

Abstract
Objectives
The Hedgehog pathway has been implicated in small cell lung cancer (SCLC) tumor initiation and progression. Pharmacologic blockade of the key Hedgehog regulator, Smoothened, may inhibit these processes. We performed a phase I study to determine the maximum tolerated dose (MTD) of sonidegib (LDE225), a selective, oral Smoothened antagonist, in combination with etoposide/cisplatin in newly diagnosed patients with extensive stage SCLC.
Materials and methods
Patients received 4–6 21-day cycles of etoposide/cisplatin with daily sonidegib. Patients with response or stable disease were continued on sonidegib until disease progression or unacceptable toxicity. Two dose levels of sonidegib were planned: 400 mg and 800 mg daily, with 200 mg daily de-escalation if necessary. Next generation sequencing was performed on available specimens. Circulating tumor cells (CTCs) were quantified at baseline and with disease evaluation.
Results
Fifteen patients were enrolled. 800 mg was established as the recommended phase II dose of sonidegib in combination with etoposide/cisplatin. Grade 3 or greater toxicities included: anemia (n = 5), neutropenia (n = 8), CPK elevation (n = 2), fatigue (n = 2), and nausea (n = 2). Toxicity led to removal of one patient from study. Partial responses were confirmed in 79% (11/14; 95% CI: 49–95%). One patient with SOX2 amplification remains progression-free on maintenance sonidegib after 27 months. CTC count, at baseline, was associated with the presence of liver metastases and after 1 cycle of therapy, with overall survival.
Conclusions
Sonidegib 800 mg daily was the MTD when administered with EP. Further genomic characterization of exceptional responders may reveal clinically relevant predictive biomarkers that could tailor use in patients most likely to benefit.
Keywords: Small cell lung cancer, LDE225, Hedgehog pathway, Hedgehog inhibitor, Circulating tumor cells, SOX2 amplification
1. Introduction
Aberrant activation of the Hedgehog (Hh) pathway has been implicated in the initiation, maintenance and proliferation of SCLC. Hh signaling is essential in early stromal development and branching morphogenesis of the embryonic airways [1]. SCLC appears to be a relatively undifferentiated airway epithelial tumor that may recapitulate aspects of early lung development [2]. Expression of the ligand, Sonic Hh, and transcriptional factor, Gli-1, are upregulated relative to normal airway epithelium in SCLC [3]. Analogous to its role in early lung formation, Hh signaling has been implicated in SCLC initiation [4]. Constitutive activation of the pathway promotes tumor progression, and deletion of Smo, the seven-transmembrane receptor, suppresses initiation and progression in murine models of SCLC [3, 4]. Pharmacologic blockade of Smo inhibits the growth of both mouse and human SCLC [3, 4]. Following chemotherapy, Hh pathway inhibition may delay or prevent recurrence of residual disease in multiple murine SCLC models [4, 5].
Hh signaling has a role in regulating stem cell maintenance and differentiation, which has been suggested to parallel its role in tumorigenesis. Inhibition of Hh signaling in both in vitro and in vivo models has been associated with a loss of tumorigenic potential and improved survival across multiple tumor models [6–10]. In SCLC cell lines, Hh inhibition reportedly decreases cell growth primarily via a progenitor population [11]. These data support a model in which clonogenic recurrence of SCLC is dependent on a subset of chemotherapy-resistant progenitor cells and which may depend on the Hh developmental pathway.
The addition of a Hh inhibitor to cisplatin and etoposide (EP) may promote a more sustained treatment response and ultimately improve clinical outcomes in SCLC. As a test of this hypothesis, we undertook this phase I study to evaluate sonidegib [LDE225, N-[6-(cis-2,6-dimethylmorpholin-4-yl) pyridine-3-yl]-2-methyl-4′-(trifluoromethoxy)-1,1′-[biphenyl]-3-carboxamidediphosphate], an oral Smo antagonist, and to determine its maximum tolerated dose (MTD) in combination with EP in newly diagnosed extensive stage SCLC (ES-SCLC) patients.
2. Materials and methods
This was a single institution, open label Phase I study [NCT01579929], reviewed and approved by the Institutional Review Board. All patients provided written informed consent.
2.1. Eligibility criteria
All patients had pathologically confirmed untreated ES-SCLC. Those with asymptomatic disease in the brain were eligible. Inclusion required age ≥18, Karnofsky performance status of ≥70%, measurable disease as per Response Evaluation Criteria in Solid Tumors (RECIST) 1.1 [12], adequate bone marrow and organ function, and if applicable, a negative pregnancy test in women.
2.2. Study design and treatment
A standard, 3+3 dose escalation phase I design was used to determine the MTD of sonidegib given in conjunction with standard chemotherapy of EP. Two successive cohorts of three evaluable patients each were planned: 400 mg daily and 800 mg daily. One additional dose (200 mg daily) was reserved for de-escalation. To establish the MTD, a minimum of 6 evaluable patients must have been treated at applicable dose level.
A patient was considered evaluable for the dose-determining cohort if he/she finished the first two cycles of treatment (42 days) and received the minimum requirement of at least 80% (34 out of 42 days) of the planned doses of sonidegib. To complete minimum safety evaluations, a patient was observed for dose limiting toxicities (DLT) throughout the first 2 cycles (42 days following the first dose on cycle 1, day 1). Patients who did not complete the first 2 cycles for reasons other than a DLT were replaced.
