

Abstract
Genomic imprinting is a complex epigenetic process that contributes substantially to embryogenesis, reproduction, and gametogenesis. Only small fraction of genes within the whole genome undergoes imprinting. Imprinted genes are expressed in a monoallelic parent-of-origin-specific manner, which means that only one of the two inherited alleles is expressed either from the paternal or maternal side. Imprinted genes are typically arranged in clusters controlled by differentially methylated regions or imprinting control regions. Any defect or relaxation in imprinting process can cause loss of imprinting in the key imprinted loci. Loss of imprinting in most cases has a harmful effect on fetal development and can result in neurological, developmental, and metabolic disorders. Since DNA methylation and histone modifications play a key role in the process of imprinting. This review focuses on the role of DNA methylation in imprinting process and describes DNA methylation aberrations in different imprinting disorders.
Keywords: DNA methylation, Genomic imprinting, Prader–Willi syndrome, Silver–Russell syndrome, Beckwith–Wiedemann syndrome, Type Ib pseudohypoparathyroidism
Introduction
Genomic imprinting (GI) is a unique phenomenon that occurs in placental mammals, marsupials, and a subset of flowering plants. Genomic imprinting plays a key role in maintaining normal embryogenesis, and prenatal and postnatal growth. In genomic imprinting, several epigenetic processes are involved to result in a unique epigenetic signature observed at a subset of loci in the genome [1, 2]. DNA methylation is the mainstay of establishing imprinting marks on either paternal or maternal alleles. Besides, histone modifications and non-coding RNAs contribute greatly to imprinting process [3, 4]. The predominant feature of genomic imprinting is the monoallelic gene expression in parent-of-origin-specific manner. It means that only one of the two inherited alleles is expressed either from the paternal or maternal side. When paternal allele is expressed, maternal copy is silenced, and vice versa [5].
Genomic imprinting is a complex form of epigenetic inheritance. One feature of the complexity of genomic imprinting is that it works in tissue-specific manner. For example, UBE3A is only imprinted in human brain where it is maternally expressed, while biallelic expression of UBE3A was reported in many tissues and cell lines apart from the brain [6, 7]. Another example is the tissue-specific imprinting of Gs-alpha from GNAS locus on chromosome 20q13. Gs-alpha is only maternally expressed in the renal tubules while it is expressed biallelically from most tissues [8]. Time-based or temporal dependence is an additional significant feature of imprinting, i.e., certain genes are biallelically expressed during embryogenesis while these genes are imprinted in parent-of-origin-specific manner after adulthood. For example, IGF2 is biallelically expressed in the fetal brain during the first trimester while IGF2 is expressed predominantly from the paternal allele in other tissues. However, during adulthood, certain parts of the human brain, such as the hypothalamus and globus pallidus, show paternal monoallelic expression of IGF2, whereas the pons shows biallelic expression of IGF2 [9]. Furthermore, individual genes often produce several transcripts, which can show different expression patterns; for example, some transcripts are imprinted and other transcripts are non-imprinted. PLAGL1 locus on chromosome 6q24 gives a good demonstration of this complex behavior of imprinting. PLAGL1 is composed of 12 exons with four different promoters. PLAGL1 transcripts initiated from promoters 2, 3, and 4 are biallelically expressed in most tissues, whereas only PLAGL1 transcripts initiated from promoter 1 (ICR/P1) are paternally expressed [10].
Interestingly, several imprinted genes are conserved among different species (human, mice, pigs, etc.). According to the most recent studies, about 127 imprinted genes in mice and around 94 imprinted genes in human have been reported and identified [11, 12]. Most of these imprinted genes are grouped together in clusters and loci. Each locus has at least one differentially methylated region (DMR). DMRs in most imprinted loci comprise CpG-rich regions called imprinting control regions (ICRs), which work as cis-acting regulatory elements controlling imprinted genes. ICRs are characterized by their monoallelic DNA methylation and distinct histone marks that achieve monoallelic parent-of origin-specific expression [13, 14].
Epigenetic reprogramming and imprinting
After fertilization, male and female pronuclei go through several changes before their fusion. One of these eminent changes is epigenetic reprogramming (ER) which plays a crucial role in early embryogenesis and gametogenesis. ER is a dynamic process of erasing and reestablishing epigenetic marks in embryonic genome during the early embryonic development. ER occurs in two cycles or waves. The first cycle occurs during the blastocyst stage and the second cycle occurs in the primordial germ cell development. There are specific regions of the genome that undergo different conditions of ER, e.g., imprinted genes and most repetitive elements [15, 16].
Most repetitive elements maintain their DNA methylation patterns throughout the embryonic development without change. Similarly, imprinted genes evade the first cycle of epigenetic reprogramming by maintaining their epigenetic marks. However, imprinted genes undergo resetting of these epigenetic marks during the stage of primordial germ cell development. Reestablishing these marks occurs in parent-of-origin-specific manner [17, 18]. Therefore, any error or defect in the process of epigenetic reprogramming of ICRs can result in loss of imprinting [19, 20].
DNA methylation and genomic imprinting
Imprinting is of epigenetic nature rather than DNA sequence dependent. Imprinting marks are established in gametes during primordial germ cell development in a parent-of-origin-specific manner. In mammals, DNA methylation and histone modifications play crucial role in establishing the imprinting marks [14, 21]. In imprinted loci, ICRs control expression of imprinted genes in unique mechanisms depending on ICR position. ICRs are found either at the intergenic region (known as intergenic ICRs) or at the promoter region of certain gene within the imprinted locus (known as promoter ICRs) [22, 23]. Many promoter ICRs control the expression of non-coding RNA that regulates imprinting of other genes in the imprinted locus. For example, ICR2 is located in the promoter region of KCNQ1OT1 and regulates the CDKN1C/KCNQ1 locus. KCNQ1OT1 is a long non-coding RNA, which silences genes located in cis and functions in a similar manner to Xist RNA [24–26]. Typically, ICR2 is maternally methylated and paternally unmethylated. Therefore, KCNQ1OT1 RNA expression is maternally silenced, whereas KCNQ1OT1 is paternally expressed. As a result, imprinted genes of the CDKN1C/KCNQ1 locus are maternally expressed and paternally silenced [27].
Intergenic ICRs generally regulate imprinting in a different way through functioning as a transcription insulator in the imprinted cluster. For example, ICR1, which controls H19/IGF2 cluster, exists in the intergenic region between H19 and IGF2 genes. DNA hypomethylation of ICR1 assists the binding of the transcriptional repressor protein called CCCTC-binding factor (CTCF) that insulates IGF2 from downstream enhancers. Without CTCF binding, the downstream enhancers can access IGF2 promoter resulting in expression of IGF2 [28, 29]. Typically, paternal ICR1 of the H19/IGF2 cluster is hypermethylated, whereas maternal ICR1 is hypomethylated. Therefore, insulin-like growth factor 2 (IGF2) is monoallelically expressed from the paternal allele and silenced in the maternal allele. Interestingly, most promoter ICRs are maternally methylated while several intergenic ICRs are methylated on the paternal side [30, 31].
Imprinting disorders
Imprinting disorders always result from loss of imprinting in the key imprinted loci, whereas loss of imprinting does not always result in imprinted disorders. For example, loss of imprinting of H19 and IGF2 has been reported in phenotypically healthy infants, and this reflects a broad spectrum of phenotypes that can be observed in association with loss of imprinting [32]. Loss of imprinting results from specific four causes, which are deletions, mutations, uniparental disomy, and epimutations. These causes affect the imprinted loci and result in aberrant silencing of the active allele or expression of the inactive allele. Mutations or deletions are common causes of imprinting disorders and can affect genes, promoters, intergenic regions, and ICRs within the imprinted loci [33, 34]. Uniparental disomy (UPD) represents another cause of imprinting disorders. UPD refers to the abnormal receiving of a chromosome pair from one parent and no chromosome for this pair from the other parent. UPD results in inheriting two paternal copies of chromosome or two maternal copies [35]. Epigenetic disruption or epimutation is also a significant cause for loss of imprinting. Epimutations can result from aberrations of DNA methylation in ICRs or disruptions in histone modifications within the imprinted clusters [36, 37].
Since environmental factors can affect epigenetic marks throughout the genome altering gene expression. Similarly, using assisted reproductive technology (ART) can affect epigenetic marks in the imprinted loci disrupting genomic imprinting and altering expression of imprinted genes. ART has been associated with an increased incidence of imprinted disorders, e.g., Angleman, Prader–Willi, and Beckwith–Wiedemann syndromes, compared to natural conception [38–40]. However, the absolute risk of developing imprinting disorders in ART-conceived children was low, and the combined odds ratio of any imprinting disorder in ART-conceived children was 3.67 (95% confidence interval, 1.39–9.74). Moreover, there was no significant association between ART and the methylation status of imprinted genes in the same meta-analysis [41]. Therefore, further investigations are warranted to unravel the correlation of ART with epigenetic changes and dysregulation of genomic imprinting, and to find out the absolute risk of developing imprinting disorders using different protocols of ART.
Imprinting disorders were firstly identified as separate disorders with their unique phenotypes and (epi)genetic causes (Table 1). However, recent studies reported that some patients with specific ID show multi-locus imprinting disturbances (MLID) in other imprinted loci [42, 43]. MLID were first reported in transient neonatal diabetes mellitus (TNDM) patients, who showed mosaic DNA hypomethylation in other imprinting regions other than the PLAGL1 locus on chromosome 6q24.2 [44]. About 50% of TNDM patients, who had hypomethylation of ICR/P1, reported another epimutation on chromosome 11p15 (i.e., hypomethylation of ICR2 or KvDMR1 which is very common epimutation in Beckwith–Wiedemann syndrome (BWS) patients) [45]. Similarly, MLID have been reported in other imprinting disorders but with lower frequency than TNDM, for example, Silver–Russell syndrome (SRS), pseudohypoparathyroidism type 1b (PHP1b), Angelman syndrome (AS), and Beckwith–Wiedemann syndrome [46–49]. Intriguingly, MLID can occur on either maternal or paternal imprinted regions with either hypomethylation or hypermethylation of DMRs. In a special case, TNDM patients, who showed MLID, reported only hypomethylation in other imprinting loci with no reports on hypermethylation [42, 50]. This review discusses different imprinting disorders and the underlying (epi)genetic causes in detail.
