

SUMMARY
The dynamic and reversible N6-methyladenosine (m6A) RNA modification installed and erased by N6-methyltransferases and demethylases regulates gene expression and cell fate. We show that the m6A demethylase ALKBH5 is highly expressed in glioblastoma stem-like cells (GSCs). Silencing ALKBH5 suppresses the proliferation of patient-derived GSCs. Integrated transcriptome and m6A-seq analyses revealed altered expression of certain ALKBH5 target genes, including the transcription factor FOXM1. ALKBH5 demethylates FOXM1 nascent transcripts, leading to enhanced FOXM1 expression. Further, a long noncoding RNA antisense to FOXM1 (FOXM1-AS) promotes the interaction of ALKBH5 with FOXM1 nascent transcripts. Depleting ALKBH5 and FOXM1-AS disrupted GSC tumorigenesis through the FOXM1 axis. Our work uncovers a critical function for ALKBH5 and provides insight into critical roles of m6A methylation in glioblastoma.
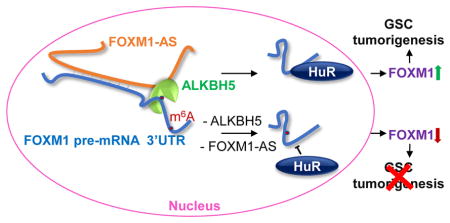
INTRODUCTION
N6-methyl-adenosine (m6A) is the most prevalent internal chemical modification of mRNAs in eukaryotes (Desrosiers et al., 1974; Dominissini et al., 2012; Meyer et al., 2012). In mammals, m6A installed by the m6A methyltransferases METTL3 and METTL14 is erased by FTO (fat-mass and obesity-associated protein) or ALKBH5 (α-ketoglutarate-dependent dioxygenase alkB homologue 5) (Jia et al., 2011; Liu et al., 2014; Wang et al., 2014b; Zheng et al., 2013). The effects of mRNA m6A modification on cellular processes include alterations in RNA stability (Wang et al., 2014a; Wang et al., 2014b), translation efficiency (Meyer et al., 2015; Wang et al., 2015), secondary structure (Liu et al., 2015), subcellular localization (Fustin et al., 2013; Zheng et al., 2013), alternative polyadenylation and splicing (Molinie et al., 2016). m6A methyltransferases are crucial for the speed of circadian clock and differentiation of mouse embryonic stem cells (Batista et al., 2014; Fustin et al., 2013; Geula et al., 2015; Wang et al., 2014b). FTO is known to regulate adipogenesis and energy homeostasis (Fischer et al., 2009; Zhao et al., 2014). ALKBH5, which is most highly expressed in the testes but has low expression in the heart and brain, affects nuclear RNA export and metabolism, gene expression and mouse fertility (Zheng et al., 2013). Alkbh5 knockout mice were viable but showed compromised spermatogenesis due to significantly altered expression of key genes required for spermatogenic maturation although the increase of overall m6A levels in testes is modest (Zheng et al., 2013). These studies suggest that altered expression of key genes, which are sensitive to the function of m6A modulator, can give rise to a significant phenotype change. However, to date, the biological significance and the key target genes of these m6A modulators in human cancer remain elusive.
Glioblastoma (GBM; World Health Organization grade IV glioma) is the most common and devastating primary malignant brain tumor. Even with surgical resection and the use of highly aggressive therapies, recurrence is inevitable, and the median survival duration of GBM patients is only one year (Wen and Kesari, 2008). GBMs are characterized by marked intra- and intertumoral heterogeneity and contain cells that are at the apex of cellular hierarchies with stem-like properties. These GBM stem-like cells (GSCs) can self-renew, are resistant to conventional therapy, and give rise to tumor recurrence by sustaining long-term tumor growth (Lathia et al., 2015). Therefore, studying the mechanisms employed by GSC for self-renewal and proliferation may provide a better understanding of GBM tumorigenesis and therapeutic response.
Numerous studies have shown that the transcription factor FOXM1 plays a pivotal role in regulating GSC proliferation, self-renewal and tumorigenicity (Kim et al., 2015; Schonberg et al., 2015; Zhang et al., 2011). FOXM1 is a key cell cycle molecule required for G1/S and G2/M transition, and M phase progression (Li et al., 2012). FOXM1 is overexpressed in GBM and informs poor survival of GBM patients (Liu et al., 2006). FOXM1 maintains GSC properties by promoting β-catenin activation (Zhang et al., 2011), interacting with MELK (Joshi et al., 2013), inducing SOX2 (Lee et al., 2015), and activating STAT3 (Gong et al., 2015). However, the molecular mechanism underlying FOXM1 upregulation in GSCs remains unclear.
Dysregulated DNA methylation by cancer epigenetic regulators is a hallmark of glioblastoma (Noushmehr et al., 2010), whereas RNA m6A-methylation in cancers including glioblastoma are largely understudied. METTL3 is suggested to promote lung adenocarcinoma whereas whether it acts as an m6A modulator or effector is unclear (Lin et al., 2016). Another study reported that ALKBH5 expression is induced by hypoxia in breast cancer cells (Zhang et al., 2016), yet its clinical relevance is unknown. These unanswered questions prompted us to investigate the role and underlying mechanisms of the m6A modulators in cancer. Most recently, FTO has been reported to play an oncogenic role in acute myeloid leukemia (Li et al., 2016), suggesting the functional importance of the mRNA m6A methylation and its modulators in cancer.
RESULTS
ALKBH5 Is Elevated in GSCs and Is a Negative Prognostic Factor for GBM Patients
To study the m6A modulators that may result in poor clinical outcome in GBM patients, we first queried The Cancer Genome Atlas (TCGA; http://www.cbioportal.org) (Brennan et al., 2013), R2 (http://hgserver1.amc.nl/cgi-bin/r2/main.cgi), Freije, Phillips, and REMBRANDT data sets. In all data sets, elevated expression of ALKBH5 predicts poor patient prognosis (Figures 1A and S1A-D).
Figure 1. ALKBH5 is Required for GSC Self-Renewal and Predicts Poor Survival of GBM Patients.

(A) Correlation between ALKBH5 mRNA expression and survival of GBM patients in the TCGA data set. Overall patient survival in groups of high, intermediate, and low expression was analyzed by Kaplan-Meier survival curve. The median overall survival duration of patients with high ALKBH5 expression (9.9 months) versus with low ALKBH5 expression (16.6 months) was compared by log-rank test (p=0.037).
(B) Western blotting of ALKBH5 in NHAs, glioma cells, and GSCs. Actin served as a loading control.
(C) Correlation between ALKBH5 and SOX2 protein expression in GBM specimens. Tumor sections from 15 GBM specimens were immunofluorescence (IF)-stained with anti-ALKBH5 and anti-SOX2 antibodies. Left, representative images are shown. Right, in 5 random selected microscope fields of each tumor, the percentage of ALKBH5 positive cells among SOX2 positive versus SOX2 negative cells was compared by t-test. Lines show mean and SD.
(D) The Pearson correlation between ALKBH5 and SOX2 mRNA expression (RNAseq V2 RSEM [log2]) in the TCGA GBM data set.
(E) ALKBH5 mRNA expression in GSCs and paired matching tumors by RNA-seq analysis.
(F) In vitro limiting dilution assays plating decreasing number of GSCs with or without ALKBH5 knockdown calculated with extreme limiting dilution assay analysis (top left), representative images of tumorspheres in dose of 100 cells/well were shown in top right panel. Scale bar, 20 μm. Stem cell frequencies from GSCs with or without ALKBH5 knockdown were estimated as the ratio 1/x with the upper and lower 95% confidence intervals, where 1 = stem cell and x = all cells (bottom). See also Figure S1 and Table S1
We then examined ALKBH5 expression in established and primary glioma cell lines representing different stages of malignancy. ALKBH5 was weakly or moderately expressed in immortalized normal human astrocytes (NHAs), Hs683 and SW1783 cell lines (lower grade gliomas), and U87MG and LN229 GBM cell lines, but was highly expressed in U251MG GBM cells and in patient-derived primary GBM cultures that enrich for cells with self-renewal and tumor propagation potential, i.e., GSCs (Figure 1B and Table S1). However, the levels of ALKBH5 were similar across the Proneural, Neural, Classical and Mesenchymal molecular subtypes of GBM (Figure S1E).
The elevated ALKBH5 expression in the GSCs led us to hypothesize that ALKBH5 is associated with cancer stem cell (CSC) niches. Thus, we first used immunohistochemical analysis to assess ALKBH5 protein expression in GBM patient samples. We found inter- and intra-tumoral heterogeneity of ALKBH5 expression in primary bulk GBM samples, with some niches being highly positive for nuclear ALKBH5 and others with low levels of ALKBH5 expression (Figure S1F). Then, using immunofluorescence analysis, we found that ALKBH5 co-expressed with SOX2, a transcription factor indicative of CSC self-renewal, and Nestin, an intermediate filament protein recognized as the marker of neural stem cells, in bulk GBM samples (Figures 1C, S1G and S1H). Moreover, gene expression analysis using the TCGA GBM data set validated the positive correlation between ALKBH5 and SOX2 or Nestin expression (Figures 1D and S1I). Furthermore, comparing ALKBH5 levels between GSCs and their matching bulk tumors indicated a significant increase of ALKBH5 expression in the GSC population (Figures 1E and S1J). ALKBH5 expression is not restricted to GSCs; however, the high ALKBH5 expression in the GBM subpopulation characterized by a CSC niche suggests an association of ALKBH5 with CSCs within the bulk tumor, and this became the focus of our investigation.
Targeting ALKBH5 Expression Impairs GSC Self-Renewal
To determine whether ALKBH5 is important to GSC self-renewal, we used two distinct short hairpin RNAs (shRNA1 and shRNA2; hereafter referred to collectively as shALKBH5) to ablate ALKBH5 expression. Compared with a non-targeting control shRNA (shCtrl), both shALKBH5 significantly reduced ALKBH5 protein expression (Figure S1K). Expression of ALKBH5 shRNAs significantly decreased the tumorsphere formation frequency of recurrent GBM-derived GSC11, GSC17 and GSC23 as examined by in vitro limiting dilution assay, a method widely used to determine self-renewal capacity (Figure 1F). Moreover, immunofluorescence analysis and Western blotting of dissociated tumorspheres revealed that shALKBH5 reduced the expression of Nestin as well as SOX2, Nanog, Oct4, the core transcription factors that endow tumor cells with self-renewal ability, confirming the impact of ALKBH5 on GSC self-renewal (Figures S1K and S1L).
GSCs are characterized by multipotency. The addition of serum deprives GSCs of their self-renewal ability and induces astrocytic or neuronal commitment defined by GFAP or Tuj-1 expression (Gong et al., 2015). In GSC11 cells cultured with serum for 3 days, shALKBH5 increased the GFAP- and Tuj-1- positive populations (Figure S1L). Taken together, these data suggest that ALKBH5 is required for GSC self-renewal.
ALKBH5 Inhibition Decreases the Proliferation and Tumorigenesis of GSCs
Next, we examined the effects of ALKBH5 knockdown on GSC proliferation. In GSC11, GSC17, and GSC23 cells, loss of ALKBH5 inhibited cell growth (Figure 2A) and decreased DNA replication, as revealed by 5-bromo-2′-deoxyuridine (BrdU) incorporation assay (Figures 2B and 2C). However, depletion of ALKBH5 had no effect on the growth of SW1783 non-CSC glioma cells (Figure S2A). Moreover, cell cycle analysis showed that knockdown of ALKBH5 in GSCs increased the proportions of cells in G0/G1 phase whereas decreased the proportions of cells in S and G2/M phase (Figure S2B). Collectively, these results suggest that ALKBH5 critically regulates GSC proliferation.