Upon completion of a minimum of four and a maximum of six 21-day cycles of sonidegib with EP, patients in each cohort with at least stable disease received maintenance sonidegib until disease progression or unacceptable toxicity. Etoposide was dosed at 120 mg/m2 daily on days 1–3; cisplatin was dosed at 60 mg/m2 on day 1; sonidegib was started on cycle 1, day 1, and patients were required to take the drug at approximately the same time daily while fasting.
2.3. Study evaluation
Throughout the administration of sonidegib with EP, patients were assessed on days 1 and 8 of cycles 1 and 2 and on day 1 of cycles 3–6 (cycles 5 and 6, if applicable). Patients were evaluated every three weeks during maintenance sonidegib, with the exception of the first cycle of sonidegib alone, when they were assessed on days 1 and 8. At each visit, a history, physical examination, toxicity assessment, complete blood count, and comprehensive metabolic panel, including creatinine phosphokinase (CPK), were completed. All toxicities were graded using National Cancer Institute Common Terminology Criteria for Adverse Events version 4.0.
Tumor assessments at baseline included a computed tomography (CT) of the chest, as well as other known or suspected sites of disease, and a contrast-enhanced MRI or CT of the brain. Follow-up scans to assess response were obtained after cycles 2, 4 and 6 (if applicable) and every 9 weeks while patients received maintenance sonidegib. Responses were determined using RECIST 1.1 [12], and confirmation was required on repeat imaging at least four weeks later. Study-associated radiologists (AIH and JBT) reviewed all imaging studies.
2.4. Pharmacokinetics
Blood samples were collected prior to sonidegib administration on day 1 of cycles 1 through 9 to determine sonidegib pharmacokinetics. Plasma concentrations of sonidegib were measured using a validated high-performance liquid chromatography method with tandem mass spectrometry. Pre-dose concentrations were summarized and graphically displayed for each dose.
2.5. Circulating tumor cells (CTCs)
Peripheral blood was collected for CTC enumeration utilizing the Veridex CellSearch System™. CTCs were quantified at the following times: (i) prior to initiating the sonidegib; (ii) after cycles 1, 2, 4, and 6 of sonidegib and concomitant chemotherapy; (iii) every 3 cycles during maintenance sonidegib therapy; and (iv) at disease progression.
2.6. Mutation analysis of actionable cancer genes using next generation sequencing (NGS)
DNA was extracted from available biopsied tissue and cytology specimens (and patient-matched normal tissue). Using our MSK-IMPACT assay (Integrated Mutation Profiling of Actionable Cancer Targets), as previously described, DNA was sequenced and analyzed to identify single nucleotide variants, indels, and copy number alterations involving 341 genes [13, 14].
2.7. Statistical analysis
Overall response rate was estimated as a proportion along with the exact 95% confidence interval (CI). Progression-free survival (PFS) and overall survival (OS) were estimated using the Kaplan-Meier method. Patients were followed from start of treatment until progression or death. Patients still alive at the end of the study period and who did not experience the event of interest were censored at the date of last available follow-up. CTC levels were correlated with presence of metastases using the Wilcoxon rank sum test and with response to treatment by RECIST 1.1 using Fisher’s exact test. All statistical tests were two-sided, and p<0.05 was considered significant. Statistical analysis was performed in R 3.2.2 (R Development Core Team), including the “survival” and “Hmisc” packages.
3. Results
3.1. Patient characteristics
Between March 2012 and September 2014, 15 patients with untreated ES-SCLC were enrolled on this phase I study. Baseline characteristics are listed in Table 1.
Table 2.
Treatment emergent adverse events.
| Grade 1 | Grade 2 | Grade 3 | Grade 4 | Total (%) | |
|---|---|---|---|---|---|
| Hematologic | |||||
| Anemia | 3 | 6 | 5 | 0 | 14 (93) |
| Neutropenia | 1 | 0 | 1 | 7 | 9 (60) |
| Thrombocytopenia | 4 | 1 | 0 | 1 | 6 (40) |
| Non-Hematologic | |||||
| Fatigue | 6 | 4 | 2 | 0 | 12 (80) |
| Anorexia | 3 | 5 | 0 | 0 | 8 (53) |
| Nausea | 4 | 2 | 2 | 0 | 8 (53) |
| Hypomagnesemia | 7 | 0 | 1 | 0 | 8 (53) |
| Dysgeusia | 5 | 2 | 0 | 0 | 7 (47) |
| CPK Increase | 4 | 0 | 1 | 1 | 6 (40) |
| Vomiting | 4 | 1 | 0 | 0 | 5 (33) |
| Myalgias | 1 | 2 | 1 | 0 | 4 (27) |
| Tinnitus/Hearing Loss | 1 | 2 | 0 | 0 | 3 (20) |
| Hypokalemia | 1 | 0 | 2 | 0 | 3 (20) |
| Pain | 0 | 3 | 0 | 0 | 3 (20) |
| Dry Skin | 2 | 0 | 0 | 0 | 2 (13) |
| Diarrhea | 1 | 0 | 1 | 0 | 2 (13) |
3.2. Dose escalation and maximum tolerated dose
The dose escalation scheme is summarized in Fig. 1. At the first dose level, two patients experienced a DLT, including one of grade 3 nausea and one of grade 3 febrile neutropenia. This led to a dose de-escalation to 200 mg daily of sonidegib, as pre-specified in the protocol, with no further DLTs observed in the two patients enrolled in this level. A study oversight committee reviewed these data, and as febrile neutropenia appeared likely attributable to EP and not to the investigational agent, a protocol amendment was made to exclude it from the DLT definition. The 400 mg dose level was expanded with four additional patients without any further DLTs. Six patients were then enrolled on dose level 2 (sonidegib 800 mg daily) without any DLTs. Thus, the MTD was established as 800 mg once daily combined with EP administered every 3 weeks. Dose escalation beyond 800 mg sonidegib was not pursued, as this dose has been previously defined as a recommended phase 2 single agent dose [15].