Table 1.
Overview of different imprinting disorders in humans
| Imprinting disorder | Imprinted domain | Cytogenetic abnormality | Uniparental disomy | Imprinting center epimutation | Deletions/duplications involving imprinting center | Gene mutation | MLID | References |
|---|---|---|---|---|---|---|---|---|
| Transient neonatal diabetes 1 (TNDM1, OMIM 601410) | 6q24 | Visible paternal duplication of 6q24 (2%) | Paternal UPD6 (35–40%) | Hypomethylation of the maternal ICR/P1 (20%) | Submicroscopic duplication of the 6q24 region on the paternal allele (35–40%) | Reported | [45, 51–53] | |
| Silver–Russell syndrome (SRS, OMIM 180860) | 11p15.5 | Maternal duplication of 11p15 (<2%) | Maternal UPD11 (1 case) | Hypomethylation of the paternal IC1 (up to 70%) | IC2 duplication on maternal allele (1 case) | Reported | [54–59] | |
| 7p12.2, 7q32.2 | Chromosomal rearrangement (<2%) | Maternal UPD7 (5%) | ||||||
| Beckwith–Wiedemann syndrome (BWS, OMIM 130650) | 11p15.5 | Chromosomal rearrangement (1–2%) | Paternal UPD11 (20%) | • Hypermethylation of the maternal IC1 (5%) • Hypomethylation of the maternal IC2 (50%) |
• IC1 deletion on maternal allele (<5%) • IC2 deletion on maternal allele (<2%) |
CDKN1C on maternal allele (5% sporadic, 50% in familial cases) | Reported | [29, 46, 60–65] |
| Prader–Willi Syndrome (PWS, OMIM 176270) | 15q11–q13 | • Deletions of 15q11.2–q13 on maternal allele (65–75%) • Chromosomal rearrangement (<1%) |
Maternal UPD15 (25–30%) | • 1% of PWS is due to imprinting defects (epimutations) • Hypermethylation of paternal PWS/ICR (75–80% of imprinting defects cases) |
Deletions in the regulatory region located around PWS/ICR on the paternal allele (15% of imprinting defect cases) | Not reported | [66–70] | |
| Angelman syndrome (AS, OMIM 105830) | 15q11–q13 | Deletions of 15q11.2–q13 on maternal allele (70%) Chromosomal rearrangement (<1%) |
Paternal UPD15 (7%) | • 2–5% of AS is due to imprinting defects (epimutations) • Hypomethylation of the SNRPN DMR on the maternal allele (80–90% of imprinting defects cases |
Deletions in the regulatory region located around AS/ICR on the maternal allele (10–20% of imprinting defect cases) | UBE3A on maternal allele (11%) | Reported once | [66, 67, 71–75] |
| Pseudohypoparathyroidism type 1b (PHP1B, OMIM 603233) | 20q13 | Microdeletion in STX16 (in about 20% of PHP1B patients with exon 1A ICR hypomethylation) | Paternal UPD20 (few cases reported) | Hypomethylation of ICR of exon 1A on the maternal allele (sporadic cases) | Deletion in NESPAS and NESP55 (very few familial cases) | Reported | [47, 48, 76–82] |
Transient neonatal diabetes mellitus
Transient neonatal diabetes mellitus (TNDM) is a growth retardation syndrome that is associated with persistent hyperglycemia during the first 6 weeks of life because of insulin deficiency. Recovery occurs in approximately 50% of these neonates after 18 months of birth. The only treatment of TNDM is exogenous insulin during persistent hyperglycemia. TNDM has a very low incidence (about 1 in 400,000 births in UK) [83–85]. Besides, neonates, who had TNDM, are more likely to develop type 2 diabetes (T2DM) later in life. TNDM has three types according to the responsible genes. TNDM1 is associated with defect in the expression of pleomorphic adenoma gene-like 1 (PLAGL1) on 6q24.2 locus and to lesser extent, the expression of zinc finger protein 57 homolog (ZFP57) on 6p22.1 locus. Meanwhile, TNDM2 and TNDM3 are associated with mutations in ABCC8 on chromosome 11p15.1 and KCNJ11 on chromosome 11p15.1 locus, respectively. TNDM1 is an imprinted form of TNDM, whereas TNDM2 and TNDM3 are non-imprinted forms [86–89]. Paternal UPD of chromosome 6 (35–40% of TNDM1) and duplication of 6q24 (35–40% of TNDM1) are the main mechanisms of TNDM1 [85, 90, 91]. However, aberrant DNA methylation in the ICR/P1 of 6q24 locus occurs in about 20% of TNDM1 patients.
ZFP57 encodes a transcription repressor which recognizes specific DNA sequences and forms a complex with co-repressor protein called KRAB-associated protein-1 (KAP1) [92, 93]. Kap1 recruits other proteins, for example, histone H3-K9 methyltransferase-4 (SETDB1) and nuclear protein 95 (NP95), which in turn recruit DNA methyltransferases (DNMTs). Hence, this complex plays a crucial role in regulating and maintaining DNA methylation at different ICRs [94, 95]. Either mutation or 1-bp deletion in ZFP57 gene can give rise to a truncated protein, which losses its function as a regulator of DNA methylation at ICRs. As a result, MLID and hypomethylation occur in certain imprinted loci leading to loss of imprinting. However, ZFP57 mutations are rare and were reported in only few cases of TNDM1 [43, 47, 50, 53].
PLAGL1 is a transcription regulator and tumor suppressor candidate gene encoding a zinc finger protein, which controls cell cycle and apoptosis. Zinc finger protein PLAGL1 has another role in inducing the transcription of pituitary adenylate cyclase-activating polypeptide (PACAP) receptor, which enhances insulin secretion and works as a regulator of pancreatic β-cells [96, 97]. Therefore, overexpression of PLAGL1 results in upregulation of PACAP receptor expression, which in turn deregulates the function of pancreatic β-cells and causes the development of TNDM1 [98–100]. Recent reports, which studied intrauterine growth restriction (IUGR) in human and mice, have revealed the novel role of PLAGL1 as a main regulator to various imprinted and non-imprinted genes network involved in cellular growth and metabolism. Therefore, up- or downregulation of PLAGL1 has been associated with altered expression levels of certain genes regulating metabolism, e.g., IGF2, H19, SLC2A4, CDKN1C, and PPARγ1 [101, 102]. This suggests an alternative mechanism of developing TNDM1 due to overexpression of PLAGL1.
PLAGL1 comprises of 12 exons controlled by four distinct promoters producing different transcripts and isoforms. The biallelic transcripts of PLAGL1 arise from promoters P2, P3, and P4, while only the PLAGL1 transcripts from the ICR/P1 promoter are paternally expressed [10]. Another important gene on PLAGL1 locus is HYMAI, which encodes a long non-coding RNA (lncRNA). HYMAI is one-exon gene that has an overlapping transcription start site with the PLAGL1 transcripts from the ICR/P1 promoter. Therefore, the methylation status of ICR/P1 promoter regulates the expression of HYMAI and certain PLAGL1 transcripts (Fig. 1) [103, 104]. Typically, the paternal ICR/P1 is hypomethylated while the maternal ICR/P1 is hypermethylated. Therefore, HYMAI and PLAGL1 transcripts, which are regulated by ICR/P1 promoter, are paternally expressed and maternally silenced [105]. Several studies have reported the loss of methylation in the maternal allele in TNDM1 patients and its correlation with higher weight and body mass index [106, 107]. Aberrant hypomethylation of ICR/P1 on the maternal allele results in overexpression of PLAGL1 and HYMAI, which accounts for about 20% of TNDM1 patients [51, 53]. Moreover, recent studies have shown the association between the methylation status of PLAGL1 promoter on chromosome 6 and different types of cancer, e.g., ovarian cancer and soft-tissue sarcomas [108, 109].
Fig. 1.

Imprinting at the PLAGL1 locus. a PLAGL1 is composed of 12 exons with four different promoters; PLAGL1 transcripts from promoter 1 (ICR/P1) are only expressed paternally. In contrast, PLAGL1 transcripts from P2, P3, and P4 are biallelically expressed in most tissues. HYMAI is one-exon gene encoding a long non-coding RNA (lncRNA). HYMAI exon is also expressed paternally due to sharing promoter 1 (ICR/P1) with PLAGL1. Typically, HYMAI and ICR/P1 transcripts of PLAGL1 are silenced on the maternal allele due to the methylated ICR/P1. On the other hand, the paternal ICR/P1 is unmethylated allowing HYMAI and ICR/P1 transcripts of PLAGL1 to be expressed. b In transient neonatal diabetes mellitus (TNDM1), ICR/P1 transcripts of PLAGL1 and HYMAI are biallelically expressed due to loss of methylation in the maternal allele or duplication of 6q24 on the paternal allele or paternal UPD6
Silver–Russell syndrome
Silver–Russell syndrome (SRS) is a very rare growth retardation disorder characterized by dwarfism, triangular face, congenital hemihypertrophy (asymmetric body), and low birth weight. Malnutrition and hypoglycemia commonly occur in SRS patients. Incidence rate of SRS is approximately 1 in 100,000 births in the USA [110]. SRS is a distinct genomic imprinting disorder because the pathogenesis of SRS depends primarily on epimutation and methylation status of ICR1 at H19/IGF2 cluster. In addition to epimutation, maternal uniparental disomy of chromosome 7 (UPD7) has been reported in some patients showing less severe phenotype of SRS [111–113]. Intriguingly, ICR1 hypomethylation of H19/IGF2 cluster on chromosome 11p15 occurs in SRS while hypermethylation of ICR1 is found in roughly 10% of Beckwith–Wiedemann syndrome (BWS) patients [30, 114].