Figure 2. Inhibition of ALKBH5 Impairs GSC Proliferation and Tumorigenecity.

(A) Proliferation of GSCs with or without ALKBH5 knockdown as determined by cell counting. n=3. *p<0.05, **p<0.01, ***p<0.001.
(B) Proliferation of GSCs with or without ALKBH5 knockdown as assessed by BrdU (5-bromo-2′-deoxyuridine) incorporation for 3 hr. Scale bar, 20 μm.
(C) Random selected microscope fields of (B) were quantified for BrdU-positive GSCs. n=5.
(D) Survival analysis of mice intracranially implanted with GSCs with or without ALKBH5 knockdown. GSCs were implanted intracranially into nude mice, and tumor formation was determined by histology.
(E) Top, histograms show tumor volumes formed by indicated GSC17 cells in mouse brains. Bottom, representative images of H&E staining for tumor formation. n=8.
All bar plot data are the means ± SEMs.
See also Figure S2
Then we examined the effect of ALKBH5 depletion on GSC tumorigenicity by intracranial injection into mice with 10,000 viable GSC11 or GSC17 cells transduced with shCtrl or shALKBH5. Compared with the mice injected with shCtrl-GSC cells, those injected with shALKBH5-GSC cells displayed extended survival with a lower rate of tumor formation (Figure 2D). Moreover, the inhibition of brain tumor formation by shALKBH5 could be effectively rescued by overexpression of the wild-type ALKBH5 but not the reported catalytic inactive mutant ALKBH5 H204A (Figure 2E). In vitro limiting dilution assay suggested that the wild-type ALKBH5, not the H204A mutant, was able to rescue the loss of self-renewal capacity caused by ALKBH5 knockdown (Figure S2C). These results demonstrate that the demethylation activity of ALKBH5 is critical to GSC tumorigenicity.
Analysis of Downstream Targets of ALKBH5 in GSCs
To understand the regulatory role of ALKBH5 in comprehensive gene expression, we employed microarray analysis to compare the gene expression profile following ALKBH5 knockdown by different shRNAs in GSC11 and GSC17 cells. A total of 206 genes that were differentially expressed by at least 2-fold in both cell lines were selected for ingenuity pathway analysis (IPA) (Figure 3A and Table S2). We found that the largest subset of genes is primarily involved with “Cell Cycle”, followed by “Cellular Assembly and Organization” and “DNA Replication, Recombination, and Repair” (Table S2). Notably, the roles of many genes, which regulate microtubule organization, spindle assembly, chromosome congregation and segregation, or DNA replication, overlap across these pathways.
Figure 3. Characterization of m6A Modification and Gene Expression Changes in the ALKBH5-Deficient Cells.

(A) Venn diagrams show 206 genes with over 2-fold expression change in shALKBH5 compared with shCtrl in both GSC11 and GSC17 (top). IPA identified the enriched gene ontology processes of the 206 genes (bottom).
(B) Top consensus motif identified by HOMER with m6A-seq peaks in GSC11 cells with or without ALKBH5 knockdown.
(C) Number of m6A peaks identified in m6A-seq in shCtrl and shALKBH5 GSC11 cells.
(D) Number of m6A-modified genes identified in m6A-seq. Common m6A genes contain at least 1 common m6A peak, while unique m6A genes contain no common m6A peaks.
(E) Graphs of m6A peak distribution showing the proportion of total m6A peaks in the indicated regions in control and ALKBH5-deficient cells (top) and the appearance of new m6A peaks (unique peaks in shALKBH5) or loss of existing m6A peaks (unique peaks in shCtrl) after ALKBH5 knockdown (bottom).
(F) Schematic of ALKBH5 downstream analysis.
(G) Relative mRNA levels of FOXM1 downstream targets from microarray analysis. Data are the means ± SEMs for GSC11 and GSC17 cells.
Combined analysis of 2 independent biological replicates in (B–E).
See also Figure S3 and Table S2–S5
To test whether the altered gene expression could be a consequence of ALKBH5-mediated m6A demethylation, we compared the m6A distribution in control or ALKBH5 knockdown GSCs. First, we found increased global m6A levels in ALKBH5-deficient GSCs, confirming the demethylation activity of ALKBH5 in GSCs (Figure S3A). Next, we mapped the m6A methylomes of shCtrl and shALKBH5-1 GSC11 cells by m6A-seq with independent biological replicates. The GGACU motif was identified to be highly enriched within m6A sites in the GSC11 cells (Figure 3B). In total, m6A-seq identified 39,288 and 34,415 m6A peaks from 12,358 and 12,196 m6A-modified transcripts in control and ALKBH5-deficient cells, respectively (Figures 3C and 3D). In the GSC11 cells with ALKBH5 stable knockdown by shRNA, 5,053 new peaks appeared with disappearance of 9,650 peaks. The other 29,362 peaks were found in both knockdown and control cells (Figure 3C). Since ALKBH5 is an m6A demethylase, the 5,053 unique peaks are expected to contain genuine targets of ALKBH5. We further investigated the m6A distribution patterns within both total and unique peaks. A similar pattern of total and common m6A distribution in control and ALKBH5-deficient cells was observed when the RNA species were divided into 5′UTR, coding sequence (CDS), 3′UTR regions of mRNAs and non-coding RNAs (Figures 3E and S3B). Interestingly, in contrast to the 9,650 lost peaks which displayed identical distribution to the total peaks, the 5,053 unique ALKBH5-dependent peaks showed a distinct pattern in which a relative increase of m6A deposit appeared in 5′UTR and non-coding RNAs (Figure 3E). These unique peaks included 1,302 peaks from 5′UTR, 2,724 from CDS and 678 from 3′UTR of mRNAs and 349 peaks from 281 non-coding RNAs.
Then, we asked whether these peaks were associated with differentially expressed genes in the microarray analysis. Among the 206 differentially regulated transcripts identified by microarray, 148 genes were found in the RNA-seq data set (m6A-seq input library), including 85 genes repeatedly showing over 2-fold quantitative difference in the same direction (Table S3). Subsequent IPA analysis of the 85 genes revealed identical functional clusters as the 206 genes (Figure S3C). Next, filtering the 5053 unique m6A peaks with the 85 genes with 2-fold expression changes resulted in the identification of 13 peaks harbored by 12 genes, including ABHD4, ANP32E, CENPF, FANCI, FOXM1, MKI67, BCL2, UBE2C, NT5E, IGFBP5, TSPYL2 and CDCA2 (Figure 3F and Table S4). It is likely that a few key candidate genes are responsible for the observed phenotype and altered expression of the majority of proliferation-related genes.
Next, we performed ingenuity’s upstream regulator analysis for the 206 genes as well as the 85-gene subset, each resulting in a similar list of regulators predicted to be activated or inhibited following ALKBH5 knockdown (Table S5). Among these potential regulators, only FOXM1 was one of the 12 candidate genes and had a high predicted activation Z-score, suggesting that FOXM1 downregulation might explain the loss of the GSC proliferation signature. Moreover, in the TCGA GBM data set, we found that the expression of ABHD4, ANP32E, CENPF, FANCI and FOXM1 were correlated with SOX2 and Nestin (data not shown). However, the functions of ABHD4, ANP32E, CENPF and FANCI in GBM remain unclear.
FOXM1 is a pivotal transcription factor in cell cycle regulation and plays critical roles in the self-renewal and tumorigenesis of GSCs. The IPA-defined and other reported FOXM1 transcriptional targets were all decreased following ALKBH5 knockdown in both our microarray and RNA-seq data set (Figure 3G and Table S3). Given that FOXM1 could mediate the expression of a number of ALKBH5-responsive pro-proliferative genes in GSCs, our following research was focused on this potentially important target gene.
ALKBH5 Regulates FOXM1 Expression in GSCs
Since FOXM1 could be a direct substrate of ALKBH5 and a driver for the altered proliferative gene profile, we investigated the regulatory mechanism of FOXM1 expression by ALKBH5. We first measured the change of total mRNA expression and different isoform (FOXM1A, -B, and -C) levels of FOXM1 upon ALKBH5 knockdown. Consistent with the gene expression data, both GSC11 and GSC17 with ALKBH5 knockdown displayed approximately 70–80% lower expression of total and isoforms of FOXM1 mRNA (Figures 4A and S4A). We also measured the protein expression of FOXM1 and SOX2 following ALKBH5 knockdown by siRNA treatment or shRNA lentiviral transduction. Both transient and stable knockdown reduced FOXM1 protein abundance (Figures 4B and 4C). In contrast, SOX2 expression was only downregulated by stable ALKBH5 knockdown but almost unaffected by transient ALKBH5 knockdown, suggesting that SOX2 may not be a direct target of ALKBH5 in GSCs (Figures 4C and S1K). FOXM1 depletion abrogated the rescue effect of ALKBH5 on GSC proliferation in shALKBH5 cells, suggesting the key role of FOXM1 (Figure S4B).
Figure 4. ALKBH5 Maintains FOXM1 Expression by Demethylating FOXM1 Nascent Transcripts.

(A) qPCR analysis of FOXM1 mRNA expression in GSC11 with or without ALKBH5 knockdown. Samples were normalized to GAPDH mRNA.
(B) Western blotting of FOXM1 in GSCs with or without ALKBH5 knockdown.
(C) Western blotting of FOXM1 and SOX2 in GSC17 cells treated with control or ALKBH5 siRNAs.
(D) Correlation between FOXM1 and ALKBH5 mRNA expression (RNAseq V2 RSEM [log2]) in the TCGA GBM data set.
(E) Correlation between ALKBH5 and FOXM1 protein expression in GBM specimens. Left, representative IF images of 15 GBM specimens. Scale bar, 20 μm. Right, in 5 random selected microscope fields of each tumor, the percentage of FOXM1 positive cells among ALKBH5 positive versus ALKBH5 negative cells was compared by t-test. Lines show mean and SD.
(F) qPCR analysis of FOXM1 pre-mRNA in GSCs with or without ALKBH5 knockdown.
(G) Cytoplasmic (C), nucleoplasmic (N), and insoluble (I) fractions of GSC11 cells treated with or without 5 μg/ml ActD (actinomycin D) for 2 hr were subjected to Western blotting for indicated proteins.
(H) Distribution of FOXM1 transcripts in subcellular fractions assessed by qPCR.
(I) RIP (RNA immunoprecipitation) analysis of transcripts from the nuclear extracts of GSC17 cells expressing exogenous ALKBH5 with or without FLAG tag. Enrichment of FOXM1 mature and pre-mRNA with FLAG was measured by qPCR and normalized to input. Western blotting of ALKBH5 showing equal expression of tagged or untagged proteins.
(J) MeRIP-qPCR analysis of m6A levels of FOXM1 pre-mRNA in GSC17 cells with or without ALKBH5 knockdown.
(K) Western blotting of FOXM1 in GSC17 with or without ALKBH5 knockdown and transfected with control, wild-type or mutant ALKBH5 plasmid.
All bar plot data are the means ± SEMs of 3 independent biological replicates. *p<0.05, **p<0.01, ***p<0.001.
See also Figure S4
Next, we examined the correlation between ALKBH5 and FOXM1 in GBM patient samples. We queried the TCGA data set and found a significant positive correlation (Pearson’s r=0.4723914, p=6.197×10−10) between ALKBH5 and FOXM1 mRNA expression (Figure 4D). This result was confirmed by immunofluorescence analysis of the expression and correlation of ALKBH5 and FOXM1 proteins in primary GBM samples (Figure 4E). Collectively, these results indicate that FOXM1 is a direct downstream target of ALKBH5 in GBM.