Fig. 1.
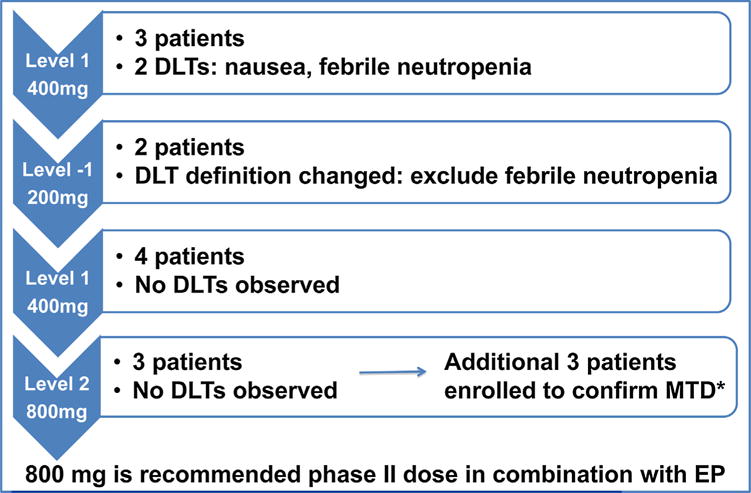
Dose Escalation Scheme and Dose Limiting Toxicities (DLT). Diagram describes the dose escalation with associated toxicities in each cohort, if any. Sonidegib was administered orally once daily with etoposide/cisplatin administered every 3 weeks. A study oversight committee reviewed the data and febrile neutropenia was determined to be secondary to etoposide/cisplatin and not sonidegib, leading to exclusion of this adverse event from the DLT definition. The maximum tolerated dose was determined to be 800 mg after treating 6 patients.
3.3. Safety and toxicity
All 15 patients were evaluable for toxicity, and all experienced at least one treatment-related adverse event on study (Table 2). The most common grade 3 or 4 related toxicities were neutropenia (53%), anemia (33%), hypokalemia (13%), nausea (13%), CPK elevation (13%), and fatigue (13%). Overall incidence of febrile neutropenia in the DLT assessment period was 7%. With the exception of one patient who experienced grade 3 CPK elevation and grade 1 acute kidney injury during maintenance of sonidegib, the majority of these toxicities occurred during the simultaneous administration of chemotherapy. These adverse events generally were reversible with temporary discontinuation of the drug, and/or appropriate dose adjustment of chemotherapy.
CPK elevation is a common adverse event noted with sonidegib and has been shown to occur in an exposure-dependent manner [15, 16]. Six patients experienced CPK elevations; four developed asymptomatic grade 1 CPK elevations that resolved without intervention and two required dose modifications of sonidegib due to symptomatic increase of CPK. One patient presented for evaluation prior to receiving his second maintenance cycle of the agent with complaints of myalgias and assessment revealed grade 3 CPK and grade 1 acute kidney injury. Sonidegib was held for 1 week and CPK returned to baseline; the patient restarted maintenance sonidegib at 200 mg daily without continued issues. Upon presenting for cycle 3 of treatment, the second patient reported grade 3 myalgias and was noted to have grade 4 CPK elevation. The third cycle was administered without sonidegib, which was restarted 3 weeks later at 400 mg daily; one further dose reduction of sonidegib was required during maintenance.
Serious adverse events were reported for ten (67%) patients, in six of whom these were considered at least possibly related to study drug. In addition to the two patients with grade 3 and 4 CPK elevations, one patient each experienced grade 3 neutropenic fever, grade 3 dehydration, and grade 4 neutropenia. Multiple serious adverse events were noted in one patient who was in the initial sonidegib cohort of 400 mg and had required a dose reduction to 200 mg for grade 3 nausea. After cycle 4 of EP and sonidegib, this patient developed recurrent grade 2 nausea, grade 2 fever, grade 3 abdominal pain, grade 3 colitis, and grade 3 acute kidney injury. Because symptoms appeared possibly related to therapy, the patient was removed from study and died of disease within 30 days. One patient died on study due to an inferior wall myocardial infarction, unrelated to therapy.
3.4. Dose modifications
As described, four patients required sonidegib dose reductions: one each for grade 3 nausea and grade 3 neutropenic fever, and two for grade 3 and 4 CPK elevations. Five patients required EP dose reductions: grade 2 acute kidney injury (n = 2), one after 3 cycles and the other after 5 cycles; grade 3 fatigue in the setting of parainfluenza infection after 3 cycles (n = 1); grade 2 fatigue and nausea after 4 cycles (n = 1), and grade 4 neutropenic fever after 1 cycle (n = 1). Two patients received maintenance sonidegib after four and five cycles of chemotherapy, respectively.
3.5. Time on study
Patients remained on study for a median of 6 months (range, 1–31 months), with 7 patients treated for 6 months or longer. The median number of cycles of chemotherapy was 6 (range 2–6) and maintenance sonidegib was 3 (range 0–39) (Fig. 2). Twelve patients discontinued the study secondary to progression of disease, with 9 patients progressing during, or within 90 days of completing, chemotherapy. One patient currently remains on maintenance sonidegib 27 months after completing chemotherapy.