Normally, paternal ICR1 in H19/IGF2 cluster is hypermethylated while maternal allele is hypomethylated. Hypomethylation of the maternal allele triggers H19 expression and silences expression of maternal IGF2. Hypermethylation of ICR1 impedes its binding to the transcriptional repressor CTCF leading to IGF2 expression and silencing of H19 expression [115, 116]. Therefore, IGF2 is entirely expressed from the paternal allele while H19 is expressed predominantly from the maternal side. Insulin-like growth factor 2 (IGF2) is an important hormone contributing in cellular proliferation and growth promotion. In SRS, hypomethylation of paternal ICR1 prevents IGF2 expression triggering defective prenatal development and growth retardation. However, there is no correlation between the level of ICR1 hypomethylation and the severity of SRS symptoms [30, 113].
Beckwith–Wiedemann syndrome
Beckwith–Wiedemann syndrome (BWS) is a rare imprinting disorder classically characterized by prenatal and postnatal overgrowth. BWS patients have distinctive manifestations, e.g., macroglossia (large tongue), macrosomia (higher birth weight than average), abdominal wall defects, hemihypertrophy (asymmetric body), and neonatal hypoglycemia. BWS has low incidence rate worldwide, i.e., about one in every 13,700 birth [61, 117]. Approximately 10% of BWS patients could develop pediatric malignancies, e.g., hepatoblastoma and Wilms tumor [118, 119]. These malignancies occur mostly before age of four; therefore, routine abdominal ultrasound and serum alpha-fetoprotein (AFP) measurements are warranted from birth until this age [120]. In BWS, epimutation of ICR1 or ICR2 accounts for about 50–60% of all cases, while paternal UPD11 accounts for 20% of cases. Additionally, CDKN1C mutation is responsible for 5% of sporadic cases and about 50% of familial BWS patients [29, 65].
CDKN1C/KCNQ1 cluster and H19/IGF2 cluster within chromosome 11p15 are the key players in most BWS patients. CDKN1C/KCNQ1 cluster is an imprinted genes cluster located centromerically to the H19/IGF2 cluster and controlled by ICR2 or KvDMR1. DNA methylation status of ICR2 decides whether paternal or maternal copy of genes will be expressed [121]. As we discussed before, CDKN1C/KCNQ1 cluster is regulated by the expression of a long non-coding RNA called KCNQ1-overlapping transcript 1 (KCNQ1OT1) or long QT intronic transcript 1 (LIT1). KCNQ1OT1 functions as an antisense to KCNQ1 and other genes in the CDKN1C/KCNQ1 domain silencing their expression. When ICR2 on an allele is hypomethylated, KCNQ1OT1 is expressed and silences this allele (Fig. 2) [122, 123]. Besides, cyclin-dependent kinase inhibitor 1C (CDKN1C) functions as a tumor suppressor gene inhibiting G1 cyclin/CDK complexes [124, 125].
Fig. 2.

Imprinting at the H19–IGF2 locus and the CDKN1C/KCNQ1 locus. a On maternal allele, ICR1 is unmethylated while ICR2 is methylated. Therefore, H19, KCNQ1, and CDKN1C are maternally expressed, whereas IGF2 and KCNQ1OT1 are silenced. In contrast, ICR1 is methylated and ICR2 is unmethylated on the paternal allele. Therefore, IGF2 and KCNQ1OT1 are paternally expressed while other genes are silenced. b Beckwith–Wiedemann syndrome (BWS) occurs due to paternal UPD11 and epimutations of ICR1 or ICR2. Hence, the maternal allele act as the paternal allele with subsequent IGF2 overexpression or loss of CDKN1C expression
Normally, paternal ICR2 is hypomethylated while maternal ICR2 is methylated. Therefore, paternal KCNQ1OT1 is expressed and is blocking expression of CDKN1C/KCNQ1 domain. Whereas, methylation of maternal ICR2 prevents KCNQ1OT1 expression allowing CDKN1C to be maternally expressed. In about 50% of BWS patients, maternal ICR2 is aberrantly hypomethylated besides the normally hypomethylated paternal ICR2 [126]. Consequently, KCNQ1OT1 is expressed biallelically resulting in diminished expression of CDKN1C [127].
In addition to epimutation of CDKN1C/KCNQ1 domain, aberrant DNA methylation of ICR1 in H19/IGF2 cluster occurs in roughly 5–10% of BWS patients [61, 128]. Normally, IGF2 is paternally expressed due to hypermethylation of ICR1. Aberrantly, hypermethylation of maternal ICR1 causes overexpression of IGF2 in neonates and displays BWS phenotype. Overall, overgrowth and predisposition to tumors in BWS patients is due to the loss of CDKN1C or overexpression of IGF2 [129]. Methylation status of both ICR1 and ICR2 has been used to diagnose BWS and SRS prenatally [130, 131].
Prader–Willi syndrome
Prader–Willi syndrome (PWS) is a rare neurodevelopmental disorder characterized by hypotonia, short stature, delayed cognitive development, and behavioral complications. PWS is one of the leading genetic causes of pediatric obesity since PWS patients have insatiable appetite with concurrent hyperphagia. PWS has low incidence rate worldwide, i.e., about one in every 10,000 to 25,000 live birth [132, 133]. PWS results from the loss of expression of the paternally imprinted genes on chromosome 15q11.2–q13. The major mechanisms involved in PWS pathogenesis are 15q11.2–q13 microdeletions on the paternal allele (67–75% of PWS patients) and maternal UPD (25–30% of PWS patients). Epimutations in chromosome 15q11.2–q13 is rare in PWS and accounts only for 1% of PWS cases [66, 134].
On chromosome 15q11.2–q13, there are two adjacent ICRs, so-called Prader–Willi syndrome (PWS) ICR and Angelman syndrome (AS) ICR. AS/PWS ICRs control imprinting process in this SNURF/SNRPN imprinted cluster [135, 136]. Methylation of AS/PWS ICRs plays a very crucial role in either silencing or triggering the expression of small nuclear ribonucleoprotein polypeptide N (SNRPN) and the adjacent small nucleolar RNAs (snoRNAs) on the same allele (Fig. 3) [137]. SNRPN encodes a protein that is essential for the formation of spliceosomes, which are responsible for alternative splicing of different mRNAs [138].
Fig. 3.

Imprinting at the SNURF/SNRPN cluster. AS/PWS ICRs regulate imprinting in SNURF/SNRPN cluster. In most tissues, UBE3A is biallelically expressed, whereas UBE3A in the brain is only expressed from the maternal side. Typically, PWS ICR is methylated on the maternal allele while it is unmethylated on the paternal allele. Chromosomal deletions, UPD, UBE3A mutations, and aberrant DNA methylation at AS/PWS ICRs are the major mechanisms behind developing Angelman syndrome (AS) and Prader–Willi syndrome (PWS)
Ubiquitin protein ligase E3A (UBE3A) is another important gene in SNURF/SNRPN cluster, which is downstream to SNRPN and snoRNAs. UBE3A encodes E3 ligase, which is involved in the ubiquitination of targeted proteins and ubiquitin/proteasome signaling. Imprinting of UBE3A is only observed in the brain to express a well-adjusted level of E3 ligase to maintain the critical function of neurons and the typical synaptic development [139]. Therefore, deletion or overexpression of UBE3A results in aberrant dendritic networks, irregular synaptic connections, and abnormal levels of neurotransmitters in Drosophila and mice models [140, 141].
Normally, AS/PWS ICRs are both unmethylated on the paternal allele permitting the paternal expression of SNRPN and snoRNAs with silencing the paternal expression of UBE3A. In contrast, PWS ICRs on the maternal allele is differentially methylated resulting in absent maternal expression of SNRPN and snoRNAs with subsequent triggering expression of the maternal UBE3A. Inappropriate silencing of paternal allele or maternal UPD of SNURF/SNRPN cluster results in loss of expression of SNRPN, snoRNAs, and other genes. Failure to express these genes leads to the development of PWS [142–144].
Angelman syndrome
Angelman syndrome (AS) is a complex genomic imprinting disorder characterized by developmental delay, mental retardation, ataxia (movement disorder), and seizures. However, the most distinctive feature of AS is the behavioral symptoms such as paroxysmal laughter, excitable personality, hyperactivity, and happy facial appearance. Therefore, AS is also referred to as happy puppet syndrome [145]. AS has very low incidence rate, i.e., about 1 in every 12,000 to 24,000 live birth [146, 147]. AS is caused by the absence of expression of maternal UBE3A from SNURF/SNRPN cluster on chromosome 15q11.2–q13 [148, 149].
As previously described in PWS, AS/PWS ICRs regulate imprinting in SNURF/SNRPN cluster. In the brain, paternal expression of SNRPN and snoRNAs warrants a well-balanced expression of UBE3A only from the maternal allele, which ensures typical cognitive functions and normal development of the brain [150]. The primary mechanisms involved in AS pathogenesis are chromosomal deletions in maternal allele (65–75% of AS cases), paternal UPD (7%), and genetic mutations in UBE3A (11%). AS is rarely caused by epigenetic defect or DNA methylation aberration in AS/PWS ICRs (only 2–5% of AS cases) [151]. Quantification of DNA methylation at SNRPN locus can represent a sensitive and specific technique for screening and diagnosing PWS and AS in numerous cases [152].
Pseudohypoparathyroidism type 1b
Pseudohypoparathyroidism (PHP) is an extremely rare group of genetic disorders, which result from resistance to parathyroid hormone (PTH). Increased serum PTH, hypocalcaemia, and hyperphosphatemia are the major biochemical features of PHP. PHP type 1b (PHP1b) is a subtype of PHP which displays PTH resistance only in the renal tissue. Most cases of PHP1b do not show the clinical features of Albright hereditary osteodystrophy (AHO) while AHO is dominant in PHP1a [153]. PHP1b is considered an imprinting disorder caused by defect or epimutation in ICRs which control the imprinting of GNAS locus on chromosome 20q13.3 [77, 82]. GNAS locus is an intricate imprinted locus, which encodes four transcripts (i.e., Gs-alpha, NESP55, A/B transcript, and XLAS) and the antisense NESPAS by alternative mRNA splicing or using different promoters and first exons [8, 154]. Imprinting of GNAS locus is controlled by three distinct ICRs or DMRs that are next to the promoters of exon 1A, XLAS, and NESP55. When these ICRs are methylated, the corresponding promoters are repressed and genes expression silenced. Therefore, NESP55 is maternally expressed due to non-methylated ICR of NESP55 promoter on the maternal allele (Fig. 4) [155]. Similarly, A/B transcript, XLAS, and NESPAS are paternally expressed in most tissues. GNAS locus expresses Gs-alpha by using exons 1–13 whereas it can alternatively express A/B transcript by using exon 1A (also referred to as exon A/B) as a substitute first exon. A/B transcript acts in cis as a negative regulator to Gs-alpha expression tuning the tissue-specific imprinting of Gs-alpha particularly in the renal tubules [156].