FOXM1 Nascent Transcripts Are Substrates of ALKBH5
Since alternative splicing cannot explain the reduced FOXM1 expression in ALKBH5-knockdown cells and ALKBH5 knockdown may affect mRNA export and RNA metabolism (Zheng et al., 2013), we assessed the stability and subcellular localization of FOXM1 mRNA. Surprisingly, measuring the loss of FOXM1 mRNA after blocking new RNA synthesis with actinomycin D revealed that shCtrl and shALKBH5 cells had similar FOXM1 mRNA stability (Figure S4C). On the other hand, ALKBH5 knockdown did not appear to affect the nuclear retention or export of FOXM1 RNA (Figure S4D), probably because our experiments could not distinguish the small amount of increased FOXM1 RNA export from the abundant cytoplasmic FOXM1 mRNA. Furthermore, ALKBH5 knockdown did not affect FOXM1 promoter activity as determined using promoter reporter assay (Figure S4E). These findings raise the possibility that ALKBH5 deficiency results in a decrease in FOXM1 nascent transcripts and then the detectable changes in the expression of mature RNA or protein. Indeed, FOXM1 precursor mRNA expression was decreased by 40% following ALKBH5 siRNA treatment and further decreased in GSCs stably expressing shALKBH5 (Figures 4F and S4F).
ALKBH5 may colocalize with nuclear speckles and promote proper assembly and/or modification of certain mRNA processing factors (Zheng et al., 2013). However, whether ALKBH5 can act on nascent transcripts is unknown. To test this, we first separated nuclei from the cytoplasmic fraction of GSC11 cells and fractionated them into a soluble nucleoplasmic fraction containing nuclear speckles and an insoluble fraction containing nascent RNAs and chromatin-associated components. Western blotting for the compartment-specific proteins α-tubulin, U1-70K, and Histone H3 confirmed organelle subfractionation (Figure 4G). We found that ALKBH5 was present in both the nucleoplasmic and insoluble fractions (Figure 4G). However, unlike the nuclear RNA-binding protein HuR (ELAVL1), the majority of ALKBH5 was enriched in the insoluble fraction in a transcription-dependent manner, because its nuclear abundance decreased after actinomycin D treatment (Figures 4G and S4G). In addition, FOXM1 pre-mRNA was predominantly in the insoluble fractions (Figure 4H), which can be explained by the widespread co-transcriptional splicing model in human brain (Ameur et al., 2011). Moreover, the results of native RNA immunoprecipitation (RIP) assay revealed that FOXM1 pre-mRNA interacts with FLAG-ALKBH5 (Figure 4I). Together, our findings indicate that ALKBH5 colocalizes and interacts with FOXM1 nascent transcripts.
To ascertain whether FOXM1 nascent transcript is a substrate for ALKBH5, we used methylated RNA immunoprecipitation (MeRIP)–quantitative polymerase chain reaction (qPCR) to determine the FOXM1 m6A methylation levels following ALKBH5 knockdown. Our analysis confirmed that m6A methylation was readily detectable on FOXM1 pre-mRNA (Figure S4H). Moreover, ALKBH5 knockdown increased the m6A level of FOXM1 pre-mRNA compared with the control (Figure 4J). Overexpression of the wild-type ALKBH5 but not the mutant H204A could restore FOXM1 expression (Figures 4K and S4I), indicating that ALKBH5 mainly affects FOXM1 expression through its demethylation activity.
FOXM1 3′UTR Mediates ALKBH5 Regulation
From our m6A-seq data of shALKBH5 cells, we have identified 4 statistically significant m6A peaks in FOXM1 mRNA, with one peak near the stop codon and one unique peak on 3′UTR (Figures 5A and S5A). This unique peak was specifically demethylated by ALKBH5, suggesting functional relevance. The m6A status of each peak was further measured by MeRIP of fragmented RNA. Using specific primers to detect each peak region, we found that m6A enrichment of the unique peak region was ~0.9% and ~1.4% in control GSC17 and GSC11, respectively (Figures 5B and 5C). However, m6A level consistently elevated by 1~2 fold on this region in shALKBH5 cells (Figures 5B and 5C). In the unfragmented RNA samples, we also found the increase of m6A on intact pre-mRNA, which had a lower m6A fraction compared with mature RNA, suggesting m6A-marked pre-mRNA is less stable (Figure 5B). To further study the importance of FOXM1 3′UTR region, we generated FLAG-tagged expression constructs containing the entire FOXM1 including the coding sequence (CDS) with the 3′UTR (CDS-3′UTR) or the CDS alone. Upon ALKBH5 knockdown, FLAG-FOXM1 expression was significantly decreased in GSCs transfected with the FOXM1 CDS-3′UTR construct but not the FOXM1 CDS-alone construct (Figures 5D and S5B), which suggested that the 3′UTR is critically involved in ALKBH5-FOXM1 regulation. It also implied that the reversible m6A incorporation could be splicing-independent, which could be supported by the detected methylation on the FLAG-FOXM1 transgene (Figure S5C). This effect of the FOXM1 3′UTR could be explained by 1 of 2 distinct models: 1) a tandem cis-regulation model in which the 3′UTR functions as a sensor and the FOXM1 CDS m6A sites are the effectors, or 2) a model in which the 3′UTR is sufficient to initiate the regulation regardless of the m6A status in the coding sequence. To test these possibilities, we performed the FOXM1 3′UTR-reporter luciferase assay and found that ALKBH5 knockdown decreased the activity of the luciferase construct containing the FOXM1 3′UTR, in favor of the second model (Figure 5E). Moreover, high resolution m6A transcriptome study identified five m6A sites covered by the unique peak region in human cells (Chen et al., 2015a). Mutation at all these sites (A to T) rendered resistance to the effect of ALKBH5 knockdown (Figure 5E).
Figure 5. The FOXM1 3′UTR Mediates ALKBH5 Regulation.

(A) Left, peaks show the relative abundance of m6A sites along FOXM1 mRNA in GSC11-shALKBH5 cells. The blue and red reads are from the non-IP control input and MeRIP of GSC11-shALKBH5, respectively. Right, identification of a unique peak at 3′UTR.
(B) MeRIP-qPCR analysis of fragmented or intact FOXM1 RNA from GSC11 with or without ALKBH5 knockdown. Left: peak region enrichment in fragmented RNA. Right: enrichment of m6A-marked FOXM1 mature and pre-mRNA in intact RNA. Up: half samples were fragmented.
(C) MeRIP-qPCR analysis of fragmented FOXM1 RNA in GSC17 with or without ALKBH5 knockdown.
(D) Western blotting of FLAG-FOXM1 in GSCs seeded in 12-well plates and transfected with 50 ng FOXM1 CDS-3′UTR or FOXM1 CDS plasmid and treated with control or ALKBH5 siRNAs.
(E) Relative activity of the wild-type or mutant FOXM1 3′UTR firefly luciferase reporter in GSC17 cells treated with control or ALKBH5 siRNAs.
(F) RIP analysis of the interaction of FLAG with FOXM1 pre-mRNA using total cell lysates of GSC17 cells expressing exogenous HuR with or without FLAG tag and treated with control or ALKBH5 siRNAs. Enrichment of FOXM1 pre-mRNA with FLAG was measured by qPCR and normalized to input. Western blotting of HuR showing equal expression of tagged or untagged proteins.
(G) qPCR analysis of FOXM1 in GSCs treated with control or HuR siRNAs.
(H) Western blotting of FOXM1 RNA expression in GSCs treated with control or HuR siRNAs.
All bar plot data are the means ± SEMs of 3 independent experiments except (B) and (C) where error bars denote SD of technical triplicates. *p<0.05, **p<0.01, ***p<0.001.
See also Figure S5
ALKBH5 Affects HuR Association with FOXM1 Nascent Transcripts
To further explain the negative correlation between methylation and FOXM1 abundance, we investigated the carrier of the biological consequences of m6A: RNA-binding proteins. The nuclear RNA-binding protein HuR, which reportedly regulates both pre-mRNA splicing and expression (Lebedeva et al., 2011; Mukherjee et al., 2011), has been shown to disfavor binding with m6A-modified RNA and exert stabilizing effects on its bound RNA (Wang et al., 2014b). To examine the role of HuR in our system, we first tested whether HuR interacted with FOXM1 nascent transcripts. RIP-qPCR revealed that HuR interacts with FOXM1 pre-mRNA, and this interaction decreased following ALKBH5 knockdown (Figures 5F and S5D). Moreover, transient HuR knockdown suppressed FOXM1 expression (Figures 5G and 5H), reduced FOXM1 3′UTR luciferase activity (Figure S5E), and decreased FLAG-FOXM1 expression in GSCs transfected with the FOXM1 CDS-3′UTR construct but not the CDS construct (Figure S5F). These data suggest that HuR plays an important role in the regulation of FOXM1 by ALKBH5 via promoting expression of FOXM1 nascent transcripts through binding to the unmethylated 3′UTR.
FOXM1-AS Is a Nuclear lncRNA that Facilitates the Interaction between ALKBH5 and FOXM1 Nascent Transcripts
Human cells have similar m6A distributions but dynamic m6A levels on specific genes at different conditions, possibly caused by trans factors (Batista et al., 2014; Chen et al., 2015b; Zhou et al., 2015). Whether removing m6A also involves gene- or cell-specific mechanisms is unknown. We investigated the local elements that may contribute to ALKBH5′s preferential regulation of the FOXM1 3′UTR. Intriguingly, we found a long non-coding RNA (lncRNA) LOC100507424 located on chromosome 12 (chr12: 2945982-2968961, GRCh37/hg19); this lncRNA is transcribed in the opposite direction to FOXM1 and has 457 nucleotides complementary to the last exon of FOXM1 mRNA (Figure 6A). We focused on LOC100507424 (hereafter referred to as FOXM1-AS) for several reasons. First, based on their sequence complementarity, FOXM1-AS may regulate FOXM1 expression in cis. Second, as determined by qPCR using primers for the FOXM1-AS non-overlapping region, most GSC cells with high levels of FOXM1-AS also express FOXM1 at high levels (Figures 6B and S6A), which suggests a concordant pattern of regulation. Third, FOXM1-AS is enriched in the insoluble fraction (Figure 6C), suggesting that FOXM1-AS is localized in the same cellular fraction as ALKBH5 and FOXM1 nascent transcripts.
Figure 6. FOXM1-AS Is a Nuclear lncRNA That Facilitates the Interaction between ALKBH5 and FOXM1 Nascent Transcripts.

(A) Physical map of FOXM1 and FOXM1-AS (F-AS) transcription units. Arrows indicate the direction of transcription. Vertical bars indicate exons.
(B) qPCR analysis of indicated RNA expression in glioma cells and GSCs.
(C) Top, RNA distribution in subcellular fractions assessed by qPCR. Bottom, Western blotting of indicated proteins in 2 independent experiments.
(D and E) Analysis of the interaction between F-AS and ALKBH5 by an RIP assay using anti-FLAG antibodies in the nuclear extracts of GSC17 cells expressing exogenous ALKBH5 with or without FLAG tag (D) and an RNA pull-down assay using biotinylated probes to capture F-AS in the nuclear extracts of GSC17 cells (E). The RNA and protein in the RNA-protein complex were then detected by qPCR and Western blotting, respectively. HuR and antisense probes consisted of complementary sequence to F-AS probes served as negative controls.
(F) RNA pull-down assay of F-AS–associated RNA from GSC17 cells. Biotinylated F-AS probes or antisense probes were incubated with AMT-crosslinked or untreated RNA and collected with streptavidin beads. Interaction with F-AS was quantified by qPCR.
(G) RIP analysis of the interaction of FOXM1 pre-mRNA with FLAG in the nuclear extracts of GSC17 cells expressing exogenous ALKBH5 with or without FLAG tag and treated with control or F-AS siRNAs. Enrichment of FOXM1 pre-mRNA with FLAG was measured by qPCR and normalized to input.