Fig. 2.

Spider Diagram. Treatment course of all 15 patients documented (identified as numbers 1–15 along the y-axis) including dose reductions. One patient remains on study at the time of the manuscript preparation.
3.6. Efficacy
Fourteen patients were evaluable for response (Fig. 3), as one patient died on study prior to assessment. Eleven patients (79%) achieved a partial response (95% CI 49–95%), and 3 patients (21%) had stable disease. Ten patients completed chemotherapy and received maintenance sonidegib, with 3 patients having continued tumor reduction on maintenance therapy. Median PFS was 5.5 months (95% CI of 4.1–8.4) and median OS was 19.7 months (95% CI 11.2-NA).
Fig. 3.

Waterfall plot. Only 14 patients are evaluable (one patient died of an unrelated cause one week after cycle 2). The individual patient identification number, along the x-axis, labels its respective bar and corresponds to patients 1–15 in Fig. 2. The response rate was 79% (11 out of 14, 95% CI: 49–95%).
3.7. Pharmacokinetics
Mean sonidegib plasma concentration-time curves on study are presented in Fig. 4. Pharmacokinetics data observed on this study were generally consistent with the previous monotherapy experience. Based on the trough plasma concentration over time, steady state seemed variable, but was reached by cycle 7 with a concentration trough of 1080 ng/ML. As the drug has a long half-life, its exposure accumulated, with median accumulation ratio from cycle 7 to cycle 2 of 5.6. The two patients with the highest sonidegib plasma concentrations were treated at the 800 mg dose; neither suffered dose limiting toxicities nor required dose reductions.
Fig. 4.

Mean sonidegib plasma concentration-time curve for each patient on study. Steady state was reached by cycle 7 with a concentration trough of 1080 ng/ML. Two patients treated at the 800 mg dose had the highest sonidegib plasma concentrations; neither required dose reductions.
3.8. Circulating tumor cells
Baseline CTCs were evaluated on 14 of the 15 patients. At baseline, CTCs ranged from 0 to >200, with 10 patients having ≥5 CTCs and 4 patients with >200 CTCs. An exploratory analysis showed that while baseline CTCs correlated with liver metastasis (median CTC of >200 vs 3.5 for those with liver metastasis vs without liver metastasis, p = 0.007), CTCs did not correlate with brain or bone metastasis. In univariate analysis, elevated baseline CTCs appeared to be associated with worse OS: the median OS for patients with baseline CTC >200 vs ≤200 was 6.2 months compared to 25.7 months. Persistently elevated CTC number at cycle 2 day 1 (N = 13) also appeared to be associated with worse OS in univariate analysis (median OS of 25.0 months if CTC = 0 (N = 7) vs 5.5 months for CTC ≥1 (N = 6)). Furthermore, 10 of 11 patients with a partial response had CTC = 0 at time of first follow-up scan, as compared to none of the three patients with stable disease. At progression, 5 of 13 patients had a rise in the CTC from nadir.
3.9. Tumor mutational profiling
Only five patients, including the one who died, had adequate tissue and matched normal available for NGS. The remaining patients received sonidegib 400 mg (n = 2) and 800 mg (n = 2) for a median of 8 months including maintenance therapy and each had RB1 and TP53 loss. Notably, only in the one patient continuing on sonidegib over 27 months since completing etoposide/cisplatin, NGS analysis revealed tumor-specific amplification of SOX2 and PIK3CA, both on chromosome 3q26.3–27.
4. Discussion
The primary goal of this Phase I study was to establish the safety profile and recommended phase II dose of the Hh inhibitor sonidegib when given with standard of care first-line cisplatin and etoposide in patients with newly diagnosed ES-SCLC, which proved to be cisplatin 60 mg/m2 on day 1 with etoposide 120 mg/m2 daily on days 1–3, and sonidegib 800 mg daily on days 1–21 of a 21-day cycle. Pharmacokinetic analyses did not suggest that EP had a substantial effect on sonidegib exposure. Based on prior single agent experience with sonidegib, this dose level is within the pharmacologically effective range, associated with objective responses in pathway-dependent tumors including basal cell carcinoma and medulloblastoma [15, 16].
The hypothesis supporting the exploration of this combination in SCLC patients was that addition of a potent and specific Hh pathway inhibitor would lead to enhanced durability of response due to targeting of a critical tumor progenitor population dependent on Hh signaling. Although a 15-patient Phase I dose-finding study may be too small to generate definitive conclusions regarding this hypothesis, the patient outcome data, including an estimated median PFS and OS of 5.5 and 19.7 months, respectively, generally are consistent with expected outcomes with EP in ES-SCLC patients [17–19].
This observation suggests a discrepancy between the data in multiple murine models of SCLC and the initial data in patients [3, 4], likely due to various factors, including tumor burden. Mouse models, whether evaluating de novo murine tumors or transplanted human tumors, generally are treated at tumor sizes below 200 mm3, generally below the level of clinical detection in humans. ES-SCLC patients often have exceptionally large tumor burden at the time of diagnosis. Substantially larger tumor bulk presumably will be associated with a larger and more heterogeneous population of tumor progenitor cells, and also may lead to protection of tumor progenitors due to anatomic considerations, including oncotic and hydrostatic pressures that limit intratumoral drug delivery. Smo inhibition has been associated with effective responses in basal cell carcinoma and medulloblastoma, which are known to harbor activating Hh pathway mutations [20–23]. However, in malignancies such as SCLC, with ligand-dependent activation of the Hh pathway, Smo inhibitors have not yet demonstrated clinically meaningful activity. The randomized phase II study that evaluated vismodegib with EP compared to chemotherapy alone in newly diagnosed ES-SCLC patients failed to demonstrate an improvement in PFS or OS [24]. Therefore, in SCLC and cancers with overexpression of the Hh signaling pathway, other oncogenic signaling pathways may primarily drive their proliferation [25].