Fig. 4.

Imprinting at the GNAS locus. Gs-alpha is only expressed from the maternal allele in the renal tubules while it is biallelically expressed from most tissues
The stimulatory G protein alpha-subunit (Gs-alpha) is one of GNAS domain transcripts, which is of vital importance in cell signaling cascades. When a specific ligand binds to G protein-coupled receptor (GPCR), this activates Gs-alpha that in turn triggers cAMP-dependent pathway [157]. Parathyroid hormone 1 receptor (PTH1R) is a member of the GPCR family, which requires Gs-alpha for signal transduction and physiological response to PTH [158]. Biallelic expression of Gs-alpha is observed in most cell lines and body tissues. However, Gs-alpha shows prominently tissue-specific imprinting controlled by the ICR of exon 1A where Gs-alpha is only maternally imprinted in the kidney, pituitary gland, and gonads. In the kidney, ICR of exon 1A on the maternal allele is methylated while this ICR of the paternal allele is non-methylated. DNA methylation of this ICR inhibits the expression of the A/B transcript with the subsequent typical expression of Gs-alpha. Hence, Gs-alpha is maternally expressed while the A/B transcript is paternally expressed [159, 160]. In PHP1b, DNA methylation defect in ICRS and loss of imprinting result in A/B transcript expression from the maternal allele, which suppresses the maternal Gs-alpha expression (Fig. 5). As a result, decreased or absent expression of Gs-alpha in the renal tubules confers renal resistance to PTH with subsequent biochemical changes, for example, hypocalcaemia and hyperphosphatemia. Furthermore, recent reports showed that maternally inherited microdeletions in STX16 locus, which is not imprinted locus and exists upstream to GNAS locus, are associated with the loss of imprinting in GNAS locus displaying PHP1b phenotype [76, 161, 162].
Fig. 5.
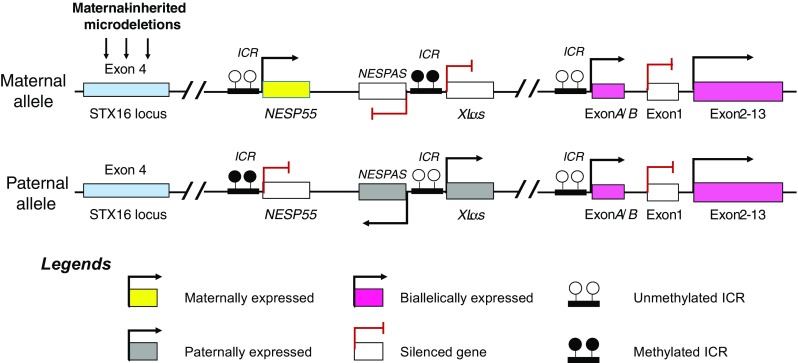
Loss of imprinting at the GNAS locus resulting in pseudohypoparathyroidism type 1b (PHP1b). In addition, maternally inherited microdeletions at the STX16 locus are associated with phenotypic PHP1b
Conclusion
Genomic imprinting is an epigenetic phenomenon that is involved in normal growth, viability, and embryonic development. Only small fraction of genes within the whole genome undergoes imprinting. These imprinted genes are typically organized in clusters controlled by imprinting control regions. The expression of these genes occurs in a parent-of-origin-specific manner. Therefore, imprinted genes are expressed only from one parent, while the other parent’s copies of these genes are silenced. However, tissue-specific expression of some imprinted genes was reported due to the complexity of genomic imprinting. DNA methylation and histone modifications play the key role in the process of imprinting.
Imprinting clusters are susceptible to several mutations, deletions, and epigenetic aberrations that result in loss of imprinting and imprinting disorders. Besides, the extensive use of assisted reproductive technology (ART) in conception is associated with an increased incidence of imprinting disorders. Imprinting disorders are complex syndromes of neurodevelopmental disabilities, cognitive problems, and metabolic diseases. DNA sequencing and DNA methylation analysis of ICRs/DMRs in different clusters represent a standard method for diagnosing imprinting disorders.
Abbreviations
- DMRs
differentially methylated regions
- ICR
imprinting control region
- IGF2
insulin-like growth factor 2
- UPD
uniparental disomy
- ER
epigenetic reprogramming
- ART
assisted reproductive technology
- MLID
multi-locus imprinting disturbances
- TNDM
transient neonatal diabetes mellitus
- PWS
Prader–Willi syndrome
- SRS
Silver–Russell syndrome
- PHP1b
pseudohypoparathyroidism type 1b
- AS
Angelman syndrome
References
- 1.Feil R, Berger F. Convergent evolution of genomic imprinting in plants and mammals. Trends Genet. 2007;23(4):192–199. doi: 10.1016/j.tig.2007.02.004. [DOI] [PubMed] [Google Scholar]
- 2.Chao W. Genomic imprinting. Handb Epigenetics. 2011; p. 353–79.
- 3.Mcewen KR, Ferguson-smith AC. Genomic imprinting—a model for roles of histone modifications in epigenetic control. Epigenomics. 2009; p. 235–58. Available from: http://www.springerlink.com/index/10.1007/978-1-4020-9187-2
- 4.Maupetit-Méhouas S, Montibus B, Nury D, Tayama C, Wassef M, Kota SK, et al. Imprinting control regions (ICRs) are marked by mono-allelic bivalent chromatin when transcriptionally inactive. Nucleic Acids Res. 2016;44:621–635. doi: 10.1093/nar/gkv960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Renfree MB, Suzuki S, Kaneko-Ishino T. The origin and evolution of genomic imprinting and viviparity in mammals. Philos Trans R Soc Lond B Biol Sci. [Internet]. 2013;368:20120151. Available from: http://europepmc.org/articles/PMC3539366 [DOI] [PMC free article] [PubMed]
- 6.Rougeulle C, Glatt H, Lalande M. The Angelman syndrome candidate gene, UBE3A/E6-AP, is imprinted in brain. Nat Genet. 1997;17:14–15. doi: 10.1038/ng0997-14. [DOI] [PubMed] [Google Scholar]
- 7.Lalande M, Calciano MA. Molecular epigenetics of Angelman syndrome. Cell Mol Life Sci. 2007; p. 947–60. [DOI] [PMC free article] [PubMed]
- 8.Bastepe M. The GNAS locus and pseudohypoparathyroidism. Adv Exp Med Biol. 2008; p. 27–40. [DOI] [PubMed]
- 9.Wilkinson LS, Davies W, Isles AR. Genomic imprinting effects on brain development and function. Nat Rev Neurosci. 2007;8:832–843. doi: 10.1038/nrn2235. [DOI] [PubMed] [Google Scholar]
- 10.Iglesias-Platas I, Court F, Camprubi C, Sparago A, Guillaumet-Adkins A, Martin-Trujillo A, et al. Imprinting at the PLAGL1 domain is contained within a 70-kb CTCF/cohesin-mediated non-allelic chromatin loop. Nucleic Acids Res. 2013;41:2171–2179. doi: 10.1093/nar/gks1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Baran Y, Subramaniam M, Biton A, Tukiainen T, Tsang EK, Rivas MA, et al. The landscape of genomic imprinting across diverse adult human tissues. Genome Res. 2015;25:927–936. doi: 10.1101/gr.192278.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Babak T, DeVeale B, Tsang EK, Zhou Y, Li X, Smith KS, et al. Genetic conflict reflected in tissue-specific maps of genomic imprinting in human and mouse. Nat Genet. [Internet]. NIH Public Access; 2015; [cited 2017 Feb 13];47:544–9. Available from: http://www.ncbi.nlm.nih.gov/pubmed/25848752 [DOI] [PMC free article] [PubMed]
- 13.Messerschmidt DM, Knowles BB, Solter D. DNA methylation dynamics during epigenetic reprogramming in the germline and preimplantation embryos. Genes Dev. 2014:812–28. [DOI] [PMC free article] [PubMed]
- 14.Ferguson-Smith AC. Genomic imprinting: the emergence of an epigenetic paradigm. Nat Rev Genet. 2011;12:565–575. doi: 10.1038/nrg3032. [DOI] [PubMed] [Google Scholar]
- 15.Cantone I, Fisher AG. Epigenetic programming and reprogramming during development. Nat Struct Mol Biol. 2013;20:282–9. [DOI] [PubMed]
- 16.Morgan HD, Santos F, Green K, Dean W, Reik W. Epigenetic reprogramming in mammals. Hum. Mol. Genet. 2005. [DOI] [PubMed]
- 17.Seisenberger S, Peat JR, Hore TA, Santos F, Dean W, Reik W. Reprogramming DNA methylation in the mammalian life cycle: building and breaking epigenetic barriers. Philos Trans R Soc Lond Ser B Biol Sci. 2013;368:20110330. doi: 10.1098/rstb.2011.0330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Marcho C, Cui W, Mager J. Epigenetic dynamics during preimplantation development. Reproduction. 2015;150:R109–R120. doi: 10.1530/REP-15-0180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Paczkowski M, Schoolcraft WB, Krisher RL. Dysregulation of methylation and expression of imprinted genes in oocytes and reproductive tissues in mice of advanced maternal age. J Assist Reprod Genet. 2015;32:713–723. doi: 10.1007/s10815-015-0463-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Elhamamsy AR. DNA methylation dynamics in plants and mammals: overview of regulation and dysregulation. Cell Biochem Funct. 2016;34(5):289–298. doi: 10.1002/cbf.3183. [DOI] [PubMed] [Google Scholar]
- 21.Sha K. A mechanistic view of genomic imprinting. Annu Rev Genomics Hum Genet. 2008;9:197–216. doi: 10.1146/annurev.genom.122007.110031. [DOI] [PubMed] [Google Scholar]
- 22.Sleutels F, Zwart R, Barlow DP. The non-coding Air RNA is required for silencing autosomal imprinted genes. Nature. 2002;415:810–813. doi: 10.1038/415810a. [DOI] [PubMed] [Google Scholar]
