 1 μg/ml brefeldin A (Sigma-Aldrich) for the last hour, washed twice in ice-cold PBS, supplemented with 20 mm NEM when samples were to be run on non-reducing SDS-PAGE gels. Cell lysis was carried out in RIPA buffer (50 mm Tris/HCl, pH 7.5, 150 mm NaCl, 1.0% Nonidet P40 substitute, 0.5% sodium deoxycholate, 0.1% SDS, 1x Roche complete Protease Inhibitor w/o EDTA; Roche Diagnostics). 20 mm NEM was added to the lysis buffer for non-reducing SDS-PAGE gels. To analyze secreted proteins, the medium was centrifuged for 5 min at 300 × g and 4 °C. Subsequently, samples were supplemented with 0.1 volumes of 500 mm Tris/HCl (pH 7.5), 1.5 m NaCl (and 200 mm NEM in the case of non-reducing SDS-PAGE), and protease inhibitor and centrifuged for 15 min at 20,000 × g and 4 °C. Samples were supplemented with 0.2 volumes of 5× Laemmli containing either β-Me for reducing SDS-PAGE or 100 mm NEM for non-reducing SDS-PAGE. For the stability assay, samples were treated like described above for secretion experiments, incubated at 37 °C for the times indicated in the figures, and recentrifuged (15 min, 20,000 × g, 4 °C), and the pellet as well as the supernatant were supplemented with equal amounts of 1× Laemmli containing β-Me.
For immunoblots, samples were run on 12% SDS-PAGE gels, transferred to PVDF membranes, and blotted with anti-IL-12α (Abcam, ab133751; 1:1000 in TBS, 0.05% Tween, 5% milk), anti-IL-12β (Abcam, ab133752; 1:500 in TBS, 0.05% Tween, 5% milk), anti-Myc tag (Millipore, 05-724; 1:500 in TBS, 0.05% Tween, 5% milk), anti-FLAG (Sigma-Aldrich, F1804; 1:1000 in TBS, 0.05% Tween, 5% milk), or anti-Hsc70 (Santa Cruz Biotechnology, sc-1059; 1:1000 in gelatin buffer (0.1% gelatin, 15 mm Tris/HCl (pH 7.5), 130 mm NaCl, 1 mm EDTA, 0.1% Triton X-100, 0.002% NaN3)). Species-specific HRP-conjugated secondary antibodies (in TBS, 0.05% Tween, 5% milk or gelatin buffer) were used to detect the proteins (Santa Cruz Biotechnology). Blots were detected using Amersham Biosciences ECL Prime (GE Healthcare) and a Fusion Pulse 6 imager (Vilber Lourmat).
Metabolic labeling
Metabolic labeling, immunoprecipitation, and autoradiography were performed as described previously (12) using the same antibodies as for immunoblotting. Endo H/PNGase F (New England Biolabs) deglycosylation experiments were carried out according to the protocols of the manufacturer.
Quantification and statistics
Western blots were quantified using the Bio-1D software (Vilber Lourmat). Statistical analyses were performed using Prism (GraphPad Software). Where indicated, data were analyzed with two-tailed, unpaired Student's t tests. Differences were considered statistically significant when p < 0.05. Where no statistical data are shown, all experiments were performed at least three times, and one representative experiment was selected.