Two patients had exceptionally durable responses: one in the 800 mg dose cohort who remained on sonidegib for over a year, and the other in the 400 mg dose cohort who remains progression-free on maintenance sonidegib after 27 months. Such durable responses would be unusual for EP alone, and suggests that a subset of patients may benefit from targeted Hh pathway inhibition. Sufficient tissue was available for NGS for five of the patients, including the patient with the longest response whose NGS analysis revealed amplification of SOX2 and PIK3CA, both on chromosome 3q26.3. This finding was not demonstrated in any of the other patients with NGS analysis in this series. Notably, coamplification and cooverexpression of PRKCI and SOX2 on 3q26 have been reported to cooperate to drive cell autonomous Hh signaling in cancer stem cells of squamous cell carcinoma of the lung [26]. Treatment of these cell lines with sonidegib led to downregulation of the transcriptional target, GLI1, consistent with pathway activity [26]. As SOX2 amplification has been reported in up to 27% of SCLC patients [27, 28], further analysis of this signaling axis is warranted and could reveal clinically relevant predictive biomarkers that may possibly tailor the use of Hh inhibitors to SCLC patients most likely to benefit.
This study reinforces prior observations regarding the prognostic and predictive value of CTC enumeration in this disease [29–32]: baseline CTC count appeared to be associated with liver metastases and worse OS; CTC count after one cycle of therapy appeared to be associated with OS, and the elimination of detectable CTC after two cycles was strongly associated with response. Recent studies have demonstrated that CTC in SCLC have clear tumorigenic capacity, further underlying the importance of this tumor compartment as a readily accessible predictor of tumor behavior [33].
This study establishes a well-tolerated three-drug regimen for first-line treatment of SCLC, with incorporation of targeted inhibition of the Hedgehog signaling pathway. Further evaluation of whether subsets of patients preferentially benefit from inclusion of sonidegib are warranted given that one patient continues to respond to the agent, with focal 3q26 amplification [26]. We also would consider evaluation of Hedgehog inhibition in the context of limited stage SCLC, where with chemoradiotherapy much deeper responses are achieved, including cures in some patients: it is perhaps in the minimal disease state after chemoradiation that the efficacy of an agent targeting the progenitor population giving rise to disease recurrence could most be definitively assessed.
Table 1.
Baseline patient characteristics (N = 15).
| All Patients (N = 15) | Dose Level 1 400 mg (N = 7) | Dose level–1 200 mg (N = 2) | Dose Level 2 800 mg (N = 6) | |
|---|---|---|---|---|
| Median Age, Range | 55 (46–69) | 53 (47–55) | 56 (52–59) | 65 (46–69) |
| Sex | ||||
| Male | 9 (60%) | 4 (57%) | 2 (100) | 3 (50%) |
| Female | 6 (40%) | 3 (43%) | – | 3 (50%) |
| Karnofsky Performance Status | ||||
| ≥90% | 6 (40%) | 3 (43%) | 1 (50%) | 2 (33%) |
| 80% | 6 (40%) | 2 (28.5%) | 1 (50%) | 3 (50%) |
| 70% | 3 (20%) | 2 (28.5%) | – | 1 (17%) |
| Smoking History | ||||
| Current | 13 (87%) | 6 (86%) | 2 (100%) | 5 (83%) |
| Former | 2 (13%) | 1 (14%) | – | 1 (17%) |
| Median No of Pack Years (range) | 40 (20–135) | 37 (30–100) | 66 (40–93) | 61 (28–135) |
| Brain Metastases at Study Initiation | ||||
| Brain | 4 (27%) | 3 (43%) | 1 (50%) | – |
| WBRTa Prior to Enrollment | 3 (75%) | 1 (33%) | 1 (100%) | – |
Whole brain radiation treatment.
Acknowledgments
Funding
This research was funded, in part, through Novartis and the National Institute of Health(NIH)/National Cancer Institute (NCI) Cancer Center Support Grant (P30CA008748).
M. Catherine Pietanza—Dr. Pietanza reports employment from Merck. Previously reports personal fees from Genentech, personal fees from CelGene Corp, personal fees from Abbvie, personal fees from Clovis Oncology, grants and personal fees from Novartis, grants and personal fees from Bristol Myers Squibb, grants from Stemcentrx, Inc, grants from OncoMed Pharmaceuticals, Inc.
Lee M. Krug—Dr. Krug reports employment from Bristol-Myers Squibb.
Cami S. Sima—Dr. Sima reports employment from Roche/ Genentech.
Mark G. Kris—Dr. Kris reports grants from National Institute of Health (NIH)/National Cancer Institute (NCI) Cancer Center Support Grant P30CA 008748, personal fees from AstraZeneca, personal fees from Genentech/Roche, personal fees from ARIAD, personal fees from Daiich Sankyo, personal fees from Array, personal fees from Threshold Pharmaceuticals.