- 23.Royo H, Cavaillé J. Non-coding RNAs in imprinted gene clusters. Biol Cell. 2008;100:149–166. doi: 10.1042/BC20070126. [DOI] [PubMed] [Google Scholar]
- 24.Pandey RR, Mondal T, Mohammad F, Enroth S, Redrup L, Komorowski J, et al. Kcnq1ot1 antisense noncoding RNA mediates lineage-specific transcriptional silencing through chromatin-level regulation. Mol Cell. 2008;32:232–246. doi: 10.1016/j.molcel.2008.08.022. [DOI] [PubMed] [Google Scholar]
- 25.Chiesa N, De Crescenzo A, Mishra K, Perone L, Carella M, Palumbo O, et al. The KCNQ1OT1 imprinting control region and non-coding RNA: new properties derived from the study of Beckwith-Wiedemann syndrome and Silver-Russell syndrome cases. Hum Mol Genet. 2012;21:10–25. doi: 10.1093/hmg/ddr419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brockdorff N. Noncoding RNA and Polycomb recruitment. RNA. 2013;19:429–442. doi: 10.1261/rna.037598.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Higashimoto K, Soejima H, Saito T, Okumura K, Mukai T. Imprinting disruption of the CDKN1C/KCNQ1OT1 domain: the molecular mechanisms causing Beckwith-Wiedemann syndrome and cancer. Cytogenet Genome Res. 2006; p. 306–12. [DOI] [PubMed]
- 28.Ulaner GA, Yang Y, Hu JF, Li T, Vu TH, Hoffman AR. CTCF binding at the insulin-like growth factor-II (IGF2)/H19 imprinting control region is insufficient to regulate IGF2/H19 expression in human tissues. Endocrinology. 2003;144:4420–4426. doi: 10.1210/en.2003-0681. [DOI] [PubMed] [Google Scholar]
- 29.Beygo J, Citro V, Sparago A, De Crescenzo A, Cerrato F, Heitmann M, et al. The molecular function and clinical phenotype of partial deletions of the IGF2/H19 imprinting control region depends on the spatial arrangement of the remaining CTCF-binding sites. Hum Mol Genet. 2013;22:544–557. doi: 10.1093/hmg/dds465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Demars J, Gicquel C. Epigenetic and genetic disturbance of the imprinted 11p15 region in Beckwith-Wiedemann and Silver-Russell syndromes. Clin Genet. 2012; p. 350–61. [DOI] [PubMed]
- 31.Jacob K, Robinson WP, Lefebvre L. Beckwith-Wiedemann and Silver-Russell syndromes: opposite developmental imbalances in imprinted regulators of placental function and embryonic growth. Clin Genet. 2013;84:326–334. doi: 10.1111/cge.12143. [DOI] [PubMed] [Google Scholar]
- 32.Rancourt RC, Harris HR, Barault L, Michels KB. The prevalence of loss of imprinting of H19 and IGF2 at birth. FASEB J. 2013;27:3335–3343. doi: 10.1096/fj.12-225284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Horsthemke B. In brief: genomic imprinting and imprinting diseases. J Pathol. 2014;232:485–487. doi: 10.1002/path.4326. [DOI] [PubMed] [Google Scholar]
- 34.Wilkins JF, Úbeda F. Diseases associated with genomic imprinting. Prog Mol Biol Transl Sci. 2011;101:401–445. doi: 10.1016/B978-0-12-387685-0.00013-5. [DOI] [PubMed] [Google Scholar]
- 35.Yamazawa K, Ogata T, Ferguson-Smith AC. Uniparental disomy and human disease: an overview. Am J Med Genet Part C Semin Med Genet. 2010; p. 329–34. [DOI] [PubMed]
- 36.Horsthemke B. Mechanisms of imprint dysregulation. Am. J. Med. Genet. Part C Semin. Med. Genet. 2010. p. 321–8. [DOI] [PubMed]
- 37.Delaval K, Wagschal A, Feil R. Epigenetic deregulation of imprinting in congenital diseases of aberrant growth. BioEssays. 2006; p. 453–9. [DOI] [PubMed]
- 38.Amor DJ, Halliday J. A review of known imprinting syndromes and their association with assisted reproduction technologies. Hum. Reprod. 2008. p. 2826–34. [DOI] [PubMed]
- 39.Iliadou AN, Janson PCJ, Cnattingius S. Epigenetics and assisted reproductive technology. J Intern Med. 2011; p. 414–20. [DOI] [PubMed]
- 40.Owen CM, Segars JH. Imprinting disorders and assisted reproductive technology. Semin Reprod Med. 2010;27:417–428. doi: 10.1055/s-0029-1237430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lazaraviciute G, Kauser M, Bhattacharya S, Haggarty P, Bhattacharya S. A systematic review and meta-analysis of DNA methylation levels and imprinting disorders in children conceived by IVF/ICSI compared with children conceived spontaneously. Hum Reprod Update. 2014;20:840–852. doi: 10.1093/humupd/dmu033. [DOI] [PubMed] [Google Scholar]
- 42.MacKay DJG, Eggermann T, Buiting K, Garin I, Netchine I, Linglart A, et al. Multilocus methylation defects in imprinting disorders. Biomol Concepts. 2015:47–57. [DOI] [PubMed]
- 43.Sanchez-Delgado M, Riccio A, Eggermann T, Maher ER, Lapunzina P, Mackay D, et al. Causes and consequences of multi-locus imprinting disturbances in humans. Trends Genet. 2016. p. 444–55. [DOI] [PubMed]
- 44.Arima T, Kamikihara T, Hayashida T, Kato K, Inoue T, Shirayoshi Y, et al. ZAC, LIT1 (KCNQ1OT1) and p57KIP2 (CDKN1C) are in an imprinted gene network that may play a role in Beckwith–Wiedemann syndrome. Nucleic Acids Res. 2005;33:2650–60. [DOI] [PMC free article] [PubMed]
- 45.Mackay DJG, Boonen SE, Clayton-Smith J, Goodship J, Hahnemann JMD, Kant SG, et al. A maternal hypomethylation syndrome presenting as transient neonatal diabetes mellitus. Hum Genet. 2006;120:262–269. doi: 10.1007/s00439-006-0205-2. [DOI] [PubMed] [Google Scholar]
- 46.Bliek J, Verde G, Callaway J, Maas SM, De Crescenzo A, Sparago A, et al. Hypomethylation at multiple maternally methylated imprinted regions including PLAGL1 and GNAS loci in Beckwith-Wiedemann syndrome. Eur J Hum Genet. 2009;17:611–619. doi: 10.1038/ejhg.2008.233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Court F, Martin-Trujillo A, Romanelli V, Garin I, Iglesias-Platas I, Salafsky I, et al. Genome-wide allelic methylation analysis reveals disease-specific susceptibility to multiple methylation defects in imprinting syndromes. Hum Mutat. 2013;34:595–602. doi: 10.1002/humu.22276. [DOI] [PubMed] [Google Scholar]
- 48.Perez-Nanclares G, Romanelli V, Mayo S, Garin I, Zazo C, Fernandez-Rebollo E, et al. Detection of hypomethylation syndrome among patients with epigenetic alterations at the GNAS locus. J Clin Endocrinol Metab. 2012;97. [DOI] [PubMed]
- 49.Rossignol S, Steunou V, Chalas C, Kerjean A, Rigolet M, Viegas-Pequignot E, et al. The epigenetic imprinting defect of patients with Beckwith-Wiedemann syndrome born after assisted reproductive technology is not restricted to the 11p15 region. J Med Genet. 2006;43:902–907. doi: 10.1136/jmg.2006.042135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Boonen SE, Mackay DJG, Hahnemann JMD, Docherty L, Gronskov K, Lehmann A, et al. Transient neonatal diabetes, ZFP57, and hypomethylation of multiple imprinted loci. Diabetes Care. 2013;36:505–512. doi: 10.2337/dc12-0700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mackay DJG, Hahnemann JMD, Boonen SE, Poerksen S, Bunyan DJ, White HE, et al. Epimutation of the TNDM locus and the Beckwith-Wiedemann syndrome centromeric locus in individuals with transient neonatal diabetes mellitus. Hum Genet. 2006;119:179–184. doi: 10.1007/s00439-005-0127-4. [DOI] [PubMed] [Google Scholar]
- 52.Gardner RJ, Mackay DJ, Mungall AJ, Polychronakos C, Siebert R, Shield JP, et al. An imprinted locus associated with transient neonatal diabetes mellitus. Hum Mol Genet. 2000;9:589–596. doi: 10.1093/hmg/9.4.589. [DOI] [PubMed] [Google Scholar]
- 53.Mackay DJ, Callaway JL, Marks SM, White HE, Acerini CL, Boonen SE, et al. Hypomethylation of multiple imprinted loci in individuals with transient neonatal diabetes is associated with mutations in ZFP57. Nat Genet. 2008;40:949–951. doi: 10.1038/ng.187. [DOI] [PubMed] [Google Scholar]
- 54.Gicquel C, Rossignol S, Cabrol S, Houang M, Steunou V, Barbu V, et al. Epimutation of the telomeric imprinting center region on chromosome 11p15 in Silver-Russell syndrome. Nat Genet. 2005;37:1003–1007. doi: 10.1038/ng1629. [DOI] [PubMed] [Google Scholar]
- 55.Netchine I, Rossignol S, Dufourg MN, Azzi S, Rousseau A, Perin L, et al. Brief report: 11p15 imprinting center region 1 loss of methylation is a common and specific cause of typical Russell-Silver syndrome: clinical scoring system and epigenetic-phenotypic correlations. J Clin Endocrinol Metab. 2007;92:3148–3154. doi: 10.1210/jc.2007-0354. [DOI] [PubMed] [Google Scholar]
- 56.Bullman H, Lever M, Robinson DO, Mackay DJG, Holder SE, Wakeling EL. Mosaic maternal uniparental disomy of chromosome 11 in a patient with Silver-Russell syndrome. J Med Genet. 2008;45:396–399. doi: 10.1136/jmg.2007.057059. [DOI] [PubMed] [Google Scholar]