PBMC stimulation assays
Whole blood was collected from six healthy volunteers at the Center of Allergy and Environment Munich after informed written consent and ethical approval by the internal ethics review board at the University Hospital of the Technical University of Munich (internal reference number 5156/11) in S-Monovette® tubes with EDTA (Sarstedt). PBMCs were isolated by gradient centrifugation using Polymophoprep (Axis Shield) and depleted of CD14-positive monocytes by using human CD14 MicroBeads (Miltenyi Biotec) according to the protocol of the manufacturer. CD14-negative PBMCs were cryopreserved until further use. Frozen PBMCs were rapidly thawed at 37 °C and resuspended in RPMI 1640 medium (Thermo Fisher Scientific) supplemented with 10% heat-inactivated FBS (GE Healthcare) and 100 units/ml penicillin, 100 μg/ml streptomycin, 1 μg/ml gentamicin, and 2 mm l-glutamine (Thermo Fisher Scientific). The different IL-12 constructs were expressed in 293T cells as described above. IL-12-containing 293T supernatants were centrifuged for 30 min at 2000 × g and 4 °C before use. Thawed PBMCs were seeded at a density of 1 × 106/ml and stimulated with 20 μl of supernatant from 293T cells expressing the different IL-12 constructs or 293T medium as a control and incubated for 24 h at 37 °C and 5% CO2. After centrifugation (2000 rpm, 3 min, 4 °C), PBMCs were rinsed with cold PBS and lysed in RLT buffer (Qiagen) supplemented with 1% β-mercaptoethanol at room temperature. Cell lysates were stored at −70 °C or directly used for RNA isolation. Total RNA was isolated using QuickRNATM MicroPrep (Zymo Research) following the instructions of the manufacturer. cDNA was synthesized using the high-capacity cDNA reverse transcription kit (Thermo Fisher Scientific) following the instructions of the manufacturer. cDNA was diluted in diethylpyrocarbonate (DEPC)-treated water (Thermo Fisher Scientific) to a concentration of 1.25 ng/μl. Quantitative PCR was performed in 384-well plates (4titude) in duplicates using FastStart Universal SYBR Green Master (Rox) (Roche) and forward and reverse target gene primers in a final concentration of 640 nm (18S forward, 5′-GTA ACC CGT TGA ACC CCA TT-3′; 18S reverse, 5′-CCA TCC ATT CGG TAG TAG CG-3′; IFNγ forward, 5′-TCA GCC ATC ACT TGG ATG AG-3′; IFNγ reverse, 5′-CGA GAT GAC TTC GAA AAG CTG-3′; Metabion). Data were collected in a ViiATM 7 real-time PCR system (Thermo Fisher Scientific) under the following conditions: 50 °C for 2 min (once) and 95 °C for 10 min (once), followed by 40 cycles of 95 °C for 15 s and 60 °C for 1 min. Samples exceeding a cycle threshold (Ct) of 35 were excluded from data processing. The relative gene expression of IFNγ was calculated using the comparative Ct method (2−ΔΔCt).
Structural modeling
Missing loops in the IL-12 structure were modeled with Yasara Structure, and the final structure was energy-minimized.
Author contributions
S. R., P. H., J. E. v. B., and M. J. F. designed the experiments. S. R., P. H., I. A., and M. J. F. performed the experiments. The data were analyzed by S. R., P. H., I. A., J. E. v. B., and M. J. F. The paper was written by S. R. and M. J. F. and edited by all authors. All authors approved the final version to be published.
Supplementary Material
Acknowledgments
We thank Johannes Buchner (Technical University of Munich) and Linda M. Hendershot (St. Jude Children's Research Hospital) for constructive comments on the manuscript. We also thank Linda M. Hendershot for the opportunity to perform metabolic labeling experiments in her laboratory.
This work was supported by CIPSM, the Fonds der Chemischen Industrie, and the Daimler and Benz Foundation and was performed in the framework of SFB 1035 (German Research Foundation DFG, Sonderforschungsbereich 1035, Project B11). The authors declare that they have no conflicts of interest with the contents of this article.
This article contains supplemental Figs. 1–3.
- ER
- endoplasmic reticulum
- HC
- heavy chain
- LC
- light chain
- Endo H
- endo-β-N-acetylglucosaminidase H
- HMW
- high-molecular-weight
- LMW
- low-molecular-weight
- ERAD
- endoplasmic reticulum-associated protein degradation
- CHX
- cycloheximide
- hPBMC
- human peripheral blood mononuclear cell
- NEM
- N-ethylmaleimide
- β-Me
- β-mercaptoethanol
- Ct
- cycle threshold
- PNGase F
- peptide:N-glycosidase F.