(H) MeRIP-qPCR analysis of m6A levels of FOXM1 pre-mRNA in GSC17 cells with or without F-AS knockdown.
(I) Western blotting of FOXM1 and ALKBH5 in GSC17 cells and transfected with FOXM1 CDS-3′UTR or FOXM1 CDS plasmid before treatment with indicated siRNAs.
(J) Relative activity of the FOXM1 3′UTR firefly luciferase reporter in GSC17 cells treated with control or F-AS siRNAs.
(K) RIP analysis of the interaction of FOXM1 pre-mRNA with HuR in GSC17 cells with or without F-AS knockdown. Enrichment of FOXM1 pre-mRNA with HuR was measured by qPCR.
All bar plot data are the means ± SEMs of 3 independent experiments. *p<0.05, **p<0.01, ***p<0.001. N.S., no significance.
See also Figure S6
Based on the above findings, we hypothesized that FOXM1-AS interacts with ALKBH5 and FOXM1 nascent transcripts and is involved in the ALKBH5-FOXM1 regulation. To test the hypothesis, we used native RIP to assess the interaction between FOXM1-AS and ALKBH5 and found that FOXM1-AS was enriched in FLAG-ALKBH5 (Figure 6D). The interaction was further confirmed by the RNA pull-down assay, where a set of biotinylated RNA probes corresponding to the FOXM1-AS without the overlapping region with FOXM1, retrieved ALKBH5 proteins from the GSC nuclear extracts (Figure 6E). However, the abundant nuclear RNA-binding protein, HuR, was not retrieved in this condition, suggesting FOXM1-AS specifically interacts with ALKBH5 (Figure 6E).
Next, we investigated whether FOXM1-AS interacts with FOXM1 transcripts. We fixed RNA-RNA hybrids in GSCs with 4′-aminomethyltrioxalen (AMT), a psoralen-derivative crosslinker. Direct in vivo RNA-RNA interactions were captured by AMT crosslink in the RNA pull-down experiment, as AMT penetrates intact cells and generates inter-strand crosslinks between uridine bases in RNA upon ultraviolet irradiation but does not react with proteins (Calvet and Pederson, 1979). FOXM1 pre-mRNA but not mature mRNA could be retrieved from the pull-down fractions of AMT-treated GSCs using the biotinylated FOXM1-AS RNA probes, whereas neither FOXM1 pre-mRNA nor mature mRNA could be retrieved from the pull-down fractions of untreated cells or from the pull-down fractions using the antisense probes (Figure 6F). These results suggest that FOXM1 nascent transcripts but not mature mRNA are associated with FOXM1-AS in vivo. Collectively, these data indicate that FOXM1-AS interacts with both FOXM1 nascent transcripts and ALKBH5.
Having discovered that FOXM1-AS interacts with FOXM1 nascent transcripts and ALKBH5 protein, and knowing that ALKBH5 binds to FOXM1 RNA, we investigated whether the former was required for the latter. First, we assessed the efficacy of 5 different siRNAs against FOXM1-AS with unique sequences at non-overlapping regions and found that siRNA#2 and #3 were the most effective (Figure S6B). Next, we constructed two shRNAs based on the sequences of siRNA#2 and #3. Notably, RNA recovery from RIP of FLAG-ALKBH5- protein complex revealed that knocking down FOXM1-AS using pooled siRNA (siRNA#2 and #3) resulted in a decrease in ALKBH5-associated FOXM1 nascent transcripts, suggesting a positive regulatory role of FOXM1-AS (Figure 6G). In consistent with the above findings, FOXM1-AS knockdown increased the m6A modification of FOXM1 pre-mRNA (Figure 6H). Furthermore, FOXM1-AS knockdown decreased the FLAG-FOXM1 expression in a similar manner as ALKBH5 knockdown (Figure 6I); reduced FOXM1 3′UTR luciferase activity (Figure 6J); and decreased HuR and FOXM1 RNA association (Figure 6K).
Next, we sought to determine whether FOXM1-AS has a biological function in GSC. Knockdown of FOXM1-AS led to a significant decrease in the expression of FOXM1 RNA and protein (Figures 7A and 7B). To determine the significance of this regulation in GBM patients, we measured FOXM1-AS and FOXM1 levels in tumors from different patients and in different regions within same tumors using laser-capture microdissection. Both transcripts displayed inter- and intra-tumoral heterogeneity expression with a positive correlation of each other (Figure 7C). Moreover, shFOXM1-AS substantially reduced GSC growth and DNA replication (Figures 7D and 7E). In addition, shFOXM1-AS impaired tumorsphere formation and suppressed GSC marker expression, which was rescued by forced re-expression of FOXM1-AS (Figures 7F, 7G and S7 A and B). Thus, we conclude that FOXM1-AS stimulates FOXM1 expression and contributes to GSC maintenance.
Figure 7. Knocking Down FOXM1-AS Impairs FOXM1 Expression and Self-renewal in GSCs.

(A) qPCR analysis of indicated RNA in GSC17 cells with or without F-AS knockdown.
(B) Western blotting of FOXM1 in GSCs with or without F-AS knockdown.
(C) The Pearson correlation between F-AS and FOXM1 RNA expression in 45 tumor regions by laser-capture microdissection from 15 GBM specimens. 3 different tumor regions from the same tumor were represented in one color.
(D) Proliferation of GSCs with or without F-AS knockdown, as determined by cell counting.
(E) Representative IF images of GSCs incorporated with BrdU overnight.
(F) Tumorsphere formation efficiency of GSC11 cells with or without F-AS knockdown after 7 days of incubation in a 96-well plate, as calculated by the number of spheres per 100 single cells.
(G) Fluorescence and bright-field micrographs of GSCs with or without F-AS knockdown.
All bar plot data are the means ± SEMs of 3 independent experiments, except those in (F), where n=5. *p<0.05, **p<0.01, ***p<0.001. Scale bar, 20 μm.
See also Figure S7
FOXM1 Reinstates the Tumor Growth of GSCs with Depleted ALKBH5 or FOXM1-AS
To ascertain if FOXM1 is a major contributor to the function of ALKBH5 in GSC proliferation, we asked whether adding back FOXM1 could reverse the effects of ALKBH5 inhibition. Forced expression of the FOXM1 CDS alone robustly stimulated the re-expression of proliferation-related genes (Figure 8A). Moreover, the mice injected with GSC11- and GSC17- control cells developed brain tumors resembling human GBM, whereas depletion of ALKBH5 or FOXM1-AS substantially inhibited brain tumor formation (Figure 8B). We confirmed the control xenografts co-expressed ALKBH5 and FOXM1 in the tumor lesions (Figure 8C) and depletion of ALKBH5 or FOXM1-AS decreased the expression of SOX2 and the proliferation marker Ki67 in addition to reduced FOXM1 (Figure S8). Further, ectopic expression of FOXM1 CDS largely abolished tumor growth inhibition and gave rise to brain tumors with high expression of SOX2 and Ki67 (Figures 8B and S8). These results suggest that ALKBH5 and FOXM1-AS cooperate in the regulation of FOXM1 and play important roles in GSC tumorigenesis.
Figure 8. FOXM1 Reinstates the Tumor Growth of GSCs with Depleted ALKBH5 or FOXM1-AS.

(A) qPCR analysis of the indicated mRNAs in GSC11 cells with or without ALKBH5 knockdown and in GSC11 cells with ALKBH5 knockdown rescued by exogenous FOXM1. Error bars: ± SEM of 3 independent experiments. *p<0.05, **p<0.01 (shALKBH5 + FOXM1 versus + Vector)
(B) Top, representative images of H&E staining for tumor formation. Bottom, histograms show tumor volumes. Error bars: ± SEM. n=8.
(C) Representative IF images of the indicated proteins in GSC17 xenografts. Scale bar, 50 μm.
See also Figure S8
DISCUSSION
Alkbh5 knockout mice are viable but have compromised spermatogenesis (Zheng et al., 2013), suggesting that the ALKBH5 contribution is limited to a subset of cells rather than being involved in the systemic regulation of vertebrate development. The disease-associated expression and function of ALKBH5 have remained unclear. We found that ALKBH5 function is important to GSCs. Interestingly, GSCs are addicted to the overexpression of transferrin, transferrin receptor, and ferritin, the depletion of which disrupts the mitotic progression of GSCs (Schonberg et al., 2015). ALKBH5 belongs to the AlkB family of nonheme Fe (II)/α-ketoglutarate-dependent dioxygenases, whose activity is iron-dependent (Zheng et al., 2013). ALKBH5 is not GSC-specific, yet GSCs rely on ALKBH5 expression and preferential iron trafficking. These molecular components of GSCs may be integrated synergistically, as evidenced by the gene expression analysis from the iron study by Schonberg et al. that also identified FOXM1 as the central molecular mediator of GSC proliferation. Schonberg et al. proposed that FOXM1 acts downstream from STAT3 in the ferritin-STAT3-FOXM1 feedback loop, which may be true for the cells that express phosphorylated STAT3. However, unlike the widespread expression of FOXM1 across all subtypes of GBM, phosphorylated STAT3 is mostly restricted to mesenchymal GBMs and absent in proneural GBMs (Bhat et al., 2013). This generally observed iron-dependent expression of FOXM1 now may be partially explained by the possibility that iron metabolism modulates ALKBH5 activity in GSCs and consequently affects FOXM1 expression.
ALKBH5 was reported to mediate the hypoxia-induced and hypoxia inducible factor (HIF)-dependent breast CSC phenotype (Zhang et al., 2016), suggesting the important tumorigenic role. ALKBH5 is a direct target of HIF-1α (Thalhammer et al., 2011) and might be regulated by HIF-2α (Zhang et al., 2016). GSCs are addicted to HIFs under both normoxic and hypoxic conditions (Li et al., 2009). Additional studies are needed to define the extent to which HIFs and other potential regulators stimulate ALKBH5 expression in GSCs.
The impaired male fertility phenotype of Alkbh5 knockout mice is caused by altered expression of spermatogenesis-related mRNAs, despite a widespread existence of mRNA methylation (Zheng et al., 2013). These results suggest perhaps not all modified sites are equally important, and only a subset of them are critically involved in some biological processes. In the current study, we found that ALKBH5 deficiency in GSCs resulted in unique m6A modification on select transcripts, and altered gene expression. We demonstrated that among these genes, FOXM1 and its downstream cell cycle genes are important to the functions of ALKBH5 in GSCs. Nevertheless, although we focused on the regulation of FOXM1, it is likely that other ALKBH5 target genes can also contribute to the ALKBH5 function.
Unlike FOXM1 orthologs, which are expressed across eukaryotic tree of life, ALKBH5 arose with the advent of vertebrates. This suggests that the ALKBH5-FOXM1 regulatory interaction reflects a late-occurring process associated with certain cell identities or tumor development. Moreover, recent studies suggested that the specificity of m6A methylation exists for some targets and can be achieved by trans factors (Aguilo et al., 2015; Chen et al., 2015b). In this study, we found that a nuclear antisense lncRNA promotes the ALKBH5-FOXM1 interaction. It is a warrant to further study the detailed mechanism of FOXM1-AS’s action on the ALKBH5-FOXM1 interaction.
More than 70% of mammalian transcriptomes have antisense transcription (Katayama et al., 2005). The current view of methylation regulation is incomplete, and determining whether there are other antisense lncRNAs that give rise to the specificity of m6A methylation would help to complete this view. A related question that warrants investigation is how these lncRNAs associate with m6A “writers,” “erasers,” and/or “readers.”