- 57.Schönherr N, Meyer E, Roos A, Schmidt A, Wollmann HA, Eggermann T. The centromeric 11p15 imprinting centre is also involved in Silver-Russell syndrome. J Med Genet. 2007;44:59–63. doi: 10.1136/jmg.2006.044370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Eggermann T, Begemann M, Binder G, Spengler S. Silver-Russell syndrome: genetic basis and molecular genetic testing. Orphanet J Rare Dis. 2010;5:19. doi: 10.1186/1750-1172-5-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Turner CLS, Mackay DM, Callaway JLA, Docherty LE, Poole RL, Bullman H, et al. Methylation analysis of 79 patients with growth restriction reveals novel patterns of methylation change at imprinted loci. Eur J Hum Genet. 2010;18:648–655. doi: 10.1038/ejhg.2009.246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Habib WA, Azzi S, Brioude F, Steunou V, Thibaud N, Das Neves C, et al. Extensive investigation of the IGF2/H19 imprinting control region reveals novel OCT4/SOX2 binding site defects associated with specific methylation patterns in Beckwith-Wiedemann syndrome. Hum Mol Genet. 2014;23:5763–5773. doi: 10.1093/hmg/ddu290. [DOI] [PubMed] [Google Scholar]
- 61.Weksberg R, Shuman C, Beckwith JB. Beckwith-Wiedemann syndrome. Eur J Hum Genet. 2010;18:8–14. [DOI] [PMC free article] [PubMed]
- 62.Cooper WN, Luharia A, Evans GA, Raza H, Haire AC, Grundy R, et al. Molecular subtypes and phenotypic expression of Beckwith–Wiedemann syndrome. Eur J Hum Genet. 2005;13:1025–1032. doi: 10.1038/sj.ejhg.5201463. [DOI] [PubMed] [Google Scholar]
- 63.Lam WW, Hatada I, Ohishi S, Mukai T, Joyce JA, Cole TR, et al. Analysis of germline CDKN1C (p57KIP2) mutations in familial and sporadic Beckwith-Wiedemann syndrome (BWS) provides a novel genotype-phenotype correlation. J Med Genet. 1999;36:518–523. [PMC free article] [PubMed] [Google Scholar]
- 64.Netchine I, Rossignol S, Azzi S, Brioude F, Bouc YL. Imprinted anomalies in fetal and childhood growth disorders: the model of Russell-Silver and Beckwith-Wiedemann syndromes. Dev Biol GH Secretion, Growth Treat. 2012; p. 60–70. [DOI] [PubMed]
- 65.Brioude F, Lacoste A, Netchine I, Vazquez MP, Auber F, Audry G, et al. Beckwith-Wiedemann syndrome: growth pattern and tumor risk according to molecular mechanism, and guidelines for tumor surveillance. Horm Res Paediatr. 2014;80:457–465. doi: 10.1159/000355544. [DOI] [PubMed] [Google Scholar]
- 66.Ramsden SC, Clayton-Smith J, Birch R, Buiting K. Practice guidelines for the molecular analysis of Prader-Willi and Angelman syndromes. BMC Med Genet. 2010;11:70. doi: 10.1186/1471-2350-11-70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Buiting K, Saitoh S, Gross S, Dittrich B, Schwartz S, Nicholls RD, et al. Inherited microdeletions in the Angelman and Prader-Willi syndromes define an imprinting centre on human chromosome 15. Nat Genet. 1995;9:395–400. doi: 10.1038/ng0495-395. [DOI] [PubMed] [Google Scholar]
- 68.Sahoo T, del Gaudio D, German JR, Shinawi M, Peters SU, Person RE, et al. Prader-Willi phenotype caused by paternal deficiency for the HBII-85 C/D box small nucleolar RNA cluster. Nat Genet. 2008;40:719–721. doi: 10.1038/ng.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Saitoh S, Buiting K, Rogan PK, Buxton JL, Driscoll DJ, Arnemann J, et al. Minimal definition of the imprinting center and fixation of chromosome 15q11-q13 epigenotype by imprinting mutations. Proc Natl Acad Sci U S A. 1996;93:7811–7815. doi: 10.1073/pnas.93.15.7811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.de Smith AJ, Purmann C, Walters RG, Ellis RJ, Holder SE, Van Haelst MM, et al. A deletion of the HBII-85 class of small nucleolar RNAs (snoRNAs) is associated with hyperphagia, obesity and hypogonadism. Hum Mol Genet. 2009;18:3257–3265. doi: 10.1093/hmg/ddp263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Horsthemke B, Wagstaff J. Mechanisms of imprinting of the Prader-Willi/Angelman region. Am J Med Genet Part A. 2008; p. 2041–52. [DOI] [PubMed]
- 72.Malzac P, Webber H, Moncla A, Graham JM, Kukolich M, Williams C, et al. Mutation analysis of UBE3A in Angelman syndrome patients. Am J Hum Genet. 1998;62:1353–1360. doi: 10.1086/301877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Matsuura T, Sutcliffe JS, Fang P, Galjaard RJ, Jiang YH, Benton CS, et al. De novo truncating mutations in E6-AP ubiquitin-protein ligase gene (UBE3A) in Angelman syndrome. Nat Genet. 1997;15:74–77. doi: 10.1038/ng0197-74. [DOI] [PubMed] [Google Scholar]
- 74.Kishino T, Lalande M, Wagstaff J. UBE3A/E6-AP mutations cause Angelman syndrome. Nat Genet. 1997;15:70–73. doi: 10.1038/ng0197-70. [DOI] [PubMed] [Google Scholar]
- 75.Baple EL, Poole RL, Mansour S, Willoughby C, Temple IK, Docherty LE, et al. An atypical case of hypomethylation at multiple imprinted loci. Eur J Hum Genet. 2011;19:360–362. doi: 10.1038/ejhg.2010.218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Linglart A, Gensure RC, Olney RC, Jüppner H, Bastepe M. A novel STX16 deletion in autosomal dominant pseudohypoparathyroidism type Ib redefines the boundaries of a cis-acting imprinting control element of GNAS. Am J Hum Genet. 2005;76:804–814. doi: 10.1086/429932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Bastepe M, Fröhlich LF, Linglart A, Abu-Zahra HS, Tojo K, Ward LM, et al. Deletion of the NESP55 differentially methylated region causes loss of maternal GNAS imprints and pseudohypoparathyroidism type Ib. Nat Genet. 2005;37:25–27. doi: 10.1038/ng1560. [DOI] [PubMed] [Google Scholar]
- 78.Bastepe M, Lane AH, Jüppner H. Paternal uniparental isodisomy of chromosome 20q—and the resulting changes in GNAS1 methylation—as a plausible cause of pseudohypoparathyroidism. Am J Hum Genet. 2001;68:1283–1289. doi: 10.1086/320117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Linglart A, Bastepe M, Jüppner H. Similar clinical and laboratory findings in patients with symptomatic autosomal dominant and sporadic pseudohypoparathyroidism type Ib despite different epigenetic changes at the GNAS locus. Clin Endocrinol. 2007;67:822–831. doi: 10.1111/j.1365-2265.2007.02969.x. [DOI] [PubMed] [Google Scholar]
- 80.Liu J, Litman D, Rosenberg MJ, Yu S, Biesecker LG, Weinstein LS. A GNAS1 imprinting defect in pseudohypoparathyroidism type IB. J Clin Invest. 2000;106:1167–1174. doi: 10.1172/JCI10431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Bastepe M, Altug-Teber O, Agarwal C, Oberfield SE, Bonin M, Jüppner H. Paternal uniparental isodisomy of the entire chromosome 20 as a molecular cause of pseudohypoparathyroidism type Ib (PHP-Ib) Bone. 2011;48:659–662. doi: 10.1016/j.bone.2010.10.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Izzi B, Van Geet C, Freson K. Recent advances in GNAS epigenetic research of pseudohypoparathyroidism. Curr Mol Med. 2012. p. 566–73. Available from: http://www.ncbi.nlm.nih.gov/pubmed/22300135 [DOI] [PubMed]
- 83.Shield JP, Gardner RJ, Wadsworth EJ, Whiteford ML, James RS, Robinson DO, et al. Aetiopathology and genetic basis of neonatal diabetes. Arch Dis Child Fetal Neonatal Ed. 1997;76:F39–42. [DOI] [PMC free article] [PubMed]
- 84.Arthur EI, Zlotogora J, Lerer I, Dagan J, Marks K, Abeliovich D. Transient neonatal diabetes mellitus in a child with invdup(6)(q22q23) of paternal origin. Eur J Hum Genet. 1997;5:417–419. [PubMed] [Google Scholar]
- 85.Temple IK, Shield JPH. Transient neonatal diabetes, a disorder of imprinting. J Med Genet. 2002;39:872–875. doi: 10.1136/jmg.39.12.872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Babenko AP, Polak M, Cavé H, Busiah K, Czernichow P, Scharfmann R, et al. Activating mutations in the ABCC8 gene in neonatal diabetes mellitus. N Engl J Med. 2006;355:456–66. [DOI] [PubMed]
- 87.Vaxillaire M, Dechaume A, Busiah K, Cave H, Pereira S, Scharfmann R, et al. New ABCC8 mutations in relapsing neonatal diabetes and clinical features. Diabetes. 2007;56:1737–1741. doi: 10.2337/db06-1540. [DOI] [PubMed] [Google Scholar]
- 88.Hattersley AT, Ashcroft FM. Activating mutations in Kir6.2 and neonatal diabetes: new clinical syndromes, new scientific insights, and new therapy. Diabetes. 2005; p. 2503–13. [DOI] [PubMed]
- 89.Anik A, Çatli G, Abaci A, Yiş U, Ören H, Güleryüz H, et al. A novel activating ABCC8 mutation underlying neonatal diabetes mellitus in an infant presenting with cerebral sinovenous thrombosis. J Pediatr Endocrinol Metab. 2014;27:533–537. doi: 10.1515/jpem-2013-0263. [DOI] [PubMed] [Google Scholar]