References
- 1. Ellgaard L., McCaul N., Chatsisvili A., and Braakman I. (2016) Co- and post-translational protein folding in the ER. Traffic 17, 615–638 [DOI] [PubMed] [Google Scholar]
- 2. Christis C., Lubsen N. H., and Braakman I. (2008) Protein folding includes oligomerization: examples from the endoplasmic reticulum and cytosol. FEBS J 275, 4700–4727 [DOI] [PubMed] [Google Scholar]
- 3. Feige M. J., Groscurth S., Marcinowski M., Shimizu Y., Kessler H., Hendershot L. M., and Buchner J. (2009) An unfolded CH1 domain controls the assembly and secretion of IgG antibodies. Mol. Cell 34, 569–579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hendershot L., Bole D., Köhler G., and Kearney J. F. (1987) Assembly and secretion of heavy chains that do not associate posttranslationally with immunoglobulin heavy chain-binding protein. J. Cell Biol. 104, 761–767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lee Y. K., Brewer J. W., Hellman R., and Hendershot L. M. (1999) BiP and immunoglobulin light chain cooperate to control the folding of heavy chain and ensure the fidelity of immunoglobulin assembly. Mol. Biol. Cell 10, 2209–2219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Corper A. L., Sohi M. K., Bonagura V. R., Steinitz M., Jefferis R., Feinstein A., Beale D., Taussig M. J., and Sutton B. J. (1997) Structure of human IgM rheumatoid factor Fab bound to its autoantigen IgG Fc reveals a novel topology of antibody-antigen interaction. Nat. Struct. Biol. 4, 374–381 [DOI] [PubMed] [Google Scholar]
- 7. Müller R., Gräwert M. A., Kern T., Madl T., Peschek J., Sattler M., Groll M., and Buchner J. (2013) High-resolution structures of the IgM Fc domains reveal principles of its hexamer formation. Proc. Natl. Acad. Sci. U.S.A. 110, 10183–10188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sitia R., Neuberger M., Alberini C., Bet P., Fra A., Valetti C., Williams G., and Milstein C. (1990) Developmental regulation of IgM secretion: the role of the carboxy-terminal cysteine. Cell 60, 781–790 [DOI] [PubMed] [Google Scholar]
- 9. Vavassori S., Cortini M., Masui S., Sannino S., Anelli T., Caserta I. R., Fagioli C., Mossuto M. F., Fornili A., van Anken E., Degano M., Inaba K., and Sitia R. (2013) A pH-regulated quality control cycle for surveillance of secretory protein assembly. Mol. Cell 50, 783–792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Schamel W. W., Kuppig S., Becker B., Gimborn K., Hauri H. P., and Reth M. (2003) A high-molecular-weight complex of membrane proteins BAP29/BAP31 is involved in the retention of membrane-bound IgD in the endoplasmic reticulum. Proc. Natl. Acad. Sci. U.S.A. 100, 9861–9866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Feige M. J., Behnke J., Mittag T., and Hendershot L. M. (2015) Dimerization-dependent folding underlies assembly control of the clonotypic αβT cell receptor chains. J. Biol. Chem. 290, 26821–26831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Feige M. J., and Hendershot L. M. (2013) Quality control of integral membrane proteins by assembly-dependent membrane integration. Mol. Cell 51, 297–309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Fayadat L., and Kopito R. R. (2003) Recognition of a single transmembrane degron by sequential quality control checkpoints. Mol. Biol. Cell 14, 1268–1278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Suzuki C. K., Bonifacino J. S., Lin A. Y., Davis M. M., and Klausner R. D. (1991) Regulating the retention of T-cell receptor α chain variants within the endoplasmic reticulum: Ca2+-dependent association with BiP. J. Cell Biol. 114, 189–205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Manolios N., Letourneur F., Bonifacino J. S., and Klausner