Charles M. Rudin—Dr. Rudin reports personal fees from Abbvie, Boehringer Ingelheim, Celgene, GSK, Genentech, Merck, Novartis, grants from Biomarin and National Institute of Health (NIH)/National Cancer Institute (NCI) Cancer Center Support Grant P30CA 008748.
Footnotes
Conflict of interest
Other authors have nothing to disclose.
References
- 1.Pepicelli CV, Lewis PM, McMahon AP. Sonic hedgehog regulates branching morphogenesis in the mammalian lung. Curr Biol. 1998;8:1083–1086. doi: 10.1016/s0960-9822(98)70446-4. [DOI] [PubMed] [Google Scholar]
- 2.Liu H, Kho AT, Kohane IS, Sun Y. Predicting survival within the lung cancer histopathological hierarchy using a multi-scale genomic model of development. PLoS Med. 2006;3:e232. doi: 10.1371/journal.pmed.0030232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Watkins DN, Berman DM, Burkholder SG, Wang B, Beachy PA, Baylin SB. Hedgehog signalling within airway epithelial progenitors and in small-cell lung cancer. Nature. 2003;422:313–317. doi: 10.1038/nature01493. [DOI] [PubMed] [Google Scholar]
- 4.Park KS, Martelotto LG, Peifer M, Sos ML, Karnezis AN, Mahjoub MR, Bernard K, Conklin JF, Szczepny A, Yuan J, Guo R, Ospina B, Falzon J, Bennett S, Brown TJ, Markovic A, Devereux WL, Ocasio CA, Chen JK, Stearns T, Thomas RK, Dorsch M, Buonamici S, Watkins DN, Peacock CD, Sage J. A crucial requirement for Hedgehog signaling in small cell lung cancer. Nat Med. 2011 doi: 10.1038/nm.2473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Travaglione V, Peacock CD, MacDougall J, McGovern K, Cushing J, Yu LC, Trudeau M, Palombella V, Adams J, Hierman J, Rhodes JT, Devereux WL, Watkins DN, A novel HH pathway inhibitor IPI-926, delays recurrence post-chemotherapy in a primary human SCLC xenograft model; 99th AACR Annual Meeting; San Diego, CA. 2008. p. 4611. [Google Scholar]
- 6.Watkins DN, Berman DM, Baylin SB. Hedgehog signaling: progenitor phenotype in small-cell lung cancer. Cell Cycle. 2003;2:196–198. [PubMed] [Google Scholar]
- 7.Merchant AA, Matsui W. Targeting Hedgehog—a cancer stem cell pathway. Clin Cancer Res. 2010;16:3130–3140. doi: 10.1158/1078-0432.CCR-09-2846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dierks C, Beigi R, Guo GR, Zirlik K, Stegert MR, Manley P, Trussell C, Schmitt-Graeff A, Landwerlin K, Veelken H, Warmuth M. Expansion of Bcr-Abl-positive leukemic stem cells is dependent on Hedgehog pathway activation. Cancer Cell. 2008;14:238–249. doi: 10.1016/j.ccr.2008.08.003. [DOI] [PubMed] [Google Scholar]
- 9.Zhao C, Chen A, Jamieson CH, Fereshteh M, Abrahamsson A, Blum J, Kwon HY, Kim J, Chute JP, Rizzieri D, Munchhof M, VanArsdale T, Beachy PA, Reya T. Hedgehog signalling is essential for maintenance of cancer stem cells in myeloid leukaemia. Nature. 2009;458:776–779. doi: 10.1038/nature07737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu S, Dontu G, Mantle ID, Patel S, Ahn NS, Jackson KW, Suri P, Wicha MS. Hedgehog signaling and Bmi-1 regulate self-renewal of normal and malignant human mammary stem cells. Cancer Res. 2006;66:6063–6071. doi: 10.1158/0008-5472.CAN-06-0054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tian F, Mysliwietz J, Ellwart J, Gamarra F, Huber RM, Bergner A. Effects of the Hedgehog pathway inhibitor GDC-0449 on lung cancer cell lines are mediated by side populations. Clin Exp Med. 2012;12:25–30. doi: 10.1007/s10238-011-0135-8. [DOI] [PubMed] [Google Scholar]
- 12.Eisenhauer EA, Therasse P, Bogaerts J, Schwartz LH, Sargent D, Ford R, Dancey J, Arbuck S, Gwyther S, Mooney M, Rubinstein L, Shankar L, Dodd L, Kaplan R, Lacombe D, Verweij J. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1) Eur J Cancer. 2009;45:228–247. doi: 10.1016/j.ejca.2008.10.026. [DOI] [PubMed] [Google Scholar]