- 90.Temple IK, Gardner RJ, Robinson DO, Kibirige MS, Ferguson AW, Baum JD, et al. Further evidence for an imprinted gene for neonatal diabetes localised to chromosome 6q22-q23. Hum Mol Genet. 1996;5:1117–1121. doi: 10.1093/hmg/5.8.1117. [DOI] [PubMed] [Google Scholar]
- 91.Das S, Lese CM, Song M, Jensen JL, Wells LA, Barnoski BL, et al. Partial paternal uniparental disomy of chromosome 6 in an infant with neonatal diabetes, macroglossia, and craniofacial abnormalities. Am J Hum Genet. 2000;67:1586–1591. doi: 10.1086/316897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Baglivo I, Esposito S, De CL, Sparago A, Anvar Z, Riso V, et al. Genetic and epigenetic mutations affect the DNA binding capability of human ZFP57 in transient neonatal diabetes type 1. FEBS Lett. 2013;587:1474–1481. doi: 10.1016/j.febslet.2013.02.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Temple IK, Shield JPH. 6q24 transient neonatal diabetes. Rev Endocr Metab Disord. 2010;11:199–204. doi: 10.1007/s11154-010-9150-4. [DOI] [PubMed] [Google Scholar]
- 94.Takikawa S, Wang X, Ray C, Vakulenko M, Bell FT, Li X. Human and mouse ZFP57 proteins are functionally interchangeable in maintaining genomic imprinting at multiple imprinted regions in mouse ES cells. Epigenetics. 2013;8:1268–1279. doi: 10.4161/epi.26544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Ideraabdullah FY, Bartolomei MS. ZFP57: KAPturing DNA methylation at imprinted loci. Mol Cell. 2011; p. 341–2. [DOI] [PubMed]
- 96.Yada T, Sakurada M, Ishihara H, Nakata M, Shioda S, Yaekura K, et al. Pituitary adenylate cyclase-activating polypeptide (PACAP) is an islet substance serving as an intra-islet amplifier of glucose-induced insulin secretion in rats. J Physiol. 1997;505:319–328. doi: 10.1111/j.1469-7793.1997.319bb.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Ciani E, Hoffmann A, Schmidt P, Journot L, Spengler D. Induction of the PAC1-R (PACAP-type I receptor) gene by p53 and Zac. Mol Brain Res. 1999;69:290–294. doi: 10.1016/S0169-328X(99)00116-3. [DOI] [PubMed] [Google Scholar]
- 98.Kamiya M, Judson H, Okazaki Y, Kusakabe M, Muramatsu M, Takada S, et al. The cell cycle control gene ZAC/PLAGL1 is imprinted—a strong candidate gene for transient neonatal diabetes. Hum Mol Genet. 2000;9:453–460. doi: 10.1093/hmg/9.3.453. [DOI] [PubMed] [Google Scholar]
- 99.Mackay DJG, Temple IK. Transient neonatal diabetes mellitus type 1. Am. J. Med. Genet. Part C Semin. Med. Genet. 2010. p. 335–42. [DOI] [PubMed]
- 100.Ma D, Shield JP, Dean W, Leclerc I, Knauf C, Burcelin RR, et al. Impaired glucose homeostasis in transgenic mice expressing the human transient neonatal diabetes mellitus locus, TNDM. J Clin Invest. 2004;114:339–348. doi: 10.1172/JCI200419876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Iglesias-Platas I, Martin-Trujillo A, Petazzi P, Guillaumet-Adkins A, Esteller M, Monk D. Altered expression of the imprinted transcription factor PLAGL1 deregulates a network of genes in the human IUGR placenta. Hum Mol Genet. 2014;23:6275–6285. doi: 10.1093/hmg/ddu347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Varrault A, Gueydan C, Delalbre A, Bellmann A, Houssami S, Aknin C, et al. Zac1 regulates an imprinted gene network critically involved in the control of embryonic growth. Dev Cell. 2006;11:711–722. doi: 10.1016/j.devcel.2006.09.003. [DOI] [PubMed] [Google Scholar]
- 103.Kameswaran V, Kaestner KH. The missing lnc(RNA) between the pancreatic β-cell and diabetes. Front Genet. 2014;5. [DOI] [PMC free article] [PubMed]
- 104.Hoffmann A, Spengler D. Role of ZAC1 in transient neonatal diabetes mellitus and glucose metabolism. World J Biol Chem. 2015;6:95–109. doi: 10.4331/wjbc.v6.i3.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Valleley EM, Cordery SF, Bonthron DT. Tissue-specific imprinting of the ZAC/PLAGL1 tumour suppressor gene results from variable utilization of monoallelic and biallelic promoters. Hum Mol Genet. 2007;16:972–981. doi: 10.1093/hmg/ddm041. [DOI] [PubMed] [Google Scholar]
- 106.Piras G, El Kharroubi A, Kozlov S, Escalante-Alcalde D, Hernandez L, Copeland NG, et al. Zac1 (Lot1), a potential tumor suppressor gene, and the gene for epsilon-sarcoglycan are maternally imprinted genes: identification by a subtractive screen of novel uniparental fibroblast lines. Mol Cell Biol. 2000;20:3308–3315. doi: 10.1128/MCB.20.9.3308-3315.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Mackay DJG, Coupe AM, Shield JPH, Storr JNP, Temple IK, Robinson DO. Relaxation of imprinted expression of ZAC and HYMAI in a patient with transient neonatal diabetes mellitus. Hum Genet. 2002;110:139–144. doi: 10.1007/s00439-001-0671-5. [DOI] [PubMed] [Google Scholar]
- 108.Peille AL, Brouste V, Kauffmann A, Lagarde P, Le Morvan V, Coindre JM, et al. Prognostic value of PLAGL1-specific CpG site methylation in soft-tissue sarcomas. PLoS One. 2013;8. [DOI] [PMC free article] [PubMed]
- 109.Kamikihara T, Arima T, Kato K, Matsuda T, Kato H, Douchi T, et al. Epigenetic silencing of the imprinted gene ZAC by DNA methylation is an early event in the progression of human ovarian cancer. Int J Cancer. 2005;115:690–700. doi: 10.1002/ijc.20971. [DOI] [PubMed] [Google Scholar]
- 110.Wakeling EL. Silver Russell syndrome. Arch Dis Child. 2011;96:1156–1161. doi: 10.1136/adc.2010.190165. [DOI] [PubMed] [Google Scholar]
- 111.Rossignol S, Netchine I, Le Bouc Y, Gicquel C. Epigenetics in Silver-Russell syndrome. Best Pract Res Clin Endocrinol Metab. 2008; p. 403–14. [DOI] [PubMed]
- 112.Monk D, Bentley L, Hitchins M, Myler RA, Clayton-Smith J, Ismail S, et al. Chromosome 7p disruptions in Silver Russell syndrome: delineating an imprinted candidate gene region. Hum Genet. 2002;111:376–387. doi: 10.1007/s00439-002-0777-4. [DOI] [PubMed] [Google Scholar]
- 113.Wakeling EL, Amero SA, Alders M, Bliek J, Forsythe E, Kumar S, et al. Epigenotype-phenotype correlations in Silver-Russell syndrome. J Med Genet. 2010;47:760–768. doi: 10.1136/jmg.2010.079111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Eggermann T. Epigenetic regulation of growth: lessons from Silver-Russell syndrome. Endocr Dev. 2009; p. 10–9. [DOI] [PubMed]
- 115.Eggermann T, Schönherr N, Meyer E, Obermann C, Mavany M, Eggermann K, et al. Epigenetic mutations in 11p15 in Silver-Russell syndrome are restricted to the telomeric imprinting domain. J Med Genet. 2006;43:615–616. doi: 10.1136/jmg.2005.038687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Bliek J, Terhal P, van den Bogaard M-J, Maas S, Hamel B, Salieb-Beugelaar G, et al. Hypomethylation of the H19 gene causes not only Silver-Russell syndrome (SRS) but also isolated asymmetry or an SRS-like phenotype. Am J Hum Genet. 2006;78:604–614. doi: 10.1086/502981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Pettenati MJ, Haines JL, Higgins RR, Wappner RS, Palmer CG, Weaver DD. Wiedemann-Beckwith syndrome: presentation of clinical and cytogenetic data on 22 new cases and review of the literature. Hum Genet. 1986;74:143–154. doi: 10.1007/BF00282078. [DOI] [PubMed] [Google Scholar]
- 118.Ward A. Beckwith-Wiedemann syndrome and Wilms’ tumour. Mol Hum Reprod. 1997;3. [DOI] [PubMed]
- 119.Weksberg R, Nishikawa J, Caluseriu O, Fei YL, Shuman C, Wei C, et al. Tumor development in the Beckwith-Wiedemann syndrome is associated with a variety of constitutional molecular 11p15 alterations including imprinting defects of KCNQ1OT1. Hum Mol Genet. 2001;10:2989–3000. [DOI] [PubMed]
- 120.Clericuzio CL, Martin RA. Diagnostic criteria and tumor screening for individuals with isolated hemihyperplasia. Genet Med. 2009;11:220–222. doi: 10.1097/GIM.0b013e31819436cf. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Choufani S, Shuman C, Weksberg R. Molecular findings in Beckwith-Wiedemann syndrome. Am J Med Genet Part C Semin Med Genet. 2013;163:131–140. doi: 10.1002/ajmg.c.31363. [DOI] [PubMed] [Google Scholar]
- 122.Kanduri C. Kcnq1ot1: a chromatin regulatory RNA. Semin. Cell Dev Biol. 2011; p. 343–50. [DOI] [PubMed]
- 123.Choufani S, Shuman C, Weksberg R. Beckwith-Wiedemann syndrome. Am J Med Genet Part C Semin Med Genet. 2010; p. 343–54. [DOI] [PubMed]
- 124.Besson A, Dowdy SF, Roberts JM. CDK inhibitors: cell cycle regulators and beyond. Dev Cell. 2008; p. 159–69. [DOI] [PubMed]
- 125.Lim S, Kaldis P. Cdks, cyclins and CKIs: roles beyond cell cycle regulation. Development [Internet] 2013;140:3079–3093. doi: 10.1242/dev.091744. [DOI] [PubMed] [Google Scholar]
- 126.Eggermann T. Silver-Russell and Beckwith-Wiedemann syndromes: opposite (epi)mutations in 11p15 result in opposite clinical pictures. Horm Res. 2009; p. 30–5. [DOI] [PubMed]