R. D. (1991) Pairwise, cooperative and inhibitory interactions describe the assembly and probable structure of the T-cell antigen receptor. EMBO J. 10, 1643–1651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Call M. E., Pyrdol J., Wiedmann M., and Wucherpfennig K. W. (2002) The organizing principle in the formation of the T cell receptor-CD3 complex. Cell 111, 967–979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Geisler C., Kuhlmann J., and Rubin B. (1989) Assembly, intracellular processing, and expression at the cell surface of the human α β T cell receptor/CD3 complex. Function of the CD3-ζ chain. J. Immunol. 143, 4069–4077 [PubMed] [Google Scholar]
- 18. Vignali D. A., and Kuchroo V. K. (2012) IL-12 family cytokines: immunological playmakers. Nat. Immunol. 13, 722–728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hasegawa H., Mizoguchi I., Chiba Y., Ohashi M., Xu M., and Yoshimoto T. (2016) Expanding diversity in molecular structures and functions of the IL-6/IL-12 heterodimeric cytokine family. Front. Immunol. 7, 479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Gearing D. P., and Cosman D. (1991) Homology of the p40 subunit of natural killer cell stimulatory factor (NKSF) with the extracellular domain of the interleukin-6 receptor. Cell 66, 9–10 [DOI] [PubMed] [Google Scholar]
- 21. Yoon C., Johnston S. C., Tang J., Stahl M., Tobin J. F., and Somers W. S. (2000) Charged residues dominate a unique interlocking topography in the heterodimeric cytokine interleukin-12. EMBO J. 19, 3530–3541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Dixon K. O., van der Kooij S. W., Vignali D. A., and van Kooten C. (2015) Human tolerogenic dendritic cells produce IL-35 in the absence of other IL-12 family members. Eur. J. Immunol. 45, 1736–1747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Teng M. W., Bowman E. P., McElwee J. J., Smyth M. J., Casanova J. L., Cooper A. M., and Cua D. J. (2015) IL-12 and IL-23 cytokines: from discovery to targeted therapies for immune-mediated inflammatory diseases. Nat. Med. 21, 719–729 [DOI] [PubMed] [Google Scholar]
- 24. Trinchieri G., Pflanz S., and Kastelein R. A. (2003) The IL-12 family of heterodimeric cytokines: new players in the regulation of T cell responses. Immunity 19, 641–644 [DOI] [PubMed] [Google Scholar]
- 25. Kobayashi M., Fitz L., Ryan M., Hewick R. M., Clark S. C., Chan S., Loudon R., Sherman F., Perussia B., and Trinchieri G. (1989) Identification and purification of natural killer cell stimulatory factor (NKSF), a cytokine with multiple biologic effects on human lymphocytes. J. Exp. Med. 170, 827–845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Gubler U., Chua A. O., Schoenhaut D. S., Dwyer C. M., McComas W., Motyka R., Nabavi N., Wolitzky A. G., Quinn P. M., and Familletti P. C. (1991) Coexpression of two distinct genes is required to generate secreted bioactive cytotoxic lymphocyte maturation factor. Proc. Natl. Acad. Sci. U.S.A. 88, 4143–4147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Stern A. S., Podlaski F. J., Hulmes J. D., Pan Y. C., Quinn P. M., Wolitzky A. G., Familletti P. C., Stremlo D. L., Truitt T., and Chizzonite R. (1990) Purification to homogeneity and partial characterization of cytotoxic lymphocyte maturation factor from human B-lymphoblastoid cells. Proc. Natl. Acad. Sci. U.S.A. 87, 6808–6812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Langrish C. L., McKenzie B. S., Wilson N. J., de Waal Malefyt R., Kastelein R. A., and Cua D. J. (2004) IL-12 and IL-23: master regulators of innate and adaptive immunity. Immunol. Rev. 202, 96–105 [DOI] [PubMed] [Google Scholar]