- 13.Won HH, Scott SN, Brannon AR, Shah RH, Berger MF. Detecting somatic genetic alterations in tumor specimens by exon capture and massively parallel sequencing. J Vis Exp ( 2013:e50710. doi: 10.3791/50710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cheng DT, Mitchell TN, Zehir A, Shah RH, Benayed R, Syed A, Chandramohan R, Liu ZY, Won HH, Scott SN, Brannon AR, O’Reilly C, Sadowska J, Casanova J, Yannes A, Hechtman JF, Yao J, Song W, Ross DS, Oultache A, Dogan S, Borsu L, Hameed M, Nafa K, Arcila ME, Ladanyi M, Berger MF. Memorial sloan kettering-integrated mutation profiling of actionable cancer targets (MSK-IMPACT): a hybridization capture-based next-generation sequencing clinical assay for solid tumor molecular oncology. J Mol Diagn. 2015;17:251–264. doi: 10.1016/j.jmoldx.2014.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rodon J, Tawbi HA, Thomas AL, Stoller RG, Turtschi CP, Baselga J, Sarantopoulos J, Mahalingam D, Shou Y, Moles MA, Yang L, Granvil C, Hurh E, Rose KL, Amakye DD, Dummer R, Mita AC. A phase I, multicenter, open-label, first-in-human, dose-escalation study of the oral smoothened inhibitor Sonidegib (LDE225) in patients with advanced solid tumors. Clin Cancer Res. 2014;20:1900–1909. doi: 10.1158/1078-0432.CCR-13-1710. [DOI] [PubMed] [Google Scholar]
- 16.Migden MR, Guminski A, Gutzmer R, Dirix L, Lewis KD, Combemale P, Herd RM, Kudchadkar R, Trefzer U, Gogov S, Pallaud C, Yi T, Mone M, Kaatz M, Loquai C, Stratigos AJ, Schulze HJ, Plummer R, Chang AL, Cornelis F, Lear JT, Sellami D, Dummer R. Treatment with two different doses of sonidegib in patients with locally advanced or metastatic basal cell carcinoma (BOLT): a multicentre, randomised, double-blind phase 2 trial. Lancet Oncol. 2015;16:716–728. doi: 10.1016/S1470-2045(15)70100-2. [DOI] [PubMed] [Google Scholar]
- 17.Hanna N, Bunn PA, Jr, Langer C, Einhorn L, Jr, Guthrie T, Beck T, Ansari R, Ellis P, Byrne M, Morrison M, Hariharan S, Wang B, Sandler A. Randomized phase III trial comparing irinotecan/cisplatin with etoposide/cisplatin in patients with previously untreated extensive-stage disease small-cell lung cancer. J Clin Oncol. 2006;24:2038–2043. doi: 10.1200/JCO.2005.04.8595. [DOI] [PubMed] [Google Scholar]
- 18.Lara PN, Jr, Natale R, Crowley J, Lenz HJ, Redman MW, Carleton JE, Jett J, Langer CJ, Kuebler JP, Dakhil SR, Chansky K, Gandara DR. Phase III trial of irinotecan/cisplatin compared with etoposide/cisplatin in extensive-stage small-cell lung cancer: clinical and pharmacogenomic results from SWOG S0124. J Clin Oncol. 2009;27:2530–2535. doi: 10.1200/JCO.2008.20.1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Spigel DR, Townley PM, Waterhouse DM, Fang L, Adiguzel I, Huang JE, Karlin DA, Faoro L, Scappaticci FA, Socinski MA. Randomized phase II study of bevacizumab in combination with chemotherapy in previously untreated extensive-stage small-cell lung cancer: results from the SALUTE trial. J Clin Oncol. 2011;29:2215–2222. doi: 10.1200/JCO.2010.29.3423. [DOI] [PubMed] [Google Scholar]
- 20.Hoff DD Von, LoRusso PM, Rudin CM, Reddy JC, Yauch RL, Tibes R, Weiss GJ, Borad MJ, Hann CL, Brahmer JR, Mackey H, Lum BL, Darbonne WC, Marsters JC, Sauvage FJ De, Low JA. Inhibition of the hedgehog pathway in advanced basal-cell carcinoma. New Engl J Med. 2009;361:1–9. doi: 10.1056/NEJMoa0905360. [DOI] [PubMed] [Google Scholar]
- 21.Rudin CM, Hann CL, Laterra J, Yauch RL, Callahaun CA, Fu L, Holcomb T, Stinson J, Gould SE, Coleman B, LoRusso PM, Hoff DD Von, Sauvage FJ De, Low JA. Treatment of medulloblastoma with hedgehog pathway inhibitor GDC-0449. New Engl J Med. 2009;361:1–6. doi: 10.1056/NEJMoa0902903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Williams JA, Guicherit OM, Zaharian BI, Xu Y, Chai L, Wichterle H, Kon C, Gatchalian C, Porter JA, Rubin LL, Wang FY. Identification of a small molecule inhibitor of the hedgehog signaling pathway: effects on basal cell carcinoma-like lesions. Proc Natl Acad Sci U S A. 2003;100:4616–4621. doi: 10.1073/pnas.0732813100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shou Y, Robinson DM, Amakye DD, Rose KL, Cho YJ, Ligon KL, Sharp T, Haider AS, Bandaru R, Ando Y, Geoerger B, Doz F, Ashley DM, Hargrave DR, Casanova M, Tawbi HA, Rodon J, Thomas AL, Mita AC, MacDonald TJ, Kieran MW. A five-gene hedgehog signature developed as a patient preselection tool for hedgehog inhibitor therapy in medulloblastoma. Clin Cancer Res. 2015;21:585–593. doi: 10.1158/1078-0432.CCR-13-1711. [DOI] [PubMed] [Google Scholar]
- 24.Belani CP, Dahlberg SE, Rudin CM, Fleisher F, Chen HX, Takebe N, Ramalingman SS, Schiller JH. Three arm randomized phase II study of cisplatin and etoposide (CE) versus CE with either vismodegib or cixutumumab for patients with extensive stage small cell lung cancer (ECOG1508) J Clin Oncol. 2013;31(suppl) abstr 7508. [Google Scholar]