- 127.Lewis A, Green K, Dawson C, Redrup L, Huynh KD, Lee JT, et al. Epigenetic dynamics of the Kcnq1 imprinted domain in the early embryo. Development. 2006;133:4203–4210. doi: 10.1242/dev.02612. [DOI] [PubMed] [Google Scholar]
- 128.Demars J, Shmela ME, Rossignol S, Okabe J, Netchine I, Azzi S, et al. Analysis of the IGF2/H19 imprinting control region uncovers new genetic defects, including mutations of OCT-binding sequences, in patients with 11p15 fetal growth disorders. Hum Mol Genet. 2010;19:803–814. doi: 10.1093/hmg/ddp549. [DOI] [PubMed] [Google Scholar]
- 129.Azzi S, Abi Habib W, Netchine I. Beckwith-Wiedemann and Russell-Silver syndromes: from new molecular insights to the comprehension of imprinting regulation. Curr Opin Endocrinol Diabetes Obes. 2014;21:30–38. doi: 10.1097/MED.0000000000000037. [DOI] [PubMed] [Google Scholar]
- 130.Calvello M, Tabano S, Colapietro P, Maitz S, Pansa A, Augello C, et al. Quantitative DNA methylation analysis improves epigenotype-phenotype correlations in Beckwith-Wiedemann syndrome. Epigenetics. 2013;8:1053–1060. doi: 10.4161/epi.25812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Paganini L, Carlessi N, Fontana L, Silipigni R, Motta S, Fiori S, et al. Beckwith-Wiedemann syndrome prenatal diagnosis by methylation analysis in chorionic villi. Epigenetics. 2015;10:643–649. doi: 10.1080/15592294.2015.1057383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Burd L, Vesely B, Martsolf J, Kerbeshian J. Prevalence study of Prader-Willi syndrome in North Dakota. Am J Med Genet. 1990;37:97–99. doi: 10.1002/ajmg.1320370122. [DOI] [PubMed] [Google Scholar]
- 133.Ehara H, Ohno K, Takeshita K. Frequency of the Prader-Willi syndrome in the San-in district, Japan. Brain Dev. 1995;17:324–326. doi: 10.1016/0387-7604(95)00060-O. [DOI] [PubMed] [Google Scholar]
- 134.Cassidy SB, Schwartz S, Miller JL, Driscoll DJ. Prader-Willi syndrome. Genet. Med. 2012;14:10–26. doi: 10.1038/gim.0b013e31822bead0. [DOI] [PubMed] [Google Scholar]
- 135.Rodriguez-Jato S, Nicholls RD, Driscoll DJ, Yang TP. Characterization of cis- and trans-acting elements in the imprinted human SNURF-SNRPN locus. Nucleic Acids Res. 2005;33:4740–4753. doi: 10.1093/nar/gki786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Perk J, Makedonski K, Lande L, Cedar H, Razin A, Shemer R. The imprinting mechanism of the Prader-Willi/Angelman regional control center. EMBO J. 2002;21:5807–5814. doi: 10.1093/emboj/cdf570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Runte M, Hüttenhofer A, Groß S, Kiefmann M, Horsthemke B, Buiting K. The IC-SNURF–SNRPN transcript serves as a host for multiple small nucleolar RNA species and as an antisense RNA for UBE3A. Hum Mol Genet. 2001;10:2687–700. [DOI] [PubMed]
- 138.Will CL, Lührmann R. Spliceosome structure and function. Cold Spring Harb Perspect Biol. 2011;3:1–2. doi: 10.1101/cshperspect.a003707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Dindot SV, Antalffy BA, Bhattacharjee MB, Beaudet AL. The Angelman syndrome ubiquitin ligase localizes to the synapse and nucleus, and maternal deficiency results in abnormal dendritic spine morphology. Hum. Mol. Genet. 2008;17:111–118. doi: 10.1093/hmg/ddm288. [DOI] [PubMed] [Google Scholar]
- 140.Greer PL, Hanayama R, Bloodgood BL, Mardinly AR, Lipton DM, Flavell SW, et al. The Angelman syndrome protein Ube3A regulates synapse development by ubiquitinating arc. Cell. 2010;140:704–716. doi: 10.1016/j.cell.2010.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Ferdousy F, Bodeen W, Summers K, Doherty O, Wright O, Elsisi N, et al. Drosophila Ube3a regulates monoamine synthesis by increasing GTP cyclohydrolase I activity via a non-ubiquitin ligase mechanism. Neurobiol Dis. 2011;41:669–677. doi: 10.1016/j.nbd.2010.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Buiting K. Prader-Willi syndrome and Angelman syndrome. Am J Med Genet Part C Semin Med Genet. 2010; p. 365–76. [DOI] [PubMed]
- 143.Elena G, Bruna C, Benedetta M, Stefania DC, Giuseppe C. Prader-Willi syndrome: clinical aspects. J. Obes. 2012. [DOI] [PMC free article] [PubMed]
- 144.Butler MG. Genomic imprinting disorders in humans: a mini-review. J Assist Reprod Genet. 2009; p. 477–86. [DOI] [PMC free article] [PubMed]
- 145.Sarkar PA, Shigli A, Patidar C. Happy Puppet syndrome. BMJ Case Rep. 2011;2011:9–11. doi: 10.1136/bcr.09.2011.4747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Mertz LGB, Christensen R, Vogel I, Hertz JM, Nielsen KB, Grønskov K, et al. Angelman syndrome in Denmark. Birth incidence, genetic findings, and age at diagnosis. Am J Med Genet Part A. 2013;161:2197–2203. doi: 10.1002/ajmg.a.36058. [DOI] [PubMed] [Google Scholar]
- 147.Steffenburg S, Gillberg CL, Steffenburg U, Kyllerman M. Autism in Angelman syndrome: a population-based study. Pediatr Neurol. 1996;14:131–136. doi: 10.1016/0887-8994(96)00011-2. [DOI] [PubMed] [Google Scholar]
- 148.Runte M, Kroisel PM, Gillessen-Kaesbach G, Varon R, Horn D, Cohen MY, et al. SNURF-SNRPN and UBE3A transcript levels in patients with Angelman syndrome. Hum Genet. 2004;114:553–561. doi: 10.1007/s00439-004-1104-z. [DOI] [PubMed] [Google Scholar]
- 149.Daily J, Smith AG, Weeber EJ. Spatial and temporal silencing of the human maternal UBE3A gene. Eur J Paediatr Neurol. 2012;16:587–591. doi: 10.1016/j.ejpn.2012.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Margolis SS, Sell GL, Zbinden MA, Bird LM. Angelman syndrome. Neurotherapeutics. 2015;12:641–50. Available from: http://www.ncbi.nlm.nih.gov/pubmed/26040994 [DOI] [PMC free article] [PubMed]
- 151.Judson MC, Sosa-Pagan JO, Del Cid WA, Han JE, Philpot BD. Allelic specificity of Ube3a expression in the mouse brain during postnatal development. J Comp Neurol. 2014;522:1874–1896. doi: 10.1002/cne.23507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.White HE, Hall VJ, Cross NCP. Methylation-sensitive high-resolution melting-curve analysis of the SNRPN gene as a diagnostic screen for Prader-Willi and Angelman syndromes. Clin Chem. 2007;53:1960–1962. doi: 10.1373/clinchem.2007.093351. [DOI] [PubMed] [Google Scholar]
- 153.Mantovani G. Clinical review: pseudohypoparathyroidism: diagnosis and treatment. J Clin Endocrinol Metab. 2011;96:3020–30. Available from: http://www.ncbi.nlm.nih.gov/pubmed/21816789 [DOI] [PubMed]
- 154.Bastepe M. The GNAS locus: quintessential complex gene encoding Gsalpha, XLalphas, and other imprinted transcripts. Curr Genomics. 2007;8:398–414. doi: 10.2174/138920207783406488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Hayward BE, Bonthron DT. An imprinted antisense transcript at the human GNAS1 locus. Hum Mol Genet. 2000;9:835–841. doi: 10.1093/hmg/9.5.835. [DOI] [PubMed] [Google Scholar]
- 156.Turan S, Bastepe M. The GNAS complex locus and human diseases associated with loss-of-function mutations or epimutations within this imprinted gene. Horm Res Paediatr. 2013; p. 229–41. [DOI] [PMC free article] [PubMed]
- 157.Liu J, Erlichman B, Weinstein LS. The stimulatory G protein alpha-subunit Gs alpha is imprinted in human thyroid glands: implications for thyroid function in pseudohypoparathyroidism types 1A and 1B. J Clin Endocrinol Metab. 2003;88:4336–4341. doi: 10.1210/jc.2003-030393. [DOI] [PubMed] [Google Scholar]
- 158.Vilardaga JP, Romero G, Friedman PA, Gardella TJ. Molecular basis of parathyroid hormone receptor signaling and trafficking: a family B GPCR paradigm. Cell Mol Life Sci. 2011; p. 1–13. [DOI] [PMC free article] [PubMed]
- 159.Li T, Vu TH, Zeng ZL, Nguyen BT, Hayward BE, Bonthron DT, et al. Tissue-specific expression of antisense and sense transcripts at the imprinted Gnas locus. Genomics. 2000;69:295–304. doi: 10.1006/geno.2000.6337. [DOI] [PubMed] [Google Scholar]
- 160.Plagge A, Kelsey G. Imprinting the Gnas locus. Cytogenet Genome Res. 2006; p. 178–87. [DOI] [PubMed]
- 161.Turan S, Ignatius J, Moilanen JS, Kuismin O, Stewart H, Mann NP, et al. De novo STX16 deletions: an infrequent cause of pseudohypoparathyroidism type Ib that should be excluded in sporadic cases. J Clin Endocrinol Metab. 2012;97. [DOI] [PMC free article] [PubMed]
- 162.Elli FM, De Sanctis L, Peverelli E, Bordogna P, Pivetta B, Miolo G, et al. Autosomal dominant pseudohypoparathyroidism type Ib: a novel inherited deletion ablating STX16 causes loss of imprinting at the A/B DMR. J Clin Endocrinol Metab. 2014;99. [DOI] [PubMed]