- 29. Ma X., Yan W., Zheng H., Du Q., Zhang L., Ban Y., Li N., and Wei F. (2015) Regulation of IL-10 and IL-12 production and function in macrophages and dendritic cells. F1000 Res. 4, 10.12688/f1000research.7010.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kulig P., Musiol S., Freiberger S. N., Schreiner B., Gyülveszi G., Russo G., Pantelyushin S., Kishihara K., Alessandrini F., Kündig T., Sallusto F., Hofbauer G. F., Haak S., and Becher B. (2016) IL-12 protects from psoriasiform skin inflammation. Nat. Commun. 7, 13466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wolf S. F., Temple P. A., Kobayashi M., Young D., Dicig M., Lowe L., Dzialo R., Fitz L., Ferenz C., and Hewick R. M. (1991) Cloning of cDNA for natural killer cell stimulatory factor, a heterodimeric cytokine with multiple biologic effects on T and natural killer cells. J. Immunol. 146, 3074–3081 [PubMed] [Google Scholar]
- 32. Ling P., Gately M. K., Gubler U., Stern A. S., Lin P., Hollfelder K., Su C., Pan Y. C., and Hakimi J. (1995) Human IL-12 p40 homodimer binds to the IL-12 receptor but does not mediate biologic activity. J. Immunol. 154, 116–127 [PubMed] [Google Scholar]
- 33. Carra G., Gerosa F., and Trinchieri G. (2000) Biosynthesis and posttranslational regulation of human IL-12. J. Immunol. 164, 4752–4761 [DOI] [PubMed] [Google Scholar]
- 34. Jalah R., Rosati M., Ganneru B., Pilkington G. R., Valentin A., Kulkarni V., Bergamaschi C., Chowdhury B., Zhang G. M., Beach R. K., Alicea C., Broderick K. E., Sardesai N. Y., Pavlakis G. N., and Felber B. K. (2013) The p40 subunit of interleukin (IL)-12 promotes stabilization and export of the p35 subunit: implications for improved IL-12 cytokine production. J. Biol. Chem. 288, 6763–6776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. D'Andrea A., Rengaraju M., Valiante N. M., Chehimi J., Kubin M., Aste M., Chan S. H., Kobayashi M., Young D., and Nickbarg E. (1992) Production of natural killer cell stimulatory factor (interleukin 12) by peripheral blood mononuclear cells. J. Exp. Med. 176, 1387–1398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Oppmann B., Lesley R., Blom B., Timans J. C., Xu Y., Hunte B., Vega F., Yu N., Wang J., Singh K., Zonin F., Vaisberg E., Churakova T., Liu M., Gorman D., et al. (2000) Novel p19 protein engages IL-12p40 to form a cytokine, IL-23, with biological activities similar as well as distinct from IL-12. Immunity 13, 715–725 [DOI] [PubMed] [Google Scholar]
- 37. Pflanz S., Timans J. C., Cheung J., Rosales R., Kanzler H., Gilbert J., Hibbert L., Churakova T., Travis M., Vaisberg E., Blumenschein W. M., Mattson J. D., Wagner J. L., To W., Zurawski S., et al. (2002) IL-27, a heterodimeric cytokine composed of EBI3 and p28 protein, induces proliferation of naive CD4+ T cells. Immunity 16, 779–790 [DOI] [PubMed] [Google Scholar]
- 38. Collison L. W., Workman C. J., Kuo T. T., Boyd K., Wang Y., Vignali K. M., Cross R., Sehy D., Blumberg R. S., and Vignali D. A. (2007) The inhibitory cytokine IL-35 contributes to regulatory T-cell function. Nature 450, 566–569 [DOI] [PubMed] [Google Scholar]
- 39. Aparicio-Siegmund S., Moll J. M., Lokau J., Grusdat M., Schröder J., Plöhn S., Rose-John S., Grötzinger J., Lang P. A., Scheller J., and Garbers C. (2014) Recombinant p35 from bacteria can form interleukin (IL-)12, but not IL-35. PLoS ONE 9, e107990, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Murphy F. J., Hayes M. P., and Burd P. R. (2000) Disparate intracellular processing of human IL-12 preprotein subunits: atypical processing of the P35 signal peptide. J. Immunol. 164, 839–847 [DOI] [PubMed] [Google Scholar]
- 41. Braakman I., and Hebert D. N. (2001) Analysis of disulfide bond formation. Curr. Protoc. Protein Sci. Chapter 14, Unit 14.1 [DOI] [PubMed] [Google Scholar]