- 25.Justilien V, Fields AP. Molecular pathways: novel approaches for improved therapeutic targeting of Hedgehog signaling in cancer stem cells. Clin Cancer Res. 2015;21:505–513. doi: 10.1158/1078-0432.CCR-14-0507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Justilien V, Walsh MP, Ali SA, Thompson EA, Murray NR, Fields AP. The PRKCI and SOX2 oncogenes are coamplified and cooperate to activate Hedgehog signaling in lung squamous cell carcinoma. Cancer Cell. 2014;25:139–151. doi: 10.1016/j.ccr.2014.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rudin CM, Durinck S, Stawiski EW, Poirier JT, Modrusan Z, Shames DS, Bergbower EA, Guan Y, Shin J, Guillory J, Rivers CS, Foo CK, Bhatt D, Stinson J, Gnad F, Haverty PM, Gentleman R, Chaudhuri S, Janakiraman V, Jaiswal BS, Parikh C, Yuan W, Zhang Z, Koeppen H, Wu TD, Stern HM, Yauch RL, Huffman KE, Paskulin DD, Illei PB, Varella-Garcia M, Gazdar AF, de Sauvage FJ, Bourgon R, Minna JD, Brock MV, Seshagiri S. Comprehensive genomic analysis identifies SOX2 as a frequently amplified gene in small-cell lung cancer. Nat Genet. 2012;44:1111–1116. doi: 10.1038/ng.2405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Peifer M, Fernandez-Cuesta L, Sos ML, George J, Seidel D, Kasper LH, Plenker D, Leenders F, Sun R, Zander T, Menon R, Koker M, Dahmen I, Muller C, Cerbo V Di, Schildhaus HU, Altmuller J, Baessmann I, Becker C, Wilde B de, Vandesompele J, Bohm D, Ansen S, Gabler F, Wilkening I, Heynck S, Heuckmann JM, Lu X, Carter SL, Cibulskis K, Banerji S, Getz G, Park KS, Rauh D, Grutter C, Fischer M, Pasqualucci L, Wright G, Wainer Z, Russell P, Petersen I, Chen Y, Stoelben E, Ludwig C, Schnabel P, Hoffmann H, Muley T, Brockmann M, Engel-Riedel W, Muscarella LA, Fazio VM, Groen H, Timens W, Sietsma H, Thunnissen E, Smit E, Heideman DA, Snijders PJ, Cappuzzo F, Ligorio C, Damiani S, Field J, Solberg S, Brustugun OT, Iversen M Lund, Sanger J, Clement JH, Soltermann A, Moch H, Weder W, Solomon B, Soria JC, Validire P, Besse B, Brambilla E, Brambilla C, Lantuejoul S, Lorimier P, Schneider PM, Hallek M, Pao W, Meyerson M, Sage J, Shendure J, Schneider R, Buttner R, Wolf J, Nurnberg P, Perner S, Heukamp LC, Brindle PK, Haas S, Thomas RK. Integrative genome analyses identify key somatic driver mutations of small-cell lung cancer. Nat Genet. 2012;44:1104–1110. doi: 10.1038/ng.2396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hou JM, Greystoke A, Lancashire L, Cummings J, Ward T, Board R, Amir E, Hughes S, Krebs M, Hughes A, Ranson M, Lorigan P, Dive C, Blackhall FH. Evaluation of circulating tumor cells and serological cell death biomarkers in small cell lung cancerpatients undergoing chemotherapy. Am J Pathol. 2009;175:808–816. doi: 10.2353/ajpath.2009.090078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Naito T, Tanaka F, Ono A, Yoneda K, Takahashi T, Murakami H, Nakamura Y, Tsuya A, Kenmotsu H, Shukuya T, Kaira K, Koh Y, Endo M, Hasegawa S, Yamamoto N. Prognostic impact of circulating tumor cells in patients with small cell lung cancer. J Thorac Oncol. 2012;7:512–519. doi: 10.1097/JTO.0b013e31823f125d. [DOI] [PubMed] [Google Scholar]
- 31.Hou JM, Krebs MG, Lancashire L, Sloane R, Backen A, Swain RK, Priest LJ, Greystoke A, Zhou C, Morris K, Ward T, Blackhall FH, Dive C. Clinical significance and molecular characteristics of circulating tumor cells and circulating tumor microemboli in patients with small-cell lung cancer. J Clin Oncol. 2012;30:525–532. doi: 10.1200/JCO.2010.33.3716. [DOI] [PubMed] [Google Scholar]
- 32.Hiltermann TJ, Pore MM, van den Berg A, Timens W, Boezen HM, Liesker JJ, Schouwink JH, Wijnands WJ, Kerner GS, Kruyt FA, Tissing H, Tibbe AG, Terstappen LW, Groen HJ. Circulating tumor cells in small-cell lung cancer: a predictive and prognostic factor. Ann Oncol. 2012;23:2937–2942. doi: 10.1093/annonc/mds138. [DOI] [PubMed] [Google Scholar]
- 33.Hodgkinson CL, Morrow CJ, Li Y, Metcalf RL, Rothwell DG, Trapani F, Polanski R, Burt DJ, Simpson KL, Morris K, Pepper SD, Nonaka D, Greystoke A, Kelly P, Bola B, Krebs MG, Antonello J, Ayub M, Faulkner S, Priest L, Carter L, Tate C, Miller CJ, Blackhall F, Brady G, Dive C. Tumorigenicity and genetic profiling of circulating tumor cells in small-cell lung cancer. Nat Med. 2014;20:897–903. doi: 10.1038/nm.3600. [DOI] [PubMed] [Google Scholar]