In conclusion, we show that inhibition of ALKBH5 represses GSC tumorigenesis and FOXM1 is a key component in mediating ALKBH5-dependent GSC proliferation. These findings open up avenues for developing effective therapeutic strategies in the treatment of glioblastoma.
STAR Methods
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Suyun Huang (suhuang@mdanderson.org).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animals
Mice and Animal Housing
Male Foxn1nu/nu athymic nude mice at 6–8 week age were purchased from MD Anderson Cancer Center ERO Breeding Core. Mice were grouped by 5 animals in large plastic cages and were maintained under pathogen-free conditions according to the NIH Guide for the Care and Use of Laboratory Animals. All mouse experiments were approved by MD Anderson’s Institutional Animal Care and Use Committee.
Intracranial Tumor Assay
Mice were randomly assigned to experimental groups for all the experiments. For the animal survival analysis, mice were intracranially injected with 10,000 GSCs and maintained until moribund or 80 days after injection. For the rescue studies, GSCs with ALKBH5 or FOXM1-AS shRNAs were co-transfected with a FOXM1, ALKBH5 wild-type or mutant expression construct. A total of 50,000 GSCs were intracranially injected into mice (n = 8 mice per group). At 30 days after GSC11 injection and 20 days after GSC17 injection, the mice were humanely killed, and their brains were harvested. Each mouse brain was fixed in 4% formaldehyde, embedded in paraffin, and examined for tumor formation by histologic analysis of hematoxylin and eosin–stained sections. Tumor volume was calculated by the formula V = ab2/2, where a and b are the tumor’s length and width, respectively.
Human Subjects
All tumor collection and analysis were approved by the University of Texas MD Anderson Cancer Center Institutional Review Board with informed consent.
Cell Lines and Primary Cell Cultures
Human glioma Hs683 and SW1783 cell lines and GBM LN229 and U87MG cell lines were from the American Type Culture Collection. U251MG cell line was from Sigma. The immortalized NHA-E6/E7/hTERT cell line has been described previously (Sonoda et al., 2001). These cell lines were cultured in Dulbecco’s modified Eagle’s medium (DMEM) with 10% fetal bovine serum. GSCs were obtained from fresh surgical specimens of human primary and recurrent GBMs and cultured as tumorspheres in DMEM/F12 medium supplemented with B27 supplement (Life Technologies), bFGF and EGF (20 ng/ml each). Only early-passage GSCs were used for the study. The characteristics of the GSCs were presented in Table S1. All patient-related studies were reviewed and approved by an Institutional Review Board at The University of Texas MD Anderson Cancer Center with informed consent.
METHOD DETAILS
Immunofluorescence and Immunohistochemistry
For immunohistochemical (IHC) and immunofluorescence (IF) analysis, GBM xenografts or surgical specimens tissue slides were deparaffinized, rehydrated through an alcohol series followed by antigen retrieval with sodium citrate buffer. Tumor sections were blocked with 5% normal goat serum (Vector) with 0.1% Triton X-100 and 3% H2O2 in PBS for 60 min at room temperature and then incubated with appropriate primary antibodies 43°C overnight. IHC staining was performed with horseradish peroxidase (HRP) conjugates using DAB detection. IF staining was performed with appropriate Alexa Fluor 488 or Alexa Fluor 594 secondary antibodies (Invitrogen, dilution 1:1000).
For IF analysis of cultured cells, GSCs were fixed with 4% formaldehyde (Fisher) for 15 min and then blocked with 5% normal goat serum (Vector) with or without 0.1% Triton X-100 in PBS for 60 min at room temperature. Immunostaining was performed using the appropriate primary and secondary antibodies. Nuclei were counterstained with Hoechst. Images were taken with a ZEISS Axio Scope.A1 Upright Microscope.
Specific antibodies against FOXM1 (K19, Western Blotting; G5, IF, IHC), Nestin, HuR, GFAP, Tuj-1, SSEA-1, SOX2 (E4, IF), Nanog, α-Tubulin, β-actin from Santa Cruz; SOX2 (D6D9, immunoblotting), Oct4, Histone H3, CD133 from Cell Signaling; U1-70K, ALKBH5 (ABE547, immunoblotting) from Millipore; FLAG, ALKBH5 (HPA007196) from Sigma were used for the IF, IHC, or immunoblot analyses.
RNA Pull-Down and RNA Immunoprecipitation
A mix of six 90- to 120-nucleotide probes antisense to the FOXM1-AS sequence was designed to capture FOXM1-AS. Antisense probes to the capture probe set omitted the FOXM1–FOXM1-AS overlapping region were also generated and served as the negative control. RNA probes (50 pmole) were transcribed in vitro with the MEGAscript® T7 Transcription Kit and then labeled with the Pierce RNA 3′ End Biotinylation Kit (Life Technologies), treated with TURBO DNase (Life Technologies), and purified with the RNeasy Mini Kit (QIAGEN). GSC17 nuclear pellet was resuspended and homogenized in 1 ml RIP buffer (150 mM KCl, 25 mM Tris pH 7.4, 0.5 mM DTT, 0.5% NP40, 1 mM PMSF and protease Inhibitor). Heat-denatured RNA probes were then incubated with the nuclear extract at RT for one hour and further incubated with 30 μl of Dynabeads MyOne Streptavidin C1 (Life Technologies) at RT for one hour. Associated proteins were detected by Western blotting.
RNA immunoprecipitation was performed with the Magna RIP™ RNA-Binding Protein Immunoprecipitation Kit (Millipore) according to the manufacturer’s instructions. Briefly, magnetic beads coated with 5 μg of normal antibodies against mouse immunoglobulin G (Millipore), HuR (Santa Cruz), or FLAG (Sigma Aldrich) were incubated with pre-frozen cell lysates or nuclear extracts overnight at 4°C. Associated RNA-protein complexes were collected and washed 6 times and then subjected to proteinase K digestion and RNA extraction by TRIzol. The relative interaction between protein and RNA was determined by qPCR and normalized to input. For FLAG RIP, GSCs were transfected with same amount of either FLAG-tagged or untagged plasmid expressing the same gene (ALKBH5 or HuR). Then both cells were subjected to the same treatment. RNA enriched by FLAG RIP of cells expressing untagged protein was served as negative control. For HuR RIP, treated cells were divided and subjected to RIP by anti-HuR and mouse normal IgG, which was served as negative control.
Subcellular Fractionation Assay
The method for subcellular fractionation assay was adapted from Wuarin and Schibler (Wuarin and Schibler, 1994). Briefly, GSCs were collected by centrifugation and washed with phosphate-buffered saline. Cell pellets were resuspended in cold NP-40 lysis buffer (10 mM Tris-HCl pH 7.5, 0.15% NP40, 150 mM NaCl) for 5 min. The lysate was then transferred onto 2.5 volumes of a chilled sucrose cushion (ice-cold sucrose buffer [10 mM Tris-HCl pH 7.5, 150 mM NaCl, 24% sucrose]), and centrifuged at 13,000 rpm for 10 min at 4°C. The supernatant (the cytoplasmic fraction) was collected for immunoblotting analysis or RNA extraction by TRIzol. The pellet (nuclear fraction) was resuspended in cytoplasmic lysis buffer without NP40 and passed through the sucrose buffer again. The washed nuclear fraction was resuspended in an ice-cold glycerol buffer (20 mM Tris pH 7.9, 75 mM NaCl, 0.5 mM EDTA, 50% glycerol) and then mixed with an equal volume of cold nuclei lysis buffer (10 mM HEPES pH 7.6, 1 mM DTT, 7.5 mM MgCl2, 0.2 mM EDTA, 0.3 M NaCl, 1 M UREA, 1% NP-40) by vortexing twice for 5 seconds each. After incubation for 2 min on ice, the supernatant (nucleoplasm fraction) was collected for immunoblot analysis or TRIzol RNA extraction by centrifuge at 13,000 rpm for 2 min at 4°C. The insuluble pellet was washed with cold phosphate-buffered saline and then dissolved in TRIzol or saved for immunoblotting analysis.
BrdU Incorporation and Cell-Cycle Analysis
For the BrdU (5-bromo-2′-deoxyuridine) incorporation assay, cells were cultured with a BrdU-labeling reagent (Life Technologies) and stained with an anti-BrdU antibody (Cell Signaling) according to the manufacturer’s instructions. Five fields of view per slide were examined for BrdU-positive cells. For the cell-cycle analysis, cells were fixed and then stained with PI/RNase Staining Buffer (BD Pharmingen) for 15 min at room temperature. Samples were acquired with a FACScan flow cytometer (BD Biosciences).
Luciferase Reporter Assay
Cells seeded in 12-well plates were transfected with the pMIR-REPORT luciferase vector (Thermo Fisher) fused with or without the wild-type or mutated FOXM1-3′UTR. Mutation of FOXM1-3′UTR is generated by Geneart (Life Technologies). Transfection efficiency was quantified by co-transfection with an actin promoter-driven Renilla luciferase reporter (Zhang et al., 2011). The activities of firefly luciferase and Renilla luciferase in each well were calculated by a dual-luciferase reporter assay system (Promega). The ratios between the FOXM1 3′UTR reporter and Renilla control were determined 48 hr after siRNA treatment. The relative luciferase activity of the FOXM1-3′UTR luciferase activities were further normalized to that in cells transfected with the firefly luciferase vector control under the same treating conditions. The FOXM1 promoter activity was also normalized by co-transfection with the β-actin-Renilla luciferase reporter.
AMT Crosslink Assay
GSCs were suspended in PBS with or without 0.5 mg/mL AMT (4′-Aminomethyltrioxsalen hydrochloride, Sigma) at a concentration of 2×107 cells/ml in 6-well tissue culture plates and incubated on ice for 15 min. Then the plates were covered with a 2-mm-thick glass plate without lid. The cells were irradiated for 15 min from a distance of 2.5 cm with a handheld 365-nm UV light and mixed every 5 min. After cross-linking, cells were pelleted in 1.5 ml microcentrifuge tubes for RNA isolation by TRIzol. RNA yield was determined with a NanoDrop spectrophotometer. To pull-down the target RNAs, 25 pmole in vitro transcribed and biotin labeled RNA probes were denatured to 90°C for 2 min, transferred immediately on ice. Then probes and 10 μg RNA were mixed in binding buffer (50 mM Tris-HCl pH 7.5, 150 mM NaCl, 0.5% NP40 and 1 mM ribonucleoside vanadyl complexes) and transferred to a 37°C Thermomixer, shaking at 1,200 r.p.m. After 2 hours, 30 μl washed Dynabeads MyOne Streptavidin C1 were added and incubated for 2 hours at room temperature. After six washes, precipitated RNAs were extracted by TRIzol.
RNA Isolation and Quantitative Real Time PCR
RNA was isolated using TRIzol (Life Technologies) following the manufacturer’s protocol. mRNA was purified by Dynabeads® purification kit (Ambion) or GenElute™ mRNA Miniprep Kit (Sigma). cDNA was generated using the iScript cDNA Synthesis Kit (BioRad). Quantitative real-time PCR using Powerup SYBR Green PCR Master Mix (Life Technologies) was performed on a 7500 Fast Real-time PCR System (Applied Biosystems). For RNA stability assay, GSCs were plated in a poly-lysine coated 6-cm dish and incubated with actinomycin D (Santa Cruz) at 5 μg/ml for indicated time. Total RNA was isolated for qPCR analysis. See Table S6 for quantitative PCR primers.
m6A Quantification
The change of global m6A levels in mRNA was measured by EpiQuik m6A RNA Methylation Quantification Kit (Colorimetric) (Epigentek) following the manufacturer’s protocol. 200 ng poly-A-purified RNA was used for each sample analysis.