- 42. Ushioda R., Hoseki J., Araki K., Jansen G., Thomas D. Y., and Nagata K. (2008) ERdj5 is required as a disulfide reductase for degradation of misfolded proteins in the ER. Science 321, 569–572 [DOI] [PubMed] [Google Scholar]
- 43. Otero J. H., Lizák B., and Hendershot L. M. (2010) Life and death of a BiP substrate. Semin. Cell Dev. Biol. 21, 472–478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Abdi K., Singh N. J., Spooner E., Kessler B. M., Radaev S., Lantz L., Xiao T. S., Matzinger P., Sun P. D., and Ploegh H. L. (2014) Free IL-12p40 monomer is a polyfunctional adaptor for generating novel IL-12-like heterodimers extracellularly. J. Immunol. 192, 6028–6036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Mattner F., Fischer S., Guckes S., Jin S., Kaulen H., Schmitt E., Rüde E., and Germann T. (1993) The interleukin-12 subunit p40 specifically inhibits effects of the interleukin-12 heterodimer. Eur. J. Immunol. 23, 2202–2208 [DOI] [PubMed] [Google Scholar]
- 46. Hurtley S. M., and Helenius A. (1989) Protein oligomerization in the endoplasmic reticulum. Annu. Rev. Cell Biol. 5, 277–307 [DOI] [PubMed] [Google Scholar]
- 47. Leitzgen K., Knittler M. R., and Haas I. G. (1997) Assembly of immunoglobulin light chains as a prerequisite for secretion: a model for oligomerization-dependent subunit folding. J. Biol. Chem. 272, 3117–3123 [DOI] [PubMed] [Google Scholar]
- 48. Jansens A., van Duijn E., and Braakman I. (2002) Coordinated nonvectorial folding in a newly synthesized multidomain protein. Science 298, 2401–2403 [DOI] [PubMed] [Google Scholar]
- 49. Kosuri P., Alegre-Cebollada J., Feng J., Kaplan A., Inglés-Prieto A., Badilla C. L., Stockwell B. R., Sanchez-Ruiz J. M., Holmgren A., and Fernández J. M. (2012) Protein folding drives disulfide formation. Cell 151, 794–806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Qin M., Wang W., and Thirumalai D. (2015) Protein folding guides disulfide bond formation. Proc. Natl. Acad. Sci. U.S.A. 112, 11241–11246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Oka O. B., Pringle M. A., Schopp I. M., Braakman I., and Bulleid N. J. (2013) ERdj5 is the ER reductase that catalyzes the removal of non-native disulfides and correct folding of the LDL receptor. Mol. Cell 50, 793–804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Tannous A., Pisoni G. B., Hebert D. N., and Molinari M. (2015) N-linked sugar-regulated protein folding and quality control in the ER. Semin. Cell Dev. Biol. 41, 79–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Hernandez-Alcoceba R., Poutou J., Ballesteros-Briones M. C., and Smerdou C. (2016) Gene therapy approaches against cancer using in vivo and ex vivo gene transfer of interleukin-12. Immunotherapy 8, 179–198 [DOI] [PubMed] [Google Scholar]
- 54. Yeku O. O., and Brentjens R. J. (2016) Armored CAR T-cells: utilizing cytokines and pro-inflammatory ligands to enhance CAR T-cell anti-tumour efficacy. Biochem. Soc. Trans. 44, 412–418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Lupardus P. J., and Garcia K. C. (2008) The structure of interleukin-23 reveals the molecular basis of p40 subunit sharing with interleukin-12. J. Mol. Biol. 382, 931–941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Devergne O., Birkenbach M., and Kieff E. (1997) Epstein-Barr virus-induced gene 3 and the p35 subunit of interleukin 12 form a novel heterodimeric hematopoietin. Proc. Natl. Acad. Sci. U.S.A. 94, 12041–12046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. McLaughlin M., and Vandenbroeck K. (2011) The endoplasmic reticulum protein folding factory and its chaperones: new targets for drug discovery? Br. J. Pharmacol. 162, 328–345 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.