MeRIP-qPCR
MeRIP assay was adapted from a reported protocol (Dominissini et al., 2013). Briefly, intact poly-A-purified RNA was denatured to 70°C for 10 minutes, transferred immediately on ice and then incubated with m6A antibody in 1ml buffer containing RNasin Plus RNase inhibitor 400 U (Promega), 50 mM Tris-HCl, 750 mM NaCl and 0.5% (vol/vol) Igepal CA-630 (Sigma Aldrich) for 2 hr at 4°C. Dynabeads Protein G (Invitrogen) were washed, added to the mixture and incubated for 2 hr at 4°C with rotation. m6A RNA was eluted twice with 6.7 mM N6-methyladenosine 5′-monophosphate sodium salt at 4°C for 1 hour and precipitated with 5 μg glycogen, one-tenth volumes of 3 M sodium acetate in 2.5 volumes of 100% ethanol at −80°C overnight. m6A enrichment was determined by qPCR analysis. Fragmented mRNA was directly incubated with m6A antibody containing buffer and treated similarly.
Laser-capture microdissection
Snap-frozen and OTC-embedded (Tissue-Tek® OCT Compound, Ted Pella, INC., Redding, CA) of 15 GBM specimens were sectioned using a Microm HM525 NX Cryostat (Thermo Fisher Scientific Inc., Grand Island, NY); one of many serial consecutive sections per case was stained with hematoxylin and eosin to confirm the localizations of tumor cells in different regions. Tumor cells in different regions were subsequently isolated by laser-capture microdissection using a Zeiss PALM (Carl Zeiss, Inc., Thornwood, NY). For each case, 3 regions of tumor cells were separately collected for RNA extraction. Total RNA was isolated from the cells using TRIzol RNA Isolation Reagent according to the manufacturer’s instructions. The concentrations and quality of the recovered RNA were measured using a NanoDrop 2000 spectrophotometer (Nanodrop Technologies, Wilmington, DE). For quantitative analysis of mRNA expressions of FOXM1-AS and FOXM1 by qPCR, mRNA levels were normalized to GAPDH mRNA.
m6A-seq
m6A-IP and library preparation were performed according to the reported protocol (Dominissini et al., 2012). Briefly, poly-A-purified RNA was fragmented and incubated with m6A primary antibody for 2 hours at 4°C. The mixture was then immunoprecipitated by incubation with Protein A beads (Thermo Fisher) for 2 hours at 4°C. Captured RNA was washed for 3 times, eluted with m6A nucleotide solution and purified by RNA Clean and Concentrator kit (Zymo). Sequencing was carried out on Illumina HiSeq 2000 according to the manufacturer’s instructions.
Analysis of high-throughput sequencing data
All samples of m6A-seq study were sequenced by Illumina Hiseq 2000 with single-end 50-bp read length. The deep sequencing data were mapped to human genome version 38 (GRCh38). Data analysis for each experiment: (1) for m6A-seq, reads were aligned to the reference genome using Tophat v2.0.14 (Kim et al., 2013) with parameter -g 1 --library-type=fr-firststrand. RefSeq Gene structure annotations were downloaded from UCSC Table Browser. The longest isoform was used if the gene had multiple isoforms. Aligned reads were extended to 150 bp (average fragments size) and converted from genome-based coordinates to isoform-based coordinates, in order to eliminate the interference from introns in peak calling. The peak calling method was modified from published work (Dominissini et al., 2012). To call m6A peaks, the longest isoform of each gene was scanned using a 100 bp sliding window with 10 bp step. To reduce bias from potential inaccurate gene structure annotation and the arbitrary usage of the longest isoform, windows with read counts less than 1/20 of the top window in both m6A-IP and input sample were excluded. For each gene, the read counts in each window were normalized by the median count of all windows of that gene. A Fisher exact test was used to identify the differential windows between IP and input samples. The window was called as positive if the FDR < 0.01 and log2 (Enrichment Score) >= 1. Overlapping positive windows were merged. The following four numbers were calculated to obtain the enrichment score of each peak (or window): reads count of the IP samples in the current peak/window (a), median read counts of the IP sample in all 100 bp windows on the current mRNA (b), reads count of the input sample in the current peak/window (c), and median read counts of the input sample in all 100 bp windows on the current mRNA (d). The enrichment score of each window was calculated as (a×d)/(b×c). (2) for mRNA-seq (the input samples of m6A-seq), reads were mapped with Tophat and Cufflink (v2.2.1) was used to calculate the FPKM of each gene to represent their mRNA expression level (Trapnell et al., 2010). Data accession: all the raw data and processed files have been deposited in the Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo) and accessible under GSE87515.
Plasmids and RNA knockdown
ALKBH5 expression plasmid was generated by cloning the full-length ORF of human ALKBH5 gene (NM_017758.3) into pcDNA3.1-DYK vector (GenScript). ALKBH5 H204A was generated by GenScript. pcDNA3.1-DYK-HuR was generated by GenScript. ALKBH5 and HuR were cloned to pcDNA3.1 vector without tag. Wild-type and H204 mutant of ALKBH5 were cloned to pLVX (Clontech) for stable expression. Coding region and 3′UTR of FOXM1 were cloned from GSC17 cells into 3xFLAG expression vector (Sigma).
Transfections were performed using X-tremeGENE HP DNA Transfection Reagent for plasmid and X-tremeGENE siRNA Transfection Reagent (Roche) for siRNA following the manufacturer’s protocols.
RNAi oligonucleotides sequences are listed in Table S7.
Lentiviral Transduction for Stable Cell Lines
Lentiviral vectors expressing non-targeting pLKO.1 control shRNA (SCH002), and two shRNA constructs targeting ALKBH5 (NM_017758), shRNA1 (TRCN0000064783) and shRNA2 (TRCN0000064787) were obtained from Sigma. shRNA for FOXM1-AS was generated according to the pLKO.1 protocol from Addgene. The lentiviral vectors were co-transfected with packaging vectors psPAX2 and pMD2G (Addgene) into 293FT cells for lentivirus production. To establish stable cell lines, GSC cells were transduced by using the above lentiviruses with polybrene (6 μg/ml, Sigma). After 72 hr of transduction, cells were selected with 2 μg/ml puromycin for 4 days. For the ALKBH5 rescue experiment, shRNA targeting 3′UTR of ALKBH5 (shRNA1) was used for knockdown.
Limiting Dilution Assay
In vitro limiting dilution assay (LDA) was performed as described previously (Zhou et al., 2016). Briefly, dissociated GSC cells were seeded in 96-well plates at density of 5, 10, 20, 50, 100 or 200 cells per well and each well was examined for formation of tumorspheres after 7 days. Stem cell frequency was calculated using extreme limiting dilution analysis (http://bioinf.wehi.edu.au/software/elda/). Tumorsphere formation efficiency in Figure 7F was determined by the spheres formed 73 days after dissociated single cells were plated at a density of 1 cell/μl.
Microarray Analysis
Total RNA from GSC11 cells with shControl or shALKBH5-2, GSC17 cells with shControl or shALKBH5-1 was collected on day 7 after transduction for microarray analysis at MD Anderson DNA Core Facility using Affymetrix Human Gene 2.0 ST array, Expression Console Software and Transcriptome Analysis Console v3.0 (Affymetrix). The genes showing altered expression (fold change > 2) compared with the control shRNA in both cell lines were selected and analyzed using Ingenuity Pathway Analysis (IPA, Ingenuity Systems). Data accession: all the raw data have been deposited in the GEO under GSE93054.
QUANTIFICATION AND STATISTICAL ANALYSIS
Data are presented as the mean ± standard error of the means (SEM), or standard deviations (SD). Differences in the mean values between 2 groups were assessed for significance with a 2-tailed Student t-test using GraphPad Prism 6.0. Kaplan-Meier survival data were analyzed using the log-rank test. The Pearson correlation test was used to assess relationships between variables in tumor tissues.
DATA AND SOFTWARE AVAILABILITY
The microarray gene expression and m6A-seq data are deposited at the GEO under the accession number GEO: GSE93054, GSE87515.
KEY RESOURCES TABLE
The table highlights the genetically modified organisms and strains, cell lines, reagents, software, and source data essential to reproduce results presented in the manuscript. Depending on the nature of the study, this may include standard laboratory materials (i.e., food chow for metabolism studies), but the Table is not meant to be comprehensive list of all materials and resources used (e.g., essential chemicals such as SDS, sucrose, or standard culture media don’t need to be listed in the Table). Items in the Table must also be reported in the Method Details section within the context of their use. The number of primers and RNA sequences that may be listed in the Table is restricted to no more than ten each. If there are more than ten primers or RNA sequences to report, please provide this information as a supplementary document and reference this file (e.g., See Table S1 for XX) in the Key Resources Table.
Please note that ALL references cited in the Key Resources Table must be included in the References list. Please report the information as follows:
REAGENT or RESOURCE: Provide full descriptive name of the item so that it can be identified and linked with its description in the manuscript (e.g., provide version number for software, host source for antibody, strain name). In the Experimental Models section, please include all models used in the paper and describe each line/strain as: model organism: name used for strain/line in paper: genotype. (i.e., Mouse: OXTRfl/fl: B6.129(SJL)-Oxtrtm1.1Wsy/J). In the Biological Samples section, please list all samples obtained from commercial sources or biological repositories. Please note that software mentioned in the Methods Details or Data and Software Availability section needs to be also included in the table. See the sample Table at the end of this document for examples of how to report reagents.
SOURCE: Report the company, manufacturer, or individual that provided the item or where the item can obtained (e.g., stock center or repository). For materials distributed by Addgene, please cite the article describing the plasmid and include “Addgene” as part of the identifier. If an item is from another lab, please include the name of the principal investigator and a citation if it has been previously published. If the material is being reported for the first time in the current paper, please indicate as “this paper.” For software, please provide the company name if it is commercially available or cite the paper in which it has been initially described.
-
IDENTIFIER: Include catalog numbers (entered in the column as “Cat#” followed by the number, e.g., Cat#3879S). Where available, please include unique entities such as RRIDs, Model Organism Database numbers, accession numbers, and PDB or CAS IDs. For antibodies, if applicable and available, please also include the lot number or clone identity. For software or data resources, please include the URL where the resource can be downloaded. Please ensure accuracy of the identifiers, as they are essential for generation of hyperlinks to external sources when available. Please see the Elsevier list of Data Repositories with automated bidirectional linking for details. When listing more than one identifier for the same item, use semicolons to separate them (e.g. Cat#3879S; RRID: AB_2255011). If an identifier is not available, please enter “N/A” in the column.
A NOTE ABOUT RRIDs: We highly recommend using RRIDs as the identifier (in particular for antibodies and organisms, but also for software tools and databases). For more details on how to obtain or generate an RRID for existing or newly generated resources, please visit the RII or search for RRIDs.
Please see the sample Table at the end of this document for examples of how reagents should be cited. To see how the typeset table will appear in the PDF and online, please refer to any of the research articles published in Cell in the August 25, 2016 issue and beyond.
Please use the empty table that follows to organize the information in the sections defined by the subheading, skipping sections not relevant to your study. Please do not add subheadings. To add a row, place the cursor at the end of the row above where you would like to add the row, just outside the right border of the table. Then press the ENTER key to add the row. You do not need to delete empty rows. Each entry must be on a separate row; do not list multiple items in a single table cell.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| anti-FOXM1 (K19) | Santa Cruz Biotechnology | Cat#sc-500 |
| anti-FOXM1 (G5) (IHC, IF) | Santa Cruz Biotechnology | Cat#sc-376471 |
| anti-Nestin | Santa Cruz Biotechnology | Cat#sc-23927 |
| anti-HuR | Santa Cruz Biotechnology | Cat#sc-5261 |
| anti-GFAP | Santa Cruz Biotechnology | Cat#sc-166481 |
| anti-Tuj-1 | Santa Cruz Biotechnology | Cat#sc-58888 |
| anti-SSEA-1 | Santa Cruz Biotechnology | Cat#sc-21702 |
| anti-SOX2 (IF) | Santa Cruz Biotechnology | Cat#sc-365823 |
| anti-OCT3/4 | Santa Cruz Biotechnology | Cat#sc-5279 |
| anti-Nanog | Santa Cruz Biotechnology | Cat#sc-33759 |
| anti-β-actin | Santa Cruz Biotechnology | Cat#sc-47778 |
| anti-Ki67 | Santa Cruz Biotechnology | Cat#sc-15402 |
| anti-α-Tubulin | Santa Cruz Biotechnology | Cat#sc-5286 |
| anti-SOX2 (D6D9) (IHC, Western) | Cell Signaling Technology | Cat#3579 |
| anti-Histone H3 | Cell Signaling Technology | Cat#4499 |
| anti-CD133 | Cell Signaling Technology | Cat#3663 |
| anti-BrdU | Cell Signaling Technology | Cat#5292 |
| anti-N6-methyladenosine | Synaptic Systems | Cat#202003 |
| anti-U1-70K | Millipore | Cat#05-1588 |
| anti-ALKBH5 | Millipore | Cat#ABE547 |
| Normal rabbit IgG antibody | Millipore | Cat#PP64B |
| Normal mouse IgG antibody | Millipore | Cat#CS200621 |
| anti-ALKBH5 (IF, IHC) | Sigma-Aldrich | Cat#HPA007196 |
| anti-FLAG | Sigma-Aldrich | Cat#F3165 |
| Biological Samples | ||
| GBM patient specimen | MD Anderson Cancer Center | |
| Chemicals, Peptides, and Recombinant Proteins | ||
| BrdU-labeling reagent | Life Technologies | Cat#000103 |
| PI/RNase Staining Buffer | BD Pharmingen | Cat#550825 |
| 4′-Aminomethyltrioxsalen hydrochloride | Sigma | Cat#A4330 |
| Dynabeads MyOne Streptavidin C1 | Life Technologies | Cat#65001 |
| Powerup SYBR Green PCR Master Mix | Life Technologies | Cat#A25742 |
| iScript cDNA Synthesis Kit | BioRad | Cat#1708891 |
| Dynabeads Protein G | Life Technologies | Cat#10003D |
| RNasin Plus RNase inhibitor | Promega | Cat#N2115 |
| X-tremeGENE HP DNA Transfection Reagent | Roche | Cat#06366236001 |
| X-tremeGENE siRNA Transfection Reagent | Roche | Cat#04476093001 |
| N6-methyladenosine 5′-monophosphate sodium | Chem-Impex International | Cat#00587 |
| actinomycin D | Santa Cruz | Cat#sc-200906 |
| Ribonucleoside Vanadyl Complex | NEB | Cat#S1402S |
| Critical Commercial Assays | ||
| The Dual-Luciferase® Reporter (DLR™) Assay System | Promega | Cat#E1910 |
| Magna RIP™ RNA-Binding Protein Immunoprecipitation Kit | Millipore | Cat#17-700 |
| TruSeq Stranded mRNA Library Prep Kit | Illumina | Cat#RS-122-2101 |
| Deposited Data | ||
| Microarray data | This paper | GEO:GSE93054 |
| RNAseq data | This paper | GEO:GSE87515 |
| Experimental Models: Cell Lines | ||
| Human glioma cell lines (Hs683, SW1783) | ATCC | |
| Human GBM cell lines (LN229, U87MG) | ATCC | |
| Human GBM U251MG cells | Sigma | |
| immortalized NHA-E6/E7/hTERT cells | Sonoda et al., 2001 | |
| 293FT packaging cells | Thermo Fisher | |
| Experimental Models: Organisms/Strains | ||
| Nude mice: Foxn1nu/nu, male | MD Anderson Cancer Center ERO Breeding Core | |
| Recombinant DNA | ||
| pcDNA3.1-DYK-ALKBH5 | This paper | |
| pcDNA3.1-ALKBH5 | This paper | |
| pcDNA3.1-DYK-ALKBH5-H204A | This paper | |
| pcDNA3.1-DYK-HuR | This paper | |
| pcDNA3.1-HuR | This paper | |
| 3xFLAG FOXM1 CDS | This paper | |
| 3xFLAG FOXM1 CDS + 3′UTR | This paper | |
| psPAX2 | Addgene | |
| pMD2G | Addgene | |
| pLKO.1 non-targeting shRNA control | Sigma | Cat#SCH002 |
| pLKO.1 shALKBH5-1 | Sigma | Clone ID: TRCN0000064783 |
| pLKO.1 shALKBH5-2 | Sigma | Clone ID: TRCN0000064787 |
| pLKO.1 FOXM1-AS targeting shRNA | This paper | |
| pMIR-REPORT luciferase vector | Thermo Fisher | Cat#AM5795 |
| pMIR-REPORT- FOXM1-3′UTR wild-type | This paper | |
| pMIR-REPORT- FOXM1-3′UTR mutant | This paper | |
| FOXM1-promoter luciferase reporter | Schonberg, et al. 2015 | |
| β-actin-Renilla luciferase reporter | Zhang, et al. 2011 | |
| Sequence-Based Reagents | ||
| Primers for Real-time PCR, see Table S6 | Sigma | |
| RNAi oligonucleotides sequences, see Table S7 | Sigma | |
| Software and Algorithms | ||
| Tophat v2.0.14 | Kim, D. et al. Genome Biol 14, R36 (2013). | https://ccb.jhu.edu/software/tophat/index.shtml |
| Cufflink v2.2.1 | Trapnell, C. et al. Nat Biotechnol 28, 511–515 (2010). | http://cole-trapnell-lab.github.io/cufflinks/ |
| Other | ||
Supplementary Material
Significance.
N6-methyladenosine is the most prevalent internal modification on mRNA, but its functions in human diseases are poorly understood. We show that the overexpression of the m6A demethylase ALKBH5 is required for the proliferation and tumorigenesis of GSCs and informs poor patient survival. Moreover, we identified FOXM1 as a key target of ALKBH5 which mediates ALKBH5’s function in GSCs. Furthermore, a long noncoding RNA facilitates demethylation of FOXM1 nascent transcripts by ALKBH5. This promotes the interaction of FOXM1 pre-mRNA with HuR, thereby maintaining FOXM1 expression. The vulnerability of GSCs to disruptions in the ALKBH5-dependent gene expression suggests that RNA m6A methylation has a critical role in tumor development and provides a rationale for therapeutically targeting epitranscriptomic modulators in patients with glioblastoma.
Highlights.
ALKBH5 expression increases in GSCs and predicts poor survival in GBM patients
ALKBH5 demethylates FOXM1 nascent transcripts and enhances FOXM1 expression
FOXM1-AS promotes the interaction of FOXM1 nascent transcripts with ALKBH5
ALKBH5-FOXM1 pathway is critical for GSC proliferation and tumorigenesis
Acknowledgments
We thank Drs. R. Behringer and Z. Fan for scientific suggestions, L. Ma for assistance with revision. We thank Joseph A. Munch for language editing. This work was supported in part by U.S. NIH grants R01CA157933, R01CA182684, R01CA201327, R01CA152309, CA16672, P50CA127001, and GM071440.
Footnotes
AUTHOR CONTRIBUTIONS
S. Zhang conceived the project, designed and performed the research, analyzed and interpreted data, and wrote the manuscript. B.Z. performed the m6A-seq and analyzed data with the help of Z.L. A.Z. helped with FOXM-AS ideas and some experiments. K.L., S.Z., and Y.C. provided assistance in some experiments. E.S. analyzed expression data from GSCs and matched tumors. K.X. provided material support. S.M. and O.B. provided scientific input. C.H. provided material support and critical scientific input. S.H. conceived and designed the GSC study, interpreted data, wrote the manuscript, and provided study supervision.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aguilo F, Zhang F, Sancho A, Fidalgo M, Di Cecilia S, Vashisht A, Lee DF, Chen CH, Rengasamy M, Andino B, et al. Coordination of m(6)A mRNA Methylation and Gene Transcription by ZFP217 Regulates Pluripotency and Reprogramming. Cell Stem Cell. 2015;17:689–704. doi: 10.1016/j.stem.2015.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ameur A, Zaghlool A, Halvardson J, Wetterbom A, Gyllensten U, Cavelier L, Feuk L. Total RNA sequencing reveals nascent transcription and widespread co-transcriptional splicing in the human brain. Nat Struct Mol Biol. 2011;18:1435–1440. doi: 10.1038/nsmb.2143. [DOI] [PubMed] [Google Scholar]
- Batista PJ, Molinie B, Wang J, Qu K, Zhang J, Li L, Bouley DM, Lujan E, Haddad B, Daneshvar K, et al. m(6)A RNA modification controls cell fate transition in mammalian embryonic stem cells. Cell Stem Cell. 2014;15:707–719. doi: 10.1016/j.stem.2014.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhat KP, Balasubramaniyan V, Vaillant B, Ezhilarasan R, Hummelink K, Hollingsworth F, Wani K, Heathcock L, James JD, Goodman LD, et al. Mesenchymal differentiation mediated by NF-kappaB promotes radiation resistance in glioblastoma. Cancer Cell. 2013;24:331–346. doi: 10.1016/j.ccr.2013.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennan CW, Verhaak RG, McKenna A, Campos B, Noushmehr H, Salama SR, Zheng S, Chakravarty D, Sanborn JZ, Berman SH, et al. The somatic genomic landscape of glioblastoma. Cell. 2013;155:462–477. doi: 10.1016/j.cell.2013.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calvet JP, Pederson T. Heterogeneous nuclear RNA double-stranded regions probed in living HeLa cells by crosslinking with the psoralen derivative aminomethyltrioxsalen. Proc Natl Acad Sci U S A. 1979;76:755–759. doi: 10.1073/pnas.76.2.755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen K, Lu Z, Wang X, Fu Y, Luo GZ, Liu N, Han D, Dominissini D, Dai Q, Pan T, He C. High-resolution N(6) -methyladenosine (m(6) A) map using photo-crosslinking-assisted m(6) A sequencing. Angew Chem Int Ed Engl. 2015a;54:1587–1590. doi: 10.1002/anie.201410647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen T, Hao YJ, Zhang Y, Li MM, Wang M, Han W, Wu Y, Lv Y, Hao J, Wang L, et al. m(6)A RNA methylation is regulated by microRNAs and promotes reprogramming to pluripotency. Cell Stem Cell. 2015b;16:289–301. doi: 10.1016/j.stem.2015.01.016. [DOI] [PubMed] [Google Scholar]
- Desrosiers R, Friderici K, Rottman F. Identification of methylated nucleosides in messenger RNA from Novikoff hepatoma cells. Proc Natl Acad Sci U S A. 1974;71:3971–3975. doi: 10.1073/pnas.71.10.3971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominissini D, Moshitch-Moshkovitz S, Salmon-Divon M, Amariglio N, Rechavi G. Transcriptome-wide mapping of N(6)-methyladenosine by m(6)A-seq based on immunocapturing and massively parallel sequencing. Nature protocols. 2013;8:176–189. doi: 10.1038/nprot.2012.148. [DOI] [PubMed] [Google Scholar]
- Dominissini D, Moshitch-Moshkovitz S, Schwartz S, Salmon-Divon M, Ungar L, Osenberg S, Cesarkas K, Jacob-Hirsch J, Amariglio N, Kupiec M, et al. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature. 2012;485:201–206. doi: 10.1038/nature11112. [DOI] [PubMed] [Google Scholar]
- Fischer J, Koch L, Emmerling C, Vierkotten J, Peters T, Bruning JC, Ruther U. Inactivation of the Fto gene protects from obesity. Nature. 2009;458:894–898. doi: 10.1038/nature07848. [DOI] [PubMed] [Google Scholar]
- Fustin JM, Doi M, Yamaguchi Y, Hida H, Nishimura S, Yoshida M, Isagawa T, Morioka MS, Kakeya H, Manabe I, Okamura H. RNA-methylation-dependent RNA processing controls the speed of the circadian clock. Cell. 2013;155:793–806. doi: 10.1016/j.cell.2013.10.026. [DOI] [PubMed] [Google Scholar]
- Geula S, Moshitch-Moshkovitz S, Dominissini D, Mansour AA, Kol N, Salmon-Divon M, Hershkovitz V, Peer E, Mor N, Manor YS, et al. Stem cells. m6A mRNA methylation facilitates resolution of naive pluripotency toward differentiation. Science. 2015;347:1002–1006. doi: 10.1126/science.1261417. [DOI] [PubMed] [Google Scholar]
- Gong AH, Wei P, Zhang S, Yao J, Yuan Y, Zhou AD, Lang FF, Heimberger AB, Rao G, Huang S. FoxM1 Drives a Feed-Forward STAT3-Activation Signaling Loop That Promotes the Self-Renewal and Tumorigenicity of Glioblastoma Stem-like Cells. Cancer research. 2015;75:2337–2348. doi: 10.1158/0008-5472.CAN-14-2800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia G, Fu Y, Zhao X, Dai Q, Zheng G, Yang Y, Yi C, Lindahl T, Pan T, Yang YG, He C. N6-methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat Chem Biol. 2011;7:885–887. doi: 10.1038/nchembio.687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi K, Banasavadi-Siddegowda Y, Mo X, Kim SH, Mao P, Kig C, Nardini D, Sobol RW, Chow LM, Kornblum HI, et al. MELK-dependent FOXM1 phosphorylation is essential for proliferation of glioma stem cells. Stem Cells. 2013;31:1051–1063. doi: 10.1002/stem.1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katayama S, Tomaru Y, Kasukawa T, Waki K, Nakanishi M, Nakamura M, Nishida H, Yap CC, Suzuki M, Kawai J, et al. Antisense transcription in the mammalian transcriptome. Science. 2005;309:1564–1566. doi: 10.1126/science.1112009. [DOI] [PubMed] [Google Scholar]
- Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013;14:R36. doi: 10.1186/gb-2013-14-4-r36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SH, Joshi K, Ezhilarasan R, Myers TR, Siu J, Gu C, Nakano-Okuno M, Taylor D, Minata M, Sulman EP, et al. EZH2 protects glioma stem cells from radiation-induced cell death in a MELK/FOXM1-dependent manner. Stem cell reports. 2015;4:226–238. doi: 10.1016/j.stemcr.2014.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lathia JD, Mack SC, Mulkearns-Hubert EE, Valentim CL, Rich JN. Cancer stem cells in glioblastoma. Genes Dev. 2015;29:1203–1217. doi: 10.1101/gad.261982.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebedeva S, Jens M, Theil K, Schwanhausser B, Selbach M, Landthaler M, Rajewsky N. Transcriptome-wide analysis of regulatory interactions of the RNA-binding protein HuR. Mol Cell. 2011;43:340–352. doi: 10.1016/j.molcel.2011.06.008. [DOI] [PubMed] [Google Scholar]
- Lee Y, Kim KH, Kim DG, Cho HJ, Kim Y, Rheey J, Shin K, Seo YJ, Choi YS, Lee JI, et al. FoxM1 Promotes Stemness and Radio-Resistance of Glioblastoma by Regulating the Master Stem Cell Regulator Sox2. PLoS One. 2015;10:e0137703. doi: 10.1371/journal.pone.0137703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Zhang S, Huang S. FoxM1: a potential drug target for glioma. Future oncology. 2012;8:223–226. doi: 10.2217/fon.12.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Bao S, Wu Q, Wang H, Eyler C, Sathornsumetee S, Shi Q, Cao Y, Lathia J, McLendon RE, et al. Hypoxia-inducible factors regulate tumorigenic capacity of glioma stem cells. Cancer Cell. 2009;15:501–513. doi: 10.1016/j.ccr.2009.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Weng H, Su R, Weng X, Zuo Z, Li C, Huang H, Nachtergaele S, Dong L, Hu C, et al. FTO Plays an Oncogenic Role in Acute Myeloid Leukemia as a N6-Methyladenosine RNA Demethylase. Cancer Cell. 2016 doi: 10.1016/j.ccell.2016.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin S, Choe J, Du P, Triboulet R, Gregory RI. The m(6)A Methyltransferase METTL3 Promotes Translation in Human Cancer Cells. Mol Cell. 2016;62:335–345. doi: 10.1016/j.molcel.2016.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Yue Y, Han D, Wang X, Fu Y, Zhang L, Jia G, Yu M, Lu Z, Deng X, et al. A METTL3-METTL14 complex mediates mammalian nuclear RNA N6-adenosine methylation. Nat Chem Biol. 2014;10:93–95. doi: 10.1038/nchembio.1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M, Dai B, Kang SH, Ban K, Huang FJ, Lang FF, Aldape KD, Xie TX, Pelloski CE, Xie K, et al. FoxM1B is overexpressed in human glioblastomas and critically regulates the tumorigenicity of glioma cells. Cancer research. 2006;66:3593–3602. doi: 10.1158/0008-5472.CAN-05-2912. [DOI] [PubMed] [Google Scholar]
- Liu N, Dai Q, Zheng G, He C, Parisien M, Pan T. N(6)-methyladenosine-dependent RNA structural switches regulate RNA-protein interactions. Nature. 2015;518:560–564. doi: 10.1038/nature14234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer KD, Patil DP, Zhou J, Zinoviev A, Skabkin MA, Elemento O, Pestova TV, Qian SB, Jaffrey SR. 5′ UTR m(6)A Promotes Cap-Independent Translation. Cell. 2015;163:999–1010. doi: 10.1016/j.cell.2015.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer KD, Saletore Y, Zumbo P, Elemento O, Mason CE, Jaffrey SR. Comprehensive analysis of mRNA methylation reveals enrichment in 3′ UTRs and near stop codons. Cell. 2012;149:1635–1646. doi: 10.1016/j.cell.2012.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molinie B, Wang J, Lim KS, Hillebrand R, Lu ZX, Van Wittenberghe N, Howard BD, Daneshvar K, Mullen AC, Dedon P, et al. m(6)A-LAIC-seq reveals the census and complexity of the m(6)A epitranscriptome. Nat Methods. 2016;13:692–698. doi: 10.1038/nmeth.3898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee N, Corcoran DL, Nusbaum JD, Reid DW, Georgiev S, Hafner M, Ascano M, Jr, Tuschl T, Ohler U, Keene JD. Integrative regulatory mapping indicates that the RNA-binding protein HuR couples pre-mRNA processing and mRNA stability. Mol Cell. 2011;43:327–339. doi: 10.1016/j.molcel.2011.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noushmehr H, Weisenberger DJ, Diefes K, Phillips HS, Pujara K, Berman BP, Pan F, Pelloski CE, Sulman EP, Bhat KP, et al. Identification of a CpG island methylator phenotype that defines a distinct subgroup of glioma. Cancer Cell. 2010;17:510–522. doi: 10.1016/j.ccr.2010.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schonberg DL, Miller TE, Wu Q, Flavahan WA, Das NK, Hale JS, Hubert CG, Mack SC, Jarrar AM, Karl RT, et al. Preferential Iron Trafficking Characterizes Glioblastoma Stem-like Cells. Cancer Cell. 2015;28:441–455. doi: 10.1016/j.ccell.2015.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonoda Y, Ozawa T, Hirose Y, Aldape KD, McMahon M, Berger MS, Pieper RO. Formation of intracranial tumors by genetically modified human astrocytes defines four pathways critical in the development of human anaplastic astrocytoma. Cancer Res. 2001;61:4956–4960. [PubMed] [Google Scholar]
- Thalhammer A, Bencokova Z, Poole R, Loenarz C, Adam J, O’Flaherty L, Schodel J, Mole D, Giaslakiotis K, Schofield CJ, et al. Human AlkB homologue 5 is a nuclear 2-oxoglutarate dependent oxygenase and a direct target of hypoxia-inducible factor 1alpha (HIF-1alpha) PLoS One. 2011;6:e16210. doi: 10.1371/journal.pone.0016210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, Pachter L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol. 2010;28:511–515. doi: 10.1038/nbt.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Lu Z, Gomez A, Hon GC, Yue Y, Han D, Fu Y, Parisien M, Dai Q, Jia G, et al. N6-methyladenosine-dependent regulation of messenger RNA stability. Nature. 2014a;505:117–120. doi: 10.1038/nature12730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Zhao BS, Roundtree IA, Lu Z, Han D, Ma H, Weng X, Chen K, Shi H, He C. N(6)-methyladenosine Modulates Messenger RNA Translation Efficiency. Cell. 2015;161:1388–1399. doi: 10.1016/j.cell.2015.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Li Y, Toth JI, Petroski MD, Zhang Z, Zhao JC. N6-methyladenosine modification destabilizes developmental regulators in embryonic stem cells. Nat Cell Biol. 2014b;16:191–198. doi: 10.1038/ncb2902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen PY, Kesari S. Malignant gliomas in adults. N Engl J Med. 2008;359:492–507. doi: 10.1056/NEJMra0708126. [DOI] [PubMed] [Google Scholar]
- Wuarin J, Schibler U. Physical isolation of nascent RNA chains transcribed by RNA polymerase II: evidence for cotranscriptional splicing. Mol Cell Biol. 1994;14:7219–7225. doi: 10.1128/mcb.14.11.7219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Samanta D, Lu H, Bullen JW, Zhang H, Chen I, He X, Semenza GL. Hypoxia induces the breast cancer stem cell phenotype by HIF-dependent and ALKBH5-mediated m6A-demethylation of NANOG mRNA. Proc Natl Acad Sci U S A. 2016 doi: 10.1073/pnas.1602883113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang N, Wei P, Gong A, Chiu WT, Lee HT, Colman H, Huang H, Xue J, Liu M, Wang Y, et al. FoxM1 promotes beta-catenin nuclear localization and controls Wnt target-gene expression and glioma tumorigenesis. Cancer Cell. 2011;20:427–442. doi: 10.1016/j.ccr.2011.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X, Yang Y, Sun BF, Shi Y, Yang X, Xiao W, Hao YJ, Ping XL, Chen YS, Wang WJ, et al. FTO-dependent demethylation of N6-methyladenosine regulates mRNA splicing and is required for adipogenesis. Cell Res. 2014;24:1403–1419. doi: 10.1038/cr.2014.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng G, Dahl JA, Niu Y, Fedorcsak P, Huang CM, Li CJ, Vagbo CB, Shi Y, Wang WL, Song SH, et al. ALKBH5 is a mammalian RNA demethylase that impacts RNA metabolism and mouse fertility. Mol Cell. 2013;49:18–29. doi: 10.1016/j.molcel.2012.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou A, Lin K, Zhang S, Chen Y, Zhang N, Xue J, Wang Z, Aldape KD, Xie K, Woodgett JR, Huang S. Nuclear GSK3beta promotes tumorigenesis by phosphorylating KDM1A and inducing its deubiquitylation by USP22. Nat Cell Biol. 2016 doi: 10.1038/ncb3396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J, Wan J, Gao X, Zhang X, Jaffrey SR, Qian SB. Dynamic m(6)A mRNA methylation directs translational control of heat shock response. Nature. 2015;526:591–594. doi: 10.1038/nature15377. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.